

Abstract
Objective:
To identify the unknown genetic cause in a nuclear family with an axonal form of peripheral neuropathy and atypical disease course.
Methods:
Detailed neurologic, electrophysiologic, and neuropathologic examinations of the patients were performed. Whole exome sequencing of both affected individuals was done. The effect of the identified sequence variations was investigated at cDNA and protein level in patient-derived lymphoblasts. The plasma sphingoid base profile was analyzed. Functional consequences of neuron-specific downregulation of the gene were studied in Drosophila.
Results:
Both patients present an atypical form of axonal peripheral neuropathy, characterized by acute or subacute onset and episodes of recurrent mononeuropathy. We identified compound heterozygous mutations cosegregating with disease and absent in controls in the SGPL1 gene, encoding sphingosine 1-phosphate lyase (SPL). The p.Ser361* mutation triggers nonsense-mediated mRNA decay. The missense p.Ile184Thr mutation causes partial protein degradation. The plasma levels of sphingosine 1-phosphate and sphingosine/sphinganine ratio were increased in the patients. Neuron-specific downregulation of the Drosophila orthologue impaired the morphology of the neuromuscular junction and caused progressive degeneration of the chemosensory neurons innervating the wing margin bristles.
Conclusions:
We suggest SPL deficiency as a cause of a distinct form of Charcot-Marie-Tooth disease in humans, thus extending the currently recognized clinical and genetic spectrum of inherited peripheral neuropathies. Our data emphasize the importance of sphingolipid metabolism for neuronal function.
Charcot-Marie-Tooth disease (CMT) represents a heterogeneous group of peripheral neuropathies affecting 1:2,500 individuals.1 CMT is characterized by progressive length-dependent degeneration of peripheral motor and sensory neurons, resulting in distal muscles weakness and wasting, motor impairment, sensory loss, and skeletal deformities. The disease is classified into CMT1, with median motor nerve conduction velocities (NCVs) <38 m/s and histologic evidence of demyelination and remyelination; CMT2, with normal or slightly reduced NCVs and axonal changes; and intermediate CMT, with NCVs ranging between 25 and 45 m/s and histopathologic signs of both demyelination and axonal degeneration.2,3
CMT shows all modes of inheritance, with autosomal recessive forms (AR-CMT) accounting for <10% of the European CMT population.4 This is likely underestimated due to the usually small sibship size, with many AR-CMT cases being considered sporadic. There are >24 AR-CMT genes explaining the disease in a small subset of patients.5 Most AR-CMT families carry private mutations in rare causal genes, challenging their molecular diagnosis. Furthermore, the encoded proteins are involved in diverse cellular processes and the cause-effect relationship is unclear. The extensive etiologic heterogeneity impedes the development of effective treatment strategies.
Here, we report a candidate gene (SGPL1) for AR-CMT2 with atypical disease course. Studies in patient-derived biosamples suggested that this phenotype is due to partial loss of SGPL1 function. Neuron-specific downregulation of the Drosophila orthologue impaired the morphology of the neuromuscular junction (NMJ) and caused progressive neurodegeneration, emphasizing the role of SGPL1 in establishing neuronal contact sites and axonal maintenance.
METHODS
Standard protocol approvals and patient consents.
Written informed consent was obtained. This study was approved by the local institutional review boards.
Clinical and electrophysiologic evaluations.
Patients and parents underwent routine neurologic examinations. Motor examination followed Medical Research Council (MRC) scale. Standard nerve conduction studies (NCS) with surface electrode and concentric needle EMG were performed using a Premier electromyograph (Medelec, Woking, UK).6
Morphologic studies.
Nerve biopsy was obtained from left sural nerve and was examined as described.7
Whole exome sequencing (WES) and homozygosity mapping.
WES was performed at Beijing Genomics Institute, China. Exomes were captured with SureSelect Human All Exon V5 array (Agilent, Santa Clara, CA) followed by paired‐end sequencing on Hiseq2000 (Illumina, San Diego, CA). Sequencing read mapping, variant calling, and annotation were done using GenomeComb.8 Homozygosity mapping was performed as described.9
Mutation analysis.
SGPL1 (NM_003901.3, NP_003892.2) was analyzed by Sanger sequencing as described.9 Pathogenicity was predicted with Condel (bg.upf.edu/fannsdb/) and MetaSnp (snps.biofold.org/meta-snp/).
Cohort screening.
The cohort contained 107 AR-CMT cases and 803 sporadic cases. Probands displayed CMT1 (267), CMT2 (375), intermediate (85), or unclassified peripheral neuropathy (183). All exons, exon–intron boundaries, and 5′UTR of SGPL1 were analyzed using custom-designed MASTRTM assay (multiplicom.com/), followed by Sanger sequencing validation.
Dosage analysis.
Multiplex amplicon quantification assay (multiplicom.com) included 15 amplicons encompassing SGPL1 exons and 5 control amplicons. A dosage quotient (DQ) was calculated by comparing normalized peak areas between patients and controls; DQ <0.5 indicated a deletion.
Lymphoblast cultures.
