

Abstract
Nonsense suppression therapy encompasses approaches aimed at suppressing translation termination at in-frame premature termination codons (PTCs, also known as nonsense mutations) to restore deficient protein function. In this review, we examine the current status of PTC suppression as a therapy for genetic diseases caused by nonsense mutations. We discuss what is currently known about the mechanism of PTC suppression as well as therapeutic approaches under development to suppress PTCs. The approaches considered include readthrough drugs, suppressor tRNAs, PTC pseudouridylation, and inhibition of nonsense-mediated mRNA decay. We also discuss the barriers that currently limit the clinical application of nonsense suppression therapy and suggest how some of these difficulties may be overcome. Finally, we consider how PTC suppression may play a role in the clinical treatment of genetic diseases caused by nonsense mutations.
Keywords: nonsense suppression therapy, premature termination codons, nonsense mutation, readthrough
INTRODUCTION
The general public recognizes the names of a relatively small number of genetic diseases, such as cystic fibrosis (CF) and Duchenne muscular dystrophy (DMD). Because of their prevalence, significant amounts of energy and resources have been devoted to finding treatments (and cures) for these diseases. However, according to the National Institutes of Health Office of Rare Diseases Research (http://rarediseases.info.nih.gov) and the National Organization for Rare Disorders (http://www.rarediseases.org), more than 7,000 distinct genetic diseases are present in the human population. The great majority are termed rare diseases because they affect fewer than 200,000 Americans, and the pharmaceutical industry frequently considers these to not be economically viable targets for drug development. This is unfortunate, because in total these maladies affect roughly 1 in 10 Americans. This translates to ∼30 million people in the United States, and ∼300 million people worldwide.
Until very recently, the specific defective genes associated with many of these diseases were unknown. However, the advent of next-generation sequencing has led to a revolution in the large-scale identification of mutations in both known and novel disease genes. A recent study used the Human Gene Mutation Database (http://www.hgmd.org) to carry out a meta-analysis of more than 7,500 nonsense mutations in 995 different genes causing inherited diseases. The results revealed that nonsense mutations [defined as single nucleotide base changes within a gene that result in an in-frame premature termination codon (PTC)] account for ∼11% of all described gene lesions that cause inherited human diseases (90). This high incidence of nonsense mutations in inherited diseases suggests that therapeutic strategies aimed at suppressing nonsense mutations (so-called nonsense suppression therapies) have the potential to provide a therapeutic benefit for a genotypic subset of patients with a broad range of genetic diseases.
RELEVANT FEATURES OF TRANSLATION ELONGATION AND TERMINATION
Eukaryotic translation can be divided into four stages: initiation, elongation, termination, and recycling. During both the elongation and termination stages of translation, a competition for codon binding occurs between aminoacyl-tRNAs and termination factors. Nonsense suppression takes place when an amino acid carried by an aminoacyl-tRNA is incorporated into the nascent polypeptide at a PTC. This mechanism suppresses translation termination at the PTC, which allows continued translation elongation in the proper reading frame and the generation of a full-length polypeptide. The amino acid inserted during nonsense suppression may not be the one normally encoded. However, as long as the substituted amino acid does not carry out an essential function (for example, as a critical active site residue), the resulting protein may have normal (or at least partial) activity. Furthermore, the amount of function restored will be proportional to the level of PTC suppression obtained. In this section, we describe the basic details of elongation and termination and the features that make these processes susceptible to drugs that stimulate nonsense suppression.
Fidelity During Eukaryotic Translation Elongation
Much of our knowledge of the mechanistic details of translational fidelity is based on prokaryotic systems, as recently reviewed (146). Given the high overall conservation of the translation process, it is assumed that the key features of fidelity are also conserved in eukaryotes, although direct evidence of many aspects of this remains lacking. Translation elongation represents the ordered addition of amino acids to the carboxy-terminal end of the growing polypeptide chain by ribosomes. The string of mRNA codons (read in the 5′→ 3′ direction) determines the order in which amino acids are added to form the primary amino acid sequence of each protein. Following each successive translocation step, the growing polypeptide chain on the peptidyl-tRNA rests in the ribosomal P site (Figure 1), while the codon located in the ribosomal A site awaits recognition by the next aminoacyl-tRNA.
Figure 1.
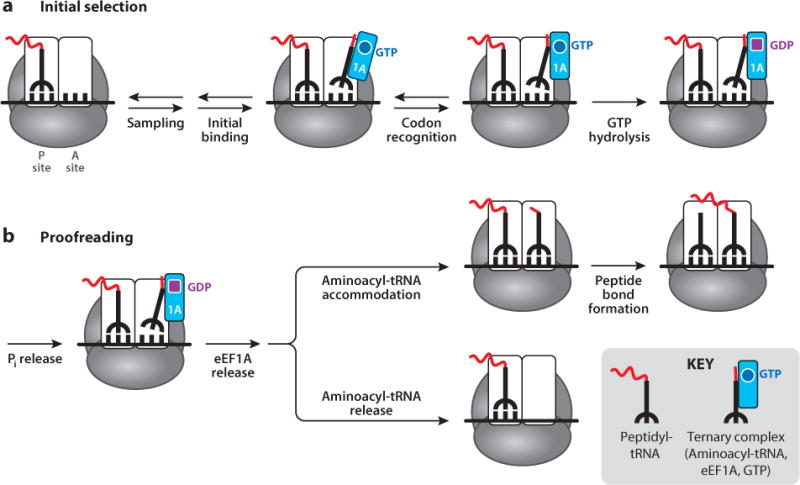
Steps in aminoacyl-tRNA discrimination during eukaryotic translation elongation: (a) initial selection and (b) proofreading.
Because the codon in the A site could represent any of the 64 candidates of the genetic code, a process of aminoacyl-tRNA (and release factor) sampling occurs during the recognition of each successive codon. Various aminoacyl-tRNAs successively enter the A site as ternary complexes with the translation elongation factor eEF1A and GTP until a cognate tRNA (carrying an anticodon with a correct match for the codon) is selected (Figure 1). This process is monitored at two distinct steps separated by the irreversible step of GTP hydrolysis by eEF1A, thus ensuring that aminoacyl-tRNA selection is achieved with a high level of accuracy (146). First, the initial selection step mediates rejection of noncognate aminoacyl-tRNAs with multiple mismatched base pairs in the codon–anticodon interaction prior to GTP hydrolysis by eEF1A. Initially (Figure 1a), structural fluctuations of the tRNA scan the codon through an interaction with the decoding center (the network of rRNA and proteins in the lower portion of the ribosomal A site that monitors proper codon–anticodon interactions). If the initial structural fluctuations of the tRNA reveal that the codon is not a proper match, the ternary complex dissociates as a unit and aminoacyl-tRNA sampling continues. If correct codon–anticodon interactions are achieved, the tRNA will initially reside in a hybrid (A/T) configuration, in which the anticodon is properly positioned in the decoding center of the A site but the aminoacyl end has not yet moved into the peptidyl transferase center. Attainment of the proper codon–anticodon base pairing allows a series of conformational changes to occur in the decoding center that establishes a complex network of hydrogen-bonding interactions between the rRNA and tRNA. Ultimately, selection of the correct aminoacyl-tRNA leads to domain closure of the small ribosomal subunit and activation of GTP hydrolysis by eEF1A (Figure 1a).
In a second step, near-cognate aminoacyl-tRNAs with only a single mismatch in the codon–anticodon interaction are rejected in the proofreading stage that follows GTP hydrolysis (Figure 1b). Phosphate release induces a major conformational change in eEF1A, which disrupts its tRNA binding site and facilitates its release from the ternary complex. eEF1A release leads to rapid accommodation of the aminoacyl end of the aminoacyl-tRNA into the peptidyl transferase center of the 60S ribosomal subunit, allowing peptide bond formation to occur. If the codon–anticodon interaction is not cognate, the aminoacyl-tRNA will dissociate from the A site along with the eEF1A following GTP hydrolysis. The differences in binding affinities between the correct and incorrect aminoacyl-tRNAs during the proofreading stage are based on the binding constants of each species and result in the discrimination of cognate from near-cognate aminoacyl-tRNAs by a kinetic proofreading mechanism. Dissociation of an incorrect aminoacyl-tRNA results in a resumption of tRNA sampling, and the entire cycle is repeated until a ternary complex with the correct aminoacyl-tRNA is selected. Once peptide bond formation is achieved, translocation occurs, and the entire process is then repeated when the next codon enters the ribosomal A site.
Eukaryotic Translation Termination
Two translation termination factors are required to mediate eukaryotic translation termination. eRF1 directly recognizes any of the three stop codons (UAA, UAG, and UGA). Notably, codon recognition during termination is the only step in translation where a protein factor—rather than a nucleic acid adaptor (the tRNA)—serves as the adaptor that decodes a codon. The second termination factor, eRF3, is a GTPase that binds eRF1 and assists in the termination process. When a stop codon enters the ribosomal A site to form the pretermination complex (Figure 2), the process of sampling to identify the appropriate binding partner again occurs. This includes both aminoacyl-tRNAs (in a ternary complex with eEF1A and GTP) and the release factor eRF1 (in a ternary complex with eRF3 and GTP). Upon initial stop codon recognition by eRF1, GTP hydrolysis by eRF3 induces conformational changes in eRF1 that finalize stop codon recognition (114). Accommodation of domain 2 of eRF1 subsequently positions its GGQ motif into the peptidyl transferase center of the ribosome (2). This allows eRF1 to stimulate hydrolysis of the ester bond of the peptidyl-tRNA, thus facilitating the release of the completed polypeptide chain.
Figure 2.
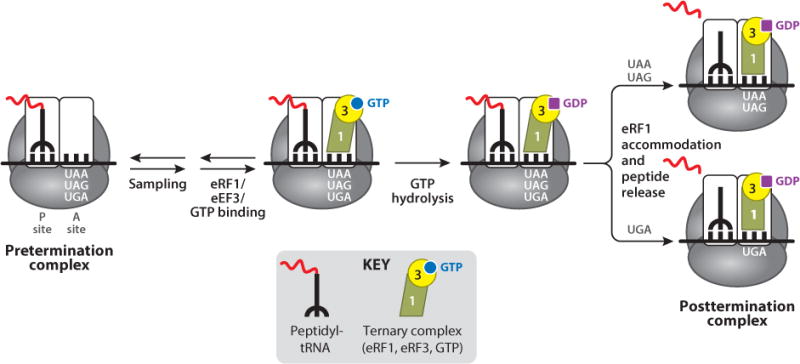
Mechanism of eukaryotic translation termination.
