

Abstract
Objective:
To retrospectively investigate whether disease-modifying therapies (DMTs) exert differential effects on rates of retinal atrophy in relapsing-remitting multiple sclerosis (RRMS), as assessed using optical coherence tomography (OCT).
Methods:
A total of 402 patients with RRMS followed at the Johns Hopkins MS Center who underwent Cirrus-HD OCT were assessed for eligibility. Inclusion criteria included at least 1 year of OCT follow-up and adherence to a single DMT during the period of follow-up. Combined thickness of the ganglion cell + inner plexiform (GCIP) and other retinal layers was computed utilizing automated macular segmentation. Retinal thickness changes were analyzed using mixed-effects linear regression.
Results:
The effects of glatiramer acetate (GA; n = 48), natalizumab (NAT; n = 46), and interferon-β-1a subcutaneously (IFNSC; n = 35) and intramuscularly (IFNIM; n = 28) were assessed. Baseline analyses revealed no significant differences between groups in terms of age, sex, optic neuritis history, or follow-up duration. During follow-up, relative to NAT-treated patients, IFNSC- and GA-treated patients exhibited 0.37 μm/y (p < 0.001) and 0.14 μm/y (p = 0.035) faster rates of GCIP thinning, respectively, adjusting for the interval between initiation of DMT and OCT monitoring (gap time), age, sex, relapses, and disease duration. In the IFNSC group, GCIP thinning was 1.53 μm/y faster during the first year of therapy vs during the time interval afterwards (p < 0.001).
Conclusions:
Rates of GCIP atrophy in patients with RRMS vary according to DMT utilization. Our findings support OCT for monitoring neurodegenerative treatment effects in the retina, an easily accessible tissue, and as a practical outcome measure in RRMS clinical trials.
Although multiple sclerosis (MS) is conventionally regarded as an autoimmune, inflammatory, demyelinating disorder of the CNS, neurodegeneration resulting from these processes is recognized as the principal substrate of long-term disability.1,2 Currently available disease-modifying therapies (DMTs) for relapsing-remitting MS (RRMS) modulate or suppress the immune system, reducing the risk for future inflammation and associated neurodegeneration.3 Accordingly, the effect of DMTs on MRI-derived estimates of brain atrophy is a common outcome in MS trials.4–7 Recent studies reveal that retinal atrophy mirrors brain atrophy over time in MS.8 However, it remains unclear whether various DMTs differentially affect retinal atrophy, similar to their varying effects upon brain atrophy.
Optical coherence tomography (OCT) is a noninvasive, inexpensive, reliable, and reproducible imaging technique that generates high-resolution images of the retina, allowing most individual retinal layers to be discerned and quantified. Prior studies demonstrate that OCT-derived ganglion cell + inner plexiform layer (GCIP) thickness has superior reliability and reproducibility and correlates better with visual function and disability in MS than peripapillary retinal nerve fiber layer thickness (pRNFL).8–10 Rates of GCIP thinning are accelerated in patients with MS exhibiting inflammatory activity,11 and correlate strongly with rates of brain, particularly gray matter, atrophy over time.8 Despite the increasing support for OCT as an outcome for assessing neurodegeneration in MS trials, whether different DMTs differentially affect retinal atrophy in MS remains to be assessed.
The goal of this retrospective and preliminary study was to determine whether commonly utilized DMTs (for which sufficient data were available) exert differential effects on rates of retinal atrophy in RRMS, as assessed using OCT.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Johns Hopkins institutional review board and written informed consent was obtained from all study participants.
Participants.
A cohort of 402 patients with RRMS followed at the Johns Hopkins MS Center who underwent Cirrus HD-OCT (Carl Zeiss Meditec, Dublin, CA) between September 2008 and December 2015 were assessed for eligibility. Eligibility criteria included at least 1 year of OCT follow-up, with adherence to a single DMT during this period. In order to have sufficient sample sizes, only DMTs with which at least 20 patients were receiving treatment were included in the final analyses. Medical records were screened to determine the following clinical characteristics: DMT use (including date of initiation), disease duration, history and date of optic neuritis (ON) including the side affected, and clinical relapses. Patients were excluded if they had other known neurologic conditions, ocular pathology, diabetes, glaucoma, or refractive errors of ±6 D, or were receiving combinations of DMTs. Patients within 6 months of ON or who developed ON during the observational follow-up period were excluded. The final analysis included 157 patients (mean follow-up 3.0 years, SD 1.4, range 1–6) (figure).
Figure. Inclusion/exclusion flow diagram for patients with relapsing-remitting multiple sclerosis (RRMS).

DMT = disease-modifying therapy; IFN = interferon; OCT = optical coherence tomography; ON = optic neuritis.
Patients with RRMS on the following DMTs were included in this study: glatiramer acetate (GA) (Copaxone; Teva Pharmaceutical Industries, Petah Tikva, Israel), subcutaneous interferon (IFN)–β-1a (IFNSC) (Rebif; Merck Serono, Merck Serono, Darmstadt, Germany), intramuscular IFN-β-1a (IFNIM) (Avonex; Biogen Idec, Cambridge, MA), and natalizumab (NAT) (Tysabri; Biogen Idec).
