

Abstract
Myeloproliferative neoplasms (MPNs) are the most common underlying prothrombotic disorder found in patients with splanchnic vein thrombosis (SVT). Clinical risk factors for MPN-associated SVTs include younger age, female sex, concomitant hypercoagulable disorders, and the JAK2 V617F mutation. These risk factors are distinct from those associated with arterial or deep venous thrombosis (DVT) in MPN patients, suggesting disparate disease mechanisms. The pathophysiology of SVT is thought to derive from local interactions between activated blood cells and the unique splanchnic endothelial environment. Other mutations commonly found in MPNs, including CALR and MPL, are rare in MPN-associated SVT. The purpose of this article is to review the clinical and molecular risk factors for MPN-associated SVT, with particular focus on the possible mechanisms of SVT formation in MPN patients.
Keywords: Budd-Chiari syndrome, essential thrombocythemia, JAK2, myeloproliferative neoplasm, polycythemia vera, portal vein thrombosis, primary myelofibrosis, splanchnic vein thrombosis
Introduction
Myeloproliferative neoplasms (MPNs) are a group of hematopoietic stem cell disorders characterized by clonal proliferation of myeloid-lineage cells. Classic MPNs include polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), and chronic myeloid leukemia (CML) [Tefferi and Vardiman, 2008]. CML is defined by a reciprocal translocation of chromosomes 9 and 22, resulting in the Philadelphia chromosome (Ph), and will not be discussed in this article. The Ph-negative MPNs, which include PV, ET, and PMF, share many common clinical and molecular characteristics, including increased risk of thrombosis and leukemic transformation.
Arterial and venous thromboses are a major cause of morbidity in Ph-negative MPNs. Venous thrombosis may occur at unusual anatomic sites, such as splanchnic vein thrombosis (SVT) or cerebral sinus thrombosis. SVT refers to thrombosis formation in the portal venous system (portal vein thrombosis, PVT), hepatic venous system Budd-Chiari syndrome (BCS), splenic venous system, or mesenteric venous system. SVTs are rare, with large epidemiological studies estimating the incidence rate to be 0.7 per 100,000 patients per year for PVTs and 0.8 per million patients per year for BCS [Rajani and Almer, 2009; Rajani et al. 2010]. This is significantly lower than the incidence rate of deep venous thromboses (DVTs), which is estimated to be roughly 100 per 100,000 patients per year in the United States [White, 2003]. Interestingly, MPNs are the most common underlying prothrombotic disorder found in patients diagnosed with SVT, in the absence of local inciting factors such as liver cirrhosis or nearby malignancy. In patients with BCS specifically, systemic factors, such as an underlying MPN, are more common than local factors [Ageno et al. 2014]. The strong association between SVT and MPN has led to recommendations to screen for MPN when SVT is diagnosed [Smalberg et al. 2012].
The reason for the association between SVT and MPN is not immediately clear. Age, sex, concomitant hypercoagulable disorders, and the presence of the JAK2 V617F mutation have all been implicated in the pathogenesis of SVT in MPN. Improving our understanding of the mechanisms that predispose to SVT formation is an area of ongoing research. The purpose of this article is to review the clinical and molecular risk factors for MPN-associated SVT, with particular focus on the possible disease mechanisms of SVT formation in MPN patients. The treatment and management of MPN-associated SVTs have been discussed extensively in two recently published reviews [Sekhar et al. 2013; De Stefano et al. 2015], and will not be discussed in this review.
Prevalence of SVT in MPNs
The prevalence of a venous thromboembolism (VTE) at the time of MPN diagnosis is estimated to be approximately 11–39% for PV, 8–29% for ET, and 3–7% for PMF [Barbui et al. 2010; Kreher et al. 2014]. SVTs represent a fraction of VTE in MPNs, with an estimated prevalence of 5–10% in PV patients and 9–13% in ET patients [Anger et al. 1989; De Stefano et al. 2008]. SVTs occur even less frequently in PMF patients, with an estimated prevalence rate of 0.6–1% [Anger et al. 1989; Barbui et al. 2010]. PVT is the most common type of SVT (40%), while BCS is the least common (5%) [De Stefano et al. 2008; Smalberg et al. 2012]. Conversely, in patients presenting with SVT, MPNs are the most common underlying prothrombotic disorder. Prevalence rates of MPN in SVT have ranged from 5–70%, with a large meta-analysis estimating that 40% of BCS patients and 30% of PVT patients are subsequently found to have an underlying MPN [Smalberg et al. 2012; Sekhar et al. 2013]. PV makes up the largest subgroup in patients with SVT (27%), and PMF makes up the smallest subgroup (13%) [Smalberg et al. 2012]. Table 1 summarizes the prevalence rates of SVTs in MPNs, and vice versa.
Table 1.
(a) SVTs in patients with MPNs compared with the general population. (b) Prevalence rates of MPNs in patients found to have an SVT.
| (a) Prevalence rates of SVTs. | ||
| Within MPNs | Within general population | |
| PVT | 6100 per 100,000 | 3.7 per 100,000 |
| BCS | 1200 per 100,000 | 0.14 per 100,000 |
| (b) Prevalence rates of MPNs | ||
| Within PVT | Within BCS | |
| 40% | 30% | |
Sources: De Stefano et al. [2008] - 6100/100,000 PVT within MPN; Anger et al. [1989] - 1200/100,000 BCS within MPN; Rajani and Almer [2009] - 0.14 per 100,000 BCS within general population; Rajani et al. [2010] - 3.7 per 100,000 PVT within general population; Smalberg et al. [2012] - prevalence rates of MPNs within PVT and BCS.
