

Abstract
Medical responders to radiological and nuclear disasters currently lack sufficient high-throughput and minimally invasive biodosimetry tools to assess exposure and injury in the affected populations. For this reason, we have focused on developing robust radiation exposure biomarkers in easily accessible biofluids such as urine, serum and feces. While we have previously reported on urine and serum biomarkers, here we assessed perturbations in the fecal metabolome resulting from exposure to external X radiation in vivo. The gastrointestinal (GI) system is of particular importance in radiation biodosimetry due to its constant cell renewal and sensitivity to radiation-induced injury. While the clinical GI symptoms such as pain, bloating, nausea, vomiting and diarrhea are manifested after radiation exposure, no reliable bioindicator has been identified for radiation-induced gastrointestinal injuries. To this end, we focused on determining a fecal metabolomic signature in X-ray irradiated mice. There is overwhelming evidence that the gut microbiota play an essential role in gut homeostasis and overall health. Because the fecal metabolome is tightly correlated with the composition and diversity of the microorganism in the gut, we also performed fecal 16S rRNA sequencing analysis to determine the changes in the microbial composition postir-radiation. We used in-house bioinformatics tools to integrate the 16S rRNA sequencing and metabolomic data, and to elucidate the gut integrated ecosystem and its deviations from a stable host-microbiome state that result from irradiation. The 16S rRNA sequencing results indicated that radiation caused remarkable alterations of the microbiome in feces at the family level. Increased abundance of common members of Lactobacillaceae and Staphylococcaceae families, and decreased abundances of Lachnospiraceae, Ruminococcaceae and Clostridiaceae families were found after 5 and 12 Gy irradiation. The metabolomic data revealed statistically significant changes in the microbial-derived products such as pipecolic acid, glutaconic acid, urobilinogen and homogentisic acid. In addition, significant changes were detected in bile acids such as taurocholic acid and 12-ketodeoxycholic acid. These changes may be associated with the observed shifts in the abundance of intestinal microbes, such as R. gnavus, which can transform bile acids.
Introduction
The efficiency of clinical tests to accurately assess radiation exposure in a high-throughput manner after a radiological event or nuclear explosion depends heavily on the robustness and predictive power of the biomarkers. Easily accessible biofluids such as urine and blood have been the obvious choices in many biomarker discovery efforts and have provided valuable insight into the mechanism of radiation-induced injury and stress signaling. In this regard, the field of metabolomics has played a particularly important role in expanding our knowledge of physiological and metabolic changes after exposure to various types of radiation. Metabolites are the end points of chemical reactions in our bodies, thus, changes in their concentrations represent systemic responses to radiation exposure. To date, most radiation metabolomic studies have focused on mammalian metabolites while widely ignoring those that are microbial in origin. Considering that in the human gut alone the microbial cells outnumber the eukaryotic cells in the entire body by ten to one and contribute over 100-fold more genetic material, it is essential to assess their contributions to phenotype and radiation injury. The gut microbiota and their metabolic products are in constant cross-talk with the host cellular metabolism. Recent studies have provided evidence on the regulatory role of microbiota in immunological responses and stress signaling, such as those observed after injury induced by ionizing radiation (1–3).
The gastrointestinal (GI) system is particularly susceptible to radiation injury due to increased apoptosis of crypt epithelial cells, mucosal cell loss and by lymphocyte infiltration of the underlying tissue (4). In addition, cytotoxic effects of radiation exposure can also lead to defects in the intestinal epithelial barrier, which offers higher permeability to luminal bacteria and triggers immune responses (5). Furthermore, while the severity of these histological changes tends to be dose dependent (4), there are also secondary adverse GI effects that are directly related to the radiation exposure such as bile salt malabsorption from terminal ileal damage (6, 7), malabsorption of lactose or other fermentable sugars (8), small bowel bacterial overgrowth (9, 10) and altered transit time (11). Therefore, to study the local biology and the perturbations in gut homeostasis, stool may be a valuable biofluid. Stool not only contains significant information on the status of the GI system, its metabolome reflects changes in the metabolism of the host and the intestinal microbiota, as gut microbiota also contribute to transformation and fermentation of dietary components and production of final metabolites, including short-chain and branched-chain fatty acids as well as phenol, indole and sulfur compounds. It is now understood that the state of GI microbial communities closely relates to the status of human health, with the population of microorganisms in the intestines being sensitive to changes in the intestinal microenvironment and dysbacteriosis of the microbiota being associated with an expanding list of diseases (12, 13).
Taken together, this evidence emphasizes the importance of the microbial contribution to maintaining the host health, and to shaping and refining the host metabolomic response to radiation exposure. While the wide diversity of species that make up the gut microbiota is difficult to characterize, it is known that disturbances in both the bacterial diversity and abundance as a result of environmental exposures are associated with gut dysbiosis (14). For instance, preclinical studies have shown that radiation exposure causes a substantial increase in the number of bacteria on intestinal villi, and small intestinal bacterial overgrowth was observed in irradiated patients (9, 15). Also, a cohort study of irradiated patients with intestinal toxicity showed that impaired motility was a cause of gastrointestinal colonization of Gram-negative bacilli (16), while a more recent study showed a progressively increasing dysbiotic pattern in six postirradiation patients with diarrhea symptoms (17). Therefore, in this study, over the course of 30 days we explored the changes in the fecal microbiome and metabolome of mice that were whole-body irradiated with a single fraction of 5 and 12 Gy of external beam X rays. We used 16S rRNA gene pyrosequencing to determine the significant differences in fecal microbiota after irradiation. We observed a remarkable decrease of Firmicute bacteria, which has also been previously reported in mice (18) and humans (19) after broad-spectrum antibacterial treatments. In addition, fecal metabolomic analysis revealed significant changes in the microbial metabolic products. Together, these results show that fecal metabolomics can provide valuable insight into the effects of radiation on the gut microbiota and ultimately the host metabolism, and that it can serve as a radiation exposure assessment biofluid.
Materials and Methods
The lipid standard, phosphatidylserine PS (14:0/14:0), was purchased from Avanti® Polar lipids (Alabaster, AL). Debrisoquine sulfate, 4-nitrobenzoic acid (4-NBA) and UPLC-grade solvents such as acetonitrile, water and isopropanol were purchased from Fisher Scientific (Hanover Park, IL). Glyceric acid, glutaconic acid, pipecolic acid, homogentisic acid, sebacic acid, serotonin, hypoxanthine, α-ketobutyric acid, hippuric acid, taurine and hydroxyphenylpyruvic acid were purchased from Sigma-Aldrich® (Seelze, Germany). In addition, the MS/MS spectra provided by Scripps Center for Metabolomics (La Jolla, CA) were used as reference spectra for hydroquinone.
Animal Experiment and Sample Collection
Animal housing
C57BL/6J eight-week-old male mice (Jackson Laboratory, Bar Harbor, ME) were maintained in a 12:12 h light-dark schedule at 22°C in 30–70% humidity and provided certified rodent diet along with filtered water ad libitum. Mice (n = 6–7 per study group) were exposed to a whole-body dose of 0, 5 or 12 Gy X rays using an X-RAD 320 X-ray irradiator (Precision X-ray Inc., North Branford, CT). We used 320 kV and 12.5 mA X-rays filtered with a 1.5 mm aluminum/0.25 mm copper/0.75 mm tin filter placed in front of the source for both 5 and 12 Gy irradiations. After irradiations mice were returned to their home cages and monitored regularly. All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committees (IACUC) at Georgetown University. For our research, we followed Guide for the Care and Use of Laboratory Animals, prepared by the Institute of Laboratory Animal Resources, National Research Council and U.S. National Academy of Sciences.
Feces collection
Feces were collected by placing the mice into customized Tecniplast® (West Chester, PA) metabolic cages for 24 h at a time. At the end of 24 h, feces were collected and stored at −80°C until analysis. Feces were collected at one day before irradiation and 3, 14 and 30 days after irradiation (Table 1).
Table 1. Experimental Design.
| Radiation dose | Number of mice per time point | Time point (days postirradiation) |
|---|---|---|
| 0 Gy (control) | 7 | −1, 3, 14, 30 |
| 5 Gy | 7 | −1, 3, 14, 30 |
| 12 Gy | 7 | −1, 3a |
Mice died before the 14- and 30-day time points.
16S rRNA Sequencing and Microbiome Data Analysis
Feces collected from mice before and after irradiations (0, 5 and 12 Gy) were subjected to 16S rRNA sequencing. For mice in the 0 and 5 Gy groups, feces were obtained at day 1 before irradiation and at day 3 and day 30 postirradiation, while for the 12 Gy irradiated group, samples were obtained only at day 1 before and day 3 postirradiation. Genomic DNA extraction, amplification of the V4 region of 16S ribosomal RNA genes and paired-end sequencing on an Illumina® HiSeq 2500 were performed as described previously (27). The 150 base-paired-end reads were processed using QIIME version 1.8.0, with default parameters to generate approximately 254 base-pair-joined reads (50). The number of reads per sample ranged from 413,213 to 953,186, with a mean of 692,987. Operational taxonomic units (OTUs) were picked against the May 2013 version of the Greengenes database (http://greengenes.secondgenome.com), prefiltered at 97% identity. The biom-format file derived from QIIME was imported into Phyloseq package on R data filtering, normalization, species richness (Observed, Chao1) and alpha diversity measurements (Shannon) were performed in Phyloseq.