Epstein-Barr virus–transformed lymphoblast cultures were maintained as described.10 To inhibit nonsense-mediated mRNA decay (NMD), lymphoblasts were incubated with 150 μg/mL cycloheximide (CHX, Sigma-Aldrich, St. Louis, MO) or vehicle (DMSO, Sigma-Aldrich) for 4 hours, followed by RNA isolation and RT-PCR analysis. To inhibit proteasome function, cells were treated with 40 μM MG-132 (Calbiochem, San Diego) or vehicle (DMSO).
RNA isolation and RT-PCR.
Total RNA of lymphoblasts was extracted using RNeasy Mini Kit (Qiagen, Germantown, MD) and treated with TURBO DNase (Applied Biosystems, Foster City, CA). SGPL1 cDNA was synthesized using SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Primers are listed in table e-1 at Neurology.org.
Immunoblotting.
Protein extracts from lymphoblasts or mouse tissues were isolated as described.10 Membranes were immunoblotted with anti–sphingosine 1-phosphate lyase (SPL) (1:200, R&D Systems, Minneapolis, MN; and Atlas Antibodies, Stockholm, Sweden) or anti‐β‐actin (1:20,000, Sigma‐Aldrich) antibodies. Results were visualized by chemiluminescence detection (GE Healthcare, Waukesha, WI).
Protein structure modeling.
Chimera11 was used to visualize SPL (pdb: 4Q6R)12 and to substitute the isoleucine184 for a threonine with the swapaa command. The conformation of the threonine side chain was modeled with the highest probability (96%) based on an internal rotamer library, the lowest clash score, and the largest amount of H-bonding interactions with the surrounding amino acids.
Lipid analysis.
For lipid analysis, refer to e-Methods 1–4.
Drosophila stocks and analysis.
For Drosophila stocks and analysis, refer to e-Methods 1–4 and table e-2.
RESULTS
Clinical findings.
We present clinical data of 2 affected siblings from a family of Serbian ethnicity (figure 1A). The proband (II-1) developed walking difficulties within several weeks at age 12 years, 1 month after intense stress and a febrile episode (table 1). Initial neurologic examination revealed weakness of distal lower limb (LL) muscles, more pronounced in extensor muscle groups, absent Achilles tendon reflexes, mildly diminished vibration sense at the level of ankles, and mild scoliosis. The rest of the examination was unremarkable. Within the following months, wasting of distal LL muscles became prominent, while their weakness plateaued (MRC 0–3). Initial NCS revealed undetectable compound muscle action potentials and sensory nerve action potentials for LL nerves and normal parameters for upper limb (UL) nerves (table 2). EMG showed spontaneous activity for distal LL muscles and a neuropathic pattern for all examined distal LL and UL muscles. At ages 18 years and 21 years, the patient experienced 2 episodes of muscle weakness in the distribution of the left ulnar nerve, without any provocative events. Full recovery occurred within 1 year after the first episode and only partial improvement after the second. NCS supported the diagnosis of axonal polyneuropathy (table 2) and the 2 episodes of left ulnar mononeuropathy were corroborated by electrophysiologic findings (data not shown). In the next 11 years, there were no changes in his symptoms and signs. At the last clinical examination (age 32 years), he had mild motor deficit of the left hand, in the distribution of the ulnar nerve, bilateral severe motor impairment of the legs, and mildly decreased vibration sense (figure 1B).
Figure 1. Pedigree and clinical features of the studied autosomal recessive Charcot-Marie-Tooth 2 family.

(A) Family tree. Square symbols represent males and circles females. Black symbols indicate affected individuals. (B) Serial images of the patients. Patient II-1 at age 32 presents with unilateral mild hypotrophy and weakness of the muscles in the first interosseal space, bilateral atrophy, and severe weakness (Medical Research Council scale 0–2) of all distal leg muscles. Patient II-2 at age 28 has normal appearance of the distal muscles of the hands, unilateral severe weakness of hallux extensor, and normal standing on tiptoe.
Table 1.