A healthy control (HC) cohort (n = 47) undergoing annual OCT scans as part of a prospective OCT study at the Johns Hopkins MS center was utilized for comparison purposes. Inclusion criteria for HCs similarly included having at least 1 year of OCT follow-up, in the absence of any known neurologic or ophthalmologic conditions, ocular pathology, or diabetes.
OCT scanning.
Retinal imaging was performed with spectral-domain Cirrus HD-OCT (model 4000, software version 6; Carl Zeiss Meditec), as described elsewhere.11 Briefly, peripapillary and macular data were obtained with the Optic Disc Cube 200×200 protocol and Macular Cube 512×128 protocol, respectively. Scans with signal strength less than 7/10 or with motion artifact were excluded. All scans were reviewed to ensure adequate quality in accordance with the OSCAR-1B quality control criteria.12
pRNFL thickness values were generated by conventional Cirrus HD-OCT software, as described elsewhere.8 An automated macular segmentation method described in detail elsewhere,13 with slight modifications of the size of regions from which measurements were derived, was used to compute thicknesses of the GCIP, inner nuclear layer (INL), outer nuclear layer (ONL), and average macular thickness (AMT). The segmentation method uses a validated algorithm14 that generates thickness measurements by averaging the thickness values within a 5×5 mm circle centered at the fovea. The foveal region consisting of a 1×1 mm circle is excluded from analysis. This reproducible segmentation method has proven capability for detecting differences between MS and HC eyes.13 We have previously shown that OCT segmentation has intervisit intraclass correlation coefficients ranging from 0.91 to 0.99 for all thickness measurements, highlighting its reproducibility.10
All macular cube scans were assessed for macular microcystoid changes (referred to by some as microcystic macular edema [MME]), as well as other retinal pathologies that could adversely affect macular segmentation, as described elsewhere.15
Statistical analyses.
Statistical analyses were performed using STATA version 13 (StataCorp, College Station, TX). Comparisons between DMT cohorts were performed using the Kruskal-Wallis test for age, disease duration, follow-up duration, and gap time. The χ2 test was used to determine differences between the groups for ON history and sex. Fisher exact test was used to assess the effect of clinical relapses, race, first treatment, and MME.
In order to analyze the course of retinal layer thickness changes over the period of follow-up, time was taken as a continuous covariate (starting at the date of the first OCT observation). Retinal layer thickness changes were analyzed using mixed-effects linear regression with random intercepts. Models accounted for within-subject intereye correlations and adjusted for age, sex, the interval between initiation of DMT and OCT monitoring (gap time), baseline retinal layer thickness (implicitly accounted for in the mixed effects regression models utilized due to the longitudinal retinal thickness measures of interest representing the dependent variable in the context of models using random intercepts), disease duration, and relapses during follow-up. Although both ON history and baseline GCIP thickness influence overall rates of GCIP atrophy, baseline GCIP thickness explains a greater proportion of slope variance in this regard.8 Therefore, accounting for the baseline retinal layer thickness of interest in regression models may be superior to simply adjusting for ON history.
Based on visual inspection of the raw data, a linear spline (allowing for a change in slope to occur) was inserted at year 1 to study differences in rates of retinal layer thinning during and after the first year of treatment. p Values <0.05 were considered statistically significant. Since this was an exploratory study,16 correction for multiple comparisons was not performed, and therefore reported p values should be read as descriptive and interpreted with caution.
RESULTS
Patients.
Final analyses included 157 patients with RRMS (308 eyes). Six eyes were excluded due to ocular pathology or motion artifacts. OCT data from sufficient numbers of patients was available to assess the effects of 4 different DMT classes: GA (n = 48), IFNSC (n = 35), IFNIM (n = 28), and NAT (n = 46). Age, sex, history of ON, disease duration, or observational follow-up duration did not differ between the groups. Clinical relapse activity during the observation period was significantly different only between the IFNSC and NAT-treated groups (p = 0.024; table 1).
Table 1.