BCS, Budd-Chiari syndrome; MPN, myeloproliferative neoplasms; PVT, portal vein thrombosis; SVT, splanchnic vein thrombosis.
Clinical factors associated with SVT in MPN
The proportion of SVT among all VTEs is disproportionately higher in MPN patients compared with the general population, as SVTs make up an estimated 7.5% of total VTE cases [De Stefano et al. 2008]. In comparison, the frequency of DVTs and PEs in the general population is 400 times greater than that of SVTs [Deitelzweig et al. 2011]. Interestingly, the risk factors associated with these unusual thromboses are different from the risk factors traditionally associated with all-cause arterial and venous thrombosis. Figure 1 compares the known clinical risk factors of SVTs with those of all-cause VTE.
Figure 1.
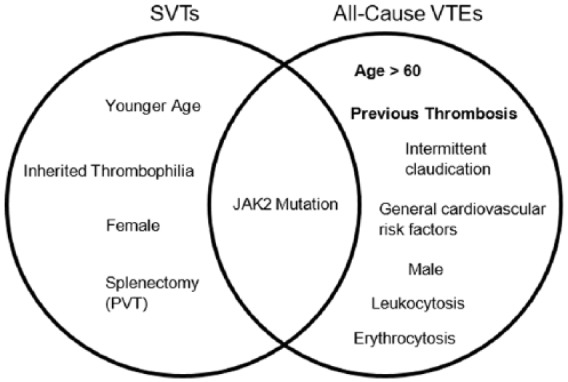
Venn diagram comparing cited clinical risk factors for SVTs and all-cause VTEs. Bold indicates the most validated risk factors.
PVT, portal vein thrombosis; SVT, splanchnic vein thrombosis; VTE, venous thromboembolism.
Age and sex
The best established risk factors for thrombosis in MPN are age >60 years and prior history of thrombosis [Marchioli et al. 2005; Kreher et al. 2014; Tefferi and Barbui, 2015]. Analysis of 1638 PV patients collected in the ECLAP (European Collaboration on Low-dose Aspirin in Polycythemia vera) trial showed that only advanced age (>60 years), previous venous thrombosis (but not arterial thrombosis), and intermittent claudication demonstrated a statistically significant increased risk of a subsequent VTE [Landolfi et al. 2007]. In a multivariate analysis of 891 patients with ET, male sex was also found to be associated with increased venous thrombosis [Carobbio et al. 2011]. However, sex was not found to have a significant effect on VTE in PV patients, nor has this finding been demonstrated in other epidemiological studies [Marchioli et al. 2005; Landolfi et al. 2007].
In contrast, MPN patients with SVTs are predominantly younger and female. Lussana and colleagues evaluated the prevalence of thrombosis in JAK2-mutated patients younger than 40 years of age. In this cohort of 538 patients, venous thrombosis was significantly higher than arterial thrombosis (47% versus 34%), and SVTs made up 79% (26 out of 33) of these VTE cases [Lussana et al. 2014]. This is in contrast with the observation that arterial thromboses rates are 2–3 times higher than venous thrombosis rates in the total MPN population [Barbui et al. 2013]. In addition, the proportion of SVTs in this younger population was much higher than in the total MPN population (79% compared with 7.5%) [De Stefano et al. 2011; Lussana et al. 2014]. Another study, by Stein and colleagues, compared disease characteristics in a cohort of younger (age < 45 years) and older (age > 65 years) MPN patients. The investigators found that although younger patients had a comparable rate of all-cause thrombosis, SVTs occurred significantly more frequently (13% compared with 2%) in patients younger than 45 years of age [Stein et al. 2013]. The younger cohort was also predominantly female, and had a significantly lower JAK2 allele burden [Stein et al. 2010, 2013]. The observation that SVTs are significantly more frequent in women compared with men was also noted in the study by Lussana and colleagues [Lussana et al. 2014]. There are limited data to suggest that other clinical or laboratory risk factors, such as a history of prior thrombosis or elevated cell counts, increase the risk of SVTs.
Thrombophilic disorders
Hypercoagulable disorders such as antiphospholipid syndrome, protein C or S deficiency, factor V Leiden mutation, or antithrombin deficiency are independent risk factors for SVT [Kiladjian et al. 2008]. In one study of 40 patients with SVTs, it was found that 38% of patients had an additional risk factor to MPN, including a hypercoagulable disorder, paroxysmal nocturnal hemoglobinuria (PNH), autoimmune disorder (such as a connective tissue disease), or ulcerative colitis [De Stefano et al. 2011]. Overall, it has been estimated that multiple concurrent risk factors are present in 10–46% of BCS patients and 10–64% of PVT patients [De Stefano et al. 2011].
Environmental risk factors
Oral contraceptive (OCP) use has been implicated as a predisposing risk factor for VTEs and cerebral sinus thrombosis [Vandenbroucke et al. 1994; Stam, 2005]. It is unclear of the extent to which OCP use increases the risk of SVT formation, given the widespread use of OCPs and the presence of concomitant hypercoagulable disorders. In one study examining the etiological factors in BCS, Smalberg and colleagues identified OCP use in 52% of the 26 female patients. However, these patients often had an additional prothrombotic disorder, and the authors did not specify which combination of etiological factors were present [Smalberg et al. 2006]. An additional study examining OCP use and VTE found that nine OCP-users had developed an SVT, of which two were JAK2 positive [Grandone et al. 2008]. Neither of these two women had a concomitant hypercoagulable disorder, although a thrombophilia was present in five of the remaining JAK2-negative patients [Grandone et al. 2008]. OCP use has been shown to increase the risk of VTE in the presence of an inherited thrombophilia, and it is possible that SVT risk may be similarly increased in MPN patients who use OCPs [Vandenbroucke et al. 1994]. This may also partially explain the observation that females are more likely to develop an MPN-associated SVT; however, larger epidemiological studies are needed to potentially establish an association between MPNs, SVTs, and OCP use.