Clinical findings
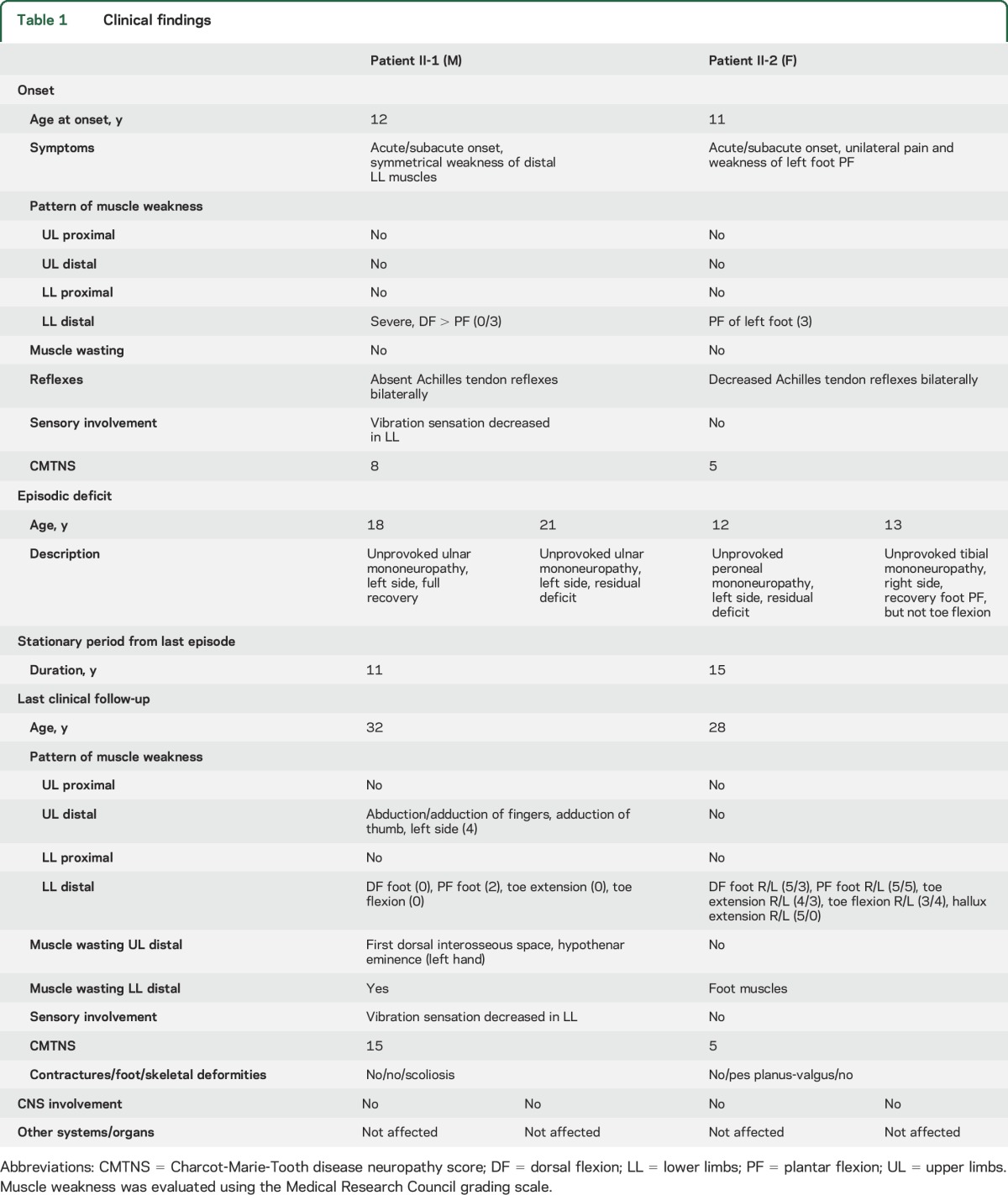
Table 2.
Electrophysiologic findings
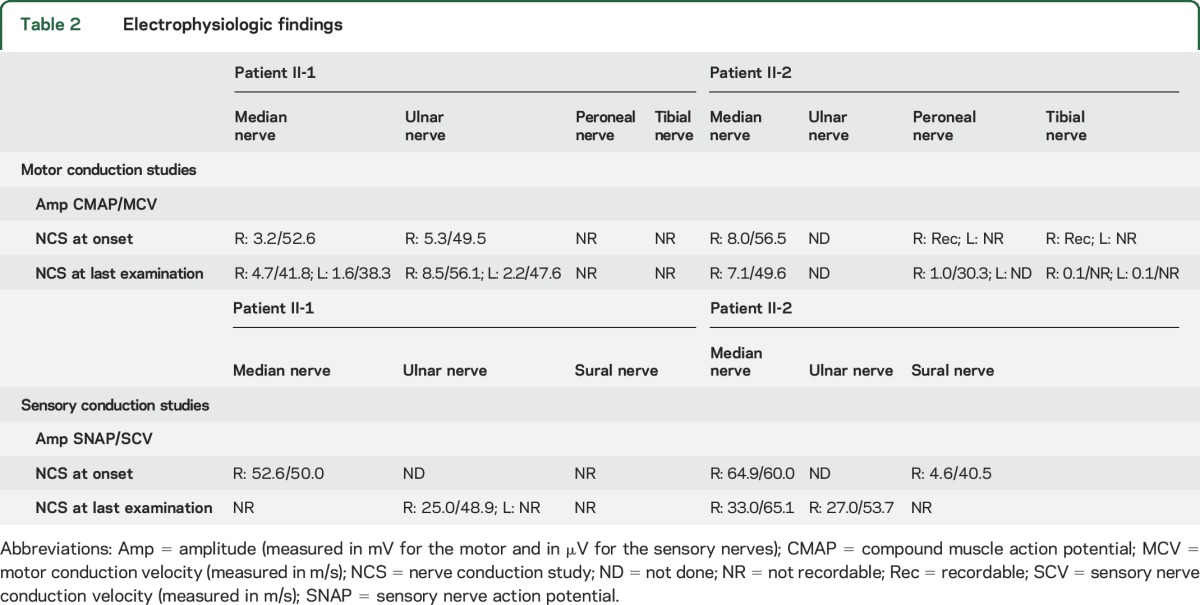
The younger sister (II-2) developed acute transient pain in the left foot without any provocative events at age 11 years, followed by a weakness of left foot plantar flexors, bilaterally decreased Achilles tendon reflexes, and no sensory deficit (table 1). NCS performed 2 months later showed normal electrophysiology for the median nerve and abnormal parameters for sural, peroneal, and tibial nerves, more pronounced on the left side (table 2). NCS findings were compatible with an axonal neuropathy. Over the next 5 months, the disease progressed and the most notable deficit was in the domain of the left tibial nerve (weakness of foot plantar flexion [PF] and toe flexion). Distal LL muscles showed moderate weakness of the left toe extensor and very mild weakness of the same muscle group on the right. Improvement was noticed for left foot PF within a few months. Two episodes of muscle weakness followed, first at age 12 years in the distribution of the left peroneal nerve and second at age 13 years in the distribution of the right tibial nerve. Improvement of right foot PF was detected in the follow-up period. No further progression appeared over the last 15 years. At the last clinical examination (age 28 years), she had asymmetrical distal LL motor deficit and no sensory impairment (figure 1B). Her functional status was very good.