Demographics and clinical characteristics
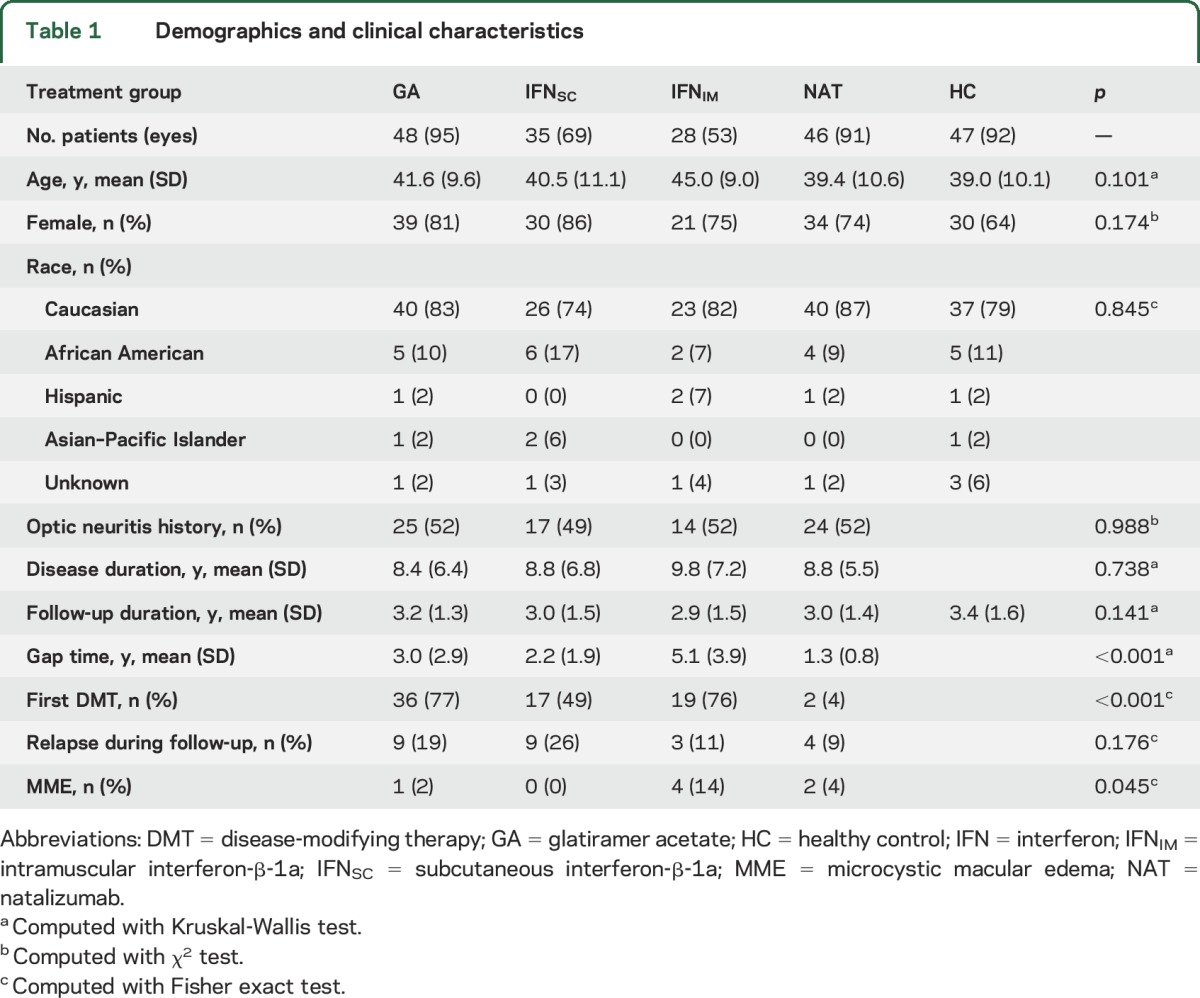
The interval between DMT initiation and commencement of OCT monitoring (gap time) was longer for the IFNIM group as compared to the IFNSC, NAT, and GA groups (mean differences: 2.9, 3.8, and 2.1 years, respectively, p < 0.001) as well as for the GA group compared to the NAT group (mean difference: 1.7 years, p < 0.001).
The HC cohort consisted of 47 individuals (92 eyes). Two eyes were excluded due to missing follow-up scans. There were no differences in age (p = 0.101), sex (p = 0.174), or follow-up duration (p = 0.141) between the HC and RRMS cohorts.
Comparisons in baseline OCT measures.
At baseline, analyses revealed significant differences between the IFNSC- and NAT-treated groups for GCIP thickness (72.7 and 67.5 μm, respectively; p = 0.003), pRNFL thickness (87.8 and 81.5 μm, respectively; p = 0.015), and AMT (306.6 and 298.4 μm, respectively; p = 0.030). INL thickness differed between the IFNIM- and GA-treated groups at baseline (43.3 and 44.6 μm, respectively; p = 0.036). Baseline analyses comparing ONL thicknesses between groups revealed increased ONL thickness in the IFNIM group (70.3 μm) compared to the GA (67.5 μm; p = 0.048) and NAT (67.2 μm; p = 0.034) groups. There were no other differences in baseline OCT measures between treatment groups.
Comparison of rates of GCIP thinning by DMTs.
During follow-up, relative to NAT-treated patients, IFNSC- and GA-treated patients exhibited 0.37 μm/y (p < 0.001) and 0.14 μm/y (p = 0.035) faster rates of GCIP thinning, respectively. Rates of GCIP thinning in IFNIM-treated patients were not significantly different from rates of GCIP thinning in NAT-treated patients (0.11 μm/y; p = 0.157). Patients treated with IFNSC had rates of GCIP thinning 0.23 μm/y (p = 0.001) and 0.26 μm/y (p = 0.002) faster compared to patients treated with GA and IFNIM, respectively (table 2). No difference in rates of GCIP thinning between GA- and IFNIM-treated patients were observed (p = 0.750).
Table 2.