Metabolomics and Data Analysis
The fecal samples were prepared by resuspending 50 mg of dry fecal pellets in 150 μl of Optima™ LC/MS grade water. The samples were then homogenized and heated in a 37°C water bath for 90 s, then chilled on ice. One microliter from each sample was removed for protein concentration measurement. Optima acetonitrile (150 μl) containing internal standards, 4-NBA and debrisoquine, were added to the samples. The samples were then spun at maximum speed for 20 min at 4°C. The supernatant was placed in a new tube, dried under a gentle stream of nitrogen and resuspended in 300 μl of solvent A (98% water, 2% ACN and 0.1% formic acid) for LC/MS. The MS analysis was performed by injecting 2 μl 1aliquots of each sample into a reverse-phase 50 × 2.1 mm H-class ACQUITY UPLC 1.7 μM BEH C18 column (Waters® Corp., Milford, MA) coupled to a time-of-flight mass spectrometry (TOFMS). The mobile phase consisted of solvent A and 100% acetonitrile (solvent B). The Xevo G2-S mass spectrometer (Waters Corp.) was operated in the positive (ESI+) and negative (ESI−) electrospray ionization modes scanning a 50–1,200 m/z range. The following 11 min gradient was used: 98%/2% solvent A/solvent B at 0.5 ml/min for 8 min; 2%/98% solvent A/solvent B for 2 min; and back to 98%/2% solvent A/solvent B for the last minute. The lock-spray consisted of leucine-enkephalin (556.2771 [M + H]+ and 554.2615 [M – H]−). The MS data were acquired in centroid mode and assessed for quality assurance using MassLynx® software (Waters Corp.). In-house bioinformatics tools and workflows were used to putatively identify ions, utilizing the Human Metabolome Database (HMDB; http://www.hmdb.ca), LipidMaps® (http://www.lipidmaps.org), the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/) and BioCyc (http://biocyc.org; SRI Intl., Menlo Park, CA). The m/z values were used to putatively assign IDs to the ions by neutral mass elucidation, which was accomplished by considering the possible adducts (H+, Na+ and/or NH4+ in the ESI+ mode; H− and Cl− in the ESI− mode). The masses were then compared to the exact mass of small molecules in the databases, from which putative metabolites were identified with a mass error of 20 parts per million (ppm) or less. KEGG-annotated pathways associated with these putative metabolites were also identified. Candidate metabolite markers were then chosen from this list for further MS/MS validation.
Progenesis® QI Informatics (Nonlinear Dynamics, Newcastle, UK) was used to deconvolute the data and align the high-energy scans (fragments) and the low-energy scans (precursors) of MSE data. The data was normalized to total protein concentration for each sample. Our in-house statistical analysis program, MetaboLyzer (20), was then used to analyze the data and identify statistically significant ions as described previously (21). The ion presence threshold was set at 0.7 in MetaboLyzer in each study group for identifying complete-presence ions (which have nonzero abundance values in >70% of the samples for both groups) and partial-presence ions (which are >70% present for only one group). Data were then log-transformed and analyzed for statistical significance via the nonparametric Kolmogorov-Smirnov test for statistical significance with false discovery rate (FDR) correction (FDR &≤ 0.10). Partial-presence ions were analyzed as categorical variables for presence status (i.e., nonzero abundance in 70% of samples) via FDR-corrected Fisher's exact test (FDR < 0.10). The data for statistically significant complete-presence ions (398 ions) in positive ESI mode were then utilized for principal component analysis (PCA) and for constructing a receiver operating characteristic (ROC) curve. The ROC curve was constructed utilizing a Gaussian kernel support vector machine (SVM)-based binary classifier coupled with a Monte Carlo cross-validation (MCCV) procedure. Approximately one-third of all samples were randomly removed from the dataset, while the remaining two-thirds were utilized to train the SVM. The removed samples were then classified with this trained SVM to evaluate the predictive ability of the statistically significant ions. This procedure was repeated 30 times for the purposes of gathering enough data to construct a ROC curve.
In addition to the aforementioned classical statistical analysis procedure, differential correlation analysis was also conducted on complete-presence ions for the purposes of examining subtler shifts in the coregulation of metabolic processes. For each group, Pearson's correlation coefficients were calculated between all possible ion pair combinations and arranged into a triangular matrix. The matrices for each group (control and irradiated) were then utilized to construct two dissimilarity heatmaps for a qualitative comparison of correlation shifts in the coregulation structure of the metabolome. Statistically significant correlation differentials were also identified via the Z test for statistical significance (P < 0.01) after a Fisher transformation and visualized via heatmap construction (20). The subset of statistically significant differential correlation pairs, whose constituent ions' putative identities both map to the same KEGG pathway (i.e. “double hit”), were also further analyzed via pathway enrichment analysis (FDR-corrected hypergeometric statistical testing). This differential correlation-based “double hit” pathway analysis was dichotomized into statistically significant correlation gains and losses, which represent increased coregulatory activity and dysregulation of metabolic pathways, respectively.
Inter-omic Correlation Analysis
Inter-omic networks allow for exploration of correlation patterns across two large datasets to reveal possible biological interactions (in this case between microbes and spectral features) (61). These networks are constructed from pairwise correlations between features of interest in each dataset (i.e. each combination of a microbe and ion). We constructed an inter-omic network from microbes and spectral features that had a statistically significant association with radiation exposure. To adjust for differences in biological states across samples, the two datasets were fitted to multivariate models incorporating radiation dose and time point. The two datasets were first filtered to remove features present in less than 10% of samples. Microbes were additionally filtered for a minimum abundance of 0.0001. DESeq2 (http://bit.ly/295KboX) was used to fit microbial abundance to negative binomial models and shrink dispersion using an empirical Bayesian approach (22). Significance was calculated with the Wald test and P values were adjusted for multiple hypothesis testing; Q values below 0.05 were considered significant (23). Metabolomics data were normalized and variance stabilized using mean-variance regularization (MVR), an approach designed for metabolomic analysis, then fitted to Gaussian models with the limma package in R (24, 25). Significance was calculated using empirical Bayes moderated t statistics and P values were adjusted to Q values to correct for multiple hypothesis testing (23, 25). Microbes and spectral features that were differentially abundant after either 5 or 12 Gy irradiation then underwent inter-omic correlation analysis. Spearman correlation was calculated for all pairs of microbes and ions using the residuals of microbiome data fitted to negative binomial models and residuals of transformed metabolomics data fitted to Gaussian models. P values were computed for Spearman correlation coefficients with the asymptotic t approximation, and were then converted to Q values. Microbe-ion correlation pairs with Q < 0.15 were used to construct a microbe-ion network in Cytoscape version 3.2.1 (http://cytoscape.org).
Results
In this study we assessed the changes in the fecal metabolome and metagenome of mice exposed to 5 and 12 Gy on external beam X rays. The LD50/30 dose for mice of this age and strain is typically about 8 Gy, thus there is no lethality at 5 Gy but the dose is sufficient to cause some GI toxicity (60); doses of ≥10 Gy show accelerated lethality typical of increasing GI toxicity (49). The metabolomics analysis was performed using a UPLC-TOFMS which enabled us to detect over 3,500 spectral features in positive and negative ESI modes. The fecal metabolomic signature of mice in the positive ESI mode showed changes in the abundances of 720 spectral features which were present in more than 70% of the samples from a total of 2,700 detected features, as shown in the volcano plot (Fig. 1A). The red circles represent the spectral features, which were found to display statistically significant changes in their abundances after 5 Gy irradiation. The x-axis of this plot represents the magnitude of change (fold change) and the y-axis shows the statistical significance (P value). The red circles on the positive scale of the x-axis show the spectral features, which had statistically significant increasing abundances after irradiation, and those on the left had a decreasing pattern in their abundances. These perturbations in the fecal ions from the irradiated mice drove a statistically significant shift in the overall fecal metabolomic signature of these mice away from that of the control mice, as shown by the separation of the signatures in the PCA plot (Fig. 1B). The heatmap shown in Fig. 1C highlights the spectral features, which contributed the most to this separation. The bottom one-fifth of this heatmap shows the spectral features whose fecal levels decreased after irradiation while the top four-fifths of the heatmap show spectral features with an increase in their fecal levels. The ROC curve in Fig. 1D shows the results to evaluate the predictive ability of the statistically significant spectral features from Fig. 1A. The ROC was constructed utilizing an SVM-based classification model coupled with Monte Carlo cross-validation and shows that the statistically significant features, upon more comprehensive evaluation, may be used to predict radiation exposure. It is, however, important to note that we utilized SVMs in our analysis for exploratory purposes. Thus a comprehensive evaluation of predictive potential of the features would require much larger sample sizes and more stringent analysis.
Fig. 1.