Sural nerve biopsy of II-2 was performed 2 months after disease onset (data not shown). On the teased preparation, about 20% of nerve fibers showed active axonal disintegration. There were no detectable regenerating axons or endoneurial fibrosis.
Additional evaluations of both patients included blood tests with complete biochemical and immunologic analysis, CSF analysis, physical examination, and cognitive testing. All tests and examinations were within normal limits. Involvement of CNS, or any other systems and organs, was not noted.
Identification of SGPL1 as AR-CMT2 candidate gene.
WES was performed on patients II-1 and II-2. All known CMT genes were initially analyzed, and sequence gaps therein were covered by Sanger sequencing; however, no causal variants were found. WES-based homozygosity mapping revealed a low number (6) and small size (maximum 6 Mb) of shared autozygous regions, suggesting a compound heterozygous recessive trait. WES data were nevertheless filtered for both homozygous and compound heterozygous shared variants, based on novelty, effect on protein, population frequency, and cosegregation (table e-3). No homozygous variants fulfilled these criteria, while 2 heterozygous sequence variations, c.551T>C (p.Ile184Thr) and c.1082C>G (p.Ser361*), were identified in SGPL1 encoding SPL (figure 2A). Both mutations were in trans, cosegregated with the phenotype, and were absent in 190 Serbian and 470 non-Serbian controls. Subsequently, 910 AR-CMT and sporadic cases of various ethnicity were screened for mutations in SGPL1. In 8 probands, single rare heterozygous variants were identified (table e-4); however, no in/dels were found on the second allele using a gene dosage assay. No homozygous single exon in/dels were found either. Thus, SGPL1 mutations are a very rare cause of AR-CMT.
Figure 2. Sphingosine 1-phosphate (S1P) lyase (SPL): Transcript, protein, and metabolite analysis.

(A) Electropherograms around the 2 mutations in SGPL1 (c.551T>C and c.1082C>G) upon analyzing genomic DNA (gDNA) or complementary DNA (cDNA) isolated from peripheral mononuclear blood cells, before and after cycloheximide (CHX) treatment. (B) Crystal structure of the dimeric human sphingosine 1-phosphate lyase, adapted from pdb 4Q6R. The 2 subunits (A, B) of the dimeric protein are displayed in tan and blue ribbons, respectively. Top right: zoom in view shows the Ile184 residue that is surrounded by 7 other hydrophobic residues (i.e., Ala163, Phe167, Leu181, Ala402, Trp405 from subunit A; Leu130, Tyr133 from subunit B) to constitute a hydrophobic core. Bottom right: the p.Ile184Thr mutation substitutes the long aliphatic side chain of isoleucine with a small and polar hydroxyl group of threonine in the middle of the hydrophobic core. (C) Immunoblotting analysis of SPL in 20 μg of protein lysates of lymphoblast cultures from all family members and 7 healthy control individuals. β-actin is used as loading control. Chemiluminescence exposure time of 60 seconds. (D) Immunoblotting analysis of SPL in 30 μg of protein lysates of lymphoblasts of patient II-2 incubated with vehicle alone (DMSO), with the MG-132 proteasome inhibitor, or nontreated (NT). SPL accumulates in a time-dependent manner in cells from the patient, compared to the control. β-actin demonstrates equal loading. Chemiluminescence exposure time of 600 seconds. (E) S1P levels in plasma of patents, patients, and unrelated controls (n = 24). (F) Total sphingosine/sphinganine (SO/SA) ratio after hydrolysis in plasma of patients, parents, and unrelated controls (n = 3). (E, F) Error bars represent the SD. ***p < 0.001; ns = nonsignificant (one-way analysis of variance with Bonferroni multiple comparison test against the control group).
SGPL1 mutational effects.