Differences in rates of retinal atrophy (μm/y) by disease-modifying therapies (DMTs)
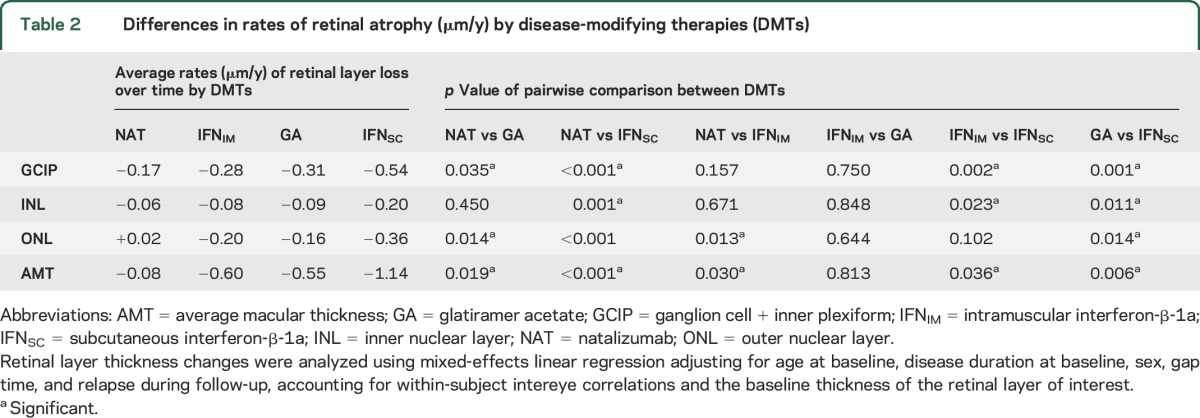
In the IFNSC group, GCIP thinning was 1.53 μm/y faster during the first year of therapy vs afterwards (p < 0.001), possibly related to retinal toxicity or less likely pseudoatrophy since the GCIP is relatively unsusceptible to edema. A significant difference between the rates of GCIP thinning during the first year of therapy vs the time interval afterwards was not observed in the GA (p = 0.478), IFNIM (p = 0.230), or NAT (p = 0.864) groups.
Comparison of GCIP thinning by DMTs excluding the first year of treatment.
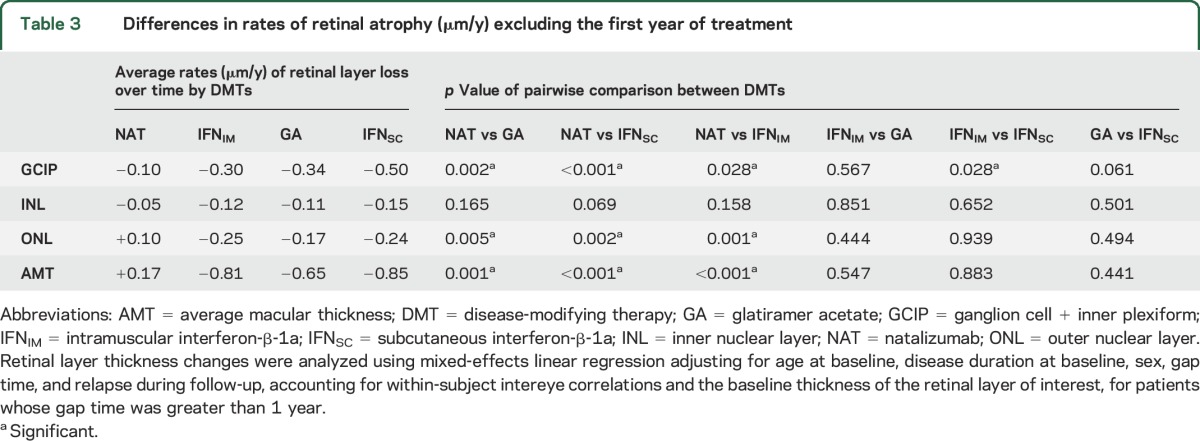
Analyses excluding patients with a gap time lower than 1 year to assess for more consistent and longer-term treatment effects revealed that NAT-treated patients had slower rates of GCIP atrophy compared to all other DMTs; 0.24 μm/y (p = 0.002), 0.40 μm/y (p < 0.001), and 0.20 μm/y (p = 0.028) slower compared to the GA, IFNSC, and IFNIM cohorts, respectively (table 3). Excluding patients with a gap time less than 1 year, IFNSC-treated patients exhibited borderline (approaching but not reaching statistical significance) faster GCIP atrophy as compared to GA (difference = 0.16 μm/y, p = 0.061). However, IFNSC-treated patients still exhibited faster rates of GCIP atrophy as compared to IFNIM-treated patients (difference = 0.20 μm/y, p = 0.028). Again, there was no difference in the rate of GCIP thinning between the GA and IFNIM cohorts (p = 0.567).
Table 3.
Differences in rates of retinal atrophy (μm/y) excluding the first year of treatment
Comparison of rates of GCIP thinning between RRMS and HCs.