The molecular details of stop codon recognition by eRF1 remain poorly understood. However, domain-swap experiments using eRF1 clones from variant-code organisms that recognize only a subset of stop codons clearly indicate that domain 1 of eRF1 is sufficient to mediate stop codon recognition (23, 116). Genetic evidence also suggests that alternative eRF1 conformational states may mediate whether polypeptide release occurs at the UAA/UAG stop codons or at the UGA codon (Figure 2). It has been suggested that this latter recognition event further discriminates between the UGA stop codon and the UGG tryptophan codon (40).
The Efficiency of Termination Differs Between Normal Stop Codons and Premature Termination Codons
Both the structure of the ribosomal decoding center and the process of aminoacyl-tRNA selection are generally conserved between prokaryotes and eukaryotes. This has allowed investigators to use high-resolution X-ray structures of prokaryotic ribosomal complexes to inform our knowledge of translational fidelity in eukaryotes. In contrast, the molecular details of translation termination are much less conserved, which has limited our understanding of this process in eukaryotes. However, we know that the process of sampling occurs at stop codons by both eRF1 and tRNAs. Accordingly, stop codon recognition by eRF1 occasionally can be superseded by a near-cognate aminoacyl-tRNA, resulting in stop codon suppression (also referred to as readthrough).
The basal level of termination suppression at naturally occurring stop codons occurs at a frequency of <0.1% (84, 131). In contrast, the basal level of termination suppression at PTCs occurs at higher frequencies (<1%) (19, 82). This difference may be due to the higher-order structure of translating mRNAs and protein–protein interactions that occur within the messenger ribonucleoprotein (mRNP) complex (Figure 3). The association of three proteins in the mRNP complex leads to a circular mRNA structure known as the closed-loop complex. First, the capbinding protein (eIF4E) is bound to the 5′-cap structure of the mRNA. Second, poly(A)-binding protein (PABP) is bound to the mRNA poly(A) tail. A third protein, eIF4G, simultaneously binds both eIF4E and PABP, resulting in circularization of the mRNP complex and formation of the closed-loop complex (55, 62, 142).
Figure 3.
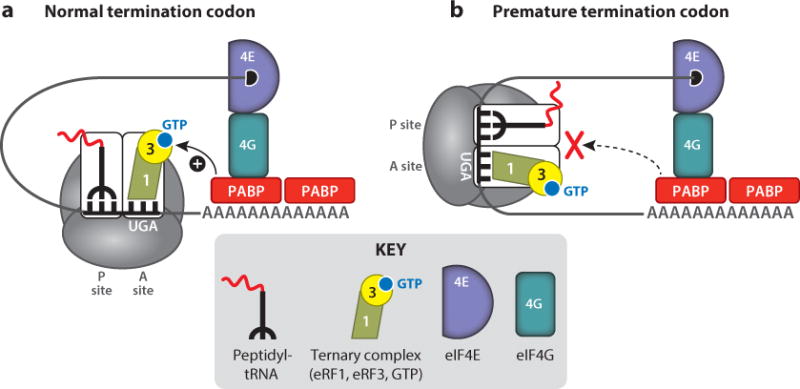
(a) Normal and (b) premature termination codons. The messenger ribonucleoprotein (mRNP) structure leads to a higher basal level of termination suppression (readthrough) at the latter compared with the former.
The closed-loop configuration serves two important functions. First, it protects the ends of the mRNA from exonucleolytic degradation by both the 5′→ 3′ and 3′→ 5′ RNA decay pathways (24). In addition, it enhances translation of the mRNA. Because the closed loop brings the ends of the mRNA together in close proximity, this structure is thought to enhance recycling of translational components from the posttermination complex back to the 5′ end of the mRNA following polypeptide chain release, thus increasing the frequency of translation initiation. In addition to its role in the formation of the closed loop, PABP plays an important role in translation termination. Because it is bound to the poly(A) tail, PABP is normally close to the termination complex at normal stop codons, allowing it to interact with eRF3 during translation termination and stimulate polypeptide chain release (27). In contrast, termination at a PTC usually does not occur in proximity to the poly(A) tail, which limits the interaction between eRF3 and PABP and leads to prolonged ribosomal pausing at PTCs (4). This has important implications for nonsense suppression, because it increases the extent of aminoacyl-tRNA sampling and makes PTCs more susceptible to readthrough. The increased level of pausing at PTCs and their enhanced susceptibility to readthrough are exacerbated by readthrough drugs, as described in more detail in the following sections.
CURRENT STATUS OF NONSENSE SUPPRESSION THERAPY
PTC suppression was first described in 1996 as a potential therapy for diseases caused by nonsense mutations in the CFTR gene (mutations in which result in the common genetic disease CF) (56). That study demonstrated that the aminoglycoside G418 suppresses nonsense mutations in the CFTR gene and restores significant levels of both CFTR protein and function in cultured cells. Since that initial report, approximately 100 studies have investigated the effectiveness of nonsense suppression as a possible treatment for nearly 40 different diseases (76). Below, we describe the status of investigations that have been conducted with current PTC suppression drugs using primarily in vivo models.
Aminoglycosides
Aminoglycosides are a class of antibiotic consisting of a 2-deoxystreptamine ring linked to multiple amino sugars. These compounds bind to the ribosomal decoding center (108). As described above, the decoding center carries out a proofreading function that monitors codon–anticodon interactions to ensure that only cognate aminoacyl-tRNAs are correctly accommodated into the peptidyl transferase center where peptide bond formation can occur. Aminoglycosides bind strongly to the bacterial decoding center, which leads to misincorporation of near-cognate aminoacyl-tRNAs at both sense and stop codons, resulting in extensive translational misreading (and inhibition of protein synthesis at higher concentrations). Differences in the eukaryotic ribosomal RNA sequence at two key nucleotides significantly reduce the affinity of aminoglycosides for the eukaryotic decoding center (78, 108). The preferential binding of aminoglycosides to the prokaryotic ribosome permits these compounds to be safely used as antibiotics without inhibiting eukaryotic translation. Although aminoglycosides do not appear to induce significant misreading at sense codons in eukaryotes (73, 115), a subset of them have been shown to bind to eukaryotic ribosomes (79, 120) and lead to misincorporation of near-cognate aminoacyl-tRNAs at PTCs (97, 98, 143).
Dozens of in vitro studies have reported that certain aminoglycosides suppress translation termination at PTCs within numerous diverse mRNAs and restore physiologically relevant levels of functional protein in mammalian cells (for a review, see 76). Here, we focus on studies that have investigated the ability of aminoglycosides to suppress PTCs and restore protein function in vivo. Gentamicin, the aminoglycoside most commonly used for nonsense suppression studies, has been shown to restore functional protein in short-term studies using mouse models of DMD (10), CF (33), nephrogenic diabetes insipidus (118), hemophilia (148), retinal degeneration (52), APC-mediated colon cancer (152), and mucopolysaccharidosis I-Hurler (MPS I-H) (65).
Pilot clinical trials to test the ability of gentamicin to mediate nonsense suppression in patients have produced variable results. Restoration of functional CFTR protein could be detected in nasal epithelia from approximately half of CF patients administered gentamicin for 1–2 weeks intravenously (25) or via nasal droplets (145). Similarly, an increase in dystrophin protein in muscle biopsies and a decrease in serum creatine kinase (an indicator of muscle breakdown) were observed in a portion of DMD patients treated with gentamicin for periods ranging from 1 week to 6 months (81, 104). Another pilot study found that intravenously administered gentamicin restored coagulation factor function in a fraction of hemophilia patients (63). In addition, topical gentamicin treatment resulted in early recovery from an episode of Hailey–Hailey disease, an inherited skin disorder, in a patient with a nonsense mutation in the ATP2C1 gene (67). However, gentamicin was unable to restore myophosphorylase activity in patients with McArdle disease after 10 days of systemic treatment (122). Another study found that gentamicin restored full-length CD18 protein in patient blood samples with the neutrophil disorder leukocyte adhesion deficiency 1, but the restored protein did not localize or function normally and did not alleviate the disease phenotype (127).
These studies indicate that aminoglycosides such as gentamicin can restore a significant amount of protein function, but only in a fraction of patients with nonsense mutations. In addition, it is unlikely that long-term administration of traditional aminoglycosides is feasible owing to their toxic side effects, which include hearing loss (60) and kidney damage (1). Importantly, the toxicity associated with aminoglycosides does not appear to be attributable to their ability to suppress PTCs. Rather, aminoglycoside-mediated toxicity is due primarily to their association with off-target sites such as lysosomal membranes (75). In addition, because of their similarities to bacterial ribosomes, aminoglycosides bind preferentially to the decoding sites of mitochondrial ribosomes (105).
Strategies to Improve Aminoglycosides for Suppression Therapy
Several strategies have been used to decrease the toxicity of aminoglycosides. Antioxidants such as D-methionine (18) and melatonin (110) have been used to reduce free-radical formation resulting from aminoglycoside-induced toxicity. Poly-L-aspartate decreases both nephrotoxicity (106) and ototoxicity (59) by reducing the interaction of aminoglycosides with lysosomal membranes. Poly-L-aspartate also increases cytoplasmic aminoglycoside concentrations, which enhances PTC suppression (34, 46).
The encapsulation of aminoglycosides into liposomes is another approach being explored to reduce aminoglycoside-induced toxicity. The intravenous administration of unilamellar, low-clearance liposomes containing amikacin (MiKasome®) resulted in effective bacterial clearance and a good clinical cure rate. Compared with conventional amikacin, MiKasome showed prolonged drug residence as well as altered tissue distribution and elimination that prevented nephrotoxicity (121). More recently, a liposomal preparation of amikacin for aerosol delivery (Arikace®) was shown to reduce Pseudomonas aeruginosa density in sputum and improve lung function in CF patients (26).
Although conventional amikacin suppresses PTCs (76), liposomal amikacin formulations have not yet been investigated for suppression therapy. Even though the administration of liposomal aminoglycoside preparations may be less toxic than conventional aminoglycosides, the slower release of these vesicle-encapsulated drugs may not be as effective for suppression therapy, because aminoglycosides appear to exhibit a peak-driven pharmacokinetic profile for PTC suppression. For example, gentamicin administered once daily via subcutaneous injection restored dystrophin protein in the mdx mouse model (which carries a PTC in the dystrophin gene), whereas no increase in dystrophin was observed when gentamicin was administered at a low, sustained level via an osmotic pump (10). However, a recent study demonstrated that the intraperitoneal administration of gentamicin delivered in hybrid liposomes restored dystrophin protein expression more effectively in skeletal muscle of mdx mice than conventional gentamicin (150). This suggests that liposomal encapsulation of aminoglycosides may provide a means to reduce aminoglycoside toxicity while maintaining or enhancing PTC suppression for long-term suppression therapy.