The c.1082C>G nonsense mutation is located in exon 12 (out of 15) of SGPL1 and therefore might trigger NMD.13 Sanger sequencing of SGPL1 cDNA derived from lymphoblasts did not reveal the c.1082 G allele in the father or the patients, resulting in an ostensible homozygosity for the c.551C mutation in the affected individuals (figure 2A). Inhibition of NMD with CHX restored the cDNA heterozygosity for both mutations, suggesting that the c.1082 G allele was post-transcriptionally degraded. In the patients, SPL function would then be secured by the remaining c.551C-allele encoding p.Ile184Thr protein. In silico structural analysis showed that isoleucine184 lies within a stretch of 8 hydrophobic residues. The p.Ile184Thr mutation substitutes an aliphatic by a polar residue, disrupting the hydrophobic core and causing protein instability (figure 2B). Accordingly, the protein was nearly undetectable in lymphoblast lysates from the patients compared to controls upon immunoblotting analysis with 2 different anti-SPL antibodies (figure 2C). MG-132 treatment of cells from patient II-2 induced SPL accumulation, indicating that the mutant protein is translated but unstable and subject to degradation (at least in part) via the proteasomal system (figure 2D). This suggests that the SGPL1 mutations affect transcription and protein expression, leading to a depletion of SPL function. SPL catalyzes the degradation of S1P in the last step of sphingolipid catabolism. Therefore, we analyzed the S1P and total sphingoid base levels in plasma of patients, parents, and unrelated controls. S1P levels appeared to be generally lower in the parents compared to the control average but were significantly elevated in patients (figure 2E). Also, the ratio of total sphingosine/sphinganine (SO/SA) was significantly increased in patient plasma (figure 2F).
SPL downregulation in Drosophila neurons.
SPL is an essential and evolutionary conserved enzyme expressed in multiple tissues14 including sciatic nerve (figure e-1). In Drosophila melanogaster, SPL has no paralogues and one orthologue (Sply, CG8946), sharing 49% identity and 68% similarity with the human protein.15 Sply-deficient flies exhibit compromised muscle development, decreased fecundity, and semilethality.15 Because our patients present with neurologic symptoms, we downregulated Sply exclusively in the nervous system, using RNAiSply expressed by the pan-neuronal driver nsyb-Gal4, and assessed NMJ morphology in third instar larvae. This in vivo paradigm has been widely used to study the effect of genes causing neurodegeneration.16,17 For control purposes, we RNAi-downregulated Myc, an essential gene for synapse development,16 and Smn, a gene associated with spinal muscular atrophy (SMA).17 To account for random RNAi effects, antisense oligos against genes not transcribed in neurons (Cup and S-Lap5) were expressed.
In control flies, we observed mature NMJs characterized by beads-on-a-string-like structures formed at the axon terminus (figures 3A and e-2). In contrast, neuronal downregulation of Sply using RNAis targeting different transcript regions reduced NMJ arborization and decreased the number of synaptic boutons (figure 3, B–C). As expected, NMJ effects were observed when downregulating both neuronally relevant control genes.16,17 No differences in NMJ length were found. These findings demonstrate a role for Sply in maintaining the morphology of axonal terminals.
Figure 3. Analysis of neuron-specific Sply deficiency in Drosophila.

(A) Immunolabeling of presynaptic (anti-HRP antibody, green) and postsynaptic (anti-DLG antibody, magenta) compartments of the neuromuscular junctions (NMJs) in third instar larvae, pan-neuronally (nsyb-Gal4) expressing RNAi constructs against Sply (Sply knockdown; RNAiSply[1], RNAiSply[2]), neuronally relevant genes (RNAiSmn, RNAiMyc), genes not expressed in neurons (RNAi controls; RNAiCup, RNAiS-Lap5[1], RNAiS-Lap[2]), or the driver alone (control genetic background; nsyb-Gal4/+). Scale bar, 20 μm. (B–D) Quantitative analysis of the NMJ phenotypes. Maximum intensity projections of Z-stacks comprising the full NMJ were used. The number of boutons per NMJ (B) was counted using the Cell Counter plugin. ImageJ was used to count the number of individual branch segments (C) and calculate the total NMJ length (D). Error bars represent the SD of at least 13 NMJs per genotype. *p < 0.05; ***p < 0.001; ns = nonsignificant (one-way analysis of variance with Bonferroni multiple comparison test against the control genetic background). (E) Nerve tract along the wing L1 vein visualized by mCherry, where RNAi constructs are expressed using the dpr-Gal4 driver. Representative images of wings are shown for day 1 and day 20+ old flies expressing the driver alone (Dpr-Gal4/+), neuronally relevant gene (RNAiSmn), or Sply (RNAiSply[1]). Scale bar, 20 μm. (F) Quantification of the number of wings showing axonal fragmentation per genotype (n = 30–60/genotype).
To determine whether Sply downregulation might cause axonal neurodegeneration, similarly to Smn, we expressed RNAiSply in the chemosensory neurons innervating the wing margin bristles (dpr-Gal4 driver) visualized by the red fluorescent protein mCherry (UAS-mCD8chRFP, figure e-3). While at day 1 posteclosion the wing neurons looked comparable in flies with different genotypes, from day 20 onward pronounced fragmentation of the mCherry signal was noted in flies expressing RNAiSply or RNAiSmn (figure 3, E–F). These results demonstrate that neuronal depletion of Sply induces progressive axonal degeneration in Drosophila.
DISCUSSION
Currently known genes explain only ∼40% of AR-CMT cases, suggesting many unidentified genetic causes.18 Here, we provide genetic and functional evidence that SGPL1 is a candidate AR-CMT gene. The associated phenotype is distinct from other AR-CMT2 subtypes and is characterized by acute/subacute onset, unilateral motor deficit in one patient, and episodes of mononeuropathy with a tendency for improvement in both patients.