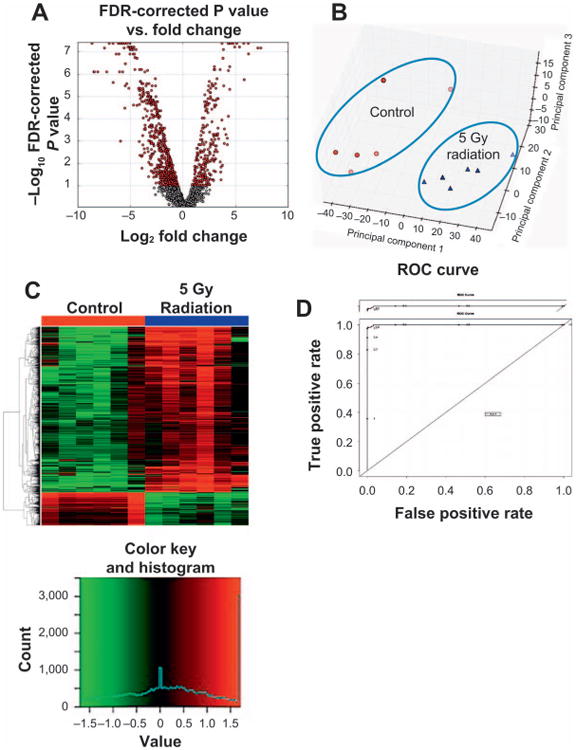
Perturbations in the fecal metabolomic signature in mice 3 days after irradiation was assessed via MetaboLyzer. Here we show these perturbations after 5 Gy irradiation with 12 Gy irradiation showing similar changes (Supplementary Fig. S2; http://dx.doi.org/10.1667/RR14306.1.S3). Panel A shows a volcano plot, where the statistically significant (Kolmogorov-Smirnov test, FDR < 0.10) spectral features are shown in red. The x-axis of the volcano plot, log2 fold change, highlights the direction of the shift in the abundance of the spectral features. On this axis the positive scale represents an increase and the negative scale represents a decrease in the abundance of spectral features at day 3 after 5 Gy irradiation. The y-axis of the volcano plot, −log P value, specifies the significance of the change, decrease or increase, in the abundance of the spectral features. Panel B shows a principle component analysis (PCA) scores plot and highlights the distinct separation between the overall fecal metabolomic profile of the control mice vs. those irradiated at 5 Gy at day 3. The statistically significant ions in panel A contribute to the separation seen in panel B. Panel C is a heatmap of individual ions with most significant contribution to the separation of the fecal metabolomic signature of mice exposed to 5 Gy external beam irradiation compared to the control mice. The top four-fifth of the heatmap shows spectral features with increasing abundances after 5 Gy irradiation, while those at the bottom one-fifth of the heatmap show a decreasing pattern in their abundances after irradiation. To evaluate the predictive ability of the statistically significant spectral features, an ROC curve (panel D) was constructed utilizing an SVM based classification model coupled with Monte Carlo cross-validation.
Results from differential correlation analysis reveal shifts in coregulation of metabolic processes. The dissimilarity heatmap generated from the Pearson's correlation coefficients calculated for the control group (Fig. 2A) exhibits a high degree of coregulation and coordination, as represented by the abundance of dark red hues. When comparing this to the heatmap generated from the irradiated group (Fig. 2B), there is a clear loss of correlation, as exhibited by the lighter red hues in the periphery of the top right corner where darker hues existed in the previous heatmap. However, there is also evidence of increased coregulation as a result of radiation exposure, as exhibited by the newly darkened areas in the bottom left corner compared to the control. These differences are emphasized in the differential correlation heatmap (Fig. 2C), where orange hues represent a gain of correlation (and thus increased coregulation), while blue hues represent a loss of correlation (i.e., dysregulation). With respect to the number of statistically significant correlation shifts, correlation losses exceed gains by approximately 2 to 1, implying that the overall effect of radiation exposure resulted in widespread dysregulation of metabolic processes. However, the correlation gains are not negligible, and represent a biologically relevant subset of metabolic processes that were observed to have increased levels of coregulation, rather than dysregulation. The results of “double hit” pathway enrichment analysis on this subset of correlation shifts (Fig. 2D), which plots the − 1*log10 (P value) for each KEGG pathway, as calculated by the uncorrected (blue bar) and FDR-corrected (red bar) hypergeometric test, yielded tyrosine metabolism, tryptophan metabolism, cyanoamino acid metabolism, bile acid secretion and phenylalanine metabolism all undergoing increased coregulatory activity.
Fig. 2.
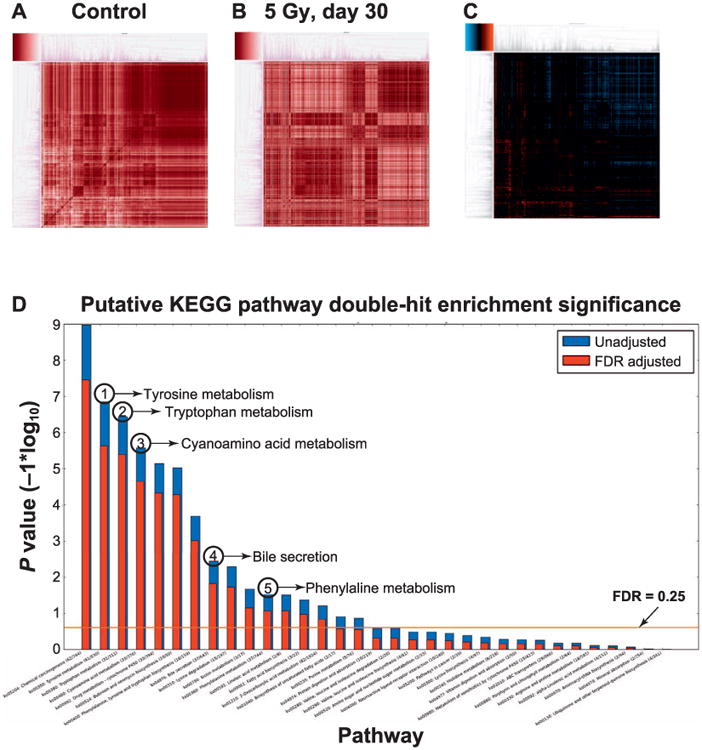
Differential correlation analysis showed shifts in the coregulation of several key pathways. Panel A: Dissimilarity heatmap generated from the Pearson's correlation coefficients calculated for the control group exhibiting a high degree of coregulation and coordination, as represented by the abundance of dark red hues. Panel B: Dissimilarity heatmap generated from the Pearson's correlation coefficients calculated for mice exposed to 5 Gy (30-day time point) showing a clear loss of correlation compared to panel A, as exhibited by the lighter red hues in the periphery of the top right corner where darker hues existed in the previous heatmap. There is also evidence of increased coregulation as a result of radiation exposure, as exhibited by the newly darkened areas in the bottom left corner when compared to panel A. These differences are emphasized in the differential correlation heatmap in panel C, where orange hues represent a gain of correlation (and thus increased coregulation), while blue hues represent a loss of correlation (i.e. dysregulation). Panel D: The results of “double hit” pathway enrichment analysis on this subset of correlation shifts. The y-axis is the −1*log10 (P value) for each KEGG pathway, as calculated by the uncorrected (blue bar) and FDR corrected (red bar) hypergeometric test. Several pathways are highlighted based on their statistical and biological significance including tyrosine metabolism, tryptophan metabolism, cyanoamino acid metabolism, bile acid secretion and phenylalanine metabolism all undergoing increased coregulatory activity.
Furthermore, we focused on identifying microbial metabolites with statistically significant changes in their fecal concentration at day 3 after 5 and 12 Gy irradiation (Fig. 3A). Glyceric acid, homogentisic acid, glutaconic acid and pipecolic acid showed decreasing levels with radiation dose, while hippuric acid, taurine and urobilinogen showed increasing levels with dose. This indicates radiation has a distinct effect on the fecal microbial metabolic products even as early as day 3 postirradiation. In addition, we assessed the changes in the fecal metabolome over the course of the 30-day study. Due to signs of morbidity, the 12 Gy irradiated mice were humanely euthanized and did not survive the 30-day study. Therefore, we followed the fecal metabolome of the 5 Gy irradiated mice from the earliest time point, day 3, through day 30, and found that their fecal metabolome at all three time points (day 3, 14 and 30) was distinct from that of control mice, as shown in the random forests-generated multidimensional scaling (MDS) plot in Fig. 3B. This plot also shows that the fecal metabolomic profiles of mice at day 14 and 30 after 5 Gy irradiation were more similar to each other, as they separate only slightly on the second dimension of the plot, than to that at day 3 separated on the first dimension. This may indicate an early response at day 3 due to irradiation and an adaptation of the response at the later time points. Furthermore, the heatmap shown in Fig. 3C details the changes in the abundances of the top 50 most important variables in a time-dependent manner as marked by yellow boxes. From these spectral features we identified serotonin and sebacic acid, which had a persistent decrease throughout the 30-day study after 5 Gy irradiation, while hypoxanthine and 2-ketobutyric acid showed persistent increases in their radiation response (Fig. 3D).
Fig. 3.
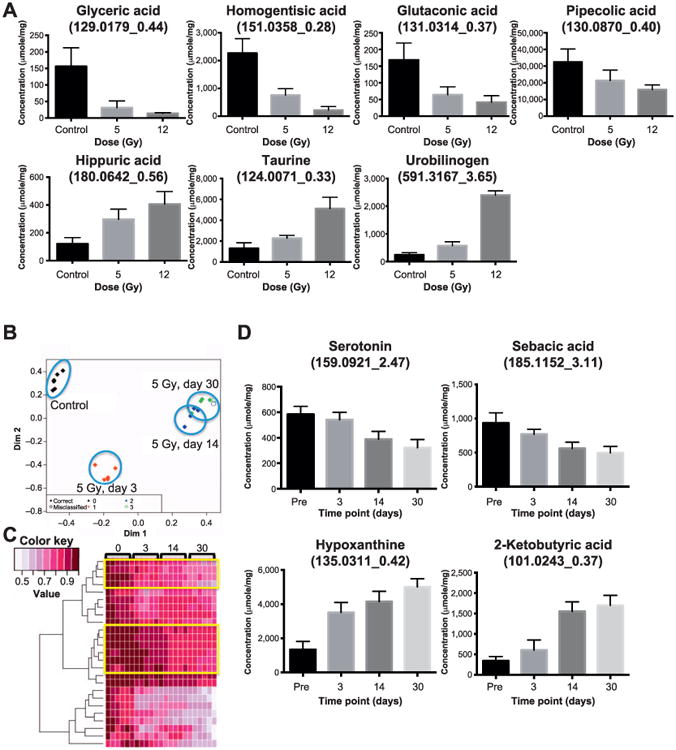
Panel A: Seven fecal metabolite markers known to be of bacterial origin show statistically significant changes in their abundances after irradiation. Glyceric acid, homogentisic acid, glutaconic acid and pipecolic acid show decreasing abundances after 5 and 12 Gy at day 3. This decrease is dose specific as the mice exposed to 12 Gy show greater decrease in the fecal abundance of these metabolites than those exposed to 5 Gy. Hippuric acid, taurin and urobilinogen show increase in their fecal abundances postirradiation in a dose specific pattern. Because these metabolites are products of the gut microbiota, significant changes in their abundances imply changes in the microbial metabolism and a shift toward gut dysbiosis. Panel B: Multidimensional scaling plot showing the separation of fecal metabolomic profile after 5 Gy irradiation throughout the 30 days. The fecal metabolomic signature of the irradiated mice are well separated from the preirradiation mice (control group). In addition, the metabolomic signatures of day 30 and 14 are closer while that of day 3 is further separated and closer to that of the control group. Panel C: The heatmap of top 50 important variables highlights the time dependence of the metabolomic response to 5 Gy irradiation. The two yellow boxes show individual spectral features with gradual decreasing abundances throughout the 30-day study while the features at the bottom of the heatmap show a rapid drop. Several ions from panel C are shown in panel D. Panel D: these individual microbial markers show time-specific responses to 5 Gy irradiation throughout the 30-day study. Sebacic acid and serotonin showed decreasing levels while 2-ketobutyric acid and hypoxanthine show increasing levels throughout the 30-day study at 5 Gy irradiation.