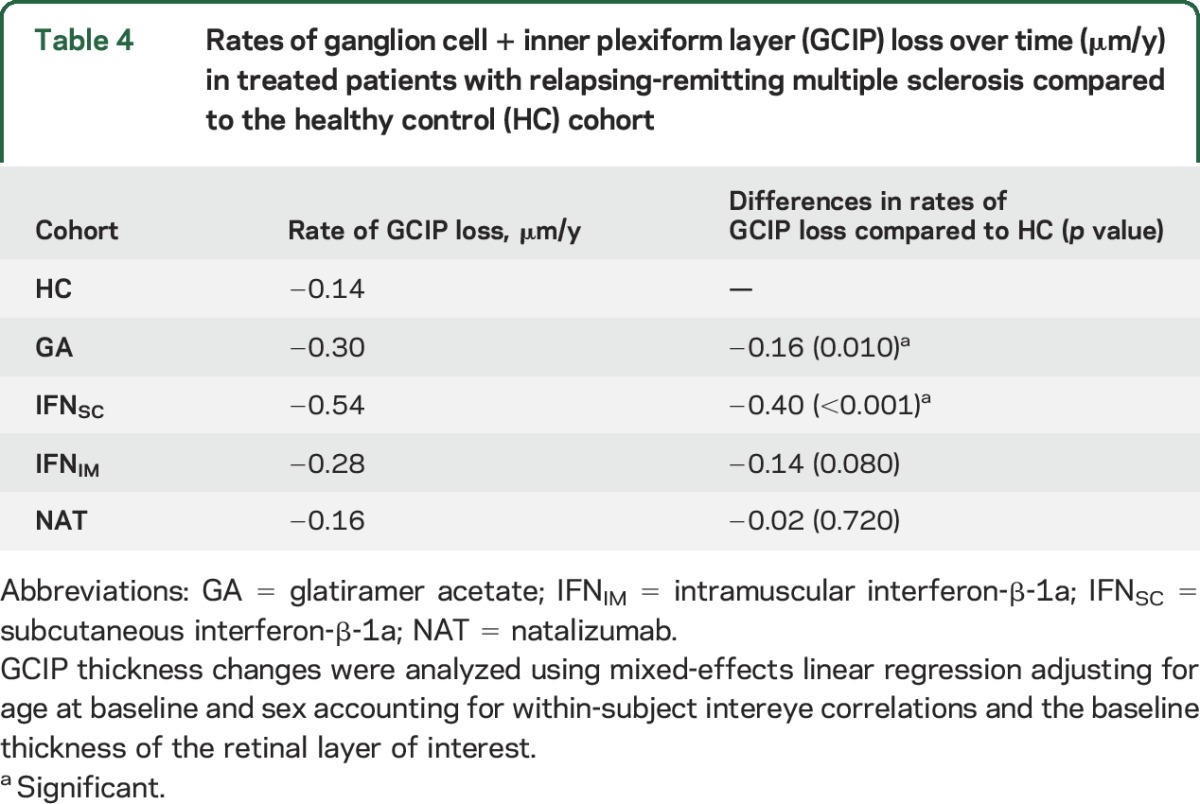
The rate of GCIP thinning in the HC cohort was 0.14 μm/y adjusting for baseline GCIP thickness, age, and sex. The GA, IFNSC, and IFNIM-treated patients exhibited 0.16 μm/y (p = 0.010), 0.40 μm/y (p < 0.001), and 0.14 μm/y (p = 0.080) greater rates of GCIP thinning compared to HCs, respectively. NAT-treated patients and HCs did not differ in their rates of GCIP loss (p = 0.720) (table 4).
Table 4.
Rates of ganglion cell + inner plexiform layer (GCIP) loss over time (μm/y) in treated patients with relapsing-remitting multiple sclerosis compared to the healthy control (HC) cohort
Comparison of rates of change in other retinal layers according to DMTs.
pRNFL thickness changes over time did not vary between the DMT cohorts studied (results not shown). Interestingly, the IFNSC cohort had a 0.14 μm/y (p = 0.001), 0.11 μm/y (p = 0.011), and 0.12 μm/y (p = 0.023) faster rate of INL thinning compared to the NAT, GA, and IFNSC cohorts, respectively. Moreover, the GA, IFNSC, and IFNIM cohorts exhibited 0.18 μm/y (p = 0.014), 0.38 μm/y (p < 0.001), and 0.22 μm/y (p = 0.013) faster rates of ONL thinning relative to NAT-treated patients, respectively. In addition, IFNSC-treated patients had a 0.20 μm/y faster rate of ONL thinning compared to GA-treated patients (p = 0.014). With respect to AMT change, the NAT cohort exhibited slower rates of thinning by 0.47 μm/y (p = 0.019), 1.06 μm/y (p < 0.001), and 0.52 μm/y (p = 0.030) relative to GA, IFNSC, and IFNIM, respectively. In addition, the GA-treated patients had 0.59 μm/y (p = 0.006) and the IFNIM-treated patients had 0.54 μm/y (p = 0.036) slower rates of AMT thinning compared to IFNSC-treated patients. No other differences were seen between groups within these layers (table 2).
DISCUSSION
The results of this study suggest that rates of retinal layer thinning, as assessed by OCT, are differentially modulated by different DMTs in RRMS. The reported findings provide objective support that more aggressive therapy with NAT slows the rate of GCIP, ONL, INL, and AMT thinning relative to more conventional MS treatments such as GA and the IFNs (particularly IFNSC). Our study findings support the potential utility of applying validated OCT measures for monitoring neurodegenerative treatment effects in the retina as a practical outcome measure in RRMS clinical trials.
NAT has been shown to strongly and effectively reduce relapse rate, new lesion formation, brain atrophy, and disease progression in patients with RRMS.6,17,18 Despite the lack of head-to-head studies directly comparing the efficacy of NAT to other DMTs in RRMS, the overall consensus based on available data and clinical experience is that NAT is one of the most effective RRMS therapies currently available. Considering the potent effects of NAT in reducing CNS inflammation, with the consequential effect of reducing brain atrophy, it is not surprising that NAT could play a similar role in reducing retinal thinning, and in particular GCIP atrophy, which has been shown to mirror global atrophy over time in MS.8 Indeed, our findings suggest that NAT has a greater effect in reducing rates of GCIP, INL, ONL, and AMT neurodegeneration, as compared to GA and the IFNs. NAT-treated patients did not differ in rates of GCIP thinning as compared to HCs, highlighting its potent effect in reducing retinal neurodegeneration in RRMS.