There are few data to suggest that other environmental exposures play a significant role in MPN-associated SVT. Some research has suggested that smoking, along with other general cardiovascular risk factors, increases the rate of arterial thrombosis, but the data are conflicting [Falanga and Marchetti, 2014]. There are few data, however, regarding whether these risk factors apply to venous thrombosis, and specifically SVT.
The JAK2 V617F mutation in MPN-associated SVT
JAK2 is a tyrosine kinase that initiates intra-cellular signaling in the JAK/STAT pathway via binding to receptors for erythropoietin, thrombopoietin, granulocyte colony-stimulating factor, and granulocyte macrophage colony-stimulating factor [James, 2008; Oh and Gotlib, 2010]. The JAK2 V617F mutation causes constitutive activation of the JAK2 kinase, resulting in subsequent phosphorylation of STAT proteins, ultimately leading to overproduction of myeloid-lineage cells [Campbell and Green, 2006; Oh and Gotlib, 2010]. The identification of the JAK2 V617F mutation in nearly 95% of PV cases and 50–60% of ET and PMF cases has led to its incorporation into the diagnostic evaluation of MPNs [Campbell and Green, 2006]. Current guidelines recommend testing for JAK2 mutations in any patient suspected to have an MPN [Ageno et al. 2016].
JAK2 mutation in SVT in the absence of overt MPN
An estimated 40% of BCS patients and 28% of PVT patients also test positive for the JAK2 V617F mutation [Smalberg et al. 2012]. For this reason, in any patient presenting with an SVT, JAK2 testing is recommended as part of the initial work-up for MPN. This has led to the observation that a significant proportion of SVT patients (15–17%) test positive for JAK2 V617F, but do not otherwise meet WHO criteria for an MPN [Smalberg et al. 2012]. Indeed, one study estimated that testing for JAK2 can identify an additional 30% of MPN patients who do not otherwise have overt evidence of disease [De Stefano et al. 2007]. The most frequently cited reason for the absence of elevated blood counts is the presence of portal hypertension and splenomegaly that result from SVT and MPN, which can lead to sequestration of blood cells as well as hemodilution, thus ‘masking’ the clinical phenotype of MPN. Investigators have found that while ‘masked’ patients display lower hemoglobin levels, they also have increased plasma volumes secondary to splenomegaly or SVT formation [Lamy et al. 1997].
It is also possible that SVT patients with JAK2 V617F, but no overt clinical features of MPN, may represent a distinct subtype of MPN or an early phase of the disease. Up to 70% of patients presenting with SVT do not carry a prior diagnosis of MPN, and it has been estimated that SVTs represent the first thrombotic event in 7.5% of patients with PV and ET [De Stefano et al. 2008; Hoekstra et al. 2011]. SVTs may thus occur during an early stage of MPN, prior to the development of the clinical phenotype. For instance, one meta-analysis found that 21 out of 41 JAK2-positive patients without an MPN diagnosis at the time of SVT presentation subsequently developed an MPN during follow up [Dentali et al. 2009]. A more recent study looking at long-term outcomes of SVT patients also observed that 9 out of 13 JAK2-positive patients were found to have an MPN after 64 months [Colaizzo et al. 2013]. These findings suggest that in these patients, SVTs represent an early manifestation of their disease. In Colaizzo and colleagues’ study, six out of the eight SVT patients who were initially JAK2-negative were found to be JAK2-positive with clinical symptoms of an MPN by 21 months [Colaizzo et al. 2013]. Such observations suggest either that a disease process was already in existence before the occurrence of the JAK2 mutation, or that the initial JAK2 V617F allele burden had not reached a detectable level by conventional laboratory testing at the time of SVT diagnosis. The hypothesis that SVTs are early manifestations of MPN may partially account for the finding that MPN-associated SVT patients tend to be younger with lower JAK2 V617F allele burdens.
The hypothesis that SVT patients represent an earlier stage of MPN disease has important implications in evaluating the underlying molecular pathogenesis of MPNs. It has been hypothesized that blood cancers arise from stepwise genetic mutations that confer survival and growth advantages to hematopoietic stem cells, eventually resulting in clonal hematopoiesis and malignancy [Xie et al. 2014]. While early mutations may allow clonal expansion, cooperating mutations that occur later enable development of a hematopoietic cancer. JAK2-positive patients without clinical findings of an MPN may lack these cooperating mutations that result in blood cell proliferation. For instance, a significant percentage of MPN-associated mutations, including JAK2 V617F, have been found in elderly individuals without overt evidence of an MPN [Genovese et al. 2014; Xie et al. 2014]. These individuals, however, are at an increased risk of developing a subsequent hematologic malignancy and cardiovascular mortality [Jaiswal et al. 2014]. These data suggest that mutations such as JAK2 V617F can be initiating events that may ultimately lead to the development of overt malignancy. Similarly, in SVT patients who subsequently develop MPNs, it is possible that the development of thrombosis reflects pathology related to an initiating clone. The progression to overt MPN, including elevated cell counts, may only become apparent upon acquisition of subsequent mutations. These patients with positive JAK2 mutation status may thus be considered a ‘pre-malignant’ state, and as such represent a unique population for studying the clonal evolution of MPNs.