Among the fecal metabolites affected the most by exposure were three bile acids: taurocholic acid, 7-sulfocholic acid and 12-ketodeoxycholic acid, (Fig. 4). The taurine and the sulfur-conjugated primary bile acids, taurocholic acid and 7-sulfocholic acid showed increasing levels in their fecal abundances at day 3 after 5 and 12 Gy irradiation while the secondary bile acid, 12-ketodeoxycholic acid, showed decreased abundance. The increase in the levels of primary bile acids may indicate a reduction of their intestinal absorption and a lower rate of their deconjugation by the gut microbiota caused by radiation.
Fig. 4.

Three bile acids were detected to have statistically significant changing levels postirradiation. Secondary bile acid ketodeoxycholic acid shows decreasing levels postirradiation while the conjugated bile acids, taurocholic acid and sulfocholic acid, show increasing levels at day 3 postirradiation.
Fecal bacterial diversity and comparative community structure were determined by 16S rRNA sequencing assay followed by bioinformatics analysis using QIIME and Phyloseq packages. The samples had similar sequencing depth and library sizes regardless of treatment, and the range of the number of reads were consistent with other mouse fecal microbiome studies (26, 48). To assess the effects of radiation on gut bacterial community complexity, the filtered 16S rRNA sequencing results were subjected to alpha diversity measures, which estimated the number of types of bacteria in a single sample. Figure 5A shows three common measures; observed richness indicates the actual number of different taxa observed in a sample, chao1 index shows the predicted number of taxa in a sample by extrapolating the number of rare bacteria that may have been missed due to undersampling, and Shannon index combines the richness of a sample and the evenness of taxa in the sample. Both chao1 and Shannon measures indicated a lower diversity on day 3 after 5 Gy irradiation compared to preirradiation (5 Gy, day 0; i.e., the samples collected before irradiation), which recovers by the end of the 30-day study. The 12 Gy dose did not cause significant changes to the alpha diversity compared to preirradiation (12 Gy, day 0). As shown in Fig. 5B, the bacteria in mouse feces were dominated by three main phyla: Bacteroidetes, Firmicutes and Verrucomicrobia. Radiation also caused a decrease in the abundance of Firmicutes and an increase in the abundance of Verrucomicrobia, which recovered by day 30.
Fig. 5.
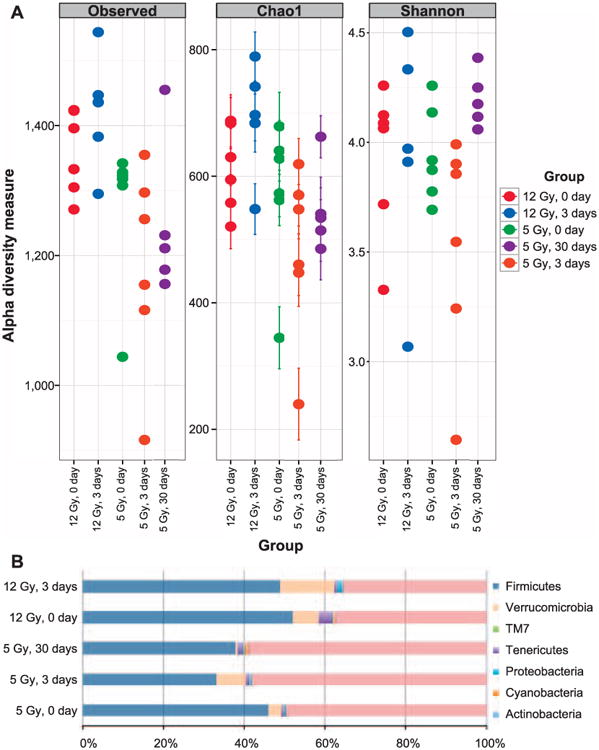
Microbiome diversity analysis of fecal samples of pre- and postirradiation. (panel A) Alpha diversity measures of fecal microbiome, observed richness, Chao1 measure and Shannon index are shown. In this plot each closed circle represents an individual mouse. The “0 day” denotes preirradiation (panel B). Proportion of OTUs present in mouse feces classified at the phylum level.
To determine if radiation caused shifts in the microbiome, paired differential analysis was performed on the OTUs' abundance from mice before and after irradiation. At day 3 after 5 and 12 Gy irradiation, 90 and 82 OTUs, respectively, were differentially abundant compared to day 1 before irradiation, and 12 of these OTUs were in common. At day 30 after 5 Gy irradiation, 91 OTUs had significantly different abundance compared to the preirradiation group. Furthermore, 24 of the differential OTUs were in common at day 3 and 30 postirradiation. The differential OTUs are listed in Supplementary Table S1 (http://dx.doi.org/10.1667/RR14306.1.S1). The common changes at the genus level observed in more than one condition were marked in blue for the ones with decreased abundances compared to preirradiation and in red for the increased abundances. We observed that both dose exposures resulted in a decline in the abundances of Lachnospiraceae and Ruminococcaceae. Decreased abundance of Clostridiaceae was also observed at day 3 postirradiation and it persisted until the end of the 30-day study. On the other hand, the abundance of members of the Lactobacillaceae, Staphylococcaceae, Bacteroidaceae families and two members of the Ruminococcaceae family increased after irradiation. There were a handful of differential OTUs belonging to the family of S24-7, but we could not determine their genus assignment.
Previously reported studies have shown that radiation causes bile salt malabsorption in the intestine due to terminal ileal damage (6, 7). The metabolomics results above also indicated altered levels of bile acids in feces after irradiation. It is of interest to see changes of gut bacteria with bile acid transforming activities under the same conditions. As shown in Fig. 6, the levels of species Ruminococcus gnavus (R. gnavus), an abundant commensal that converts deoxycholic acid (DCA) to less toxic isoDCA, declined significantly after irradiation. To display the intragroup variation, the abundance data of individual mouse samples for the species R. gnavus are shown in the Supplementary Fig. S1 (http://dx.doi.org/10.1667/RR14306.1.S3). While histological results did not show visible changes in intestinal tissues (data not shown) at 30 days postirradiation, the sensitive mass spectrometry approaches used here in addition to 16S rRNA sequencing highlighted the significant alterations in the metabolome and microbiome after exposure.
Fig. 6.
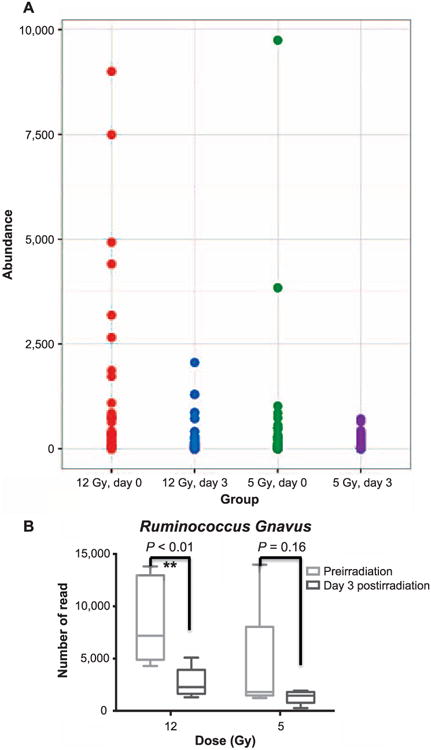
Panel A: The abundance of Lachnospiraceae (Ruminococcus) detected in the experimental groups. Panel B: The box and whiskers plot of the abundance of the species R. gnavus observed at day 3 post irrradiation (5 and 12 Gy). The data for individual samples are shown in Supplementary Fig. S1 (http://dx.doi.org/10.1667/RR14306.1.S3).