Aminoglycoside antibiotic derivatives have also been developed that are less toxic than conventional aminoglycosides. Derivatives of kanamycin and neomycin that improved antibiotic function were also found to suppress nonsense mutations in the SMN1 and SMN2 genes that cause spinal muscular atrophy (SMA) (22). Direct administration to the central nervous system of the most promising derivative, TC007, restored SMN protein, reduced neuropathology, and increased life span in an SMA mouse model (83). In contrast, systemic administration of TC007 to SMA mice for 2 weeks via subcutaneous injections improved gross motor function but did not increase the abundance of SMN protein in brain, spine, or muscle or prolong life span.
More recently, a rational design approach was used to generate aminoglycoside derivatives with enhanced nonsense suppression efficiency and reduced toxicity. Through this strategy, structural moieties predicted to enhance aminoglycoside binding to cytoplasmic ribosomes while reducing aminoglycoside binding to mitochondrial ribosomes were identified and incorporated onto an aminoglycoside chemical scaffold (95). In an early application of this approach, the synthetic aminoglycoside NB30 was found to suppress nonsense mutations associated with Usher syndrome in cultured cells. Although the level of PTC suppression achieved with NB30 was not as robust as the level mediated by gentamicin, NB30 was found to be approximately 10–15-fold less toxic than gentamicin in mammalian cells (49, 107).
Further application of this rational design strategy produced drugs that not only were much less toxic than conventional aminoglycosides, but also suppressed PTCs more efficiently. For example, the NB54 aminoglycoside derivative suppressed nonsense mutations associated with CF, DMD, MPS I-H, Rett syndrome, and Usher syndrome in cultured cells more efficiently than gentamicin (13, 47, 94, 111, 139). In addition, NB54 suppressed PTCs in mouse models of CF and MPS I-H more efficiently than gentamicin (111, 139). Subsequent generations of synthetic aminoglycosides have shown even greater improvement in PTC suppression efficiency (93, 126). For example, NB84 suppresses a nonsense mutation associated with MPS I-H more effectively than gentamicin or any of the previous generations of synthetic aminoglycosides both in cultured cells and in an MPS I-H mouse model (65, 139). These results validate the utility of this rational design strategy to generate safer, more effective aminoglycosides for suppression therapy. This approach may eventually lead to the development of synthetic aminoglycosides that are suitable for long-term suppression therapy.
Non-Aminoglycoside Antibiotics
A number of non-aminoglycoside antibiotics have also been shown to suppress PTCs in mammalian cells. Studies using in vitro models showed that negamycin, a peptide antibiotic that binds to the eukaryotic small ribosomal subunit (6, 128), suppresses nonsense mutations and restores protein function in the APC gene associated with colon cancer (42), in the laminin α-2 gene associated with congenital muscular dystrophy (3), and in the dystrophin gene associated with DMD (5, 6). In addition, mdx mice administered negamycin showed a restoration of dystrophin protein in skeletal and cardiac muscles with reduced toxicity compared with gentamicin (5, 6).
Finally, several macrolide antibiotics, such as spiramycin, josamycin, and tylosin, have been shown to suppress APC nonsense mutations and restore APC protein function in cultured cells. In addition, these drugs reduced tumor size in nude mice and reduced intestinal polyp number and size in Min mice that carry a nonsense mutation in the Apc gene (152).
PTC124 (Ataluren)
PTC124, also known as ataluren, is an oxadiazole compound discovered by PTC Therapeutics that suppresses termination at PTCs in mammalian cells without affecting translation termination at natural stop codons (141). Comprehensive preclinical studies showed that PTC124 is safe, has minimal off-target side effects, has no antibacterial activity, and is orally bioavailable (54, 141). However, the ability of PTC124 to suppress nonsense mutations has been questioned based on the results of some in vitro studies. PTC124 was initially identified as a nonsense suppression agent from a high-throughput screen of approximately 800,000 compounds using a firefly luciferase-based readthrough reporter, where PTC suppression resulted in an increase of firefly luciferase activity (141). However, subsequent studies found that PTC124 directly binds firefly luciferase, leading to stabilization of the luciferase protein (7, 8). It was therefore proposed that the initial identification of PTC124 was based on increased firefly luciferase activity resulting from the stabilization of the small amount of firefly protein produced by basal readthrough, rather than from PTC suppression. Another study also questioned the ability of PTC124 to suppress PTCs because it was unable to mediate readthrough using multiple readthrough reporter constructs in cultured cells (85). Furthermore, PTC124 was unable to restore functional protein expressed from endogenous mutant transcripts in cell models associated with obesity (melanocortin 4 receptor) (14), peroxisome biogenesis disorders (30), and long-QT syndrome (53).
In contrast to those negative results, numerous other in vitro studies found that PTC124 suppresses nonsense mutations and restores deficient protein function associated with numerous disorders, including MPS VI (9), MPS I-H (100), Usher syndrome (47, 48), infantile neuronal ceroid lipofuscinosis (119), propionic acidemia (117), carnitine palmitoyltransferase 1A deficiency (129), Miyoshi myopathy (100, 138), DMD (51, 141), methylmalonic aciduria (15), pulmonary arterial hypertension (29), pseudoxanthoma elasticum (151), and CF (51). Based on the functional diversity of the deficient proteins associated with these various diseases, it is highly unlikely that PTC124 stabilizes each of these proteins in a manner similar to its proposed effect on firefly luciferase. The inconsistencies observed may be attributable to subtle differences in treatment or assay conditions (8, 100, 101). For example, PTC124 efficacy exhibited a bell-shaped dose–response curve in some in vitro studies, which results in a therapeutic response only within a narrow dose range. This effect was observed both in cultured myoblasts taken from DMD patients and in cultured mouse embryonic fibroblasts derived from an MPS I-H mouse model (100). Alternatively, the local mRNA sequence context surrounding the PTCs may have influenced the effectiveness of PTC124 to promote readthrough, because context effects significantly influence the efficiency of aminoglycoside-mediated PTC suppression (19, 57, 82).
In vivo investigations with PTC124 have produced more consistent positive results. PTC124 suppressed nonsense mutations and restored approximately 20% of normal protein function in mouse models of DMD (141) and CF (36). Promising safety and efficacy data from phase 1 and phase 2 clinical trials with DMD (41, 102) and CF (69, 125, 144) patients led to the initiation of double-blind, placebo-controlled phase 3 clinical trials for both diseases. The phase 3 DMD trial is still ongoing (ClinicalTrials.gov identifier NCT01826487), but the phase 3 CF trial was recently completed (72). Administration of PTC124 to CF patients for 48 weeks led to trends toward improvement relative to control subjects in several parameters, including lung function and exacerbation frequency, but statistical significance was not reached in the overall patient population. Intriguingly, a significant treatment effect was observed in a subset of patients that were not concurrently receiving an inhaled aminoglycoside (Tobi®), suggesting that aminoglycosides may confound the benefits of PTC124 treatment. However, the overall results led to the conclusion that PTC124 did not restore enough CFTR function to reach the threshold required for a therapeutic benefit. Previous studies have estimated that as much as 35% of wild-type CFTR activity may be needed to alleviate the CF phenotype (68). It will be interesting to see whether better therapeutic results are obtained in the ongoing clinical trial of PTC124 in DMD patients, because it has been reported that 20–30% of normal dystrophin may be sufficient to provide a clinical benefit for DMD patients (20, 91).
Other Non-Aminoglycoside Compounds
High-throughput drug screens have also identified other low-molecular-weight lead compounds that suppress nonsense mutations in the ATM gene that cause ataxia telangiectasia (31, 32). These drugs include RT13, RT14, GJ071, and GJ072 as well as their derivatives. All of these drugs restore the expression of full-length, functional ATM protein.
Another drug screen was carried out to identify compounds that inhibit nonsense-mediated mRNA decay (NMD), a pathway that degrades mRNAs containing PTCs. Amlexanox was identified as a compound that both inhibits NMD and suppresses PTCs (51). Amlexanox is an anti-inflammatory compound that is FDA approved for the topical treatment of oral canker sores (11). In Japan, it is used orally to treat asthmatic bronchitis and allergic rhinitis (80). Recently, oral administration of amlexanox to an obesity mouse model was also found to alleviate insulin resistance (109), and it is currently in phase 2 clinical trials for the treatment of diabetes (ClinicalTrials.gov identifier NCT01842282).
OTHER APPROACHES TO MEDIATE (OR ENHANCE) PREMATURE TERMINATION CODON READTHROUGH
In addition to pharmacological suppression of PTCs, other methods to suppress PTCs (or to enhance their suppression) are also being explored. Some of these approaches are described below.
Suppressor tRNAs
A nonsense suppressor tRNA is a tRNA derivative whose anticodon has been altered to recognize a stop codon, thus allowing the incorporation of an amino acid at the stop codon and a bypass of translation termination. Because suppressor tRNAs mediate cognate codon–anticodon interactions, aminoacyl-tRNA rejection by ribosomal proofreading is avoided and efficient suppression of stop codons can be attained. Suppressor tRNAs are transcribed and processed in a manner similar to natural tRNAs and are recognized and charged by endogenous aminoacyl-tRNA synthetases. Because nucleotides within the anticodon serve as both binding and identity elements during the selection of cognate tRNAs for charging by tRNA synthetases (61), suppressor tRNAs may be charged with lower efficiency.
Several studies have demonstrated that suppressor tRNAs can potentially be used for human somatic gene therapy to treat diseases caused by nonsense mutations. As early as 1982, a functional human suppressor tRNA was successfully constructed and shown to suppress a UAG nonsense mutation in the human β-globin gene associated with β-thalassemia (132). The suppressor tRNA was constructed by modifying the anticodon of a tRNALys to recognize the UAG nonsense codon instead of the normal AAA (lysine) codon. When the suppressor tRNA and the mutant β-globin mRNAs were coinjected into Xenopus oocytes, full-length β-globin chain synthesis was observed. In another study using xeroderma pigmentosum group A mutant cells containing an R207X nonsense mutation in the XPA gene, transfection of these cells with a human arginine UGA suppressor tRNA restored DNA repair activity, indicating that a partial restoration of the XPA protein had been achieved (99).