Prevalence of the JAK2 V617F in MPN-associated SVTs
There is a high prevalence of the JAK2 V617F mutation found in SVT, and the percentage is even higher when restricted to patients meeting MPN criteria with SVT [Patel et al. 2006]. An estimated 71–100% of patients with overt MPN and SVT test positive for JAK2 V617F, which is significantly higher than the prevalence of the JAK2 mutation in MPN patients with other VTEs [Austin and Lambert, 2008]. A systematic review of 24 studies and 3123 patients found that JAK2 V617F conferred a significantly increased risk of SVT formation, with an odds ratio of 54 [Dentali et al. 2009]. In addition, while the mean prevalence of the JAK2 mutation in SVTs was high (33%, similar to rates reported in the literature), the mean prevalence of JAK2 V617F in other VTEs was low (0.88–2.57%) [Dentali et al. 2009]. Despite these associations, one study observed that young females tended to have a lower JAK2 mutant allele burden, even though they are at higher risk for SVTs [Stein et al. 2010]. The findings of increased incidence of the JAK2 mutation but lower allele burden may suggest an ‘all or nothing’ effect of the JAK2 mutation, which is perhaps subsequently modified by additional clinical risk factors such as concomitant hypercoagulable disorders, as discussed in the previous section.
JAK2 V617F mutation as a molecular driver of SVT formation
The majority of SVTs (with the exception of BCS) are due to local perturbations in the venous system, such as cirrhosis or nearby malignancy. It is possible that the JAK2 mutation itself may have local effects on the splanchnic venous system. This venous system is unique in that blood flow velocity is much slower, leading to prolonged interactions between blood and sinusoidal endothelial cells [Aird, 2007a]. In vitro models have shown that PV red blood cells demonstrate increased adhesion to endothelial laminin, and this adhesiveness is particularly increased at low shear rates [Wautier and Wautier, 2013]. Furthermore, the portal venous system is exposed to gut-derived inputs, altering its immunogenicity such that vessels are more vulnerable to activated platelets and states of high viscosity such as increased cell count [Aird, 2007b]. These characteristics may put the splanchnic venous system at particular risk to perturbations caused by JAK2 V617F, which affect not only cell quantity, but cell quality.
The JAK2 mutation has a widespread effect on the hematologic system. JAK2-mutant megakaryocytes display hypersensitive signaling and increased mobility and aggregation [Hobbs et al. 2013]. Studies with JAK2 V617F knock-in mice have demonstrated that megakaryocytes carrying the JAK2 mutation show greater chemotaxis along migration assays, as well as increased pro-platelet formation in response to low-level thrombopoietin signaling [Hobbs et al. 2013]. JAK2-positive platelets also display increased in vitro thrombus formation when exposed to collagen-coated surfaces, although Lamrani and colleagues found decreased platelet reactivity when tested in a different mouse model [Hobbs et al. 2013; Lamrani et al. 2014]. However, accelerated platelet aggregation occurred when studied in vivo in artificially injured blood vessels [Lamrani et al. 2014]. Blood samples taken from JAK2-positive patients with MPN also show significantly higher levels of soluble P-selectin, a marker of platelet activation, compared with JAK2-negative patients with MPN [Robertson et al. 2007].
Leukocyte activation also contributes to the pro-coagulant milieu, as studies have shown that markers of polymorphonuclear activation correlate with markers of hypercoagulability in ET and PV patients [Falanga et al. 2005]. It is thus believed that activated leukocytes can cause secretion of pro-coagulant factors, leading to further platelet activation and aggregation. In JAK2-positive patients, the level of both leukocyte and platelet activation increases with higher JAK2 mutational burden, and an epidemiological study by Barbui and colleagues found that the highest incidence of thrombosis was seen in PMF patients with both leukocytosis and the JAK2 mutation [Arellano-Rodrigo et al. 2006; Barbui et al. 2013]. Red blood cells also display increased adhesion to the endothelium in PV patients [Wautier and Wautier, 2013]. This effect may be mediated by aberrations in JAK/STAT signaling, as JAK2 V617F can induce phosphorylation of glycoproteins critical for red blood cell adhesion [De Grandis et al. 2013]. Taken together, these findings suggest that the JAK2 mutation leads to distinct changes in hematopoietic cells that increase the likelihood of thrombus formation.
The propensity for SVT development in MPN is also potentially due to differential regulation of hemostasis in specific vascular beds. Endothelial cells are integrally involved in regulating thrombosis, and are capable of expressing both pro- and anticoagulant factors depending on both the environmental milieu and cell type [Rosenberg and Aird, 1999]. This is supported by observations that MPNs are associated with specific anatomical locations within the splanchnic venous system. In MPN patients with BCS, for example, thrombosis tends to occur in the large, hepatic veins, while BCS patients with other hypercoagulable disorders tend to have thrombosis involving the inferior vena cava [Valla, 2015]. Signaling pathways specific to the splanchnic venous system can then cause unique interactions with mutated blood cells. For instance, in PNH, a complement-mediated disorder that also presents with increased susceptibility to SVT, the presence of gut-derived antigens in the splanchnic veins may cause higher local activation of complement, with subsequent higher rates of hemolysis and thrombotic activation [van Bijnen et al. 2012].