We investigated whether the abundance of intestinal microbes could be correlated with levels of the validated metabolites affected by radiation. For this we performed an inter-omic network analysis, which allowed for exploration of correlation patterns across the two large datasets to reveal possible biological interactions [in this case, between microbes and spectral features (Supplementary Table S2: http://dx.doi.org/10.1667/RR14306.1.S2)]. The networks were constructed from pairwise correlations between features of interest in each dataset (i.e., each combination of a microbe and ion). We constructed an inter-omic network from microbes and 20 validated metabolites with a statistically significant association with radiation exposure. Members of the S24-7 family had a central position in the resulting inter-omic network, with positive and negative correlations to the majority of the validated metabolites (Fig. 7). This suggests the importance of this poorly characterized phylogenetic group in the meta-metabolome of the mouse intestine. Metabolites correlated in most cases with multiple phylogenetic groups, highlighting the need to consider metabolic activity in the intestine as a community phenotype rather than as an attribute of single microbes. For instance, urobilinogen levels were positively correlated with Ruminococcaceae, Lachnospiraceae and Rikenellaceae. Other subnetworks were more complex such as that for serotonin, which was positively correlated with Ruminococcaceae and two Lachnospiraceae OTUs, but was negatively correlated with Lactobacillus and a separate Lachnospiraceae OTU. These correlations could reflect either direct metabolic activity of the microbes or their response to radiation exposure associated with the co-correlated metabolites. These metabolites may be produced by microbes or the epithelium, or may be derived from a combination of host mucosal and microbial metabolic activities.
Fig. 7.
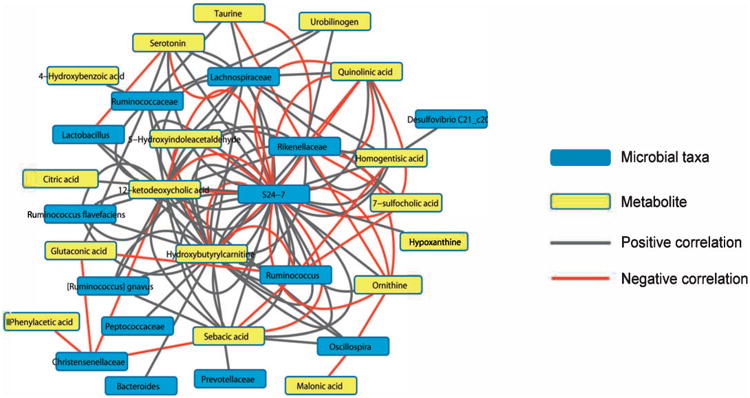
Inter-omic network between metabolites and microbes. The network was created from correlations between 20 validated metabolites and OTUs with differential abundance after irradiation that were identified at the family to species level. These correlations were based on the residuals of fitted models to account for radiation status. The metabolites are in yellow and the taxa in blue, with red lines showing negative correlation and gray showing positive correlation. Each line represents a correlation between a single OTU within the taxa and a validated metabolite.
Discussion
In this study, metabolomics and 16S rRNA sequencing were employed to assess the changes in the fecal metabolome and phylogenome in mice after irradiation. While 5 Gy irradiated mice did not show any alterations at day 30 in the crypt count and villi structure (data not shown) and survived the 30-day study, the 12 Gy irradiated mice experienced lethal GI toxicity and possibly early bacteremia within the first week of the experiment as shown in a previous study (49). Radiation is known to cause apoptotic or necrotic cell death in the GI tract (58, 59), which not only leads to discontinuity of the epithelial barrier and increased intestinal permeability, but also triggers inflammation. Recent clinical (53, 54) and preclinical (55) studies have also shown significant alterations of the intestinal micro-biome after radiotherapy. For instance, germ-free mice were found to be markedly more resistant to lethal radiation enteritis than the conventionally-housed animals (56). While this highlighted the importance of microbiota in radiation-induced intestinal pathogenesis, little information is available on the mechanism of action and the regulatory signaling through which microorganisms and host factors interact to maintain intestinal homeostasis (57). While a healthy gut harbors thousands of different species of microorganisms, it is dominated by three phyla: Bacteroidetes, Firmicutes and Verrumicrobia. Here we showed that radiation exposure caused remarkable changes at the phylum level, which are consistent with previous studies performed either on rodents after irradiation (43) or patients undergoing chemotherapy (44). Alterations of some genera such as Lachnospiraceae (Ruminococcus), Clostridium and Lactobacillus that were seen at day 3 postirradiation persisted until the later time points, suggesting that radiation exposure may lead to a prolonged change in the gut microbiome (Fig. 5). These alterations can be the consequence of radiation-induced disturbance of the intestinal environment including a shift in the levels of a variety of metabolites as revealed by the metabolomics investigations. It is important to note the intragroup variations observed in 5 and 12 Gy preirradiation samples (day 0) in Fig. 5A, where each dot represents a sample from an individual mouse. These intragroup variations did not affect the results of our analyses presented here, as we compared the sample from each mouse after irradiation to before irradiation (day 0) to discount the observed intragroup variation and the potential cage effects. Our investigations revealed statistically significantly increased coregulatory activity of several metabolic pathways, such as tyrosine metabolism, tryptophan metabolism, cyanoamino acid metabolism, bile acid secretion and phenylalanine metabolism, as shown in the differential metabolomic correlation-based “double hit” pathway analysis in Fig. 2D.
Gut microbiota have a role in metabolic activity and utility of amino acids, and thus regulate energy and lipid metabolism in the host. The amino acid-derived products of bacterial fermentation also have a role in regulating host immune responses and cell function. In addition, amino acids, particularly branched-chain amino acids such as alloisoleucine, provide a reliable food source for the gut bacteria, thus influencing their diversity and composition, metabolism and gut ecology (28). In this study, we observed statistically significant changes in the fecal levels of several amino acids postirradiation. For instance, while we observed a strong and persistent decrease in the levels of alloisoleucine and tyrosine, we observed an increase in the postirradiation levels of taurine and ornithine (Table 2 and Fig. 3). Taurine is synthesized in the pancreas via cysteine sulfinic acid pathway and is readily utilized as a substrate by the gut microbiota in the synthesis of hydrogen sulfide. The taurine degradation pathway is essential to maintain the beneficial colonic sulfate-reducing bacteria, Clostridia (52). An increase in the levels of colonic content of taurine may indicate a decrease in the rate of the taurine degradation pathway due to disturbances in the abundance and metabolism of Clostridia (Lachnospiraceae family) postir-radiation. Phylogenetic analysis further revealed a decrease in the abundance of Clostridia postirradiation (Supplementary Table S1; http://dx.doi.org/10.1667/RR14306.1.S1), which negatively correlates with the increase in the colonic content of the free substrate, taurine. Ornithine, on the other hand, is used by the colonic bacteria to form polyamines such as putrescine. We observed a slight increase in the fecal levels of ornithine after irradiation (Table 2), which may be due to the mucosal stress response after radiation-induced injury (41). Ornithine is decarboxylated by the bacterial ornithine decarboxylase to form polyamines, which play an important role in the growth and maturation of intestinal epithelium. The intestinal mucosa undergoes constant cell regeneration, which involves cell division maturation and loss into the lumen. This process is altered upon injury to the mucosa. Ornithine decarboxylase has been found at increasing levels during the cell maturation phase in the intestinal mucosa. However, after injury such as that induced by radiation, the activity of the enzyme is reversibly yet markedly reduced, leading to an increase in the substrate ornithine and a drop in the polyamine products. This in turn alters the distribution of epidermal growth factor receptor, modulates its signal transduction and decreases its association with the actin filaments in the IEC-6 cells, ultimately leading to reduced mucosal cell proliferation and migration (42). Thus, the slight increase in the fecal levels of ornithine observed in this study may be indicative of a decrease in the activity of this bacterial metabolic enzyme postirradiation.
Table 2. Additional Statistically Significant Fecal Metabolites Postirradiation.
| m/z_retention time | ESI mode | Adduct, error (ppm) | BioCyc ID | P value (K-S test, FDR corrected) | Log2 fold change | |
|---|---|---|---|---|---|---|
|
| ||||||
| 5 Gy | 12 Gy | |||||
| 153.0547_2.64 | Positive | [M + H]+, 0.47 | p-Hydroxyphenylacetic acida | 0.023 | –1.84 | –2.83 |
| 176.0704_2.72 | Positive | [M + H]+, 1.17 | 5-Hydroxyindoleacetaldehydea | 0.025 | 1.34 | 1.06 |
| 137.0589_1.08 | Positive | [M + H]+, 5.95 | Phenylacetic acida | 0.033 | 3.53 | 1.32 |
| 132.1026_0.62 | Positive | [M + H]+, 5.23 | L-alloisoleucine | 0.045 | –0.48 | –0.29 |
| 182.0812_1.12 | Positive | [M + H]+, 1.98 | Tyrosine | 0.036 | –0.54 | –0.75 |
| 131.0824_0.33 | Negative | [M – H]−, 1.56 | Ornithine | 0.00031 | 0.61 | 0.34 |
| 139.0052_0.38 | Negative | [M 1+ Cl]−, 14.50 | Malonic acida | 0.0014 | 0.41 | 0.66 |
| 166.0178_0.37 | Negative | [M – H]−, 12.56 | Quinolinic acid | 0.0022 | 0.75 | 2.21 |
| 227.0232_0.37 | Negative | [M + Cl]−, 13.16 | Citric acid | 0.0076 | 0.95 | 0.79 |
| 471.2418_4.82 | Negative | [M– H]−, 0.91 | Chenodeoxycholic acid sulfate | 0.041 | –0.95 | –1.51 |
Notes. ESI = electrospray ionization; ppm = parts per million; K-S = Kolmogorov-Smirnov; FDR = false discovery rate.
Putative identification.