The deterioration pattern seen in both patients is atypical for axonal CMT. Rather, it is observed in acquired neuropathies,19 hereditary neuropathies with liability to pressure palsies (HNPP), or hereditary neuralgic amyotrophy (HNA).20 As opposed to the AR-CMT in our family, HNPP and HNA are dominantly inherited. HNPP presents as recurrent painless focal neuropathy that, contrary to our patients, is demyelinating in nature. Sural nerve biopsies of patients with HNPP show segmental demyelination and remyelination, which are not found in the nerve specimen of II-2. HNA is characterized by triggered transient painful episodes of brachial or less frequently lumbar plexus neuropathy with muscle weakness and atrophy. Apart from axonal neuropathy in the implicated nerve, electrophysiology is normal. Contrastingly, our patients exhibit clear neurophysiologic evidence of broader neuropathy than clinical signs expressed. The biopsy findings also lack some characteristic histopathologic features of CMT2; however, this might be due to the unusual onset and sampling too early in the disease course. Even though the phenotype in our patients could resemble one of inflammatory neuropathy,21 coexistence of hereditary and inflammatory neuropathy was excluded. Provocative factors were not reported in any but the first episode in the proband. Overall, the neurologic presentation of our patients extends the currently recognized clinical spectrum of inherited peripheral neuropathies.
We identified compound heterozygous SPL mutations in patients with AR-CMT. p.Ser361* triggers NMD in patients' cells. p.Ile184Thr changes a nonpolar to polar amino acid, leading to protein instability and subsequent degradation. While the degree of degradation might differ between tissues, this finding supports the hypothesis that the observed CMT phenotype is due to SPL deficiency. Notably, we detected trace amounts of SPL in patient lymphoblasts compared to controls. This result, together with the fact that Ile184 is located outside of the catalytic pocket, suggests that the mutant has some residual activity.
SPL catalyzes the irreversible degradation of S1P into phosphoethanolamine and hexadecenal in the final step of sphingolipid catabolism.22,23 Phosphoethanolamine can be used for phospholipid synthesis. Alternatively, S1P may re-enter the sphingolipid metabolism by dephosphorylation to sphingosine and subsequent conversion to ceramide and complex sphingolipids. S1P itself is a potent signaling molecule involved in cell migration, proliferation, and apoptosis.22 Thus, SPL acts at the crossroad of sphingolipid and phospholipid metabolism, and plays a pivotal role in fine-tuning the concentrations of its bioactive substrate. In patient plasma, S1P levels were significantly increased compared to parents or unrelated controls. In parallel, the patients showed a significant increase in the total SO/SA ratio, indicating that the relative proportion of individual sphingolipid subspecies is altered.
Sgpl−/− mice display growth retardation and severe congenital multisystemic abnormalities, leading to premature death.24 Brain morphology is unremarkable, but mice were not examined for neurologic symptoms. Isolated cerebellar neurons, however, have decreased viability and undergo neurodegeneration.25 They accumulate S1P, which is neurotoxic25,26 and causes calpain activation, cell cycle re-entry, and ultimately apoptosis.25 Thus, neuron-specific defects likely exist in Sgpl−/− mice, but are masked by the severe impairment of other systems. Flies with systemic Sply deficiency exhibit developmental lethality, abnormal flight muscle development and integrity, impaired locomotion, decreased fecundity, and enhanced apoptosis.15
Using our experience in modeling CMT in Drosophila,27,28 we aimed to disentangle the neuron-specific from somatic effects of SPL deficiency. Because of the dying-back neuropathy in our patients, we investigated the Drosophila NMJ, a powerful system for studying synaptic development, morphology, and function.29 Moreover, this is a valid model for genetic and molecular analysis of neuromuscular disorders like CMT27,30–32 and SMA.17 Silencing Sply in the fly nervous system induced characteristic NMJ morphologic defects. Similar abnormalities were observed when knocking down Myc or Smn, essential genes for neuronal development or involved in neuromuscular pathologies, respectively.16,17 To better distinguish developmental from neurodegeneration effects, we applied a second in vivo paradigm, where we downregulated Sply in the long chemosensory neurons in the wings of adult flies. Remarkably, Sply deficiency caused age-dependent destruction of axonal integrity, thereby demonstrating its causal involvement in axonal degeneration. In this way, our functional genomics approach provided independent evidence for the essential function of SPL in neurons in vivo. Furthermore, we demonstrate that the fruit fly is a functional tool to validate candidate genes involved in neurodegeneration.
Neurons depend on sphingolipids for axon guidance, neurotrophin signaling, and synaptic transmission.33 Within the CMT category, mutations in SPTLC1 and SPTLC2 encoding subunits of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I.34,35 Defects in sphingolipid-metabolizing enzymes lead to AR neurometabolic disorders with highly variable onset, development, and symptoms.36 This variability depends mainly on the cell-type specific pattern of sphingolipid expression or uptake, and the residual activity of the defective enzyme. A complete enzyme deficiency leads to early onset and severe course, presenting as a neurologic and somatic fatal disorder. Patients whose cells retain traces of residual activity will have an attenuated course with less somatic signs, or can be clinically undiagnosed and detected as accidental biochemical finding.36,37 Introducing next-generation sequencing into the clinical workup, together with deep phenotyping, will allow identification of such rare neurometabolic patients.38 Our study illustrates this threshold concept by demonstrating that partial loss of SPL function leads to a noncongenital disorder exclusively affecting peripheral nerves. Molecular studies of additional cases of SPL deficiency will help delineate its full clinical spectrum.