Mutant endothelial cells may also contribute to SVT pathogenesis. Liver endothelial cells isolated from two BCS patients have been found to carry the JAK2 mutation [Sozer et al. 2009]. Further studies have identified the JAK2 mutation in spleen endothelial cells and circulating endothelial progenitor cells [Teofili et al. 2011; Rosti et al. 2013]. JAK2 V617F-mutated endothelial progenitor cells also displayed increased adherence to normal mononuclear cells, and were found to abnormally activate the JAK/STAT pathway [Teofili et al. 2011]. These findings provide a framework in which potentially aberrant endothelial cells in the splanchnic venous system interact with aberrant blood cells, eventually leading to the formation of SVT. The exact mechanism of how alterations in the endothelium lead to SVT is still unclear, and warrants further investigation. Given the unique environment of the splanchnic venous system, it is likely that the mechanisms of SVT formation differ from the mechanisms of arterial and DVT, accounting for the difference in risk factors.
Other molecular drivers of MPN-associated SVT
Although the JAK2 mutation plays a significant role in MPN in SVT, approximately 14–20% of SVT patients with MPN are JAK2-negative [Kiladjian et al. 2008; Smalberg et al. 2012]. In terms of clinical risk factors, JAK2-negative patients with MPN/SVT are not statistically different from their JAK2-positive counterparts [Kiladjian et al. 2008]. This leads to the question of whether there are other mutations that can account for the prothrombotic state. Specifically, several mutations with increased incidence in MPN have been identified, and only recently have investigators evaluated these mutations in SVT.
CALR mutations
The CALR gene encodes for calreticulin, a multifunctional protein that regulates calcium signaling and protein folding in the endoplasmic reticulum and cytoplasm [Johnson et al. 2001]. Only recently has a mechanism for CALR-mutant MPN pathogenesis been proposed. A series of papers demonstrated that mutated calreticulin is able to induce MPN phenotypes in mice through interaction with the thrombopoietin receptor (MPL) [Araki et al. 2016; Chachoua et al. 2016; Elf et al. 2016; Marty et al. 2016]. Mutated calreticulin has been shown to bind preferentially to MPL, resulting in thrombopoietin-independent activation of MPL with subsequent stimulation of the JAK/STAT pathway [Araki et al. 2016; Chachoua et al. 2016; Elf et al. 2016; Marty et al. 2016]. The specificity of mutant calreticulin interaction with MPL accounts for the findings that CALR mutations are largely absent in PV, but found in 30% of all ET and PMF patients, and 70–80% of JAK2-negative ET or PMF [Klampfl et al. 2013; Nangalia et al. 2013]. Compared with JAK2, CALR-mutant patients tend to have increased platelet counts, lower hemoglobin and white cell counts, as well as decreased rates of thrombosis, with a more indolent disease course [Nangalia et al. 2013].
How or whether CALR mutations induce SVT susceptibility is less clear. Despite the relatively high frequency of CALR mutations in MPNs, recent studies have failed to find an increased incidence of CALR mutations in SVT patients. CALR mutations have been identified in 0–2% of SVT, with an estimated total prevalence of approximately 0.9% of cases [Castro et al. 2015; Colaizzo et al. 2015; Haslam and Langabeer, 2015; Iurlo et al. 2015; Marzac et al. 2015; Plompen et al. 2015; Roques et al. 2015; Turon et al. 2015]. Given its relatively low prevalence, testing for CALR is not generally recommended in patients with SVT [Colaizzo et al. 2015]. Furthermore, its rarity suggests that it is not a major contributor to SVT formation, a fact consistent with the observation that CALR-mutated patients exhibit decreased rates of thrombosis overall [Rumi et al. 2014].
MPL mutations
Mutations in the MPL gene are the third most commonly tested for mutation after JAK2 and CALR in MPN patients. MPL encodes for the thrombopoietin receptor, and mutations expectedly cause abnormalities in megakaryocyte and platelet production. The most common mutations result in an amino acid substitution (either lysine or leucine) at the 515 position (MPL W515), are found in approximately 5% of ET/PMF patients, and are mutually exclusive to JAK2 and CALR mutations [Pikman et al. 2006; Rumi et al. 2013]. MPL-mutant patients tend to be older, with lower cell counts compared with JAK2 and CALR-positive patients [Beer et al. 2008]. MPL mutations have not been shown to confer significant prognostic impact, and patients do not have differing risks of thrombosis or major hemorrhage [Beer et al. 2008]. However, similar to CALR, MPL W515 mutations are rare in SVT, with incidence rates even less than CALR [Bergamaschi et al. 2008; Kiladjian et al. 2008; Jadli et al. 2012; Smalberg et al. 2012; Colaizzo et al. 2015]. This may be related to the overall low prevalence of MPL in MPNs, but there is no evidence to suggest that MPL-positive patients have higher rates of SVTs.
The JAK2 46/1 haplotype
The JAK2 46/1 haplotype has recently been found to be associated with JAK2-positive MPNs, and it is believed that the JAK2 V617F mutation arises specifically on the 46/1 allele [Jones et al. 2009]. Its role in SVT formation is controversial, with some studies showing that the haplotype is an independent risk factor for SVT development in JAK2 V617F patients [Colaizzo et al. 2010a; Smalberg et al. 2011], and some studies showing only a weak association [Kouroupi et al. 2011]. A recent meta-analysis of 26 observational studies evaluated the risk of MPN with the JAK2 haplotype, and found that the haplotype was significantly increased in MPN and SVT patients. In JAK2-negative patients, however, the haplotype did not confer an additional risk of SVT [Li et al. 2014]. The increased risk of SVT with the 46/1 haplotype may be due to an interaction of the haplotype with the JAK2 V617F mutation, rather than the haplotype itself.
