

Abstract

High throughput screening and subsequent hit validation identified 4-isopropyl-3-(2-((1-phenylethyl)amino)pyrimidin-4-yl)oxazolidin-2-one as a potent inhibitor of IDH1R132H. Synthesis of the four separate stereoisomers identified the (S,S)-diastereomer (IDH125, 1f) as the most potent isomer. This also showed reasonable cellular activity and excellent selectivity vs IDH1wt. Initial structure–activity relationship exploration identified the key tolerances and potential for optimization. X-ray crystallography identified a functionally relevant allosteric binding site amenable to inhibitors, which can penetrate the blood–brain barrier, and aided rational optimization. Potency improvement and modulation of the physicochemical properties identified (S,S)-oxazolidinone IDH889 (5x) with good exposure and 2-HG inhibitory activity in a mutant IDH1 xenograft mouse model.
Keywords: Mutant IDH1 inhibitor, allosteric inhibition, 2-HG, preclinical in vivo activity, 3-pyrimidin-4-yloxazolidin-2-one, chirality-defined potency
Hotspot heterozygous mutations in human cytoplasmic isocitrate dehydrogenase 1 (IDH1) at Arg132 (R132*) have been identified in multiple cancer types, including acute myeloid leukemia (AML), glioma, chondrosarcoma, and cholangiocarcinoma.1 These mutations have been shown to confer a neomorphic catalytic activity to produce high levels of intracellular R-2-hydroxyglutarate (2-HG) and effect downstream epigenetic markers on DNA and proteins.2,3 Catalytic inhibitors4−10 of IDH1R132* have shown preclinical reduction of 2-HG levels in xenograft mouse models with mutant IDH1 tumors and have also shown preclinical tumor regression.4−7 Recent clinical trials in AML patients with a specific inhibitor of IDH1 has shown clinical benefit, confirming the causal link between this genetic mutation, the production of 2-HG, and cancer.11 Efforts herein focused on the identification of compounds that could potentially target all classes of mutant-IDH1 tumors, including those in the brain.
The substrate-binding site of mutant IDH1 is highly polar as defined by the amino acids lining the pocket (Figure 1), in addition to the active-site magnesium ion and NADPH cofactor. This suggests a low probability of being able to optimize a compound for potent binding to this site while also fulfilling the criteria most conducive to crossing the blood–brain barrier (BBB).12 It was decided to explore the identification of catalytic inhibitors with different mechanisms of action, which may bind distal to this polar substrate-binding site.
Figure 1.
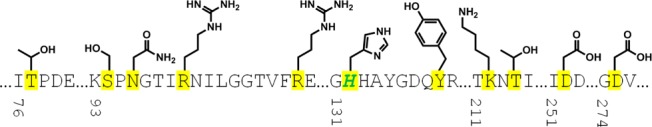
Sequence and side chain representation of the substrate-binding site for IDH1R132H. Amino acids lining the pocket are highlighted in yellow, and mutation site R132H is shown in green.
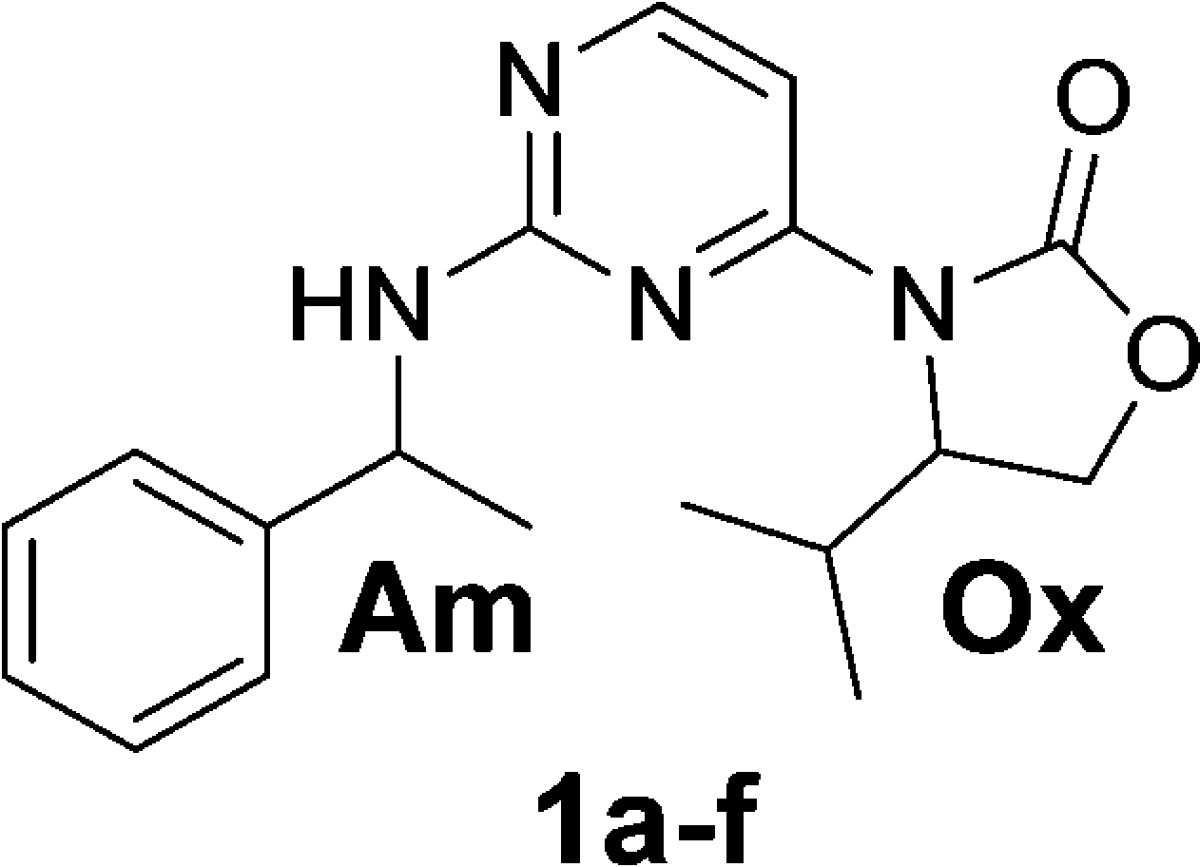
