

Abstract

JNJ-63623872 (2) is a first-in-class, orally bioavailable compound that offers significant potential for the treatment of pandemic and seasonal influenza. Early lead optimization efforts in our 7-azaindole series focused on 1,3-diaminocyclohexyl amide and urea substitutions on the pyrimidine-7-azaindole motif. In this work, we explored two strategies to eliminate observed aldehyde oxidase (AO)-mediated metabolism at the 2-position of these 7-azaindole analogues. Substitution at the 2-position of the azaindole ring generated somewhat less potent analogues, but reduced AO-mediated metabolism. Incorporation of a ring nitrogen generated 7-azaindazole analogues that were equipotent to the parent 2-H-7-azaindole, but surprisingly, did not appear to improve AO-mediated metabolism. Overall, we identified multiple 2-substituted 7-azaindole analogues with enhanced AO stability and we present data for one such compound (12) that demonstrate a favorable oral pharmacokinetic profile in rodents. These analogues have the potential to be further developed as anti-influenza agents for the treatment of influenza.
Keywords: Influenza, PB2 subunit, 7-azaindole, aldehyde oxidase, metabolic stability
Seasonal influenza causes high morbidity and mortality around the globe annually. The impact of influenza epidemics is estimated to be approximately 3.5 million cases for severe illness and 300,000 to 500,000 deaths annually.1 Transmission of novel strains of influenza from other species (mammals and birds) can cause human pandemics, such as the 2009 H1N1 swine flu pandemic as well as the H5N1 avian flu outbreaks.2 Influenza is caused by three orthomyxoviridae family viruses, influenza A, B, and C.3 The current antiviral standard of care (SOC) for treatment of influenza cases in the United States are the neuraminidase inhibitors, oseltamivir 1 (Figure 1) and zanamivir. While these agents can be effective against a variety of type A and B influenza viruses, they suffer from two main limitations. First, the neuraminidase inhibitors have only a moderate impact on the severity of symptoms as well as duration of sickness, and they must be administered within 24–48 h of the infection.4 Second, resistance to this class of antivirals has generated significant concern,5 especially with the report that the H5N1 influenza virus has shown resistance to oseltamivir,6 reinforcing the critical need for new anti-influenza therapeutics with novel mechanisms of action.
Figure 1.

Inhibitors of influenza virus: NA inhibitor oseltamivir (1), 2, and azaindole screening hit 3.
We have previously reported a series of potent 7-azaindole-based influenza inhibitors, highlighted by JNJ-63623872 (2) (formerly known as VX-787).7
The molecular target for these compounds was identified as the PB2 subunit of the influenza viral polymerase. The PB2 subunit contains a cap binding domain for 7-methyl GTP (m7-GTP) on the 5′-end of the host pre-mRNA. Once bound to PB2, the polymerase acidic protein (PA) endonuclease subunit cleaves the host RNA strand, leaving a 10–13 nucleotide primer. The PB1 subunit contains the conserved polymerase domain and utilizes the primer for viral mRNA elongation. Soon after identification of PB2 as the target for these 7-azaindole screening hits, the X-ray crystal structure of initial screening hit 3 bound to the PB2 cap-binding domain was determined.7 During early lead optimization efforts we identified a set of promising lead compounds containing a 1,3-diaminocyclohexyl functionality appended to the azaindole-pyrimidine core.7,8 Of particular early interest was the tertiary morpholine urea analogue 4 (Figure 2) that showed potency in both the cellular protection assay (CPE IC50 = 4 nM) and branched DNA assay (bDNA EC50 = 12 nM) as well as encouraging oral exposure in both rat and mouse.7 This compound was advanced into a mouse influenza model showing 75% survival in a +48 h delay to treatment model (30 mg/kg b.i.d. for 10 days).7 One of the primary metabolites observed for compound 4 in human hepatocytes was the result of oxidation at the 2-position of the 7-azaindole ring to form 2-hydroxy-7-azaindole 5 via aldehyde oxidase (AO). This was evidenced by differential stability in cytosol incubation ± addition of an AO inhibitor followed by detailed metabolite ID (data are not shown). AO is a cytosolic drug-metabolism enzyme that is not present in human liver microsomes (HLM). The emerging importance of AO to drug discovery and development has been highlighted.9 AO oxidation could lead to metabolic products that can cause liver injury and renal toxicity through formation of reactive oxygen species or toxic metabolites.9 Generally, nitrogen-containing heterocycles are more labile to AO-mediated oxidation.9 We have reported a similar AO-mediated oxidation of 2-position of the 7-azaindole recently.10
Figure 2.