We demonstrate that biallelic mutations in SGPL1 may lead to a distinct CMT phenotype and confirm the importance of the sphingolipid metabolism for neuronal function. Our findings have implications for clinical and molecular diagnosis and potential treatment strategy in patients with inherited peripheral neuropathies.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the family members for their participation, the Genomics Service Facility of the VIB Department of Molecular Genetics (VIB-DMG, Belgium) for sequencing and cell maintenance support, the VIB-DMG Bioinformatics Unit for next-generation sequencing data analysis, and Dr. Maria-Luise Erfurth and Prof. Ivo Kremensky for discussions.
GLOSSARY
- AR
autosomal recessive
- CHX
cycloheximide
- CMT
Charcot-Marie-Tooth disease
- DQ
dosage quotient
- HNA
hereditary neuralgic amyotrophy
- HNPP
hereditary neuropathies with liability to pressure palsies
- LL
lower limb
- MRC
Medical Research Council
- NCS
nerve conduction studies
- NCV
nerve conduction velocities
- NMD
nonsense-mediated mRNA decay
- NMJ
neuromuscular junction
- PF
plantar flexion
- SMA
spinal muscular atrophy
- SO/SA
sphingosine/sphinganine
- SPL
sphingosine 1-phosphate lyase
- UL
upper limb
- WES
whole exome sequencing
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Derek Atkinson, Jelena Nikodinovic Glumac: study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript for intellectual content. Biljana Ermanoska, Bob Asselbergh, Alejandro Estrada-Cuzcano, Kristien Peeters, David Blocquel, Xiang-Lei Yang, Regula Steiner, Thorsten Hornemann: acquisition of data, analysis and interpretation of data, critical revision of manuscript for intellectual content. Els De Vriendt, Tinne Ooms: acquisition of data, analysis and interpretation of data. Albena Jordanova, Vedrana Milic Rasic: study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript for intellectual content, study supervision.
STUDY FUNDING
This study was funded in part by the University of Antwerp (TOP BOF 29069 to A.J.), the Fund for Scientific Research-Flanders (FWO, G.0543.13 and G0D7713N, to A.J.), the Belgian Association against Neuromuscular Disorders (ABMM to A.J.), and the NIH (NS 085092 to X.-L.Y. and A.J.). D.A. and A.E-C. are supported by fellowships from the Fund for Scientific Research-Flanders.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Reilly MM, Murphy SM, Laura M. Charcot-Marie-Tooth disease. J Peripher Nervous Syst 2011;16:1–14. [DOI] [PubMed] [Google Scholar]
- 2.Ouvrier R. What can we learn from the history of Charcot-Marie-Tooth disease? Dev Med Child Neurol 2010;52:405–406. [DOI] [PubMed] [Google Scholar]
- 3.Davis CJ, Bradley WG, Madrid R. The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve biopsy studies: I: clinical, genetic and electrophysiological findings and classification. J Genet Hum 1978;26:311–349. [PubMed] [Google Scholar]
- 4.Dubourg O, Azzedine H, Verny C, et al. Autosomal-recessive forms of demyelinating Charcot-Marie-Tooth disease. Neuromolecular Med 2006;8:75–86. [DOI] [PubMed] [Google Scholar]
- 5.Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol 2013;9:562–571. [DOI] [PubMed] [Google Scholar]
- 6.Heckman JD. Electrodiagnosis in diseases of nerve and muscle. Orthopedics 1984;7:601–604. [DOI] [PubMed] [Google Scholar]
- 7.Drulovic J, Dozic S, Levic Z, et al. Unusual association of multiple sclerosis and tomaculous neuropathy. J Neurol Sci 1998;157:217–222. [DOI] [PubMed] [Google Scholar]
- 8.Reumers J, De Rijk P, Zhao H, et al. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat Biotechnol 2011;30:61–68. [DOI] [PubMed] [Google Scholar]
- 9.Kancheva D, Atkinson D, De Rijk P, et al. Novel mutations in genes causing hereditary spastic paraplegia and Charcot-Marie-Tooth neuropathy identified by an optimized protocol for homozygosity mapping based on whole-exome sequencing. Genet Med 2015;18:600–607. [DOI] [PubMed] [Google Scholar]
- 10.Zimon M, Baets J, Almeida-Souza L, et al. Loss-of-function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nat Genet 2012;44:1080–1083. [DOI] [PubMed] [Google Scholar]
- 11.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 12.Weiler S, Braendlin N, Beerli C, et al. Orally active 7-substituted (4-benzylphthalazin-1-yl)-2-methylpiperazin-1-yl]nicotinonitriles as active-site inhibitors of sphingosine 1-phosphate lyase for the treatment of multiple sclerosis. J Med Chem 2014;57:5074–5084. [DOI] [PubMed] [Google Scholar]
- 13.Hentze MW, Kulozik AE. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 1999;96:307–310. [DOI] [PubMed] [Google Scholar]
- 14.Bektas M, Allende ML, Lee BG, et al. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J Biol Chem 2010;285:10880–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herr DR, Fyrst H, Phan V, et al. Sply regulation of sphingolipid signaling molecules is essential for Drosophila development. Development 2003;130:2443–2453. [DOI] [PubMed] [Google Scholar]