The first in vivo suppressor tRNA study was conducted in 2000 (17). Direct injection of a suppressor tRNA into the hearts of transgenic mice expressing a chloramphenicol acetyltransferase (CAT) gene containing a UAA nonsense mutation led to partial restoration of CAT activity in ∼10% of muscle fibers. A similar approach was tested in mdx DMD mice harboring a UAA nonsense mutation in the gene encoding dystrophin. One week after a single intramuscular injection of a plasmid expressing a UAA suppressor tRNA, full-length dystrophin expression was restored in ∼2.5% of muscle fibers (71). These examples suggest that, in principle, suppressor tRNAs could be used to treat human genetic diseases caused by nonsense mutations.
Although promising, this approach has several obstacles that must be overcome before it can be developed into a clinical therapy. All gene therapy protocols have the challenges of efficient delivery to target cells, but suppressor tRNAs bring additional challenges related to their efficient use. First, all tRNAs require proper charging by specific aminoacyl synthetases, and suppressor tRNAs must compete with normal tRNAs for aminoacylation. As mentioned above, modifications in the anticodon loop of tRNAs to allow stop codon recognition may result in lower aminoacylation efficiency and thus inefficient nonsense suppression activity (45). Second, the efficiency of PTC suppression by suppressor tRNAs is dependent on the position of the PTC and the surrounding sequence context. Finally, in addition to PTCs, suppressor tRNAs can recognize and suppress normal stop codons, which could potentially produce abnormal proteins that lead to deleterious effects (74). Overall, better systems are needed to ensure the safe and efficient expression of suppressor tRNAs before this method can be developed into a therapeutic approach.
Pseudouridylation Recoding
Pseudouridylation is the isomerization of the ribonucleoside uridine to the 5′-ribosyl isomer pseudouridine (Ψ) and represents the most common single-nucleotide modification of functional RNAs. This modification of uridine creates an extra hydrogen-bond donor in Ψ that increases its polarity, enhances base-pair stacking, and alters RNA structure. Thus, Ψ has distinct structural and chemical characteristics relative to uridine (58). Pseudouridylation naturally occurs as a posttranscriptional modification in eukaryotic nuclei and affects primarily tRNAs, rRNAs, and spliceosomal small nuclear RNAs (snRNAs). However, recent studies have explored targeted pseudouridylation of stop codons within mRNAs as a means of inducing suppression of translation termination at PTCs (64).
Pseudouridylation in higher eukaryotes is carried out by box H/ACA ribonucleoproteins (RNPs). Box H/ACA RNPs encompass a family of structurally conserved small nucleolar RNAs (snoRNAs) that function to provide sequence homology for site-specific RNA binding at uridine residues targeted for pseudouridylation in rRNA and snRNA. The H/ACA guide RNA folds into a conserved hairpin-hinge-hairpin-tail conformation with two pseudouridylation sites. Four protein components—DKC1 (dyskerin), NOP10, NHP2, and GAR1—associate with the guide RNA to facilitate substrate RNA incorporation into the complex and catalysis of pseudouridylation. Dyskerin is a Ψ synthase and represents the catalytic core of the H/ACA RNP complex.
The use of an H/ACA guide RNA offers the possibility of site-specific pseudouridylation via sequence homology with any RNA sequence. Pseudouridylation of tRNA molecules results in alternative codon recognition through changes in anticodon loop structure (133). This suggests that similar pseudouridylation of mRNA codons might also alter codon–anticodon pairing. Because all three nonsense codons contain a uridine (U) in the first position (UAA, UAG, UGA), pseudouridylation of the uridine of stop codons has the potential to alter the efficiency of PTC recognition.
In support of this hypothesis, recent studies have shown that pseudouridylation of the first nucleotide of a stop codon leads to PTC readthrough. In a study using a cell-free system, readthrough constructs containing a stop codon with a Ψ at the first nucleotide generated approximately 74–100% of the reporter protein produced by a sense construct. In contrast, a non-Ψ stop codon produced only ∼1% of wild-type protein (64). An H/ACA RNA guide sequence was also tested in yeast cells by targeting a PTC in the CUP1 gene, which mediates copper resistance (64). When a copper-sensitive yeast strain was cotransfected with the H/ACA RNA and the cup1-PTC constructs, it regained the ability to grow on copper-containing media. In addition, Ψ incorporation into the mRNA was confirmed by thin-layer chromatography.
Alternative amino acid coding of Ψ-containing nonsense codons is an intriguing new approach to induce PTC readthrough. Although these preliminary results are promising, site-directed pseudouridylation as a therapeutic solution to disease-causing PTCs has significant obstacles that must be overcome. As discussed above for suppressor tRNA approaches, pseudouridylation-mediated recoding must overcome the challenges of efficient gene delivery of PTC-specific guide H/ACA RNAs. These artificially constructed guide RNAs may also have off-target effects because of unintended sequence complementarity that could result in the introduction of Ψ at other sites. For example, off-target pseudouridylation could affect cellular processes such as rRNA structure, mRNA splicing, recognition of the internal ribosome entry site, and tRNA specificity. Further studies will be needed to determine whether this concept can be developed into a successful therapeutic approach.
Inhibition of Nonsense-Mediated mRNA Decay
NMD is a conserved eukaryotic surveillance pathway that recognizes and degrades mRNAs containing PTCs (Figure 4a). The efficiency of the NMD pathway, which plays an important role in modulating the phenotypes of diseases caused by PTCs, varies by as much as fourfold among the general population (77, 124, 137). Importantly, these differences in NMD efficiency influence the inheritance pattern and modulate the clinical severity of numerous disorders (12, 70), possibly because of changes in residual mRNA abundance that influence the levels of truncated protein (or full-length protein produced by basal PTC readthrough). Furthermore, variable NMD efficiency among patients also influences the effectiveness of nonsense suppression therapy. Patients that express higher levels of PTC-containing mRNA resulting from less efficient NMD respond more robustly to suppression therapy than patients that express lower levels of PTC-containing mRNA resulting from highly efficient NMD (69, 77, 145).
Figure 4.
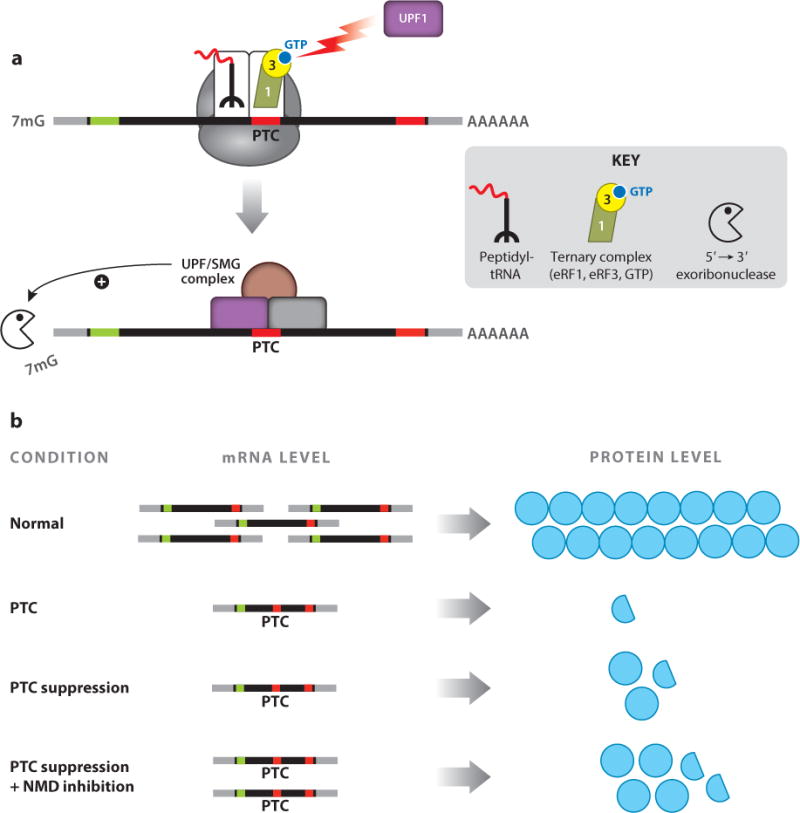
Nonsense-mediated mRNA decay (NMD) reduces premature termination codon (PTC) suppression efficiency. (a) Translation termination at a PTC induces mRNA degradation by NMD. (b) NMD inhibition enhances the amount of protein restored by PTC suppression.
Based on its role in regulating the abundance of PTC-containing transcripts, NMD represents a potential therapeutic target for diseases caused by PTCs. Numerous factors participate in NMD, including the core NMD factors UPF1, UPF2, and UPF3 as well as other factors that influence the phosphorylation status of UPF1, such as SMG-1, SMG-5, SMG-7, SMG-8, and SMG-9 (123, 147). Therefore, it may be possible to identify multiple therapeutic targets for modulating NMD function. Increasing the level of PTC-containing mRNAs by partial NMD inhibition could alleviate some disease phenotypes by increasing the expression of truncated polypeptides that retain residual function (Figure 4b). For example, in fibroblasts derived from patients with Ullrich congenital muscular dystrophy, the SMG-1 kinase inhibitors caffeine and wortmannin increased the abundance of PTC-containing COL α-2 (VI) mRNA and, subsequently, the abundance of truncated COL VI protein, which retained partial function (134). Similar results were also obtained in Ullrich patient fibroblasts when NMD was inhibited using SMG-1 or UPF1 knockdowns (135). In addition, UPF1 knockdowns restored partial hERG channel function in cells expressing hERG transcripts containing PTCs that are associated with the cardiac disorder long-QT syndrome type 2 (50).
Evidence also suggests that partial NMD inhibition can increase the abundance of full-length, functional polypeptides restored by PTC suppression. Knockdowns of UPF1 and UPF2 in cultured human bronchial epithelial cells led to a more robust recovery of CFTR protein function by suppression therapy (77). In addition, coadministration of the NMD inhibitor NMDI-1 (37) with a PTC suppression drug to MPS I-H mice carrying the Idua-W402X PTC resulted in higher levels of functional α-L-iduronidase and enhanced moderation of the MPS I-H phenotype (65).
Although NMD inhibition represents a novel concept for treating diseases caused by PTCs, altering NMD efficiency must be approached with caution because of potential deleterious side effects. In addition to regulating the expression of PTC-containing transcripts that arise from mutations, NMD regulates the expression of up to 10% of the mammalian transcriptome. These natural NMD substrates include transcripts containing upstream open reading frames in the 5′ untranslated region, transcripts with long or intron-containing 3′ untranslated regions, transcripts containing selenocysteine codons (88), and products of alternative splicing that contain PTCs. Furthermore, NMD and/or NMD factors have an essential role during embryonic development: Knockout of UPF1 (87), UPF2 (140), or SMG-1 (86) in mice results in embryonic lethality. In addition, several UPF3B mutations have been identified in patients with forms of intellectual disability (130). Associations have also been found for various forms of intellectual disability and copy-number variants of other factors that participate in NMD, including UPF2, UPF3A, SMG-6, EIF4A3, and RNPS1 (92). However, transgenic mice constitutively expressing a mutant dominant negative UPF1 allele to partially inhibit NMD developed normally except for faulty thymocyte development (43). This suggests that partial NMD inhibition after embryonic development may not lead to detrimental consequences.