An important analysis factor in the current study is gap time (interval between DMT initiation and commencement of OCT monitoring). The reason for adjusting for gap time in our regression models was to account for OCT changes that were not accounted for between the initiation of therapy and the first OCT observation. The effect a DMT has on MRI-derived brain volume loss is often focused on the period following the initial year of treatment in order to allow DMTs to become optimized and for pseudoatrophy to resolve.7,19,20 While the patient sampling in the current retrospective study may be representative of an RRMS population typically observed in clinical practice, sampling bias may still be an important factor. For instance, there is suggestion in the literature that low-dose, low-frequency IFNIM may have a tendency to be less effective than high-dose, high-frequency IFNSC.21 However, such a difference was not observed between these 2 treatment groups during follow-up in this study. It is conceivable that the IFNIM-treated group in this study may either be largely composed of patients with RRMS with a more benign disease phenotype, or alternatively IFN super-responders, as this treatment group had a longer gap time, with the vast majority being DMT-naive prior to commencing this particular treatment (76%). Collectively, these factors may contribute to why the differences in rates of GCIP atrophy did not reach significance for the IFNIM and NAT groups (although this was significant after excluding patients with a gap time less than 1 year, and differences in rates of ONL and AMT thinning were also significantly different). Similarly, this could also explain why rates of GCIP atrophy appeared to be slower in the IFNIM group as compared to the IFNSC group, and why rates of GCIP atrophy were not significantly different between the IFNIM group and HCs. Alternatively, it has been suggested that dosing IFN less frequently, even if less anti-inflammatory, may be more neuroprotective.22–24
Interestingly, the IFNSC cohort exhibited a faster rate of GCIP atrophy during the first year of treatment, as compared to afterwards. A number of possibilities may explain this finding. It is recognized that IFNs can take up to 6 months or longer before being optimized. Accordingly, rates of retinal atrophy may be higher during the first year of follow-up. However, IFNSC may optimize more rapidly than IFNIM and therefore on this basis the proposed possibility does not explain our findings well. Alternatively, reduction in volume due to suppression of inflammation (pseudoatrophy) could be a factor, although this also seems unlikely since GCIP thickness is not thought to increase due to inflammation in the optic nerves.10,25,26 Finally, high-dose, high-frequency IFNSC might exert toxic retinal effects, although this has been rarely reported and remains to be elucidated.27 This possibility could be further supported by the largely faster rates of INL and AMT thinning observed in the IFNSC group as compared to the other treatment cohorts. No difference in rates of GCIP thinning was observed before and after the first year of treatment in the NAT-treated cohort. We would expect there to be a difference between these rates if pseudoatrophy indeed affected the GCIP since pseudoatrophy in the brain of NAT-treated patients has been highly postulated.28–30
When patients with a gap time of less than 1 year were excluded from analyses, the difference in GCIP atrophy rates between IFNSC and GA diminished somewhat, becoming only borderline significant, while IFNSC-treated patients still exhibited faster rates of GCIP atrophy as compared to IFNIM-treated patients, potentially reflecting an ongoing retinotoxic effect. At the same time, differences in GCIP atrophy rates between NAT with GA and IFNs became more pronounced, indicating sustained long-term treatment effects with NAT independent of effects during the first year of treatment only. Collectively, our findings suggest that in some instances it may be most suitable to determine the effects and differences of different DMTs on retinal measures (similar to brain atrophy data) following 1 year of treatment.
With increasing evidence that GCIP thickness is superior and more reliable for tracking change over time in MS, as compared to pRNFL thickness,8–10 it is unsurprising that we did not detect significant differences between treatments when assessing pRNFL thicknesses over time. However, significant differences in INL, ONL, and AMT rates of change were observed when stratifying by DMTs. It is suggested that INL and ONL thinning in MS may also reflect global CNS processes.31 Given that retinal layer thinning reflects brain atrophy and that neurodegeneration is considered the primary basis for disability in MS, our study findings provide further support that NAT may be a highly effective therapy in RRMS.
This study has several limitations, in part related to its retrospective design. First, due to limited patients with RRMS and follow-up OCT data, only 4 DMT classes were included in the final analyses. Future studies should investigate if oral therapies and other monoclonal antibody treatments have different effects on rates of retinal atrophy. Furthermore, due to patient sampling within our RRMS cohort, we were unable to clarify if race or ethnicity alter treatment effects on rates of retinal atrophy over time. Larger, longitudinal studies in the future should address this since it has been proposed that African American patients with MS may have faster rates of retinal atrophy as compared to Caucasian American patients with MS.32 Future studies should also aim to determine whether rates of retinal atrophy vary according to therapeutics in patients with progressive MS. Finally, this study did not include MRI-derived brain atrophy data, which would help to further corroborate our findings and to confirm that the effects of DMT classes on retinal atrophy similarly reflect their effects on brain atrophy. In future prospective studies, it would be beneficial to additionally account for further baseline patient characteristics such as those derived from MRI. While OCT is emerging as an additional and complementary tool to guide clinical management in MS, the current limitation of OCT in terms of individual patient level assessment remains unclear.