High throughput screening was carried out with a NADPH fluorescence-based biochemical assay using IDH1R132H homodimer protein, and orthogonal biochemical inhibition confirmation using an LCMS readout of 2-HG levels. Compounds 1a and 1b were identified as selective and functional inhibitors of IDH1R132H from this screen. Both 1a and 1b were screened as diastereomeric mixtures at the amine (Table 1, Am), which necessitated the independent synthesis of the four separate stereoisomers in order to determine the chiral preference for ligand binding. Potency was found to be most strongly dependent upon the chirality at the amine center (Am), although still significantly affected by the chirality at the oxazolidinone (Ox) (Table 1). The preference for the S-amine isomer was maintained throughout the structure–activity relationship (SAR) on the series, but the effect of R-chirality at the oxazolidinone was sometimes only marginal. As described elsewhere,13 compound 1f (IDH125) was validated with multiple biophysical methods, and this compound enabled considerable understanding of the rationale for selectivity and mechanism of action of this series. IDH125 was also confirmed as an essentially equipotent inhibitor of both the homodimer IDH1R132H (IC50 0.22 μM) and IDH1R132C (IC50 0.15 μM) proteins and slightly less potent for the heterodimer IDH1wt–IDH1R132H (IC50 0.97 μM) protein using NADPH fluorescence-based biochemical assay.13 The cancer-specific mutation of IDH1 and lack of activity of IDH125 against wild-type IDH1 up to 50 μM presented an opportunity to develop an oncology drug with a high therapeutic index.
Table 1. IDH1R132H Biochemical Activity of HTS Hits and Pure Resynthesized Diastereomers.
| chirality at amine (Am) | chirality at oxazolidinone (Ox) | biochem. LCMS IC50 (μM) | |
|---|---|---|---|
| 1a | R/S = 1:1 | S | 0.8 |
| 1b | R/S = 1:1 | R | 8.5 |
| 1c | R | R | 19.7 |
| 1d | S | R | 4.2 |
| 1e | R | S | 4.6 |
| 1f (IDH125) | S | S | 0.22 |
It was noted that IDH125 contains an aminopyrimidine core, which is a common motif in ATP-competitive kinase inhibitor scaffolds due to the structural similarity vs the purine nucleus of ATP.14 Therefore, initial thorough kinase profiling of IDH125 in an internal kinase profiling panel, as well as an external kinase profiling panel was carried out (Supporting Information). IDH125 demonstrated virtually no inhibitory activity across the entire kinome. This structural potential for off-target activity was monitored with internal and external kinase panels, but the lack of kinase activity was maintained throughout the series.
2,4-Dichloropyrimidine underwent selective 4-chloro substitution at room temperature, by reaction with the sodium salt of (S)-4-isopropyl-oxazolidin-2-one (Scheme 1). Subsequent displacement of the 2-chloro at 140 °C with (S)-1-phenylethan-1-amine in DMSO gave (S)-4-isopropyl-3-(2-(((S)-1-phenylethyl)amino)pyrimidin-4-yl)oxazolidin-2-one (IDH125, 1f). The other diastereomers were accessed using the appropriate chiral building blocks.
Scheme 1. Synthetic Scheme for 1f (IDH125).