Our metabolomics data also showed that radiation changes the fecal bile acid profile with increases in the taurine-conjugated bile acid, taurocholic acid and the sulfur-conjugated bile acid, sulfocholic acid and a decrease in the secondary bile acid, 12-ketodeoxycholic acid (Fig. 4). This is in accordance with a previously reported study in rats showing an increase in conjugated bile acids postirradiation as the result of reduced capacity of the microbiome to deconjugate and desulphate bile acids (29). In addition, a separate study concluded that bile acid levels increased as early as 24 h postirradiation and remained persistently high three days after exposure (7). Under physiological conditions, 90% of bile acids are reabsorbed by the gut by the highly efficient enterohepatic recirculation system (38), which may indicate that the radiation-induced bile acid increases in the colonic contents is due to the reduction of intestinal absorption caused by radiation. The decrease in the secondary bile acid, ketodeoxycholic acid, is further evidence for the shift in the microbial metabolism and lower rate of deconjugation of bile salts (Fig. 4). These bile acid shifts are likely due to the development of dysbiosis in the gut postirradiation, given the considerable evidence that bile acid signaling through farnesoid X receptor (FXR) and TGR5 is critical for maintaining the epithelial barrier, promoting antibacterial defenses and regulating hepatic bile acid synthesis (39, 40). Further microbiome analysis revealed altered abundance of a bacterial species, R. gnavus, which is capable of bile acid transformation. It has been reported that R. gnavus is a prominent species in producing isodeoxycholic acid, which has less toxic detergent activity than its precursor, deoxycholic acid (45). This conversion favors the growth of the abundant genus Bacteroides, a major genus involved in deconjugation of bile acids (46, 47). These connections highlight the complexity of radiation-induced perturbations in the intestinal environment. The reciprocal interplay between gut microbiome and metabolome reflects the physiological and functional changes of the intestine after irradiation. The secondary bile acid, deoxycholic acid, which is produced by the gut microbiota, has been shown to stimulate the hepatic stellate cells to secrete pro-inflammatory factors in a senescence-associated secretory pathway under stress conditions (29). There is also evidence that bile acids serve as host factors in reshaping the gut microbiota. An increase in bile acid secretion has been shown to alter the composition of microbiota in rats (30). Such results further point to the complexity of host/microbial interactions and regulatory roles in maintaining health and homeostasis.
It is well documented that the host-microbiota interaction plays an important role in host immune development and metabolism (31, 32). For example, a fecal metabolomics study showed that antibiotic treatment disrupts intestinal homeostasis and alters the microbial metabolic byproducts (33). The microbial metabolic byproducts are essential for microbial-host co-metabolism and may be gauged to assess changes in the microbial composition. The distinct fecal metabolomic profiles observed here after 5 (Fig. 1B) and 12 Gy (Supplementary Fig. S2; http://dx.doi.org/10.1667/RR14306.1.S3) irradiations in mice were attributed to statistically significant changes in the levels of several microbial metabolic byproducts with dose-specific responses, among which were glyceric acid, homogentisic acid, glutaconic acid and pipecolic acid (Fig. 3A). The decreasing levels of sugar alcohol glyceric acid in the exposed mice may be due to a decrease in the levels of Eubacterium biforme, which are responsible for the release of glyceric acid signaling a shift in the energy metabolism. Eubacterium biforme are present in the normal flora and are thought to benefit bionts, whose metabolic products are important in maintaining the normal ecology of the large intestine. In addition, homogentisic acid is an intermediate in the catabolism of aromatic amino acids and is consumed by the gut bacteria, particularly sulfur reducing Desulfovibrio, leading to the formation of methylhydroquinone. A decrease in the levels of homogentisic acid after irradiation directly correlates with the decrease in the abundance of Desulfovibrio (Fig. 7). While the metabolites displaying statistically significant changes in their fecal levels after irradiation could be of various biological sources (i.e., produced by microbes, host epithelium), Fig. 7 focuses on the connection and co-correlation of the fecal microbial compositing and its metabolome after irradiation. Among the most statistically significant and radiation-responsive microbial metabolic byproducts as well were hippuric acid and urobilinogen, which showed increasing levels with radiation dose (Fig. 3A). This may indicate a dose-dependent shift in the metabolism of specific bacterial phyla, which catabolize these metabolites. For instance, hippuric acid is a carboxylic acid produced by the conjugation of benzoic acid with glycine, a reaction that occurs not only in the liver (34), but also directly in the intestine (35) and kidney (36). Its excretion is modulated by the composition of the intestinal microbiome (37). Thus, an increase in the fecal levels of hippuric acid after irradiation may reflect alterations in the microbiome such as those promoting the overgrowth of the pathobionts. While in this study no direct conclusions can be drawn between radiation-induced changes in the microbiome and the host inflammatory responses without complete metagenomic and transcriptomic analyses, there is evidence that the gut commensal microbiome may regulate these responses through various byproducts such as hypoxanthine, which is a purine derivative with statistically significant increasing levels after irradiation (Fig. 3D). Hypoxanthine promotes formation of reactive oxygen species (51), which has been shown to be a defense mechanism of commensal microbiota under metabolic stress against opportunistic pathogens. Thus, the increase in the colonic content of hypoxanthine may indicate a regulatory response of the gut commensal microbiome under radiation-induced inflammatory conditions.
While each of the two omics platforms in this study are useful in elucidating the changes in the gut microbiome and metabolome after irradiation, the novelty of this study is in the integration of the data from these two platforms. We used specialized in-house bioinformatics tools to achieve this integration and define the community types that are robustly preserved among study groups as well as identifying metabolites that correlate with radiation-induced phylogenetic changes (Fig. 7). The two bacterial phyla with the highest number of correlations with the metabolome are Firmicutes and Bacteroidetes. The phyla are correlated with several validated and putative metabolites. The most prominent correlations highlighted by the interomic analysis were those among the amino acids and bile acids and the Lachnospiraceae, Desulfovibrio and Ruminococcus. Because the field of microbiomics is in its infancy and the microbial metabolite databases are not well established (only 100 microbial metabolites are available in HMDB database), we could not assign identities to many of these ions. Despite the emerging importance of the local and systemic role of gut microbiome in maintaining human health, physiological homeostasis, metabolism and in regulating stress and immunological signaling, the field of microbiomics remains underdeveloped. Considering the sheer number and the vast spectrum of species living in our gut alone, there is much to be learned about their functional potential and capacity. If identified and validated, prospective microbial communities and their metabolite markers for radiation exposure may create an opportunity for new biodosimetry measures. In addition, metagenomic data may help in identifying keystone species, which may be provided in the form of probiotics to a population affected by a nuclear/radiological event to protect against GI injury by replenishing the flora. Unfortunately, there is currently very little information available on the metabolic products of the gut microbiota and their systemic influence on the host phenotype and immunological responses to radiation. Therefore, more work is needed to unravel the extent of microbiome influence on the host and whether it can be manipulated to protect the host against radiation-induced injury. This notion of cultivating the gut micro-biome to exploit their beneficial effects in the host is particularly promising as the debate on the use of probiotics heats up.
Conclusion
In this study we focused on assessing the changes in the fecal metabolome of mice exposed to 5 and 12 Gy of external X-ray beam over the course of 30 days. We also took advantage of the advances in 16S rRNA sequencing to determine the changes in microbial diversity and richness postirradiation. The metabolomics data showed a persistent response from microbial byproducts including pipecolic acid, glutaconic acid, phenylpyruvate, homogentisic acid and secondary bile acids. The 16S rRNA sequencing results showed increased abundance of common members of Lactobacillaceae and Staphylococcaceae families, and decreased abundance of Lachnospiraceae, Ruminococcaceae and Clostridiaceae families after 5 and 12 Gy irradiation. We also observed changes in the abundance of R. gnavus, a species of gut bacteria involved in bile acid transformation. Furthermore, we took advantage of specialized bioinformatics tools to bridge the gap between these two omics platforms and integrate the two datasets. This resulted in a rich inter-omic network of bacteria such as those from the Firmicute and Bacteroidete phyla with strong correlation with the fecal metabolome. Although much work is needed to advance the field of microbiome and its contributions to the host metabolism and overall health, there is much potential in the implication of such studies in radiation-induced gut injury and biodosimetry.
Supplementary Material
Table S1. A list of differential OTUs from a paired differential analysis on the OTUs' abundance from mice pre- and postirradiation. Supplementary Table S1* includes a list of OTUs, whose abundances were found to be statistically significantly different (p < 0.05) in the paired analysis of pre- and postirradiation at the specified time/dose conditions. The file contains three sheets corresponding to various time/dose conditions, namely 3 days after 5 Gy irradiation (“3D after 5 Gy IR”). In each sheet, there are three major columns: OTU ID, Change and Taxa classification. OTU ID is the unique Greengenes ID. Change indicates the direction of the change in abundance, i.e. either decrease or increase of the OTU abundance after irradiation compared to the paired preirradiation controls. A decrease in the abundance of an OTU means that lower abundance of that particular OTU was found in the postirradiation group vs. the preirradiation controls. Taxa classification column contains the taxonomy retrieved from the Greengenes database. The common changes at the genus level observed in more than one condition were marked in blue for the ones with decreased abundances compared to before irradiation, and in red for the increased abundances.
Table S2. A list of additional highly correlated (Q < 0.05) microbe-metabolite pairs. This table contains a list of highly correlated (Q < 0.05) OTU-metabolite pairs. This table includes the OTU IDs, the metabolite IDs (p stands for Positive ESI and n stands for negative ESI, the m/z and the retention time), the Q value and the correlations (p stands for positive correlation and n stands for negative correlation). The Greengene IDs and the taxonomies (column S) are also included in this table. Column J summaries all the information in columns B through G in the following order: Greengene ID OTU ID; correlation (n for negative and p for positive); metabolite ID (p for positive ESI and n for negative ESI_m/z_retention time).
Fig. S1. The abundance data of individual sample for the species R. gnavus. These data are identical to those used in the box & whiskers plot in Fig. 5B. The x-axis lists the sample ID from each individual mouse (IDs are listed) and the y-axis represents the number of OTU reads (ranging from 0 to 15000). Each group (pre- and postirradiation) re designated below each figure. For consistency reasons the color coding is kept the same as that in Fig. 6A. Although there are intragroup variations in panel A for both pre- and postirradiation, the difference observed in each sample induced by radiation is consistent and statistically significantly decreasing (P < 0.01, as shown in Fig. 6B). Panel B shows a similar decreasing trend, however, the paired analysis did not result in a significant difference (P = 0.16, as shown in Fig. 6B).