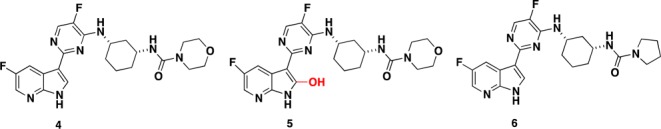
Potent anti-influenza compounds and primary metabolite (5).
This letter describes two strategies that potentially eliminate the formation of this metabolite. One strategy was to substitute the 2-position with a group that would not be a substrate for AO. The second strategy involved the incorporation of an additional nitrogen atom at the 2-position to generate an azaindazole ring, thus eliminating the metabolically labile site.
Compound 6 features a pyrrolidine urea moiety and is 12-fold more potent than compound 4 in the bDNA assay (Table 1) and was used as a reference compound for comparative studies. One of the first analogues targeted was 2-position hydroxymethyl group substituted 7-azaindole 12. The synthesis of compound 12 was accomplished in seven steps starting from commercially available 2-amino-3-iodo-5-fluoropyridine (Scheme 1). Sonagashira coupling of 2-amino-3-iodo-5-fluoropyridine with THP protected propargyl alcohol afforded compound 7 in good yield. Compound 7 was cyclized to the 7-azaindole ring system 8 by heating with potassium tert-butoxide (t-BuOK) in THF in 64% yield. Selective iodination at the 3-position of 8 with NIS generated compound 9. Following protection of the pyrrole nitrogen with benzenesulfonyl chloride, treatment of the resulting product with bis(pinacolato) diborane afforded the desired boronate 10. During this study, it was discovered that tosylate protection at the 2-position of the substituted 7-azaindole was unstable, and therefore, benzenesulfonyl group was selected as the preferred protective group. Suzuki coupling of the boronate 10 with chloropyrimidine intermediate 14 afforded compound 11 in moderate yield. Deprotection of the THP group from compound 11 with TFA followed by removal of the benzenesulfonyl protective group with 4 N HCl afforded the desired target compound 12 in 68% yield. Intermediate 14 was prepared in two steps from commercially available ethyl(1R,3S)-3-aminocyclohexanecarboxylate and 2,4-dichloro-5-fluoropyrimidine, as outlined in Scheme 2.
Table 1. SAR of 2-Position Substituted 7-Azaindole Inhibitors.
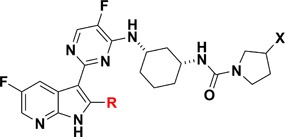
| cmpd | R | X | bDNA EC90a (μM) |
|---|---|---|---|
| 6 | H | H | 0.001 |
| 12 | CH2OH | H | 0.028 |
| 15 | CH2OH | (R)-F | 0.01 |
| 16 | CH3 | H | 0.28 |
| 17 | cyclopropyl | H | >3.3 |
| 19 | COOH | H | 2.2 |
| 20 | C=NOH | H | 0.091 |
| 21a | CH(OH)CH3(R/S) | H | 0.027 |
| 21b | CH(OH)CH3(R/S) | H | 0.46 |
The concentration of test compound resulting in viral RNA levels equal to that of 10% of the control wells was reported as EC90.
Scheme 1. Synthesis of Compound 12.

Reagents and conditions: (a) HC≡CHCH2OTHP, Pd(PPh3)2Cl2, CuI, THF, Et3N, 80 °C, 1 h, 82%; (b) t-BuOK, THF, 85 °C, 2 h, 64%; (c) NIS, DMF, DCM, RT, 1 h, 99%;(d) i. NaH, THF, RT, 30 min; ii. PhSO2Cl, 1 h, 97%; (e) bis(pinacolato)diboron, PdCl2(dppf)·DCM, KOAc, DMF, 85 °C, 3 h, 60%; (f) i. 14, Pd(PPh3)4, THF, aq. Na2CO3, 80 °C, 18 h; ii. TFA, MeOH, H2O, 1 h, 80%; (g) 4 N HCl, 1,4-dioxane, CH3CN, 65 °C, 1 h, 68%.