SAR by archive using IDH125 as the probe structure was unsuccessful in identifying additional molecules in the Novartis compound library with submicromolar activity, highlighting that this substructure was somewhat unique. Initial analogues were targeted to explore the tolerance for modification and/or removal of different parts of the molecule (compounds 2a–d, 3a–f, and 4a–d in Supporting Information). Understanding of this initial SAR was clarified by the determination of the X-ray cocrystal structure of IDH125 in the homodimer of IDH1R132H, which showed IDH125 binding into an allosteric site adjacent to the substrate binding site.13 Replacement of the nitrogen at the 3-position of the pyrimidine with a carbon, and either replacement of the 2-amino with an oxygen or N-alkylation all resulted in loss of activity due to lack of backbone/ligand hydrogen bond donor–acceptor interactions to Leu120. Small substituents such as methyl on the 4- and 5-position of the pyrimidine were tolerated. The alpha-methyl of IDH125 binds into a small nook created by the backbone of Arg109, Glu110, and Ile128 as well as the side chains of Ile130, Ser278, and Met291 resulting in a good contact for a methyl group but limited tolerance for larger groups in this region. An unsubstituted oxazolidinone does not lead to the preferred hydrophobic collapse conformation required for binding, but there is tolerance for alternate oxazolidinone substituents beyond isopropyl. Interestingly, the bound structure of IDH125(13) closely overlays with the small molecule X-ray structure, which suggests that hydrophobic collapse drives the molecule to preorder into the active binding conformation. The nature of the residues lining this allosteric pocket are also in stark contrast to those lining the substrate-binding site, and lend a more hydrophobic character to the pocket (Figure 2). This pocket has potential for potent binding of molecules, which may also be able to cross the BBB.
Figure 2.
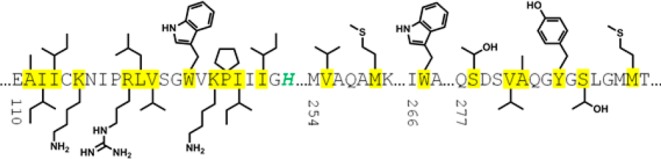
Sequence and side chain representation of the allosteric pocket for IDH1R132H. Amino acids lining the pocket are highlighted in yellow, and mutation site R132H is shown in green.
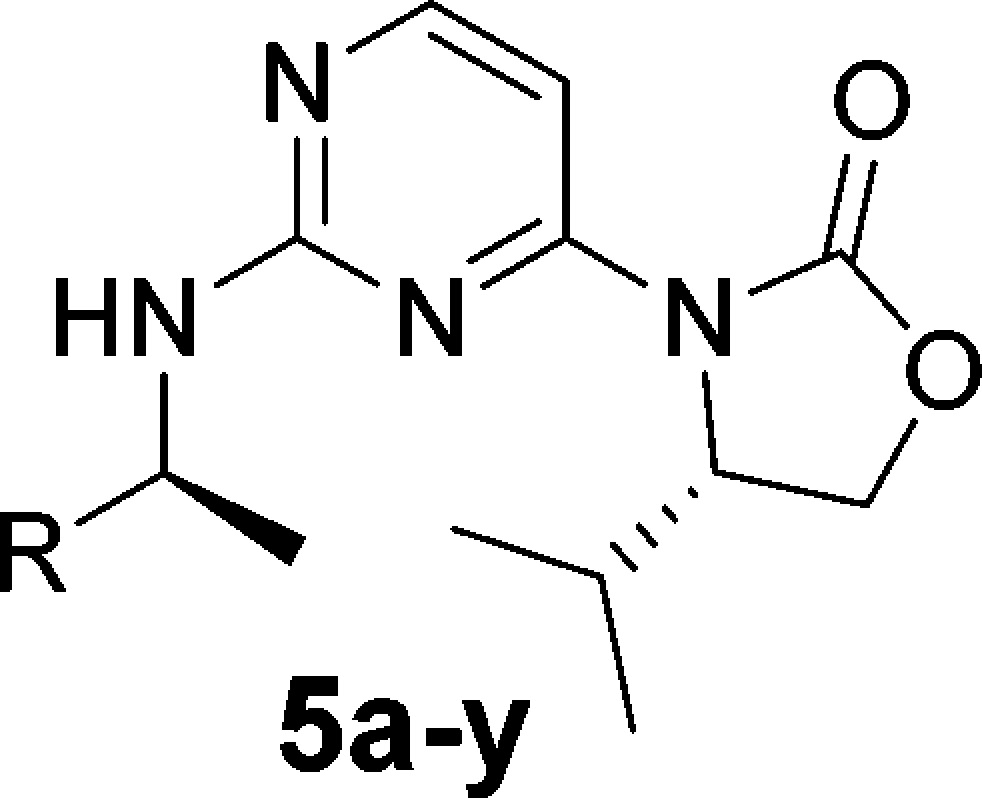
The limited ability to improve the potency with simple modifications to the oxazolidinone, alpha-methyl, or pyrimidine regions of IDH125 necessitated exploration of modifications to the phenyl group (Table 2). Saturation of the phenyl ring and simple substitutions appeared to be tolerated around the ring system (compounds 5a–m), although larger groups at the ortho position were less tolerated than the meta or para positions. In a similar fashion, 2-naphthyl 5p is also more potent than 1-naphthyl 5q. In the para-position, the fluoro and chloro groups appeared to give a slight boost in potency, but this was lost with the larger methoxy group. This could have been due to either a steric or electronic effect, but the electron-withdrawing p-cyano also showed a drop in potency, suggesting the loss of activity with electron-donating p-methoxy was not a wholly electronic effect. Despite the drop in activity for the p-methoxy and p-cyanophenyl, there was not the precipitous drop usually seen for a steric clash with an inflexible part of the protein. This suggested a low energy side chain or backbone movement of the protein in this region, which would accommodate larger side chains (i.e., for these intermediate sized groups the energy penalty for protein reorganization was not being compensated for by the interactions being made in the newly formed pocket). This hypothesis is supported by the potency seen with larger side chains such as p-biphenyl 5r and p-phenoxyphenyl 5s (IDH662). Interestingly, IDH662 also showed a significant boost in potency vs the IDHwt protein (IDHwt IC50 1.03 μM), suggesting that the allosteric pocket is also accessible in the wt protein but is preferentially accessible in the R132 mutated protein.13
Table 2. IDH1R132H Biochemical and HCT116-IDH1R132H Cellular Activity of Analogues Exploring Phenyl Substitution.