A better understanding of how the NMD pathway affects mammalian gene expression and physiology will aid in finding potential therapeutic targets for NMD inhibition. For example, current data suggest that different categories or tiers of NMD exist (38, 89, 113, 153). These different levels of NMD may be required to modulate NMD efficiency to alter the expression of specific transcripts within different cell types, during different stages of development and differentiation, or under different cellular environmental conditions. The idea of targeting only a subset of NMD substrates is supported by the identification of two distinct branches of the NMD pathway, one that functions independently of UPF2 (44) and one that functions independently of UPF3B (21).
Consistent with the premise of identifying NMD inhibitors that do not cause adverse effects, the FDA-approved drug amlexanox was recently discovered to inhibit NMD as well as suppress PTCs (51). Although amlexanox’s mechanism of NMD inhibition and PTC suppression has not been delineated, it appears to block NMD after recruitment of UPF1. Micromolar concentrations of amlexanox were found to increase the abundance of multiple mRNAs containing nonsense mutations by two- to fivefold. This corresponded to increased expression of both truncated and full-length functional polypeptides. Furthermore, amlexanox showed only minor (1.2-fold) increases in several endogenous NMD substrates, suggesting that it may not affect the expression of many natural NMD substrates. Cytotoxicity was not observed in mammalian cells treated with amlexanox.
Finally, a recent study compared the effects of individual knockdowns of 15 different NMD-factor genes in Ullrich patient fibroblasts to determine whether the depletion of certain NMD factors could mediate NMD inhibition without significant toxicity. The results showed that knockdown of SMG-8 effectively increased the abundance of truncated collagen VI α2 protein and restored extracellular matrix function without cytotoxicity (136). This suggests that it may be possible to target specific NMD factors to partially inhibit NMD without also causing detrimental side effects. This approach has the potential to provide another option for the treatment of diseases caused by PTCs as well as a method for enhancing the efficiency of nonsense suppression therapy.
SUMMARY AND FUTURE DIRECTIONS
Since the concept of nonsense suppression therapy was first introduced 18 years ago (56), many studies have demonstrated that nonsense suppression can restore deficient protein function. This has raised the intriguing possibility that this therapeutic approach could be used to treat many diverse diseases caused by PTCs. However, whether the level of protein function restored by nonsense suppression therapy can prevent or alleviate the onset of a disease phenotype has not been established.
There are several reasons for the slow advancement of nonsense suppression as a therapy. First, a dearth of safe, effective suppression drugs has prevented long-term studies. Until recently, aminoglycosides, which are known to induce toxicity with long-term administration, were the only compounds available that could efficiently suppress PTCs in mammalian cells. However, recently developed synthetic aminoglycosides exhibit reduced toxicity as well as enhanced efficiency of PTC suppression. This suggests that aminoglycoside derivatives may eventually be suitable for long-term suppression therapy. In addition, high-throughput drug screens have identified many non-aminoglycoside compounds that suppress PTCs with safety profiles much better than those of aminoglycosides. PTC124 and amlexanox are both compounds that suppress PTCs in mammalian cells and have performed exceptionally well in safety studies. The continued discovery of additional promising compounds is likely to speed the development of safe, effective nonsense suppression drugs for clinical trials.
Another factor that has hindered the progress of nonsense suppression therapy is the lack of appropriate animal models that carry nonsense mutations. Owing to the extensive time and cost required for development and maintenance, most animal models are generated using knockout strategies that create large deletions within a gene. Very few of these models have been generated using the more precise knock-in strategy to introduce a single nucleotide substitution that results in an in-frame PTC that could be used to test nonsense suppression agents. However, the recent development of new biotechnologies such as CRISPR/Cas technology (149) will make generating animal models quicker, easier, and more cost effective. The development and progression of disease pathophysiology in animal models can also be quite different from those observed in patients. For example, mouse models of CF do not develop the lung disease that is characteristic of CF patients (28). Rather, intestinal tissues represent the main site for CF disease pathology in mice. Although CF mice have been used successfully to determine the level of functional CFTR restored in intestinal tissues by suppression therapy (33–36), how well these data correlate to the airway phenotype observed in CF patients is unclear. CF pig and ferret models more faithfully recapitulate the lung phenotype observed in CF patients (66), but these animals are expensive to construct, maintain, and assay. In addition, no CF pig or ferret models are currently available that carry a nonsense mutation.
Another hindrance to the development of suppression therapy is the lack of phenotypic end points that adequately correlate therapeutic efficacy in cells, animals, and patients. The selection of meaningful end points to evaluate whether a significant clinical benefit results from suppression therapy is challenging because there can be great variability in the phenotype, even among patients with the same disease. For many diseases, the exact biochemical and pathophysiological pathways may be unknown. In addition, many traditional biochemical assays that are routinely used to diagnose genetic diseases may not be sensitive enough to reproducibly measure the low ∼0.5–20% increases in protein function restored in cells and tissues with suppression therapy.
Ultimately, the key factor that determines the effectiveness of suppression therapy is the minimum level of deficient protein function that must be restored to achieve a therapeutic improvement. This therapeutic threshold can differ significantly for different diseases. For example, it is estimated that 30–35% of normal CFTR activity is required for a therapeutic improvement in CF (68, 112), 20–30% of normal dystrophin function is required for attenuation of DMD (20, 91), and only 0.4–1% of the α-L-iduronidase enzyme activity is required to alleviate MPS I-H (16, 96). Also, the threshold for correction can differ in various tissues in individuals with the same disease and between the same tissues in different patients. For most diseases, the threshold for correction is largely unknown (other than whether heterozygotes manifest a disease phenotype). The administration of suppression therapy alone may be unlikely to alleviate the phenotype for genetic diseases that have a high threshold for correction. However, nonsense suppression therapy could still be an important component of combined therapeutic approaches for such diseases.
For example, substrate reduction therapy using the isoflavone compound genistein reduces glycosaminoglycan accumulation associated with the lysosomal storage diseases MPS I, MPS II, MPS III, and MPS VII by reducing glycosaminoglycan synthesis (103). PTC suppression therapy could complement this approach by increasing glycosaminoglycan breakdown via restoration of lysosomal enzyme function and therefore further reduce GAG accumulation in patients with nonsense mutations. The potential for a combined therapeutic approach is also possible for CF patients with nonsense mutations. The drug VX-770 (Kalydeco®) was recently shown to alleviate the CF phenotype in patients that carry the G551D mutation by acting as a potentiator that increases gating of the CFTR channel (39). Treatment of CF patients that carry nonsense mutations using both a PTC suppression drug and Kalydeco may enhance the level of CFTR function observed relative to the use of either drug alone. Suppression therapy, alone or in combination with other therapeutic approaches, may therefore be a vital component in treating many genetic diseases in patients that carry nonsense mutations.
Acknowledgments
This research was supported by grants from the University of Pennsylvania and the National Institutes of Health. We apologize to those investigators whose work was not cited owing to reference limits.
Footnotes
DISCLOSURE STATEMENT
D.M.B. serves as a scientific consultant for PTC Therapeutics, Inc. D.M.B holds a patent for a novel cystic fibrosis treatment (US Patent 5,840,702), and D.M.B. and K.M.K. hold a patent for aminoglycoside treatment for lysosomal storage diseases (US Patent 7,749,971). The authors are not aware of any other affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Ali BH, Al Za’abi M, Blunden G, Nemmar A. Experimental gentamicin nephrotoxicity and agents that modify it: a mini-review of recent research. Basic Clin Pharmacol Toxicol. 2011;109:225–32. doi: 10.1111/j.1742-7843.2011.00728.x. [DOI] [PubMed] [Google Scholar]
- 2.Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell. 2006;125:1125–36. doi: 10.1016/j.cell.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 3.Allamand V, Bidou L, Arakawa M, Floquet C, Shiozuka M, et al. Drug-induced readthrough of premature stop codons leads to the stabilization of laminin α2 chain mRNA in CMD myotubes. J Gene Med. 2008;10:217–24. doi: 10.1002/jgm.1140. [DOI] [PubMed] [Google Scholar]
- 4.Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:112–18. doi: 10.1038/nature03060. [DOI] [PubMed] [Google Scholar]
- 5.Arakawa M, Nakayama Y, Hara T, Shiozuka M, Takeda S, et al. Negamycin can restore dystrophin in mdx skeletal muscle. Acta Myol. 2001;20:154–58. [Google Scholar]
- 6.Arakawa M, Shiozuka M, Nakayama Y, Hara T, Hamada M, et al. Negamycin restores dystrophin expression in skeletal and cardiac muscles of mdx mice. J Biochem. 2003;134:751–58. doi: 10.1093/jb/mvg203. [DOI] [PubMed] [Google Scholar]