Additional mutations in MPN-associated SVTs
Approximately 10% of MPN patients are ‘triple-negative’ and do not harbor JAK2, CALR, or MPL mutations [Tefferi et al. 2014]. Additional mutations are found in varying frequencies in PV, PMF, and ET patients, although they tend not to be mutually exclusive with JAK2, CALR, MPL mutations, or with each other. These mutations include TET2, DNMT3A, EZH2 and ASXL1, and are involved in DNA methylation and chromatin structuring [Delhommeau et al. 2009; Grand et al. 2009; Ernst et al. 2010; Abdel-Wahab et al. 2011]. The specific role of these mutations in driving MPN disease formation is not fully understood. As discussed previously, SVT patients may be an ideal population for investigating the role of precursor genes and initiating mutations, as these patients may present at an early stage of MPN disease evolution. For instance, TET2 is a tumor suppressor gene which plays a key role in DNA methylation, and mutations in TET2 have been identified in up to 12% of MPN patients, including both JAK2-positive and JAK2-negative MPNs [Delhommeau et al. 2009]. In the hematopoietic stem cells of patients with both JAK2 and TET2 mutations, the TET2 mutation could often be found in the absence of the JAK2 mutation, suggesting the acquisition of TET2 before JAK2 [Delhommeau et al. 2009]. In surveying SVT patients, however, only a minority (3/23) of SVT patients tested positive for TET2, while all were JAK2 positive [Colaizzo et al. 2010b]. A larger study of 43 BCS patients identified 6 patients with a deleterious TET2 mutation; however, only 3 were JAK2-negative and none of these 3 patients developed an overt MPN [Westbrook et al. 2012]. Based on the studies by Colaizzo and colleagues and Westbrook and colleagues, the prevalence of TET2 in SVTs is roughly 14%, which is similar to the prevalence rate of TET2 in all MPNs [Delhommeau et al. 2009]. While TET2 is found more frequently than CALR and MPL in SVT, its frequent co-occurrence with JAK2 and lower prevalence rate suggests a limited role in SVT formation. Further investigations for mutations present in JAK2-negative SVT patients are limited. A small study of 25 cases of JAK2-negative BCS found no patients with IDH1 or IDH2 mutations [Jadli et al. 2012].
Table 2 summarizes the prevalence of the most common MPN-associated mutations in SVTs and all MPNs. In light of the existing literature, there seems to be a lack of evidence to suggest that these other mutations are associated with SVT formation in MPN, despite the finding that 14–20% of SVT patients with an MPN are JAK2-negative [Kiladjian et al. 2008; Smalberg et al. 2012]. The existence of a precursor mutation to JAK2 V617F critical to SVT pathogenesis has yet to be identified. Additional research examining the clinical and molecular differences between JAK2-positive and JAK2-negative patients with SVT is needed to further elucidate the driving mechanisms of SVT formation and MPN disease progression.
Table 2.
Prevalence of MPN-associated mutations in SVT and MPN patients. JAK2 makes up the highest proportion of patients, while CALR and MPL have very low prevalence rates in SVTs. TET2 is found at higher prevalence rates but is not mutually exclusive with JAK2.
| SVT patients | MPN patients | |
|---|---|---|
| JAK2 | 33% | 95% in PV; 60% in ET and PMF |
| CALR | 0.9% | 30% in ET and PMF |
| MPL | 0.7% | 5% in ET and PMF |
| Other | ||
| TET2 | 14% | 12% |
Sources: MPL- 0.7% in SVT, JAK2- 33% in SVT patients- Smalberg et al. [2012]; CALR- 0.9% in SVT patients- De Stefano et al. [2015]; MPL- 0.7% in SVT- Bergamaschi et al. [2008]; CALR- 0.9% in SVT patients- Colaizzo et al. [2015]; MPL- 0.7% in SVT- Kiladjian et al. [2008]; MPL- 0.7% in SVT, TET2- 14% in SVT- Jadli et al. [2012]; CALR- 0.9% in SVT patients- Turon et al. [2015]; TET2- 14% in SVT- Westbrook et al. [2012]; TET2- 14% in SVT- Colaizzo et al. [2010b]; 12% in MPN- Delhommeau et al. 2009; 5% in ET and PMF - Pikman et al. 2006; 95% in PV, 60% in ET/PMF- Campbell and Green 2006; 30% in ET and PMF- Klampf et al. 2013.
ET, essential thrombocythemia; MPN, myeloproliferative neoplasms; PMF, primary myelofibrosis; PV, polycythemia vera; SVT, splanchnic vein thrombosis.
Future directions and conclusion
The development of SVTs in MPN is multifactorial and likely includes both environmental and host genetic factors. Risk factors for the development of SVT in MPN patients include the JAK2 V617F mutation, young age, female sex, and the presence of concomitant hypercoagulable disorders. Intrinsic characteristics of the splanchnic venous bed may predispose to thrombotic interactions with atypical blood cells, which are modified by JAK2 V617F or unknown precursor mutations. Furthermore, the mechanisms that cause SVT formation likely differ from the mechanisms that cause arterial and DVT, accounting for the differences in risk factors. There is little evidence to suggest that other known genetic alterations implicated in MPN pathogenesis, such as CALR, MPL, or the JAK2 46/1 haplotype, are critical for SVT formation. Given that SVT patients may present at an early stage of MPN, identifying common mutations may shed light on the molecular pathogenesis of MPN disease development and progression.