A 1:1 mixture of diastereomers at amine side chain.
IDH662 was profiled vs a panel of in vitro assays and showed an overall excellent selectivity for the mutant protein. IDH662 showed potent cellular inhibition of 2-HG production in the engineered HCT116-IDH1R132H cell line (IC50 0.022 μM) and inhibition of proliferation in the engineered MCF10A cell-line (IC50 0.017 μM)13 (cell lines available from Horizon Discovery Group plc15,16). In comparison, IDH125 was considerably weaker in the HCT-116-IDH1R132H cell line (IC50 0.66 μM). Despite poor solubility (<5 μM at pH 6.8) and high in vitro clearance across species, in vivo exposure in mice (AUC 1.1 μM·h, Cmax 0.39 μM at 10 mg/kg po, AUC 7.9 μM·h, Cmax 2.5 μM at 100 mg/kg po) was deemed sufficient to explore IDH662 in an in vivo pharmacodynamic (PD) study monitoring compound effects on 2-HG. However, at doses up to 600 mg/kg there was no significant alteration of 2-HG levels in xenograft tumor tissue in the HCT116-IDH1R132H model (data not shown). The estimated free compound concentration from the measured plasma pharmacokinetics (PK) in this study was below the cellular IC50 for mutant IDH1, which suggested that the high plasma protein binding (>99%) limited productive engagement of the target in vivo.
It was decided to explore whether in vivo modulation of 2-HG inhibition was dependent on free-fraction of drug in the plasma. The high protein binding was likely contributing to a lower in vivo clearance than predicted from the in vitro microsomal assays, suggesting that lower intrinsic clearance would be needed in tandem with any increase of the free fraction in order to achieve high enough and effective exposures. In silico and in vivo metabolic studies highlighted the benzylamine as a key source of oxidative metabolic cleavage, so this was the focus of the next iteration of targets.
Heterocyclic replacements of the phenyl group should lower the oxidation potential of the benzylic carbon due to distribution of the local electron density through to the conjugated, and more electronegative, heteroatoms. Heterocycles would also increase the local polarity, and this may be less well tolerated in the binding sites of cytochrome P450 proteins, leading to reduced oxidative metabolism. This is supported by data from Ioannidis et al., who have shown excellent in vitro and in vivo PK properties with a structurally similar halopyrimidine benzylamine for a series of Janus kinase 2 (JAK2) inhibitors.17
The chemistry used to displace chloro from 2-chloro-4-oxazolidinonyl pyrimidines with benzylamine derivatives was not efficient with less nucleophilic heterocyclic amines such as 1-(5-chloropyrimidin-2-yl)ethylamine. Therefore, the more reactive 2,4-difluoropyrimidine was used to enable the synthesis of the desired products via 2-fluoro-4-oxazolidinonylpyrimidine.
The comparison of chloropyrimidine (5t) to chlorophenyl (5f) shows a significant improvement in in vitro clearance in rat microsomes (CLint 73 μL/min·mg vs CLint > 900 μL/min·mg), but this results in a 16-fold drop in biochemical activity and 50-fold drop in cellular activity. A more moderate 8 to 10-fold drop in activity is observed for the phenoxypyrimidine analogue 5v when compared to the analogous phenoxyphenyl IDH662. However, <3-fold drop in activity is seen for the phenyl-pyrimidine analogue (diastereomeric mixture 5w vs biphenyl 5r). Analogue 5w also shows an improved clearance in rat microsomes vs pyrimidine 5t (5w CLint 24 μL/min mg), whereas phenoxypyrimidine 5v was significantly higher (CLint 240 μL/min mg). Pyrimidine and para-biaryl motifs were maintained in subsequent analogues, leading to identification of IDH889 [IDH1R132C IC50 0.072 μM, IDH1wt IC50 1.38 μM].