- 7.Auld DS, Lovell S, Thorne N, Lea WA, Maloney DJ, et al. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc Natl Acad Sci USA. 2010;107:4878–83. doi: 10.1073/pnas.0909141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci USA. 2009;106:3585–90. doi: 10.1073/pnas.0813345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartolomeo R, Polishchuk EV, Volpi N, Polishchuk RS, Auricchio A. Pharmacological readthrough of nonsense ARSB mutations as a potential therapeutic approach for mucopolysaccharidosis VI. J Inherit Metab Dis. 2013;36:363–71. doi: 10.1007/s10545-012-9521-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Investig. 1999;104:375–81. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell J. Amlexanox for the treatment of recurrent aphthous ulcers. Clin Drug Investig. 2005;25:555–66. doi: 10.2165/00044011-200525090-00001. [DOI] [PubMed] [Google Scholar]
- 12.Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE. NMD: RNA biology meets human genetic medicine. Biochem J. 2010;430:365–77. doi: 10.1042/BJ20100699. [DOI] [PubMed] [Google Scholar]
- 13.Brendel C, Belakhov V, Werner H, Wegener E, Gartner J, et al. Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J Mol Med. 2010;89:389–98. doi: 10.1007/s00109-010-0704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brumm H, Muhlhaus J, Bolze F, Scherag S, Hinney A, et al. Rescue of melanocortin 4 receptor (MC4R) nonsense mutations by aminoglycoside-mediated read-through. Obesity. 2012;20:1074–81. doi: 10.1038/oby.2011.202. [DOI] [PubMed] [Google Scholar]
- 15.Buck NE, Wood LR, Hamilton NJ, Bennett MJ, Peters HL. Treatment of a methylmalonyl-CoA mutase stopcodon mutation. Biochem Biophys Res Commun. 2012;427:753–57. doi: 10.1016/j.bbrc.2012.09.133. [DOI] [PubMed] [Google Scholar]
- 16.Bunge S, Clements PR, Byers S, Kleijer WJ, Brooks DA, Hopwood JJ. Genotype-phenotype correlations in mucopolysaccharidosis type I using enzyme kinetics, immunoquantification and in vitro turnover studies. Biochim Biophys Acta. 1998;1407:249–56. doi: 10.1016/s0925-4439(98)00046-5. [DOI] [PubMed] [Google Scholar]
- 17.Buvoli M, Buvoli A, Leinwand LA. Suppression of nonsense mutations in cell culture and mice by multimerized suppressor tRNA genes. Mol Cell Biol. 2000;20:3116–24. doi: 10.1128/mcb.20.9.3116-3124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell KC, Meech RP, Klemens JJ, Gerberi MT, Dyrstad SS, et al. Prevention of noise- and drug-induced hearing loss with D-methionine. Hear Res. 2007;226:92–103. doi: 10.1016/j.heares.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Cassan M, Rousset JP. UAG readthrough in mammalian cells: effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol Biol. 2001;2:3. doi: 10.1186/1471-2199-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chamberlain J. Dystrophin levels required for genetic correction of Duchenne muscular dystrophy. Basic Appl Myol. 1997;7:251–55. [Google Scholar]
- 21.Chan WK, Huang L, Gudikote JP, Chang YF, Imam JS, et al. An alternative branch of the nonsense-mediated decay pathway. EMBO J. 2007;26:1820–30. doi: 10.1038/sj.emboj.7601628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang CW, Hui Y, Elchert B, Wang J, Li J, Rai R. Pyranmycins, a novel class of aminoglycosides with improved acid stability: the SAR of D-pyranoses on ring III of pyranmycin. Org Lett. 2002;4:4603–6. doi: 10.1021/ol0269042. [DOI] [PubMed] [Google Scholar]
- 23.Chavatte L, Kervestin S, Favre A, Jean-Jean O. Stop codon selection in eukaryotic translation termination: comparison of the discriminating potential between human and ciliate eRF1s. EMBO J. 2003;22:1644–53. doi: 10.1093/emboj/cdg146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C-YA, Shyu A-B. Mechanisms of deadenylation-dependent decay. Wiley Interdiscip Rev RNA. 2011;2:167–83. doi: 10.1002/wrna.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clancy JP, Bebok Z, Ruiz F, King C, Jones J, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med. 2001;163:1683–92. doi: 10.1164/ajrccm.163.7.2004001. [DOI] [PubMed] [Google Scholar]
- 26.Clancy JP, Dupont L, Konstan MW, Billings J, Fustik S, et al. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax. 2013;68:818–25. doi: 10.1136/thoraxjnl-2012-202230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosson B, Couturier A, Chabelskaya S, Kiktev D, Inge-Vechtomov S, et al. Poly(A)-binding protein acts in translation termination via eukaryotic release factor 3 interaction and does not influence [PSI+] propagation. Mol Cell Biol. 2002;22:3301–15. doi: 10.1128/MCB.22.10.3301-3315.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davidson DJ, Rolfe M. Mouse models of cystic fibrosis. Trends Genet. 2001;17:S29–37. doi: 10.1016/s0168-9525(01)02452-0. [DOI] [PubMed] [Google Scholar]
- 29.Drake KM, Dunmore BJ, McNelly LN, Morrell NW, Aldred MA. Correction of nonsense BMPR2 and SMAD9 mutations by ataluren in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2013;49:403–9. doi: 10.1165/rcmb.2013-0100OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dranchak PK, Di Pietro E, Snowden A, Oesch N, Braverman NE, et al. Nonsense suppressor therapies rescue peroxisome lipid metabolism and assembly in cells from patients with specific PEX gene mutations. J Cell Biochem. 2011;112:1250–58. doi: 10.1002/jcb.22979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du L, Damoiseaux R, Nahas S, Gao K, Hu H, et al. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J Exp Med. 2009;206:2285–97. doi: 10.1084/jem.20081940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du L, Jung ME, Damoiseaux R, Completo G, Fike F, et al. A new series of novel small molecular weight compounds induce readthrough of all three types of nonsense mutations in the ATM gene. Mol Ther. 2013;21:1653–60. doi: 10.1038/mt.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du M, Jones JR, Lanier J, Keeling KM, Lindsey JR, et al. Aminoglycoside suppression of a premature stop mutation in a Cftr−/− mouse carrying a human CFTR-G542X transgene. J Mol Med. 2002;80:595–604. doi: 10.1007/s00109-002-0363-1. [DOI] [PubMed] [Google Scholar]
- 34.Du M, Keeling KM, Fan L, Liu X, Bedwell DM. Poly-L-aspartic acid enhances and prolongs gentamicin-mediated suppression of the CFTR-G542X mutation in a cystic fibrosis mouse model. J Biol Chem. 2009;284:6885–92. doi: 10.1074/jbc.M806728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du M, Keeling KM, Fan L, Liu X, Kovacs T, et al. Clinical doses of amikacin provide more effective suppression of the human CFTR-G542X stop mutation than gentamicin in a transgenic CF mouse model. J Mol Med. 2006;84:573–82. doi: 10.1007/s00109-006-0045-5. [DOI] [PubMed] [Google Scholar]
- 36.Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci USA. 2008;105:2064–69. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durand S, Cougot N, Mahuteau-Betzer F, Nguyen CH, Grierson DS, et al. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J Cell Biol. 2007;178:1145–60. doi: 10.1083/jcb.200611086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durand S, Lykke-Andersen J. Nonsense-mediated mRNA decay occurs during eIF4F-dependent translation in human cells. Nat Struct Mol Biol. 2013;20:702–9. doi: 10.1038/nsmb.2575. [DOI] [PubMed] [Google Scholar]
- 39.Eckford PD, Li C, Ramjeesingh M, Bear CE. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem. 2012;287:36639–49. doi: 10.1074/jbc.M112.393637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan-Minogue H, Du M, Pisarev AV, Kallmeyer AK, Salas-Marco J, et al. Distinct eRF3 requirements suggest alternate eRF1 conformations mediate peptide release during eukaryotic translation termination. Mol Cell. 2008;30:599–609. doi: 10.1016/j.molcel.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finkel RS. Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: aminoglycosides and ataluren (PTC124) J Child Neurol. 2010;25:1158–64. doi: 10.1177/0883073810371129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Floquet C, Rousset JP, Bidou L. Readthrough of premature termination codons in the adenomatous polyposis coli gene restores its biological activity in human cancer cells. PLoS ONE. 2011;6:e24125. doi: 10.1371/journal.pone.0024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frischmeyer-Guerrerio PA, Montgomery RA, Warren DS, Cooke SK, Lutz J, et al. Perturbation of thymocyte development in nonsense-mediated decay (NMD)-deficient mice. Proc Natl Acad Sci USA. 2011;108:10638–43. doi: 10.1073/pnas.1019352108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gehring NH, Kunz JB, Neu-Yilik G, Breit S, Viegas MH, et al. Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol Cell. 2005;20:65–75. doi: 10.1016/j.molcel.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 45.Giege R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–35. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilbert DN, Wood CA, Kohlhepp SJ, Kohnen PW, Houghton DC, et al. Polyaspartic acid prevents experimental aminoglycoside nephrotoxicity. J Infect Dis. 1989;159:945–53. doi: 10.1093/infdis/159.5.945. [DOI] [PubMed] [Google Scholar]
- 47.Goldmann T, Overlack N, Möller F, Belakhov V, van Wyk M, et al. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol Med. 2012;4:1186–99. doi: 10.1002/emmm.201201438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldmann T, Overlack N, Wolfrum U, Nagel-Wolfrum K. PTC124 mediated translational read-through of a nonsense mutation causing Usher type 1C. Hum Gene Ther. 2011;22:537–47. doi: 10.1089/hum.2010.067. [DOI] [PubMed] [Google Scholar]
- 49.Goldmann T, Rebibo-Sabbah A, Overlack N, Nudelman I, Belakhov V, et al. Beneficial read-through of a USH1C nonsense mutation by designed aminoglycoside NB30 in the retina. Investig Ophthalmol Vis Sci. 2010;51:6671–80. doi: 10.1167/iovs.10-5741. [DOI] [PubMed] [Google Scholar]
- 50.Gong Q, Stump MR, Zhou Z. Inhibition of nonsense-mediated mRNA decay by antisense morpholino oligonucleotides restores functional expression of hERG nonsense and frameshift mutations in long-QT syndrome. J Mol Cell Cardiol. 2011;50:223–29. doi: 10.1016/j.yjmcc.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez S, Beghyn T, Jia J, Debreuck N, Berte G, et al. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis. 2012;7:58. doi: 10.1186/1750-1172-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guerin K, Gregory-Evans CY, Hodges MD, Moosajee M, Mackay DS, et al. Systemic aminoglycoside treatment in rodent models of retinitis pigmentosa. Exp Eye Res. 2008;87:197–207. doi: 10.1016/j.exer.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 53.Harmer SC, Mohal JS, Kemp D, Tinker A. Readthrough of long-QT syndrome type 1 nonsense mutations rescues function but alters the biophysical properties of the channel. Biochem J. 2012;443:635–42. doi: 10.1042/BJ20111912. [DOI] [PubMed] [Google Scholar]
- 54.Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007;47:430–44. doi: 10.1177/0091270006297140. [DOI] [PubMed] [Google Scholar]