The collective findings of our study suggest that thinning in OCT-derived retinal layer measures over time in RRMS vary according to DMT utilization. Specifically, more aggressive therapy, such as with NAT, appears to have a greater effect on slowing retinal atrophy, and in particular GCIP atrophy, as compared to more conventional DMTs such as GA and IFNs. While further, larger, and in particular longitudinal studies are needed to confirm our findings, this does not mitigate the current preliminary findings, which provide strong support for the potential utility of OCT-derived measures to be utilized as outcome measures for assessing effects on neurodegeneration in MS.
GLOSSARY
- AMT
average macular thickness
- DMT
disease-modifying therapy
- GA
glatiramer acetate
- GCIP
ganglion cell + inner plexiform layer
- HC
healthy control
- IFN
interferon
- IFNIM
intramuscular interferon-β-1a
- IFNSC
subcutaneous interferon-β-1a
- INL
inner nuclear layer
- MME
microcystic macular edema
- MS
multiple sclerosis
- NAT
natalizumab
- OCT
optical coherence tomography
- ON
optic neuritis
- ONL
outer nuclear layer
- pRNFL
peripapillary retinal nerve fiber layer thickness
- RRMS
relapsing-remitting multiple sclerosis
AUTHOR CONTRIBUTIONS
J. Button: data collection, drafting, and revision of manuscript. Dr. Al-Louzi: data collection and revision of manuscript for important intellectual content. Dr. Lang: data collection and revision of manuscript for important intellectual content. Dr. Bhargava: data collection and revision of manuscript for important intellectual content. Dr. Newsome: data collection, drafting and revision of manuscript for important intellectual content. Dr. Balcer: revision of manuscript for important intellectual content. Dr. Frohman: revision of manuscript for important intellectual content. Dr. Prince: drafting and revision of manuscript for important intellectual content. Dr. Calabresi: drafting and revision of manuscript for important intellectual content. Dr. Saidha: data collection, drafting and revision of manuscript for important intellectual content.
STUDY FUNDING
This study was funded by a Race to Erase MS grant to S.S. and NIH grant 5R01NS082347-02 to P.A.C.
DISCLOSURE
J. Button, O. Al-Louzi, and A. Lang report no disclosures relevant to the manuscript. P. Bhargava is supported by an Institutional Clinician Training Award from the National Multiple Sclerosis Society. S. Newsome has received consultant fees for scientific advisory boards from Biogen, Genzyme, and Novartis and has received research funding from Biogen, Novartis, and the National MS Society. T. Frohman has received speaker and consulting fees from Acorda, Genzyme, and Novartis. L. Balcer has received speaking and consulting honoraria from Biogen Idec, Bayer, and Novartis. E. Frohman has received speaker and consulting fees from Novartis, Genzyme, Acorda, and Teva. J. Prince reports no disclosures relevant to the manuscript. P. Calabresi has received personal compensation for consulting and serving on scientific advisory boards from Vertex, Vaccinex, Merck, and Abbvie and has received research funding from Biogen-Idec, MedImmune, and Novartis. S. Saidha has received consulting fees from Medical Logix for the development of CME programs in neurology, consulting fees from Axon Advisors LLC, educational grant support from Novartis & Teva Neurosciences, speaking honoraria from the National Association of Managed Care Physicians, Family Medicine Foundation of West Virginia, and Advanced Studies in Medicine, and served on scientific advisory boards for Biogen-Idec, Genzyme, and Novartis. He receives research support from the Race to Erase MS and Genentech Corporation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Fisniku LK, Chard DT, Jackson JS, et al. Gray matter atrophy is related to long-term disability in multiple sclerosis. Ann Neurol 2008;64:247–254. [DOI] [PubMed] [Google Scholar]
- 2.Popescu V, Agosta F, Hulst HE, et al. Brain atrophy and lesion load predict long term disability in multiple sclerosis. J Neurol Neurosurg Psychiatry 2013;84:1082–1091. [DOI] [PubMed] [Google Scholar]
- 3.National Multiple Sclerosis Society. Disease Management Consensus Statement. Available at: nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Brochures/ExpOp_Consensus.pdf. Accessed September 10, 2015. [Google Scholar]
- 4.Bendfeldt K, Egger H, Nichols TE, et al. Effect of immunomodulatory medication on regional gray matter loss in relapsing-remitting multiple sclerosis: a longitudinal MRI study. Brain Res 2010;1325:174–182. [DOI] [PubMed] [Google Scholar]
- 5.Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the Rebif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol 2008;7:903–914. [DOI] [PubMed] [Google Scholar]
- 6.Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354:899–910. [DOI] [PubMed] [Google Scholar]