X-ray crystallography of IDH889 (PDB 5TQH) in homodimer IDH1R132H is consistent with the structure of IDH125 (Figure 3)).13 The aminopyrimidine core moiety interacts with the backbone of Ile128 via a hydrogen bond donor–acceptor pair, the carbonyl of the oxazolidinone forms a hydrogen bond interaction with Leu120, and the alpha-methyl fits into the methyl nook. The molecule is in a hydrophobic collapse conformation with the isopropyl in van der Waals contact with the pyrimidine of the amine side chain. In addition, a hydrogen bond is observed between the pyrimidine of the amine side chain and Ser278, and this specific interaction may be compensating for the expected increase in desolvation energy for a more polar ligand (vs the biphenyl 5r).
Figure 3.
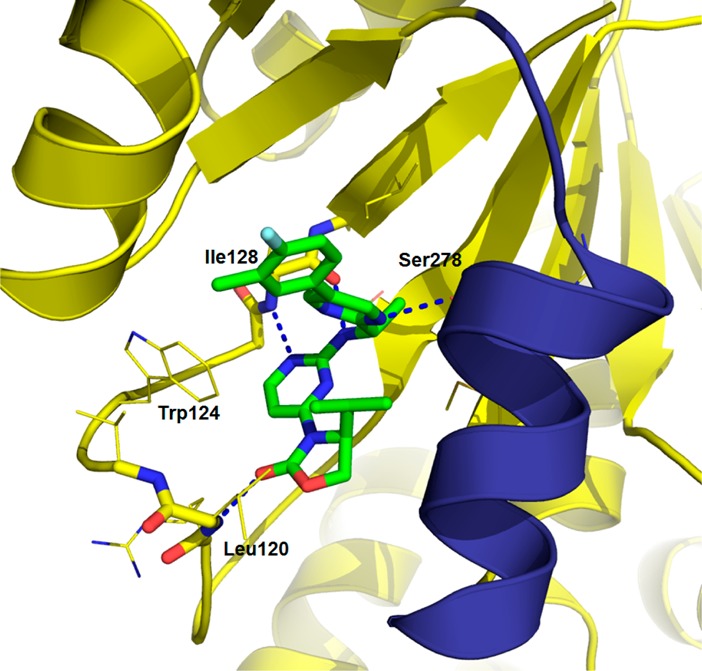
X-ray cocrystal structure of IDH889 in IDH1R132H.
IDH889 demonstrates significantly improved plasma exposure in mice vs IDH662 (AUC 3.6 μM·h, Cmax 1.7 μM at 10 mg/kg; AUC 55.5 μM·h, Cmax 14.2 μM at 100 mg/kg). This is coupled with an improved solubility (39 μM at pH 6.8) and measurable free fraction in mouse plasma (98% plasma protein binding), suggesting prolonged plasma concentration 20-fold over the cellular IC50 could be achieved at a dose of 100 mg/kg.
In the HCT116-IDH1R132H± xenograft model, IDH889 dosed orally at 200 mg/kg inhibits new 2-HG production but does not eliminate 2-HG from IDH1 mutant tissues. Thus, given the high baseline tissue concentration of 2-HG, several hours are required to clear 2-HG from the tissues. Peak reduction in tumor 2-HG levels after a single dose is observed 8–12 h postadministration. 2-HG levels begin to rebound as the free concentration of IDH889 drops below the in vitro cellular IC50 (Figure 4A). At 48 h post-treatment, 2-HG levels return to baseline. Sustained 2-HG inhibition greater than 24 h was achieved with BID (q12/12) administration of IDH889 for four doses (Figure 4B).
Figure 4.
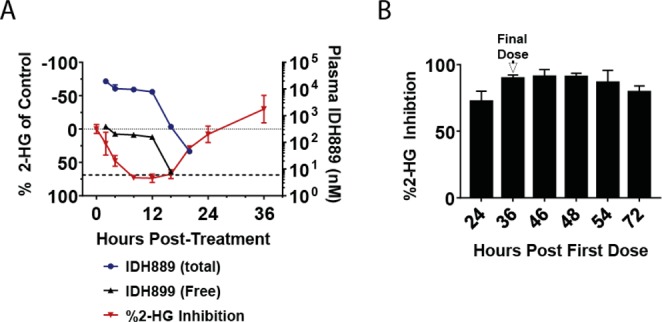
(A) Total and estimated free plasma IDH889 concentration (mean ± SD) and percent inhibition of baseline 2-HG levels (mean ± SEM) in HCT116-IDH1R132 xenograft tumor tissue following a single 200 mg/kg dose (dotted line is zero % inhibition, dashed line represents cellular EC50). (B) Percent inhibition (mean ± SEM) of 2-HG levels in tumor tissue following four doses of IDH889 at 25 mg/kg po BID (q12/12).
In a rat PK study to assess brain/plasma ratio, IDH889 demonstrates favorable distribution to the brain (30 mg/kg po, 1.4 brain/blood ratio, AUCbrain 3117 nM·h, AUCblood 2222 nM·h), albeit with a lower free fraction due to slightly higher rat brain protein binding (99.4%) vs rat plasma protein binding. IDH889 also has excellent permeability and no efflux in the Caco-2 and human MDR1-MDCK cell lines, supporting the hypothesis that potent inhibition of mutant IDH1 function by binding at the allosteric binding site is compatible with brain penetration.