Research into understanding the occurrence of SVTs in MPNs also has important implications for treatment and prognosis. Identifying patients most at risk for SVT can prevent considerable morbidity and mortality. Investigation into the use of JAK2 inhibitors to decrease the risk of future thrombosis is already under way [Keohane et al. 2015]. Future directions for research include better characterization of MPN patients with and without SVTs, and the development of a database of such phenotypes. In particular, investigating differences between JAK2-positive and JAK2-negative patients can better clarify the role of JAK2 V617F in thrombosis formation.
In summary, SVTs occur in a unique subset of MPN patients, and often present with little overt MPN disease characteristics. Understanding the association between SVTs and MPNs can provide insight not just on possible mechanisms of thrombosis, but also the pathophysiology of disease evolution.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Joan How, Division of Hematology, Washington University School of Medicine, St Louis, MO, USA.
Amy Zhou, Division of Hematology, Washington University School of Medicine, St Louis, MO, USA.
Stephen T. Oh, Division of Hematology, Washington University School of Medicine, 660 South Euclid Avenue, Campus Box 8125, St Louis, MO 63110, USA.
References
- Abdel-Wahab O., Pardanani A., Rampal R., Lasho T., Levine R., Tefferi A. (2011) DNMT3A mutational analysis in primary myelofibrosis, chronic myelomonocytic leukemia and advanced phases of myeloproliferative neoplasms. Leukemia 25: 1219–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ageno W., Beyer-Westendorf J., Garcia D., Lazo-Langner A., Mcbane R., Paciaroni M. (2016) Guidance for the management of venous thrombosis in unusual sites. J Thromb Thrombolysis 41: 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ageno W., Dentali F., Squizzato A. (2014) How I treat splanchnic vein thrombosis. Blood 124: 3685–3691. [DOI] [PubMed] [Google Scholar]
- Aird W. (2007a) Phenotypic heterogeneity of the endothelium: II. representative vascular beds. Circ Res 100: 174–190. [DOI] [PubMed] [Google Scholar]
- Aird W. (2007b) Vascular bed-specific thrombosis. J Thromb Haemost 5(Suppl. 1): 283–291. [DOI] [PubMed] [Google Scholar]
- Anger B., Seifried E., Scheppach J., Heimpel H. (1989) Budd-Chiari syndrome and thrombosis of other abdominal vessels in the chronic myeloproliferative diseases. Klin Wochenschr 67: 818–825. [DOI] [PubMed] [Google Scholar]
- Araki M., Yang Y., Masubuchi N., Hironaka Y., Takei H., Morishita S., et al. (2016) Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 127: 1307–1316. [DOI] [PubMed] [Google Scholar]
- Arellano-Rodrigo E., Alvarez-Larran A., Reverter J., Villamor N., Colomer D., Cervantes F. (2006) Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica 91: 169–175. [PubMed] [Google Scholar]
- Austin S., Lambert J. (2008) The JAK2 V617F mutation and thrombosis. Br J Haematol 143: 307–320. [DOI] [PubMed] [Google Scholar]
- Barbui T., Carobbio A., Cervantes F., Vannucchi A., Guglielmelli P., Antonioli E., et al. (2010) Thrombosis in primary myelofibrosis: incidence and risk factors. Blood 115: 778–782. [DOI] [PubMed] [Google Scholar]
- Barbui T., Finazzi G., Falanga A. (2013) Myeloproliferative neoplasms and thrombosis. Blood 122: 2176–2184. [DOI] [PubMed] [Google Scholar]
- Beer P., Campbell P., Scott L., Bench A., Erber W., Bareford D., et al. (2008) MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 112: 141–149. [DOI] [PubMed] [Google Scholar]
- Bergamaschi G., Primignani M., Barosi G., Fabris F., Villani L., Reati R., et al. (2008) MPL and JAK2 exon 12 mutations in patients with the Budd-Chiari syndrome or extrahepatic portal vein obstruction. Blood 111: 4418. [DOI] [PubMed] [Google Scholar]
- Campbell P., Green A. (2006) The myeloproliferative disorders. N Engl J Med 355: 2452–2466. [DOI] [PubMed] [Google Scholar]
- Carobbio A., Thiele J., Passamonti F., Rumi E., Ruggeri M., Rodeghiero F., et al. (2011) Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an International study of 891 patients. Blood 117: 5857–5859. [DOI] [PubMed] [Google Scholar]
- Castro N., Rapado I., Ayala R., Martinez-Lopez J. (2015) CALR mutations screening should not be studied in splanchnic vein thrombosis. Br J Haematol 170: 588–589. [DOI] [PubMed] [Google Scholar]
- Chachoua I., Pecquet C., El-Khoury M., Nivarthi H., Albu R., Marty C., et al. (2016) Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 127: 1325–1335. [DOI] [PubMed] [Google Scholar]
- Colaizzo D., Amitrano L., Guardascione M., Favuzzi G., Tiscia G., D’Andrea G., et al. (2015) Clinical utility of screening for CALR gene exon 9 mutations in patients with splanchnic venous thrombosis. Thromb Haemost 113: 1381–1382. [DOI] [PubMed] [Google Scholar]