- 7.Tsivgoulis G, Katsanos AH, Grigoriadis N, et al. The effect of disease modifying therapies on brain atrophy in patients with relapsing-remitting multiple sclerosis: a systematic review and meta-analysis. PLoS One 2015;10:e0116511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saidha S, Al-Louzi O, Ratchford JN, et al. Optical coherence tomography reflects brain atrophy in MS: a four year study. Ann Neurol 2015;78:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saidha S, Syc SB, Durbin MK, et al. Visual dysfunction in multiple sclerosis correlates better with optical coherence tomography derived estimates of macular ganglion cell layer thickness than peripapillary retinal nerve fiber layer thickness. Mult Scler 2011;17:1449–1463. [DOI] [PubMed] [Google Scholar]
- 10.Saidha S, Syc SB, Ibrahim MA, et al. Primary retinal pathology in multiple sclerosis as detected by optical coherence tomography. Brain 2011;134:518–533. [DOI] [PubMed] [Google Scholar]
- 11.Ratchford JN, Saidha S, Sotirchos ES, et al. Active MS is associated with accelerated retinal ganglion cell/inner plexiform layer thinning. Neurology 2013;80:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tewarie P, Balk L, Costello F, et al. The OSCAR-IB consensus criteria for retinal OCT quality assessment. PLoS One 2012;7:e34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhargava P, Lang A, Al-Louzi O, et al. Applying an open-source segmentation algorithm to different OCT devices in multiple sclerosis patients and healthy controls: implications for clinical trials. Mult Scler Int 2015;2015:136295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang A, Carass A, Hauser M, et al. Retinal layer segmentation of macular OCT images using boundary classification. Biomed Opt Express 2013;4:1133–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saidha S, Sotirchos ES, Ibrahim MA, et al. Microcystic macular oedema, thickness of the inner nuclear layer of the retina, and disease characteristics in multiple sclerosis: a retrospective study. Lancet Neurol 2012;11:963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bender R, Lange S. Adjusting for multiple testing: when and how? J Clin Epidemiol 2001;54:343–349. [DOI] [PubMed] [Google Scholar]
- 17.Lanzillo R, Quarantelli M, Bonavita S, et al. Natalizumab vs interferon beta 1a in relapsing-remitting multiple sclerosis: a head-to-head retrospective study. Acta Neurol Scand 2012;126:306–314. [DOI] [PubMed] [Google Scholar]
- 18.Pucci E, Giuliani G, Solari A, et al. Natalizumab for relapsing remitting multiple sclerosis. Cochrane Database Syst Rev 2011;10:CD007621. [DOI] [PubMed] [Google Scholar]
- 19.Tsivgoulis G, Katsanos AH, Grigoriadis N, et al. The effect of disease-modifying therapies on brain atrophy in patients with clinically isolated syndrome: a systematic review and meta-analysis. Ther Adv Neurol Disord 2015;8:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller DH, Soon D, Fernando KT, et al. MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology 2007;68:1390–1401. [DOI] [PubMed] [Google Scholar]
- 21.Panitch H, Goodin DS, Francis G, et al. Randomized, comparative study of interferon beta-1a treatment regimens in MS: the EVIDENCE trial. Neurology 2002;59:1496–1506. [DOI] [PubMed] [Google Scholar]
- 22.Rudick RA. Impact of disease-modifying therapies on brain and spinal cord atrophy in multiple sclerosis. J Neuroimaging 2004;14:54S–64S. [DOI] [PubMed] [Google Scholar]
- 23.Ohira M, Ito D, Shimizu T, Shibata M, Ohde H, Suzuki N. Retinopathy: an overlooked adverse effect of interferon-beta treatment of multiple sclerosis. Keio J Med 2009;58:54–56. [DOI] [PubMed] [Google Scholar]
- 24.Sommer S, Sablon JC, Zaoui M, Rozot P, Hosni A. Interferon beta-1b retinopathy during a treatment for multiple sclerosis [in French]. J Fr Ophtalmol 2001;24:509–512. [PubMed] [Google Scholar]
- 25.Syc SB, Saidha S, Newsome SD, et al. Optical coherence tomography segmentation reveals ganglion cell layer pathology after optic neuritis. Brain 2012;135:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Syc SB, Warner CV, Hiremath GS, et al. Reproducibility of high-resolution optical coherence tomography in multiple sclerosis. Mult Scler 2010;16:829–839. [DOI] [PubMed] [Google Scholar]
- 27.Gaetani L, Menduno PS, Cometa F, et al. Retinopathy during interferon-beta treatment for multiple sclerosis: case report and review of the literature. J Neurol 2015;263:422–427. [DOI] [PubMed] [Google Scholar]
- 28.Sastre-Garriga J, Tur C, Pareto D, et al. Brain atrophy in natalizumab-treated patients: a 3-year follow-up. Mult Scler 2015;21:749–756. [DOI] [PubMed] [Google Scholar]
- 29.Zivadinov R, Reder AT, Filippi M, et al. Mechanisms of action of disease-modifying agents and brain volume changes in multiple sclerosis. Neurology 2008;71:136–144. [DOI] [PubMed] [Google Scholar]
- 30.Vidal-Jordana A, Sastre-Garriga J, Pérez-Miralles F, et al. Early brain pseudoatrophy while on natalizumab therapy is due to white matter volume changes. Mult Scler 2013;19:1175–1181. [DOI] [PubMed] [Google Scholar]
- 31.Saidha S, Sotirchos ES, Oh J, et al. Relationships between retinal axonal and neuronal measures and global central nervous system pathology in multiple sclerosis. JAMA Neurol 2013;70:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimbrough DJ, Sotirchos ES, Wilson JA, et al. Retinal damage and vision loss in African American multiple sclerosis patients. Ann Neurol 2015;77:228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]