It was next tested whether the 2-HG reduction was sufficient to alter DNA methylation, a phenotype closely linked to IDH1 mutation and 2-HG production.18,19 These studies were performed using Infinium Human Methylation 450 Bead Chip arrays (Supporting Information). Separately derived HCT116-IDH1R132H and HCT116-IDH1R132C cell line clones showed consistent changes in both DNA hyper- and hypomethylation vs their isogenic wild-type counterpart cell line, which were stable over time (Figure 5A). IDH889 caused time-dependent changes in these loci, with preferential hypomethylation of the loci in mutant cells that were originally hypermethylated in mutant vs wild-type cells. Importantly, IDH889 caused no consistent changes in DNA methylation in the wild-type control cell line, highlighting that the DNA methylation changes in the mutant clones were likely due to IDH1 mutation and 2-HG production. To validate these changes in vivo, tumor samples collected at necropsy from the 25 mg/kg BID study (Figure 4B) were evaluated to compare global methylation status vs tumors obtained from untreated control animals (3 samples/group), as well as parental wildtype (wt) HCT116 tumors. Figure 5B shows that a large number of sites in the genome were either hypermethylated (red) or hypomethylated (green) in IDH1R132H± mutant HCT116 tumors vs wt control tumors. For the sites that were specifically altered in mutant tumors, IDH889 treatment caused both the hypermethylated and hypomethylated loci to trend toward reversion to their wt status (Figure 5C). The sites not altered by R132H mutation (Figure 5B, gray) showed only minor changes as a group upon IDH889 treatment (data not shown). Together, this data suggests that the level of 2-HG inhibition achieved in Figure 4B was sufficient to modulate mutant IDH1-dependent DNA methylation changes in vivo.
Figure 5.
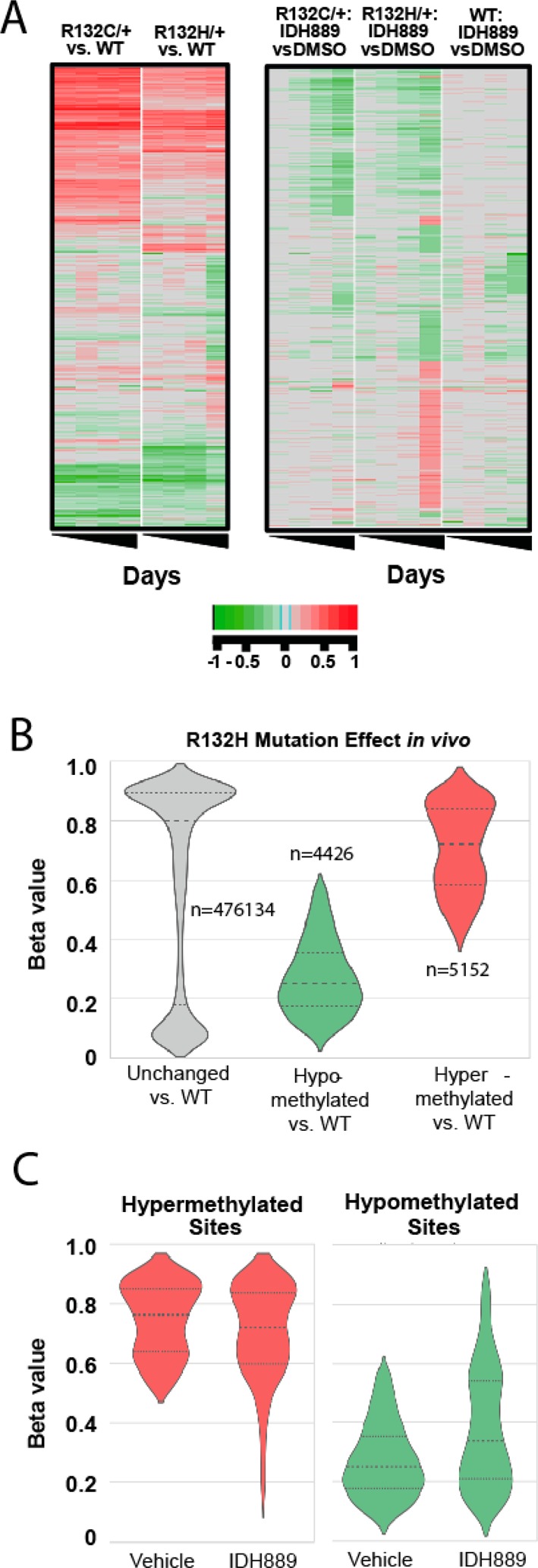
(A) In vitro DNA methylation changes upon IDH889 treatment in the indicated HCT116 cell clones. Rows represent individual sites in the genome, and columns within each cluster represent time points (days 3, 7, 14, and 28). Green shading indicates hypomethylation, and red shading indicates hypermethylation, delta values as described in Supporting Information. (B) Violin plots showing in vivo DNA methylation changes in IDH1R132H/+ mutant vs wild-type HCT116 xenograft tumors. Y axis level of methylation for each site (1 = methylated, 0 = unmethylated: beta values, as described in Supporting Information), n = number of sites within each group, large dashed line indicates median beta value, and smaller dashed lines indicate 75th (top) and 25th (bottom) percentiles. (C) Violin plot as in panel B for IDH1R132H mutant tumors after treatment with IDH889 (tumors from Figure 4B).