Scheme 2. Synthesis of Compound 14.

Reagents and conditions: (a) i. ethyl (1R,3S)-3-aminocyclohexanecarboxylate, THF, reflux, 18 h, 92%; ii. LiOH, THF, water, 95 °C, 1 h, 97%; (b) DPPA, Et3N, pyrrolidine, Et3N, THF, RT, 85 °C, 2 h, 85%.
Compounds 16 and 17 (Table 1) with methyl and cyclopropyl substitutions at the 2-position of the 7-azaindole ring were synthesized adapting the procedure described for compound 12 starting from prop-1-yne and ethynylcyclopropane as Sonagashira coupling partners.
The carboxylic acid group of compound 19 was introduced in three steps from intermediate 11 (Scheme 3). Swern oxidation of alcohol 11 afforded aldehyde 18 in 86% yield. Pinnick oxidation of 18 with a mixture of sodium hypophosphate, sodium chlorite, and 2-butene followed by the removal of the benzenesulfonyl protective group provided compound 19 in 88% yield. Reaction of aldehyde 18 with hydroxylamine followed by the removal of the benzenesulfonyl protective group afforded oxime 20. Reaction of aldehyde 18 with methyl magnesium bromide (MeMgBr) followed by removal of the benzenesulfonate protecting group afforded a 1:1 mixture of diastereomers 21a and 21b, which were separated by reverse phase preparative chromatography. The stereochemistry of the 2-position of substituent these diastereomers was not determined.
Scheme 3. Synthesis of Compounds 19, 20, and 21.

Reagents and conditions: (a) (COCl)2, DMSO, Et3N,-78 °C, 2 h, 86%; (b) i. Na2HPO4, NaClO2, 2-butene, t-BuOH, RT, 18 h, 88% ii. 4 N HCl, 1,4-dioxane, CH3CN, 65 °C, 1 h, 68%; (c) i. NH2OH, EtOH, reflux, 3 h; ii. 4 N HCl, 1,4-dioxane, CH3CN, 65 °C, 1 h, 68%; (d) i. CH3MgBr, THF, RT, 1 h; ii. 4 N HCl, 1,4-dioxane, CH3CN, 65 °C, 1 h, 68%.
The second strategy to circumvent the AO metabolism was to introduce a nitrogen at the 2-position of the 7-azaindole ring to afford 7-azaindazole scaffold. Compound 23 as an azaindazole counterpart of compound 6 was synthesized in five steps starting from commercially available 5-fluoro-7-azaindazole (Scheme 4). The synthesis of 23 began with iodination of the 3-position of commercially available 5-fluoro-7-azaindazole to afford 24 and followed by protection of NH with a Tr group to afford compound 25. Formation of boronate 26 from bis(pinacolato)diboron followed by Suzuki coupling with intermediate 14 gave compound 27 in good yield. Deprotection of the Tr group of compound 27 was accomplished with a mixture of triethylsilane and TFA to afford desired azaindazole 23 in good yield.
Scheme 4. Synthesis of Compound 23.

Reagents and conditions: (a) NIS, DCM, RT, 100 °C, 2 h, 80%; (b) trityl chloride, K2CO3, DMF, RT, 18 h, 81%; (c) bis(pinacolato)diboron, PdCl2(dppf).DCM, KOAc, DMF, 95 °C, 1 h, 100%; (d) 14, Pd2(dba)3, X-Phos, K3PO4, 2-methylTHF, H2O, 120 °C, 2 h, 48%; (e) Et3SiH, 30 mol-equiv of TFA, DCM, RT, 30 min, 92%.