- 16.Oortveld MA, Keerthikumar S, Oti M, et al. Human intellectual disability genes form conserved functional modules in Drosophila. PLoS Genet 2013;9:e1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang HC, Dimlich DN, Yokokura T, et al. Modeling spinal muscular atrophy in Drosophila. PLoS One 2008;3:e3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zimon M, Battaloglu E, Parman Y, et al. Unraveling the genetic landscape of autosomal recessive Charcot-Marie-Tooth neuropathies using a homozygosity mapping approach. Neurogenetics 2015;16:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryan MM, Tilton A, De Girolami U, Darras BT, Jones HR Jr. Paediatric mononeuritis multiplex: a report of three cases and review of the literature. Neuromuscul Disord 2003;13:751–756. [DOI] [PubMed] [Google Scholar]
- 20.Stogbauer F, Young P, Kuhlenbaumer G, De Jonghe P, Timmerman V. Hereditary recurrent focal neuropathies: clinical and molecular features. Neurology 2000;54:546–551. [DOI] [PubMed] [Google Scholar]
- 21.Rajabally YA, Adams D, Latour P, Attarian S. Hereditary and inflammatory neuropathies: a review of reported associations, mimics and misdiagnoses. J Neurol Neurosurg Psychiatry 2016;87:1051–1060. [DOI] [PubMed] [Google Scholar]
- 22.van Echten-Deckert G, Hagen-Euteneuer N, Karaca I, Walter J. Sphingosine-1-phosphate: boon and bane for the brain. Cell Physiol Biochem 2014;34:148–157. [DOI] [PubMed] [Google Scholar]
- 23.Bandhuvula P, Saba JD. Sphingosine-1-phosphate lyase in immunity and cancer: silencing the siren. Trends Mol Med 2007;13:210–217. [DOI] [PubMed] [Google Scholar]
- 24.Schmahl J, Raymond CS, Soriano P. PDGF signaling specificity is mediated through multiple immediate early genes. Nat Genet 2007;39:52–60. [DOI] [PubMed] [Google Scholar]
- 25.Hagen N, Hans M, Hartmann D, Swandulla D, van Echten-Deckert G. Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ 2011;18:1356–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagen-Euteneuer N, Lutjohann D, Park H, Merrill AH Jr, van Echten-Deckert G. Sphingosine 1-phosphate (S1P) lyase deficiency increases sphingolipid formation via recycling at the expense of de novo biosynthesis in neurons. J Biol Chem 2012;287:9128–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ermanoska B, Motley WW, Leitao-Goncalves R, et al. CMT-associated mutations in glycyl- and tyrosyl-tRNA synthetases exhibit similar pattern of toxicity and share common genetic modifiers in Drosophila. Neurobiol Dis 2014;68:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Storkebaum E, Leitao-Goncalves R, Godenschwege T, et al. Dominant mutations in the tyrosyl-tRNA synthetase gene recapitulate in Drosophila features of human Charcot-Marie-Tooth neuropathy. Proc Natl Acad Sci USA 2009;106:11782–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller DL, Ballard SL, Ganetzky B. Analysis of synaptic growth and function in Drosophila with an extended larval stage. J Neurosci 2012;32:13776–13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niehues S, Bussmann J, Steffes G, et al. Impaired protein translation in Drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nat Commun 2015;6:7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopez Del Amo V, Seco-Cervera M, Garcia-Gimenez JL, Whitworth AJ, Pallardo FV, Galindo MI. Mitochondrial defects and neuromuscular degeneration caused by altered expression of Drosophila Gdap1: implications for the Charcot-Marie-Tooth neuropathy. Hum Mol Genet 2015;24:21–36. [DOI] [PubMed] [Google Scholar]
- 32.Janssens K, Goethals S, Atkinson D, et al. Human Rab7 mutation mimics features of Charcot-Marie-Tooth neuropathy type 2B in Drosophila. Neurobiol Dis 2014;65:211–219. [DOI] [PubMed] [Google Scholar]
- 33.Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM, Jr. Lipid rafts in neuronal signaling and function. Trends Neurosci 2002;25:412–417. [DOI] [PubMed] [Google Scholar]
- 34.Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet 2001;27:309–312. [DOI] [PubMed] [Google Scholar]
- 35.Rotthier A, Auer-Grumbach M, Janssens K, et al. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am J Hum Genet 2010;87:513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolter T, Sandhoff K. Sphingolipid metabolism diseases. Biochim Biophys Acta 2006;1758:2057–2079. [DOI] [PubMed] [Google Scholar]
- 37.Rapola J. Lysosomal storage diseases in adults. Pathol Res Pract 1994;190:759–766. [DOI] [PubMed] [Google Scholar]
- 38.Tarailo-Graovac M, Shyr C, Ross CJ, et al. Exome sequencing and the management of neurometabolic disorders. N Engl J Med 2016;374:2246–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.