Fig. S2. Perturbations in the fecal metabolomic signature in mice 3 days after 12 Gy irradiation assessed via MetaboLyzer. Panel A shows a volcano plot, where the statistically significant (Kolmogorov-Smirnov test, FDR < 0.10) spectral features are shown in red. The x-axis of the volcano plot, log2 fold change, highlights the direction of the shift (decrease or increase) in the abundance of the spectral features while the y-axis of the volcano plot, −log P value, specifies the significance of the shift. Panel B is a PCA scores plot and highlights the distinct separation between the overall fecal metabolomic profile of the control mice vs. those irradiated at 12 Gy at 3 days. Panel C is a heatmap of individual ions with most significant contribution to the separation of the fecal metabolomic signature of mice exposed to 12 Gy external beam irradiation compared to the control mice. The top two thirds of the heatmap shows spectral features with increasing abundances after 12 Gy irradiation, while those at the bottom one third of the heatmap show a decreasing pattern in their abundances postirradiation.
Acknowledgments
The authors would like to thank Georgetown University's Proteomic and Metabolomics Shared Resources (NIH grant no. P30 CA51008), for providing metabolomics support. This work was also supported by a research grant from the National Institute of Allergy and Infectious Diseases (NIAID grant no. U19 A1067773-09) and from the National Natural Science Foundation of China (grant no. 81372927).
Footnotes
Editor's note. The online version of this article (DOI: 10.1667/RR14306.1) contains supplementary information that is available to all authorized users.
References
- 1.Asselin C, Gendron FP. Shuttling of information between the mucosal and luminal environment drives intestinal homeostasis. FEBS Lett. 2014;588:4148–57. doi: 10.1016/j.febslet.2014.02.049. [DOI] [PubMed] [Google Scholar]
- 2.McLaughlin MM, Dacquisto MP, Jacobus DP, Horowitz RE. Effects of the germ free state on responses of mice to whole-body irradiation. Radiat Res. 1964;23:333–49. [PubMed] [Google Scholar]
- 3.Sartor RB. Microbial-host interactions in inflammatory bowel diseases and experimental colitis. Nestle Nutr Workshop Ser Pediatr Program. 2009;64:121–32. doi: 10.1159/000235787. [DOI] [PubMed] [Google Scholar]
- 4.Polistena A, Johnson LB, Ohiami-Masseron S, Wittgren L, Bäck S, Thornberg C, et al. Local radiotherapy of exposed murine small bowel: apoptosis and inflammation. BMC Surg. 2008;8:1. doi: 10.1186/1471-2482-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nejdfors P, Ekelund M, Westrom BR, Willen R, Jeppsson B. Intestinal permeability in humans is increased after radiation therapy. Dis Colon Rectum. 2000;43:1582–7. doi: 10.1007/BF02236743. [DOI] [PubMed] [Google Scholar]
- 6.Harris V, Benton B, Sohaib A, Dearnaley D, Andreyev HJ. Bile acid malabsorption after pelvic and prostate intensity modulated radiation therapy: an uncommon but treatable condition. Int J Radiat Oncol Biol Phys. 2012;84:e601–6. doi: 10.1016/j.ijrobp.2012.07.2368. [DOI] [PubMed] [Google Scholar]
- 7.Scanff P, Souidi M, Grison S, Griffiths NM, Gourmelon P. Alteration of the enterohepatic recirculation of bile acids in rats after exposure to ionizing radiation. Can J Physiol Pharmacol. 2004;82:114–24. doi: 10.1139/y03-131. [DOI] [PubMed] [Google Scholar]
- 8.Yeoh E, Horowitz M, Russo A, Muecke T, Robb T, Maddox A, et al. Effect of pelvic irradiation on gastrointestinal function: a prospective longitudinal study. Am J Med. 1993;95:397–406. doi: 10.1016/0002-9343(93)90309-d. [DOI] [PubMed] [Google Scholar]
- 9.Wedlake L, Thomas K, McGough C, Andreyev HJ. Small bowel bacterial overgrowth and lactose intolerance during radical pelvic radiotherapy: An observational study. Eur J Cancer. 2008;44:2212–7. doi: 10.1016/j.ejca.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 10.Classen J, Belka C, Paulsen F, Budach W, Hoffmann W, Bamberg M. Radiation-induced gastrointestinal toxicity. Pathophysiology, approaches to treatment and prophylaxis. Strahlenther Onkol. 1998;174(3):82–4. [PubMed] [Google Scholar]
- 11.Otterson MF, Sarna SK, Leming SC, Moulder JE, Fink JG. Effects of fractionated doses of ionizing radiation on colonic motor activity. Am J Physiol. 1992;263:G518–26. doi: 10.1152/ajpgi.1992.263.4.G518. [DOI] [PubMed] [Google Scholar]
- 12.Khanna S, Tosh PK. A clinician's primer on the role of the microbiome in human health and disease. Mayo Clin Proc. 2014;89:107–14. doi: 10.1016/j.mayocp.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 13.Ramakrishna BS. Role of the gut microbiota in human nutrition and metabolism. J Gastroenterol Hepatol. 2013;28(4):9–17. doi: 10.1111/jgh.12294. [DOI] [PubMed] [Google Scholar]
- 14.Stecher B. The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. Microbiol Spectr. 2015;3:1–2. doi: 10.1128/microbiolspec.MBP-0008-2014. [DOI] [PubMed] [Google Scholar]
- 15.Bures J, Cyrany J, Kohoutova D, Förstl M, Rejchrt S, Kvetina J, et al. Small intestinal bacterial overgrowth syndrome. World J Gastroenterol. 2010;16:2978–90. doi: 10.3748/wjg.v16.i24.2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husebye E, Skar V, Hoverstad T, Iversen T, Melby K. Abnormal intestinal motor patterns explain enteric colonization with gram-negative bacilli in late radiation enteropathy. Gastroenterology. 1995;109:1078–89. doi: 10.1016/0016-5085(95)90565-0. [DOI] [PubMed] [Google Scholar]
- 17.Manichanh C, Varela E, Martinez C, Antolin M, Llopis M, Doré J, et al. The gut microbiota predispose to the pathophysiology of acute postradiotherapy diarrhea. Am J Gastroenterol. 2008;103:1754–61. doi: 10.1111/j.1572-0241.2008.01868.x. [DOI] [PubMed] [Google Scholar]
- 18.Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, et al. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 2010;3:148–158. doi: 10.1038/mi.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–6. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mak TD, Laiakis EC, Goudarzi M, Fornace AJ., Jr MetaboLyzer: a novel statistical workflow for analyzing Postprocessed LC-MS metabolomics data. Anal Chem. 2014;86:506–513. doi: 10.1021/ac402477z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goudarzi M, Weber WM, Mak TD, Chung J, Doyle-Eisele M, Melo DR, et al. Metabolomic and lipidomic analysis of serum from mice exposed to an internal emitter, Cesium-137, using a shotgun LC-MS approach. J Proteome Res. 2014;14:374–84. doi: 10.1021/pr500913n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550–7. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dazard JE, Rao JS. Joint adaptive mean-variance regularization and variance stabilization of high dimensional data. Comput Stat Data Anal. 2012;56:2317–33. doi: 10.1016/j.csda.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 26.Claesson MJ, O'Sullivan O, Wang Q, Nikkilä J, Marchesi JR, Smidt H, et al. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One. 2009;4:e6669. doi: 10.1371/journal.pone.0006669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McHardy IH, Goudarzi M, Tong M, Ruegger PM, Schwager E, Weger JR, et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome. 2013;1:17. doi: 10.1186/2049-2618-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai ZL, Wu G, Zhu WY. Amino acid metabolism in intestinal bacteria: links between gut ecology and host health. Front Biosci (Landmark Ed) 2011;16:1768–86. doi: 10.2741/3820. [DOI] [PubMed] [Google Scholar]
- 29.Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30:332–8. doi: 10.1097/MOG.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yokota A, Fukiya S, Islam KB, Ooka T, Ogura Y, Hayashi T, et al. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes. 2012;3:455–9. doi: 10.4161/gmic.21216. [DOI] [PubMed] [Google Scholar]
- 31.Cerf-Bensussan N, Gaboriau-Routhiau V. The immune system and the gut microbiota: friends or foes? Nat Rev Immunol. 2010;10:735–44. doi: 10.1038/nri2850. [DOI] [PubMed] [Google Scholar]
- 32.Moossavi S, Zhang H, Sun J, Rezaei N. Host-microbiota interaction and intestinal stem cells in chronic inflammation and colorectal cancer. Expert Rev Clin Immunol. 2013;9:409–22. doi: 10.1586/eci.13.27. [DOI] [PubMed] [Google Scholar]
- 33.Antunes LC, Han J, Ferreira RB, Lolic P, Borchers CH, Finlay BB. Effect of antibiotic treatment on the intestinal metabolome. Antimicrob Agents Chemother. 2011;55:1494–1503. doi: 10.1128/AAC.01664-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiba M, Poon K, Hollands J, Pang KS. Glycine conjugation activity of benzoic acid and its acinar localization in the perfused rat liver. J Pharmacol Exp Ther. 1994;268:409–16. [PubMed] [Google Scholar]
- 35.Strahl NR, Barr WH. Intestinal drug absorption and metabolism. 3. Glycine conjugation and accumulation of benzoic acid in rat intestinal tissue. J Pharm Sci. 1971;60:278–81. doi: 10.1002/jps.2600600227. [DOI] [PubMed] [Google Scholar]
- 36.Poon K, Pang KS. Benzoic acid glycine conjugation in the isolated perfused rat kidney. Drug Metab Dispos. 1995;23:255–60. [PubMed] [Google Scholar]
- 37.Williams HR, Cox IJ, Walker DG, Cobbold JF, Taylor-Robinson SD, Marshall SE, et al. Differences in gut microbial metabolism are responsible for reduced hippurate synthesis in Crohn's disease. BMC Gastroenterol. 2010;10:108. doi: 10.1186/1471-230X-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carulli N, Bertolotti M, Carubbi F, Concari M, Martella P, Carulli L, et al. Review article: effect of bile salt pool composition on hepatic and biliary functions. Aliment Pharmacol Ther. 2000;14(2):14–18. doi: 10.1046/j.1365-2036.2000.014s2014.x. [DOI] [PubMed] [Google Scholar]