Table 1 summarizes in vitro bDNA assay data for selected 2-substituted azaindole analogues. Compounds 16 and 17 that contain small alkyl substitutions such as methyl and cyclopropyl at the 2-position led to a significant loss of potency compared to the unsubstituted compound 6. However, compound 12 containing hydroxymethyl substitution at the 2-position maintained potency although not quite to the same level as 6. Substituting a fluorine atom at the 3-position of the pyrrolidine ring, compound 15 boosts the bDNA potency by 3-fold relative to 12. We explored additional functional groups that could potentially interact with neighboring amino acid residues such as Glu 361 and Arg 332. Compound 19, containing a carboxylic acid functional group at the 2-position, reduced potency compared to the hydroxymethyl compound 12 by ∼100-fold. Compound 20 containing an oxime moiety at the 2-position showed similar bDNA potency (within 3-fold) when compared to compound 12. For the secondary alcohols, one diastereomer 21a showed similar potency to 12 while the other diastereomer 21b lost more than 10-fold potency. The four most potent compounds in this series contained a hydroxyl group that may interact with neighboring amino acid residues of PB2. We obtained the X-ray crystal structure of compound 12 bound to the PB2 subunit (Figure 3A). As expected, the azaindole ring of 12 is involved in two hydrogen bonding interactions with amino acid residues, Lys 376 and Glu 361.
Figure 3.
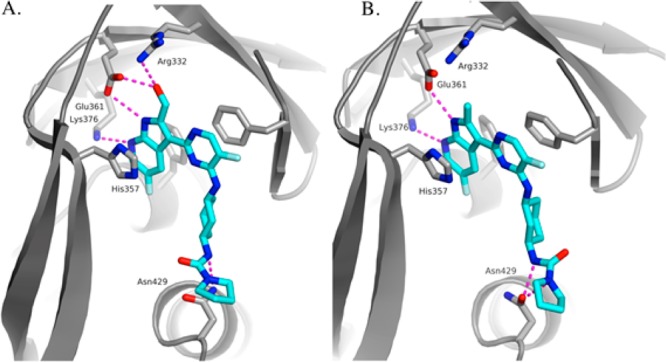
(A) X-ray crystal structure of 12 bound to PB2 (PDB ID: 5BUH). (B) X-ray crystal structure of 16 bound to PB2 (PBD ID: 5F79).
The pyrimidine ring of 12 interacts by π-stacking with the side chains of His 357, Phe 323, and Phe 404, while the cyclohexyl group occupies the neighboring hydrophobic region of the PB2 binding site.7 Additionally, the hydroxy methyl group of 12 makes hydrogen bonds with Glu 361 and Arg 332. These new hydrogen bonds could explain the potency difference between the hydroxy methyl group and the methyl and cyclopropyl groups. The crystal structure of 12 in complex with PB2 helped generate hypotheses to explain the SAR observed for the 2-substituted 7-azaindoles. Coplanarity of the pyrimidine and the 7-azaindole rings appeared to be a requirement to ensure optimal π-stacking between the pyrimidine ring and the phenyl ring of Phe 323. One hypothesis was that the loss of affinity observed for the 2-methyl and 2-cyclopropyl analogues relative to the corresponding unsubstituted compound could be due to the conformational impact of the ortho-substitution, leading to a low energy conformation where the two rings were not coplanar. As a result, the coplanar conformation required for binding would have come with an energetic penalty, resulting in a reduction in the free energy of binding. In order to test this hypothesis we ran torsional scans around the 7-azaindole-pyrimidine bond using high-level quantum mechanics. The calculations revealed that in the preferred conformations for both the 2-methyl and the 2-cyclopropyl analogues the 7-azaindole and pyrimidine rings remain coplanar, thus invalidating the hypothesis. The X-ray crystal structure of compound 16 bound to the PB2 subunit (Figure 3B) showed that in the preferred conformation for 16, the 7-azaindole and pyrimidine rings remain coplanar. The second hypothesis was that the 2-methyl group causes a partial desolvation of the carboxylic group of Glu 361, and the resulting desolvation penalty translates into a reduced free energy of binding. This hypothesis is consistent with the further decrease in affinity observed for the 2-cyclopropyl analogue 17, where the size of the 2-substitution is larger and the desolvation may extend to the side chain of the neighboring Arg 332. The reason for loss of potency of 19 is likely explained by the repulsion between the carboxylic group of the ligand and Glu 361.
A small set of 7-azaindazole analogues were designed and synthesized to explore different substituents on the cyclohexyl ring system (Table 2). The pyrrolidine and morpholine ureas, compounds 23 and 28, respectively, were both potent and showed similar potency trends to those observed in the 7-azaindole series. Additionally, the amides 30 and 31 showed good potency with the thiophene amide 31, matching the best potency of urea 29. We also synthesized the 7-azaindazole analogue 32 of 2 and retained potency relative to the 7-azaindole.7
Table 2. 7-Azaindazole-Based PB2 Inhibitors.