- 55.Hoshino S. Mechanism of the initiation of mRNA decay: role of eRF3 family G proteins. Wiley Interdiscip Rev RNA. 2012;3:743–57. doi: 10.1002/wrna.1133. [DOI] [PubMed] [Google Scholar]
- 56.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–69. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 57.Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Sequence specificity of aminoglycoside-induced stop codon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol. 2000;48:164–69. [PubMed] [Google Scholar]
- 58.Huang C, Wu G, Yu YT. Inducing nonsense suppression by targeted pseudouridylation. Nat Protoc. 2012;7:789–800. doi: 10.1038/nprot.2012.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hulka GF, Prazma J, Brownlee RE, Pulver S, Pillsbury HC. Use of poly-L-aspartic acid to inhibit aminoglycoside cochlear ototoxicity. Am J Otol. 1993;14:352–56. [PubMed] [Google Scholar]
- 60.Huth ME, Ricci AJ, Cheng AG. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. Int J Otolaryngol. 2011;2011:1–19. doi: 10.1155/2011/937861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ibba M, Hong KW, Sherman JM, Sever S, Soll D. Interactions between tRNA identity nucleotides and their recognition sites in glutaminyl-tRNA synthetase determine the cognate amino acid affinity of the enzyme. Proc Natl Acad Sci USA. 1996;93:6953–58. doi: 10.1073/pnas.93.14.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imataka H, Gradi A, Sonenberg N. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 1998;17:7480–89. doi: 10.1093/emboj/17.24.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.James PD, Raut S, Rivard GE, Poon MC, Warner M, et al. Aminoglycoside suppression of nonsense mutations in severe hemophilia. Blood. 2005;106:3043–48. doi: 10.1182/blood-2005-03-1307. [DOI] [PubMed] [Google Scholar]
- 64.Karijolich J, Yu YT. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature. 2011;474:395–98. doi: 10.1038/nature10165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keeling KM, Wang D, Dai Y, Murugesan S, Chenna B, et al. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS ONE. 2013;8:e60478. doi: 10.1371/journal.pone.0060478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keiser NW, Engelhardt JF. New animal models of cystic fibrosis: What are they teaching us? Curr Opin Pulm Med. 2011;17:478–83. doi: 10.1097/MCP.0b013e32834b14c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kellermayer R, Szigeti R, Keeling KM, Bedekovics T, Bedwell DM. Aminoglycosides as potential pharmacogenetic agents in the treatment of Hailey–Hailey disease. J Investig Dermatol. 2006;126:229–31. doi: 10.1038/sj.jid.5700031. [DOI] [PubMed] [Google Scholar]
- 68.Kerem E. Pharmacologic therapy for stop mutations: How much CFTR activity is enough? Curr Opin Pulm Med. 2004;10:547–52. doi: 10.1097/01.mcp.0000141247.22078.46. [DOI] [PubMed] [Google Scholar]
- 69.Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719–27. doi: 10.1016/S0140-6736(08)61168-X. [DOI] [PubMed] [Google Scholar]
- 70.Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006;14:1074–81. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- 71.Kiselev AV, Ostapenko OV, Rogozhkina EV, Kholod NS, Seit Nebi AS, et al. Suppression of nonsense mutations in the dystrophin gene by a suppressor tRNA gene. Mol Biol. 2002;36:43–47. [PubMed] [Google Scholar]
- 72.Konstan M, Accurso F, De Boeck K, Kerem E, Rowe S, et al. Targeting class 1 mutations: update on ataluren as a promising treatment for nonsense mutation cystic fibrosis. Pediatr Pulmonol. 2012;47(S35):108–9. [Google Scholar]
- 73.Kramer EB, Vallabhaneni H, Mayer LM, Farabaugh PJ. A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA. 2010;16:1797–808. doi: 10.1261/rna.2201210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Laski FA, Ganguly S, Sharp PA, RajBhandary UL, Rubin GM. Construction, stable transformation, and function of an amber suppressor tRNA gene in Drosophila melanogaster. Proc Natl Acad Sci USA. 1989;86:6696–98. doi: 10.1073/pnas.86.17.6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laurent G, Carlier MB, Rollman B, Van Hoof F, Tulkens P. Mechanism of aminoglycoside-induced lysosomal phospholipidosis: in vitro and in vivo studies with gentamicin and amikacin. Biochem Pharmacol. 1982;31:3861–70. doi: 10.1016/0006-2952(82)90303-3. [DOI] [PubMed] [Google Scholar]
- 76.Lee HL, Dougherty JP. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol Ther. 2012;136:227–66. doi: 10.1016/j.pharmthera.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 77.Linde L, Boelz S, Nissim-Rafinia M, Oren YS, Wilschanski M, et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Investig. 2007;117:683–92. doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lynch SR, Puglisi JD. Structural origins of aminoglycoside specificity for prokaryotic ribosomes. J Mol Biol. 2001;306:1037–58. doi: 10.1006/jmbi.2000.4420. [DOI] [PubMed] [Google Scholar]
- 79.Lynch SR, Puglisi JD. Structure of a eukaryotic decoding region A-site RNA. J Mol Biol. 2001;306:1023–35. doi: 10.1006/jmbi.2000.4419. [DOI] [PubMed] [Google Scholar]
- 80.Makino H, Saijo T, Ashida Y, Kuriki H, Maki Y. Mechanism of action of an antiallergic agent, amlexanox (AA-673), in inhibiting histamine release from mast cells. Acceleration of cAMP generation and inhibition of phosphodiesterase Int Arch Allergy Appl Immunol. 1987;82:66–71. doi: 10.1159/000234292. [DOI] [PubMed] [Google Scholar]
- 81.Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010;67:771–80. doi: 10.1002/ana.22024. [DOI] [PubMed] [Google Scholar]
- 82.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–55. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mattis VB, Ebert AD, Fosso MY, Chang CW, Lorson CL. Delivery of a read-through inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Hum Mol Genet. 2009;18:3906–13. doi: 10.1093/hmg/ddp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCaughan KK, Brown CM, Dalphin ME, Berry MJ, Tate WP. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc Natl Acad Sci USA. 1995;92:5431–35. doi: 10.1073/pnas.92.12.5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McElroy SP, Nomura T, Torrie LS, Warbrick E, Gartner U, et al. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013;11:e1001593. doi: 10.1371/journal.pbio.1001593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McIlwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, et al. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci USA. 2010;107:12186–91. doi: 10.1073/pnas.1007336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Medghalchi SM, Frischmeyer PA, Mendell JT, Kelly AG, Lawler AM, Dietz HC. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum Mol Genet. 2001;10:99–105. doi: 10.1093/hmg/10.2.99. [DOI] [PubMed] [Google Scholar]
- 88.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–78. doi: 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- 89.Metze S, Herzog VA, Ruepp MD, Muhlemann O. Comparison of EJC-enhanced and EJC-independent NMD in human cells reveals two partially redundant degradation pathways. RNA. 2013;19:1432–48. doi: 10.1261/rna.038893.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29:1037–47. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- 91.Neri M, Torelli S, Brown S, Ugo I, Sabatelli P, et al. Dystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the human. Neuromuscul Disord. 2007;17:913–18. doi: 10.1016/j.nmd.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 92.Nguyen LS, Kim HG, Rosenfeld JA, Shen Y, Gusella JF, et al. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum Mol Genet. 2013;22:1816–25. doi: 10.1093/hmg/ddt035. [DOI] [PubMed] [Google Scholar]
- 93.Nudelman I, Glikin D, Smolkin B, Hainrichson M, Belakhov V, Baasov T. Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg Med Chem. 2010;18:3735–46. doi: 10.1016/j.bmc.2010.03.060. [DOI] [PubMed] [Google Scholar]
- 94.Nudelman I, Rebibo-Sabbah A, Cherniavsky M, Belakhov V, Hainrichson M, et al. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem. 2009;52:2836–45. doi: 10.1021/jm801640k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nudelman I, Rebibo-Sabbah A, Shallom-Shezifi D, Hainrichson M, Stahl I, et al. Redesign of aminoglycosides for treatment of human genetic diseases caused by premature stop mutations. Bioorg Med Chem Lett. 2006;16:6310–15. doi: 10.1016/j.bmcl.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 96.Oussoren E, Keulemans J, van Diggelen OP, Oemardien LF, Timmermans RG, et al. Residual α-L-iduronidase activity in fibroblasts of mild to severe mucopolysaccharidosis type I patients. Mol Genet Metab. 2013;109:377–81. doi: 10.1016/j.ymgme.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 97.Palmer E, Wilhelm JM. Mistranslation in a eucaryotic organism. Cell. 1978;13:329–34. doi: 10.1016/0092-8674(78)90201-5. [DOI] [PubMed] [Google Scholar]
- 98.Palmer E, Wilhelm JM, Sherman F. Phenotypic suppression of nonsense mutants in yeast by aminoglycoside antibiotics. Nature. 1979;277:148–50. doi: 10.1038/277148a0. [DOI] [PubMed] [Google Scholar]
- 99.Panchal RG, Wang S, McDermott J, Link CJ., Jr Partial functional correction of xeroderma pigmentosum group A cells by suppressor tRNA. Hum Gene Ther. 1999;10:2209–19. doi: 10.1089/10430349950017194. [DOI] [PubMed] [Google Scholar]
- 100.Peltz SW, Morsy M, Welch EM, Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med. 2013;64:407–25. doi: 10.1146/annurev-med-120611-144851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peltz SW, Welch EM, Jacobson A, Trotta CR, Naryshkin N, et al. Nonsense suppression activity of PTC124 (ataluren) Proc Natl Acad Sci USA. 2009;106:E64. doi: 10.1073/pnas.0901936106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pichavant C, Aartsma-Rus A, Clemens PR, Davies KE, Dickson G, et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther. 2011;19:830–40. doi: 10.1038/mt.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Piotrowska E, Jakobkiewicz-Banecka J, Baranska S, Tylki-Szymanska A, Czartoryska B, et al. Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur J Hum Genet. 2006;14:846–52. doi: 10.1038/sj.ejhg.5201623. [DOI] [PubMed] [Google Scholar]
- 104.Politano L, Nigro G, Nigro V, Piluso G, Papparella S, et al. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results Acta Myol. 2003;22:15–21. [PubMed] [Google Scholar]
- 105.Qian Y, Guan MX. Interaction of aminoglycosides with human mitochondrial 12S rRNA carrying the deafness-associated mutation. Antimicrob Agents Chemother. 2009;53:4612–18. doi: 10.1128/AAC.00965-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ramsammy L, Josepovitz C, Lane B, Kaloyanides GJ. Polyaspartic acid inhibits gentamicin-induced perturbations of phospholipid metabolism. Am J Physiol. 1990;258:C1141–49. doi: 10.1152/ajpcell.1990.258.6.C1141. [DOI] [PubMed] [Google Scholar]