- 39.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920–25. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS One. 2011;6:e25637. doi: 10.1371/journal.pone.0025637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lux GD, Marton LJ, Baylin SB. Ornithine decarboxylase is important in intestinal mucosal maturation and recovery from injury in rats. Science. 1980;210:195–8. doi: 10.1126/science.6774420. [DOI] [PubMed] [Google Scholar]
- 42.McCormack SA, Blanner PM, Zimmerman BJ, Ray R, Poppleton HM, Patel TB, et al. Polyamine deficiency alters EGF receptor distribution and signaling effectiveness in IEC-6 cells. Am J Physiol. 1998;274:C192–205. doi: 10.1152/ajpcell.1998.274.1.C192. [DOI] [PubMed] [Google Scholar]
- 43.Lam V, Moulder JE, Salzman NH, Dubinsky EA, Andersen GL, Baker JE. Intestinal microbiota as novel biomarkers of prior radiation exposure. Radiat Res. 2012;177:573–83. doi: 10.1667/rr2691.1. [DOI] [PubMed] [Google Scholar]
- 44.Montassier E, Batard E, Massart S, Gastinne T, Carton T, Caillon J, et al. 16S rRNA gene pyrosequencing reveals shift in patient faecal microbiota during high-dose chemotherapy as conditioning regimen for bone marrow transplantation. Microb Ecol. 2014;67:690–9. doi: 10.1007/s00248-013-0355-4. [DOI] [PubMed] [Google Scholar]
- 45.Devlin AS, Fischbach MA. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol. 2015;11:685–90. doi: 10.1038/nchembio.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerard P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens. 2013;3:14–24. doi: 10.3390/pathogens3010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narushima S, Itoha K, Miyamoto Y, Park SH, Nagata K, Kuruma K, et al. Deoxycholic acid formation in gnobiotic mice associated with human intestinal bacteria. Lipids. 2006;41:835–43. doi: 10.1007/s11745-006-5038-1. [DOI] [PubMed] [Google Scholar]
- 48.Frank DN, Bales ES, Monks J, Jackman MJ, MacLean PS, Ir D, et al. Perilipin-2 modulated lipid adsorption and microbiome responses in the mouse intestine. PLoS One. 2015;10:e0131944. doi: 10.1371/journal.pone.0131944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Datta K, Suman S, Trani D, Doiron K, Rotolo JA, Kallakury BV, et al. Accelerated hematopoietic toxicity by high energy (56)Fe radiation. Int J Radiat Biol. 2012;88:213–22. doi: 10.3109/09553002.2012.639434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2012;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-shehri SS, Knox CL, Liley HG, Cowley DM, Wright JR, Henman MG, et al. Breastmilk-saliva interactions boost innate immunity by regulating the oral microbiome in early infancy. PLoS One. 2015;10:e0135047. doi: 10.1371/journal.pone.0135047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carbonero F, Benefiel AC, Alizadeh-Ghamsari AH, Gaskins HR. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front Physiol. 2012;3:448. doi: 10.3389/fphys.2012.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manichanh C, Varela E, Martinez C, Antolin M, LIopis M, Dore J, et al. The gut microbiota predispose to the pathophysiology of acute postradiotherapy diarrhea. Am J Gasteroenterol. 2008;103:1754–61. doi: 10.1111/j.1572-0241.2008.01868.x. [DOI] [PubMed] [Google Scholar]
- 54.Nam YD, Kim HJ, Seo JG, Kang SW, Bae JW. Impact of pelvic radiotherapy on gut microbiota of gynecological cancer patients revealed by massive pyrosequencing. PLoS One. 2013;8:e82659. doi: 10.1371/journal.pone.0082659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson LB, Riaz AA, Adawi D, Wittgren L, Back S, Thornberg C, et al. Radiation enteropathy and leucocyte-endothelial cell reactions in a refined small bowel model. BMC Surgery. 2004;13:4–10. doi: 10.1186/1471-2482-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crawford PA, Gordon JI. Microbial regulation of intestinal radiosensitivity. Proc Natl Acad Sci U S A. 2005;102:13254–9. doi: 10.1073/pnas.0504830102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Touchefeu Y, Montassier E, Nieman K, Gastinne T, Potel G, Bruley des Varannes S, et al. Systematic review: the role of the gut microbiota in chemotherapy- or radiation-induced gastrointestinal mucostitis- current evidence and potential clinical applications. Aliment Pharmacol Ther. 2014;40:409–21. doi: 10.1111/apt.12878. [DOI] [PubMed] [Google Scholar]
- 58.Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, et al. Ionizing radiation acts on cellular membranes to generate ceramide and initaite apoptosis. J Exp Med. 1994;180:525–35. doi: 10.1084/jem.180.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kolesnick RN, Haimovitz-Friedman A, Fuks Z. The sphingomyelin signal transduction pathway mediates apoptosis for tumor necrosis factor, Fas, and ionizing radiation. Biochem Cell Biol. 1994;72:471–4. doi: 10.1139/o94-063. [DOI] [PubMed] [Google Scholar]
- 60.Jarvis WD, Kolesnick RN, Fornari FA, Traylor RS, Gewirtz DA, Grant S. Induction of apoptotic DNA damage and cell death by activation of the sphingomyeline pathway. Proc Natl Acad Sci U S A. 1994;91:73–7. doi: 10.1073/pnas.91.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dong X, Yambartsev A, Ramsey SA, Thomas LD, Shulzhenko N, Morgun A. Reverse enGENEering of regulatory networks from big data: a roadmap for biologists. Bioinform Biol Insights. 2015;9:61–74. doi: 10.4137/BBI.S12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. A list of differential OTUs from a paired differential analysis on the OTUs' abundance from mice pre- and postirradiation. Supplementary Table S1* includes a list of OTUs, whose abundances were found to be statistically significantly different (p < 0.05) in the paired analysis of pre- and postirradiation at the specified time/dose conditions. The file contains three sheets corresponding to various time/dose conditions, namely 3 days after 5 Gy irradiation (“3D after 5 Gy IR”). In each sheet, there are three major columns: OTU ID, Change and Taxa classification. OTU ID is the unique Greengenes ID. Change indicates the direction of the change in abundance, i.e. either decrease or increase of the OTU abundance after irradiation compared to the paired preirradiation controls. A decrease in the abundance of an OTU means that lower abundance of that particular OTU was found in the postirradiation group vs. the preirradiation controls. Taxa classification column contains the taxonomy retrieved from the Greengenes database. The common changes at the genus level observed in more than one condition were marked in blue for the ones with decreased abundances compared to before irradiation, and in red for the increased abundances.
Table S2. A list of additional highly correlated (Q < 0.05) microbe-metabolite pairs. This table contains a list of highly correlated (Q < 0.05) OTU-metabolite pairs. This table includes the OTU IDs, the metabolite IDs (p stands for Positive ESI and n stands for negative ESI, the m/z and the retention time), the Q value and the correlations (p stands for positive correlation and n stands for negative correlation). The Greengene IDs and the taxonomies (column S) are also included in this table. Column J summaries all the information in columns B through G in the following order: Greengene ID OTU ID; correlation (n for negative and p for positive); metabolite ID (p for positive ESI and n for negative ESI_m/z_retention time).
Fig. S1. The abundance data of individual sample for the species R. gnavus. These data are identical to those used in the box & whiskers plot in Fig. 5B. The x-axis lists the sample ID from each individual mouse (IDs are listed) and the y-axis represents the number of OTU reads (ranging from 0 to 15000). Each group (pre- and postirradiation) re designated below each figure. For consistency reasons the color coding is kept the same as that in Fig. 6A. Although there are intragroup variations in panel A for both pre- and postirradiation, the difference observed in each sample induced by radiation is consistent and statistically significantly decreasing (P < 0.01, as shown in Fig. 6B). Panel B shows a similar decreasing trend, however, the paired analysis did not result in a significant difference (P = 0.16, as shown in Fig. 6B).
Fig. S2. Perturbations in the fecal metabolomic signature in mice 3 days after 12 Gy irradiation assessed via MetaboLyzer. Panel A shows a volcano plot, where the statistically significant (Kolmogorov-Smirnov test, FDR < 0.10) spectral features are shown in red. The x-axis of the volcano plot, log2 fold change, highlights the direction of the shift (decrease or increase) in the abundance of the spectral features while the y-axis of the volcano plot, −log P value, specifies the significance of the shift. Panel B is a PCA scores plot and highlights the distinct separation between the overall fecal metabolomic profile of the control mice vs. those irradiated at 12 Gy at 3 days. Panel C is a heatmap of individual ions with most significant contribution to the separation of the fecal metabolomic signature of mice exposed to 12 Gy external beam irradiation compared to the control mice. The top two thirds of the heatmap shows spectral features with increasing abundances after 12 Gy irradiation, while those at the bottom one third of the heatmap show a decreasing pattern in their abundances postirradiation.