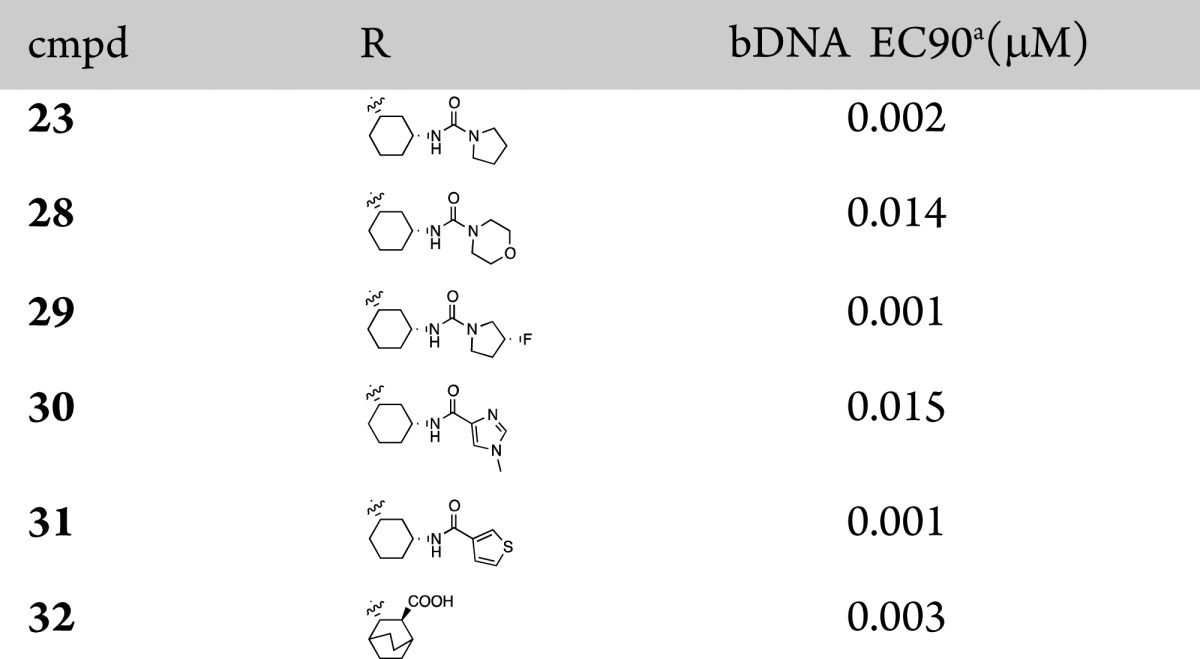
The concentration of test compound resulting in viral RNA levels equal to that of 10% of the control wells was reported as EC90.
To determine whether AO-mediated metabolism of these new 2-position substituted azaindole and azaindazole analogues had been modulated, selected analogues were incubated in triplicate at 1 μM in 1 mg/mL human liver cytosol protein at 37 °C for 4 h, with and without the AO inhibitor, Raloxifene. The results are summarized in Table 3. All of the tested compounds were stable in human liver cytosol in the presence of raloxifene. Most of the 2-position substituted azaindole analogues including 12, 15, 16, and 21b appeared to be very stable without raloxifene in cytosol. Surprisingly, most of the azaindazole analogues were unstable in cytosol, except acidic compound 32. It is possible that incorporation of the extra ring nitrogen in the azaindole ring has shifted AO-mediated metabolism to another site of the molecule and/or incurred additional metabolic pathways.
Table 3. Human Liver Cytosol Stability of 7-Azaindole and 7-Azaindazole Analogues.
| cmpd | % remaining @ 4 h without raloxifene | % remaining @4 h with raloxifene |
|---|---|---|
| 4 | 7 | 103 |
| 6 | 4 | 99 |
| 12 | 100 | 96 |
| 15 | 105 | 114 |
| 16 | 103 | 103 |
| 21b | 99 | 99 |
| 23 | 1 | 101 |
| 29 | 0.6 | 98 |
| 30 | 40 | 103 |
| 31 | 0 | 107 |
We determined rat pharmacokinetic profiles of compounds 6, 12, and 23 as shown in Table 4.