In conclusion, a novel HTS hit was optimized for potency and PK properties to generate a tool molecule (IDH889) suitable for exploring the effect of inhibition of production of 2-HG by IDH1R132H in preclinical in vitro and in vivo cancer models. IDH889 binds into an allosteric, induced-fit pocket in IDH1R132H, has good overall selectivity vs the wt protein, and inhibits both the IDH1R132H and IDH1R132C mutants, suggesting broad utility across the various known R132* mutations. Oral dosing of IDH889 in a murine IDH1 mutant tumor xenograft model shows robust reduction of tumor derived 2-HG, a PD biomarker of mutant-IDH1R132* activity. In addition to the potential treatment of AML, chondrosarcoma, cholangiocarcinoma, and other forms of mutant-IDH1 driven cancers, IDH889 demonstrates brain penetrant exposure. This suggests potential utility in preclinical orthotopic tumor models, as well as potential for the series to be optimizable for treating patients with IDH1 mutant brain cancers.
Acknowledgments
The authors would like to thank Thomas Smith, Kimberley Yue, and Daniel Baird for screen optimization and execution; Kara Herlihy, Tami Hood, and Suzanne Zhu for their help with the development of the in vitro activity assays; and Bill Sellers, Markus Warmuth, Karin Briner, Juerg Zimmerman, Tim Ramsey, and Travis Stams for project support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00334.
Synthetic procedures, analytical data, assay protocols, SAR of compounds 2a–d, 3a–f, and 4a–d, small molecule X-ray data of IDH125 and 1e, kinase profiling of IDH125, assay protocols, PK and PK/PD study protocols (PDF)
Author Present Address
† (J.R.L.) Constellation Pharmaceuticals, 215 First Street, Suite 200, Cambridge, Massachusetts 02142, United States.
Author Present Address
‡ (P.D.F. and B.B.T.) Relay Therapeutics, 215 First Street, Suite 300, Cambridge, Massachusetts 02142, United States.
Author Present Address
§ (M.X.) Dupont, 1007 Market St. Wilmington, Delaware 19898, United States.
Author Present Address
∥ (J.G.) National Cancer Institute, 9609 Medical Center Dr., Rockville, Maryland 20850, United States.
Author Present Address
⊥ (L.X.Z.) Ipsen Bioscience, Inc., 650 East Kendall Street, Cambridge, Massachusetts 02142, United States.
Author Present Address
# (B.F.) Surface Oncology, 215 First Street, Suite 400-S, Cambridge, Massachusetts 02142, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Cairns R. A.; Mak T. W. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discovery 2013, 3, 730–41. 10.1158/2159-8290.CD-13-0083. [DOI] [PubMed] [Google Scholar]
- Dang L.; White D. W.; Gross S.; Bennett B. D.; Bittinger M. A.; Driggers E. M.; Fantin V. R.; Jang H. G.; Jin S.; Keenan M. C.; Marks K. M.; Prins R. M.; Ward P. S.; Yen K. E.; Liau L. M.; Rabinowitz J. D.; Cantley L. C.; Thompson C. B.; Vander Heiden M. G.; Su S. M. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–44. 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman J. A.; Kaelin W. G. Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–52. 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovici-Muller J.; Saunders J. O.; Salituro F. G.; Travins J. M.; Yan S.; Zhao F.; Gross S.; Dang L.; Yen K. E.; Yang H.; Straley K. S.; Jin S.; Kunii K.; Fantin V. R.; Zhang S.; Pan O.; Shi D.; Biller S. A.; Su S. M. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 2012, 3, 850–5. 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks E.; Wu X.; Hanel A.; Nguyen S.; Wang J.; Zhang J.; Harrison A.; Zhang W. Identification and characterization of small-molecule inhibitors of the R132H/R132H mutant isocitrate dehydrogenase 1 homodimer and R132H/wild-type heterodimer. J. Biomol. Screening 2014, 19, 1193–1200. 10.1177/1087057114541148. [DOI] [PubMed] [Google Scholar]