- 107.Rebibo-Sabbah A, Nudelman I, Ahmed ZM, Baasov T, Ben-Yosef T. In vitro and ex vivo suppression by aminoglycosides of PCDH15 nonsense mutations underlying type 1 Usher syndrome. Hum Genet. 2007;122:373–81. doi: 10.1007/s00439-007-0410-7. [DOI] [PubMed] [Google Scholar]
- 108.Recht MI, Douthwaite S, Puglisi JD. Basis for prokaryotic specificity of action of aminoglycoside antibiotics. EMBO J. 1999;18:3133–38. doi: 10.1093/emboj/18.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013;19:313–21. doi: 10.1038/nm.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reiter RJ, Tan DX, Korkmaz A, Fuentes-Broto L. Drug-mediated ototoxicity and tinnitus: alleviation with melatonin. J Physiol Pharmacol. 2011;62:151–57. [PubMed] [Google Scholar]
- 111.Rowe SM, Sloane P, Tang LP, Backer K, Mazur M, et al. Suppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54. J Mol Med. 2011;89:1149–61. doi: 10.1007/s00109-011-0787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rowe SM, Verkman AS. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb Perspect Med. 2013;3:a009761. doi: 10.1101/cshperspect.a009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rufener SC, Muhlemann O. eIF4E-bound mRNPs are substrates for nonsense-mediated mRNA decay in mammalian cells. Nat Struct Mol Biol. 2013;20:710–17. doi: 10.1038/nsmb.2576. [DOI] [PubMed] [Google Scholar]
- 114.Salas-Marco J, Bedwell DM. GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Mol Cell Biol. 2004;24:7769–78. doi: 10.1128/MCB.24.17.7769-7778.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Salas-Marco J, Bedwell DM. Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J Mol Biol. 2005;348:801–15. doi: 10.1016/j.jmb.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 116.Salas-Marco J, Fan-Minogue H, Kallmeyer AK, Klobutcher LA, Farabaugh PJ, Bedwell DM. Distinct paths to stop codon reassignment by the variant-code organisms Tetrahymena and Euplotes. Mol Cell Biol. 2006;26:438–47. doi: 10.1128/MCB.26.2.438-447.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sanchez-Alcudia R, Perez B, Ugarte M, Desviat LR. Feasibility of nonsense mutation readthrough as a novel therapeutical approach in propionic acidemia. Hum Mutat. 2012;33:973–80. doi: 10.1002/humu.22047. [DOI] [PubMed] [Google Scholar]
- 118.Sangkuhl K, Schulz A, Rompler H, Yun J, Wess J, Schoneberg T. Aminoglycoside-mediated rescue of a disease-causing nonsense mutation in the V2 vasopressin receptor gene in vitro and in vivo. Hum Mol Genet. 2004;13:893–903. doi: 10.1093/hmg/ddh105. [DOI] [PubMed] [Google Scholar]
- 119.Sarkar C, Zhang Z, Mukherjee AB. Stop codon read-through with PTC124 induces palmitoylprotein thioesterase-1 activity, reduces thioester load and suppresses apoptosis in cultured cells from INCL patients. Mol Genet Metab. 2011;104:338–45. doi: 10.1016/j.ymgme.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scheunemann AE, Graham WD, Vendeix FA, Agris PF. Binding of aminoglycoside antibiotics to helix 69 of 23S rRNA. Nucleic Acids Res. 2010;38:3094–105. doi: 10.1093/nar/gkp1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schiffelers R, Storm G, Bakker-Woudenberg I. Liposome-encapsulated aminoglycosides in preclinical and clinical studies. J Antimicrob Chemother. 2001;48:333–44. doi: 10.1093/jac/48.3.333. [DOI] [PubMed] [Google Scholar]
- 122.Schroers A, Kley RA, Stachon A, Horvath R, Lochmüller H, et al. Gentamicin treatment in McArdle disease: failure to correct myophosphorylase deficiency. Neurology. 2006;66:285–86. doi: 10.1212/01.wnl.0000194212.31318.fc. [DOI] [PubMed] [Google Scholar]
- 123.Schweingruber C, Rufener SC, Zund D, Yamashita A, Muhlemann O. Nonsense-mediated mRNA decay: mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta. 2013;1829:612–23. doi: 10.1016/j.bbagrm.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 124.Seoighe C, Gehring C. Heritability in the efficiency of nonsense-mediated mRNA decay in humans. PLoS ONE. 2010;5:e11657. doi: 10.1371/journal.pone.0011657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med. 2010;182:1262–72. doi: 10.1164/rccm.201001-0137OC. [DOI] [PubMed] [Google Scholar]
- 126.Shalev M, Kandasamy J, Skalka N, Belakhov V, Rosin-Arbesfeld R, Baasov T. Development of generic immunoassay for the detection of a series of aminoglycosides with 6′-OH group for the treatment of genetic diseases in biological samples. J Pharm Biomed Anal. 2013;75:33–40. doi: 10.1016/j.jpba.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Simon AJ, Lev A, Wolach B, Gavrieli R, Amariglio N, et al. The effect of gentamicin-induced readthrough on a novel premature termination codon of CD18 leukocyte adhesion deficiency patients. PLoS ONE. 2010;5:e13659. doi: 10.1371/journal.pone.0013659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sutcliffe JA. Improving on nature: antibiotics that target the ribosome. Curr Opin Microbiol. 2005;8:534–42. doi: 10.1016/j.mib.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 129.Tan L, Narayan SB, Chen J, Meyers GD, Bennett MJ. PTC124 improves readthrough and increases enzymatic activity of the CPT1A R160X nonsense mutation. J Inherit Metab Dis. 2011;34:443–47. doi: 10.1007/s10545-010-9265-5. [DOI] [PubMed] [Google Scholar]
- 130.Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet. 2007;39:1127–33. doi: 10.1038/ng2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tate WP, Poole ES, Horsfield JA, Mannering SA, Brown CM, et al. Translational termination efficiency in both bacteria and mammals is regulated by the base following the stop codon. Biochem Cell Biol. 1995;73:1095–103. doi: 10.1139/o95-118. [DOI] [PubMed] [Google Scholar]
- 132.Temple GF, Dozy AM, Roy KL, Kan YW. Construction of a functional human suppressor tRNA gene: an approach to gene therapy for β-thalassaemia. Nature. 1982;296:537–40. doi: 10.1038/296537a0. [DOI] [PubMed] [Google Scholar]
- 133.Tomita K, Ueda T, Watanabe K. The presence of pseudouridine in the anticodon alters the genetic code: a possible mechanism for assignment of the AAA lysine codon as asparagine in echinoderm mitochondria. Nucleic Acids Res. 1999;27:1683–89. doi: 10.1093/nar/27.7.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Usuki F, Yamashita A, Higuchi I, Ohnishi T, Shiraishi T, et al. Inhibition of nonsense-mediated mRNA decay rescues the phenotype in Ullrich’s disease. Ann Neurol. 2004;55:740–44. doi: 10.1002/ana.20107. [DOI] [PubMed] [Google Scholar]
- 135.Usuki F, Yamashita A, Kashima I, Higuchi I, Osame M, Ohno S. Specific inhibition of nonsense-mediated mRNA decay components, SMG-1 or Upf1, rescues the phenotype of Ullrich disease fibroblasts. Mol Ther. 2006;14:351–60. doi: 10.1016/j.ymthe.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 136.Usuki F, Yamashita A, Shiraishi T, Shiga A, Onodera O, et al. Inhibition of SMG-8, a subunit of SMG-1 kinase, ameliorates nonsense-mediated mRNA decay-exacerbated mutant phenotypes without cytotoxicity. Proc Natl Acad Sci USA. 2013;110:15037–42. doi: 10.1073/pnas.1300654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Viegas MH, Gehring NH, Breit S, Hentze MW, Kulozik AE. The abundance of RNPS1, a protein component of the exon junction complex, can determine the variability in efficiency of the nonsense mediated decay pathway. Nucleic Acids Res. 2007;35:4542–51. doi: 10.1093/nar/gkm461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang B, Yang Z, Brisson BK, Feng H, Zhang Z, et al. Membrane blebbing as an assessment of functional rescue of dysferlin-deficient human myotubes via nonsense suppression. J Appl Physiol. 2010;109:901–5. doi: 10.1152/japplphysiol.01366.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang D, Belakhov V, Kandasamy J, Baasov T, Li SC, et al. The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse. Mol Genet Metab. 2012;105:116–25. doi: 10.1016/j.ymgme.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, et al. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 2008;22:1381–96. doi: 10.1101/gad.468808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 142.Wells SE, Hillner PE, Vale RD, Sachs AB. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell. 1998;2:135–40. doi: 10.1016/s1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- 143.Wilhelm JM, Jessop JJ, Pettitt SE. Aminoglycoside antibiotics and eukaryotic protein synthesis: stimulation of errors in the translation of natural messengers in extracts of cultured human cells. Biochemistry. 1978;17:1149–53. doi: 10.1021/bi00600a002. [DOI] [PubMed] [Google Scholar]
- 144.Wilschanski M, Miller LL, Shoseyov D, Blau H, Rivlin J, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J. 2011;38:59–69. doi: 10.1183/09031936.00120910. [DOI] [PubMed] [Google Scholar]
- 145.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433–41. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 146.Wohlgemuth I, Pohl C, Mittelstaet J, Konevega AL, Rodnina MV. Evolutionary optimization of speed and accuracy of decoding on the ribosome. Philos Trans R Soc Lond B. 2011;366:2979–86. doi: 10.1098/rstb.2011.0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yamashita A. Role of SMG-1-mediated Upf1 phosphorylation in mammalian nonsense-mediated mRNA decay. Genes Cells. 2013;18:161–75. doi: 10.1111/gtc.12033. [DOI] [PubMed] [Google Scholar]
- 148.Yang C, Feng J, Song W, Wang J, Tsai B, et al. A mouse model for nonsense mutation bypass therapy shows a dramatic multiday response to geneticin. Proc Natl Acad Sci USA. 2007;104:15394–99. doi: 10.1073/pnas.0610878104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–79. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yukihara M, Ito K, Tanoue O, Goto K, Matsushita T, et al. Effective drug delivery system for Duchenne muscular dystrophy using hybrid liposomes including gentamicin along with reduced toxicity. Biol Pharm Bull. 2011;34:712–16. doi: 10.1248/bpb.34.712. [DOI] [PubMed] [Google Scholar]
- 151.Zhou Y, Jiang Q, Takahagi S, Shao C, Uitto J. Premature termination codon read-through in the ABCC6 gene: potential treatment for pseudoxanthoma elasticum. J Investig Dermatol. 2013;133:2672–77. doi: 10.1038/jid.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zilberberg A, Lahav L, Rosin-Arbesfeld R. Restoration of APC gene function in colorectal cancer cells by aminoglycoside- and macrolide-induced read-through of premature termination codons. Gut. 2010;59:496–507. doi: 10.1136/gut.2008.169805. [DOI] [PubMed] [Google Scholar]
- 153.Zund D, Gruber AR, Zavolan M, Muhlemann O. Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3′ UTRs. Nat Struct Mol Biol. 2013;20:936–43. doi: 10.1038/nsmb.2635. [DOI] [PubMed] [Google Scholar]