Table 4. Rat and Mouse Pharmacokinetic Parameters (PK) for Compounds 6, 12, and 23.
| cmpd | 6 | 12 | 23 |
|---|---|---|---|
| rat IV PK (1 mg/kg) | |||
| CL (mL/min/kg) | 1.2 | 6.2 | 7.7 |
| t1/2 (h) | 3.2 | 2.3 | 1.5 |
| Vss (L/kg) | 0.3 | 0.6 | 0.4 |
| rat PO PK (3 mg/kg) dose | |||
| AUC0-inf (μg·h/mL) | 30.3 | 9.7 | 1.2 |
| Cmax (μg/mL) | 2.6 | 2.8 | 0.2 |
| %F | 82 | 100 | 15.3 |
| mouse PO PK (30 mg/kg) | |||
| AUC | 336 | 101 | 44.5 |
| Cmax (μg/mL) | 61.1 | 37.7 | 32.1 |
All three compounds showed promising PK profiles with IV clearance values of 1.2, 6.2, and 7.7 mL/min/kg, respectively. The 7-azaindole urea 6 showed the highest oral exposure and the 2-hydroxymethyl analogue 12, showed 3-fold lower AUC with a similar Cmax as 6. The 7-azaindazole 23 showed significantly reduced exposures as measured by both AUC and Cmax relative to 6 and 12. When we dosed compounds 6, 12, and 23 in mice (30 mg/kg), we also observed a significant loss in exposure from the 7-azaindole to 7-azaindazole with a 2-fold loss in Cmax and 7-fold loss in AUC in mice (30 mg/kg). While attempts at blocking AO metabolism were not demonstrated by improved pharmacokinetics in these rodent studies, AO is known to exhibit large species differences with high levels in human and monkey and low levels in rat and mouse.9 Thus, the in vitro results suggest the possibility of improved human pharmacokinetics for some of these 2-substituted 7-azaindoles.
In summary, our efforts to block the observed metabolism at the 2-position of the 7-azaindole ring involved an explorative strategy that functionalized the 2-position or incorporated a ring nitrogen atom at the 2-position. We discovered that certain functionalities containing a specifically placed H-bond donor such as 12, 20, and 21a maintained cellular potency. Moreover, 2-substituted 7-azaindole analogues showed excellent stability in human liver cytosol. The 7-azaindazole modifications resulted in equipotent analogues; however, these compounds were less stable in the human cytosol assay and are likely substrates for AO-mediated metabolism. We identified compound 12 with enhanced in vitro metabolic stability in human liver cytosol and a favorable oral pharmacokinetic profile in both rat and mouse in vivo studies. Thus, the data supports and warrants advancement of compound 12 for further evaluation for the treatment of influenza.
Acknowledgments
The authors thank Barry Davis and Frank Holland for analytical chemistry support and Jeremy Green and Simon Giroux for helpful discussions.
Glossary
ABBREVIATIONS
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- DCM
dichloromethane
- DPPA
diphenylphosphorylazide
- NIS
N-iodoscucinimide
- THP
tetrahydropyranyl acetal
- Tr
trityl
- X-phos
2-dicyclohexylphosohino-2′,4′,6′-triisopropylbiphenyl
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00487.
Experimental procedures and analytical data for compounds 7–32; X-ray crystallographic data (12 and 16); protocols for bDNA cellular assays and cytosol stability assay (PDF)
Author Present Address
† Sage Therapeutics, Inc., 215 First Street, Cambridge, Massachusetts 02142, United States.
Author Present Address
‡ Goldfinch Bio, 215 First Street, 4th Floor, Cambridge, Massachusetts 02142, United States.
Author Present Address
§ Moderna Therapeutics, Inc., 200 Technology Square, Cambridge, Massachusetts 02139, United States.
Author Present Address
∥ Contrafect Corporation, 28 Wells Avenue, Third Floor, Yonkers, New York 10701, United States.
Author Present Address
⊥ Vertex Pharmaceuticals Incorporated (Canada) Inc., 275 Armand-Frappier, Laval, Quebec H7V 4A7, Canada.