- Deng G.; Shen J.; Yin M.; McManus J.; Mathieu M.; Gee P.; He T.; Shi C.; Bedel O.; McLean L. R.; Le-Strat F.; Zhang Y.; Marquette J. P.; Gao Q.; Zhang B.; Rak A.; Hoffmann D.; Rooney E.; Vassort A.; Englaro W.; Li Y.; Patel V.; Adrian F.; Gross S.; Wiederschain D.; Cheng H.; Licht S. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J. Biol. Chem. 2015, 290, 762–74. 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoye-Okafor U. C.; Bartholdy B.; Cartier J.; Gao E. N.; Pietrak B.; Rendina A. R.; Rominger C.; Quinn C.; Smallwood A.; Wiggall K. J.; Reif A. J.; Schmidt S. J.; Qi H.; Zhao H.; Joberty G.; Faelth-Savitski M.; Bantscheff M.; Drewes G.; Duraiswami C.; Brady P.; Groy A.; Narayanagari S. R.; Antony-Debre I.; Mitchell K.; Wang H. R.; Kao Y. R.; Christopeit M.; Carvajal L.; Barreyro L.; Paietta E.; Makishima H.; Will B.; Concha N.; Adams N. D.; Schwartz B.; McCabe M. T.; Maciejewski J.; Verma A.; Steidl U. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 2015, 11, 878–86. 10.1038/nchembio.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D.; Popovici-Muller J.; Palaskas N.; Turcan S.; Grommes C.; Campos C.; Tsoi J.; Clark O.; Oldrini B.; Komisopoulou E.; Kunii K.; Pedraza A.; Schalm S.; Silverman L.; Miller A.; Wang F.; Yang H.; Chen Y.; Kernytsky A.; Rosenblum M. K.; Liu W.; Biller S. A.; Su S. M.; Brennan C. W.; Chan T. A.; Graeber T. G.; Yen K. E.; Mellinghoff I. K. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340 (6132), 626–630. 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B.; Yao Y.; Liu Z.; Deng L.; Anglin J. L.; Jiang H.; Prasad B. V. V.; Song Y. Crystallographic investigation and selective inhibition of mutant isocitrate dehydrogenase. ACS Med. Chem. Lett. 2013, 4, 542. 10.1021/ml400036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Yao Y.; Kogiso M.; Zheng B.; Deng L.; Qiu J. J.; Dong S.; Lv H.; Gallo J. M.; Li X.-N.; Song Y. Inhibition of cancer-associated mutant isocitrate dehydrogenases: synthesis, structure–activity relationship, and selective antitumor activity. J. Med. Chem. 2014, 57, 8307–8318. 10.1021/jm500660f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan B.; Le K.; Manyak E.; Liu H.; Prahl M.; Bowden C. J.; Biller S.; Agresta S.; Yang H. Longitudinal pharmacokinetic/pharmacodynamic profile of AG-120, a potent inhibitor of the IDH1 mutant protein, in a phase 1 study of IDH1-mutant advanced hematologic malignancies. Blood 2015, 126, 1310. [Google Scholar]
- Rankovic Z. CNS Drug Design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584. 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- Xie X.; Capka V.; Chen J.; Chenail G.; Cho Y. S.; Dooley J.; Farsidjani A.; Fortin P. D.; Kohl D.; Kulathila R.; Lin F.; McKay D.; Sage D.; van der Plas S.; Wright K.; Xu M.; Yin H.; Levell J.; Pagliarini R. A.. Allosteric mutant IDH1 inhibitors reveal mechanisms for IDH1 mutant and isoform selectivity. Structure 2016, submitted [DOI] [PubMed]
- Wu P.; Nielsen T. E.; Clausen M. H. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discovery Today 2016, 21 (1), 5. 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Horizon Discovery Group plc. Human IDH1(R132H/+) HCT116 cell line: HD 104–013; Human IDH1(R132H/+) MCF10A cell line: HD 101–013. www.horizondiscovery.com.
- http://www.ncbi.nlm.nih.gov/pubmed/23038259.
- Ioannidis S.; Lamb M. L.; Wang T.; Almeida L.; Block M. H.; Davies A. M.; Peng B.; Su M.; Zhang H.-J.; Hoffmann E.; Rivard C.; Green I.; Howard T.; Pollard H.; Read J.; Alimzhanov M.; Bebernitz G.; Bell K.; Ye M.; Huszar D.; Zinda M. Discovery of 5-chloro-N2-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl]-N4-(5-methyl-1H-pyrazol-3-yl)-pyrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the JAK/STAT pathway. J. Med. Chem. 2011, 54, 262. 10.1021/jm1011319. [DOI] [PubMed] [Google Scholar]
- Turcan S.; Rohle D.; Goenka A.; Walsh L. A.; Fang F.; Yilmaz E.; Campos C.; Fabius A. W.; Lu C.; Ward P. S.; Thompson C. B.; Kaufman A.; Guryanova O.; Levine R.; Heguy A.; Viale A.; Morris L. G.; Huse J. T.; Mellinghoff I. K.; Chan T. A. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479. 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan C. G.; Barwick B. G.; Jin G.; Rago C.; Kapoor-Vazirani P.; Powell D. R.; chi J. T.; Bigner D. D.; Vertino P. M.; Yan H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012, 22, 2339. 10.1101/gr.132738.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.