The authors declare no competing financial interest.
Supplementary Material
References
- Fiore A. E.; Shay D. K.; Broder K.; Iskander J. K.; Uyeki T. M.; et al. Prevention and control of influenza: recommendations of the advisory committee on immunization practices (ACIP). MMWR Recomm Rep 2008, 57, 1–60. [PubMed] [Google Scholar]
- Li Q.; Zhou L.; Zhou M.; Chen Z.; Li F.; Wu H.; Xiang N.; Chen E.; Tang F.; Wang D.; Meng L.; Hong Z.; Tu W.; Cao Y.; Li L.; Ding F.; Liu B.; Wang M.; Xie R.; Gao R.; Li X.; Bai T.; Zou S.; He J.; Hu J.; Xu Y.; Chai C.; Wang S.; Gao Y.; Jin L.; Zhang Y.; Luo H.; Yu H.; Gao L.; Pang X.; Liu G.; Shu Y.; Yang W.; Uyeki T. M.; Wang Y.; Wu F.; Feng Z. Epidemiology of the avian influenza A (H7N9) outbreak in China. N. Engl. J. Med. 2014, 370, 520–532. 10.1056/NEJMoa1304617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R. A.; Krug R. M.. Orthomyxoviridae: The viruses and their replication. In Fields Virology, 4th ed.; Knipe D. M., Howley P. M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, 2001; p 1487. [Google Scholar]
- Moscona A. Neuraminidase inhibitors for influenza. N. Engl. J. Med. 2005, 353, 1363–1373. 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- Thorlund K.; Awad T.; Boivin G.; Thabane L. Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect. Dis. 2011, 1, 134–147. 10.1186/1471-2334-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai M.; Watanabe T.; Hatta M.; Das S. C.; Ozawa M.; Shinya K.; Zhong G.; Hanson A.; Katsura H.; Watanabe S.; Li C.; Kawakami E.; Yamada S.; Kiso M.; Suzuki Y.; Maher E. A.; Neumann G.; Kawaoka Y. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortment H5 HA/H1N1 virus in ferrets. Nature 2012, 486, 420–428. 10.1038/nature10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M. P.; Ledeboer M. W.; Davies I.; Byrn R. A.; Jones S. M.; Perola E.; Tsai A.; Jacobs M.; Nti-Addae K.; Bandarage U. K.; Boyd M. J.; Bethiel R. S.; Court J. J.; Deng H.; Duffy J. P.; Dorsch W. A.; Farmer L. J.; Gao H.; Gu W.; Jackson K.; Jacobs D. H.; Kennedy J. M.; Ledford B.; Liang J.; Maltais F.; Murcko M.; Wang T.; Wannamaker M. W.; Bennett H. B.; Leeman J. R.; McNeil C.; Taylor W. P.; Memmott C.; Jiang M.; Rijnbrand R.; Bral C.; Germann U.; Nezami A.; Zhang Y.; Salituro F. G.; Bennani Y. L.; Charifson P. S. Discovery of a novel, first-in-class, orally bioavailable azaindole inhibitor (VX-787) of influenza PB2. J. Med. Chem. 2014, 57, 6668–6678. 10.1021/jm5007275. [DOI] [PubMed] [Google Scholar]
- Liang J. L.; Cochran J. E.; Dorsch W. A.; Davies I.; Clark M. P. Development of a scalable synthesis of an azaindolyl-pyrimidine inhibitor of influenza virus replication. Org. Process Res. Dev. 2016, 20, 965–969. 10.1021/acs.oprd.6b00063. [DOI] [Google Scholar]
- Pryde D. C.; Dalvie D.; Hu Q.; Jones P.; Obach R. S.; Tran T. D. Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J. Med. Chem. 2010, 53, 8441–8460. 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- Zetterberg C.; Maltais F.; Laitinen L.; Liao S.; Tsao H.; Chakilam A.; Hariparsad N. VX-509 (Decernotinib)-Mediated CYP3A time-dependent inhibition: An aldehyde oxidase metabolite as a perpetrator of drug-drug interactions. Drug Metab. Dispos. 2016, 44, 1286–1295. 10.1124/dmd.116.071100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.