- Colaizzo D., Amitrano L., Guardascione M., Tiscia G., D’Andrea G., Longo V., et al. (2013) Outcome of patients with splanchnic venous thrombosis presenting without overt MPN: a role for the JAK2 V617F mutation re-evaluation. Thromb Res 132: e99–e104. [DOI] [PubMed] [Google Scholar]
- Colaizzo D., Tiscia G., Bafunno V., Amitrano L., Vergura P., Grandone E., et al. (2010a) The JAK2 rs12343867 CC genotype frequently occurs in patients with splanchnic venous thrombosis without the JAK2V617F mutation: a retrospective study. J Thromb Haemost 8: 413–416. [DOI] [PubMed] [Google Scholar]
- Colaizzo D., Tiscia G., Pisanelli D., Bafunno V., Amitrano L., Grandone E., et al. (2010b) New TET2 gene mutations in patients with myeloproliferative neoplasms and splanchnic vein thrombosis. J Thromb Haemost 8: 1142–1144. [DOI] [PubMed] [Google Scholar]
- Deitelzweig S., Johnson B., Lin J., Schulman K. (2011) Prevalence of clinical venous thromboembolism in the USA: current trends and future projections. Am J Hematol 86: 217–220. [DOI] [PubMed] [Google Scholar]
- Delhommeau F., Dupont S., Della Valle V., James C., Trannoy S., Masse A., et al. (2009) Mutation in TET2 in myeloid cancers. N Engl J Med 360: 2289–2301. [DOI] [PubMed] [Google Scholar]
- Dentali F., Squizzato A., Brivio L., Appio L., Campiotti L., Crowther M., et al. (2009) JAK2V617F mutation for the early diagnosis of ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 113: 5617–5623. [DOI] [PubMed] [Google Scholar]
- De Grandis M., Cambot M., Wautier M., Cassinat B., Chomienne C., Colin Y., et al. (2013) JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an eporindependent Rap1/Akt pathway. Blood 121: 658–665. [DOI] [PubMed] [Google Scholar]
- De Stefano V., Fiorini A., Rossi E., Za T., Farina G., Chiusolo P., et al. (2007) Incidence of the JAK2 V617F mutation among patients with splanchnic or cerebral venous thrombosis and without overt chronic myeloproliferative disorders. J Thromb Haemost 5: 708–714. [DOI] [PubMed] [Google Scholar]
- De Stefano V., Qi X., Betti S., Rossi E. (2015) Splanchnic vein thrombosis and myeloproliferative neoplasms: molecular-driven diagnosis and long-term treatment. Thromb Haemost 115:240–249. [DOI] [PubMed] [Google Scholar]
- De Stefano V., Za T., Ciminello A., Betti S., Rossi E. (2011) Causes of adult splanchnic vein thrombosis in the mediterranean area. Mediterr J Hematol Infect Dis 3: e2011063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefano V., Za T., Rossi E., Vannucchi A., Ruggeri M., Elli E., et al. (2008) Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 93: 372–380. [DOI] [PubMed] [Google Scholar]
- Elf S., Abdelfattah N., Chen E., Perales-Paton J., Rosen E., Ko A., et al. (2016) Mutant calreticulin requires both its mutant c-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov 6: 368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst T., Chase A., Score J., Hidalgo-Curtis C., Bryant C., Jones A., et al. (2010) Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42: 722–726. [DOI] [PubMed] [Google Scholar]
- Falanga A., Marchetti M. (2014) Thrombosis in myeloproliferative neoplasms. Semin Thromb Hemost 40: 348–358. [DOI] [PubMed] [Google Scholar]
- Falanga A., Marchetti M., Vignoli A., Balducci D., Barbui T. (2005) Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp Hematol 33: 523–530. [DOI] [PubMed] [Google Scholar]
- Genovese G., Kahler A., Handsaker R., Lindberg J., Rose S., Bakhoum S., et al. (2014) Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371: 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand F., Hidalgo-Curtis C., Ernst T., Zoi K., Zoi C., Mcguire C., et al. (2009) Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 113: 6182–6192. [DOI] [PubMed] [Google Scholar]
- Grandone E., Colaizzo D., Tiscia G., Vergura P., Chinni E., Iannaccone L., et al. (2008) Venous thrombosis in oral contraceptive users and the presence of the JAK2 V617Fmutation. Thromb Haemost 99: 640–642. [DOI] [PubMed] [Google Scholar]
- Haslam K., Langabeer S. (2015) Incidence of CALR mutations in patients with splanchnic vein thrombosis. Br J Haematol 168: 459–460. [DOI] [PubMed] [Google Scholar]
- Hobbs C., Manning H., Bennett C., Vasquez L., Severin S., Brain L., et al. (2013) JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood 122: 3787–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra J., Bresser E., Smalberg J., Spaander M., Leebeek F., Janssen H. (2011) Long-term follow-up of patients with portal vein thrombosis and myeloproliferative neoplasms. J Thromb Haemost 9: 2208–2214. [DOI] [PubMed] [Google Scholar]
- Iurlo A., Cattaneo D., Gianelli U., Fermo E., Augello C., Cortelezzi A. (2015) Molecular analyses in the diagnosis of myeloproliferative neoplasm-related splanchnic vein thrombosis. Ann Hematol 94: 881–882. [DOI] [PubMed] [Google Scholar]
- Jadli A., Kulkarni B., Ghosh K., Shetty S. (2012) Non-conventional mutations associated with myeloproliferative disorders are absent in splanchnic venous thrombosis cases. Liver Int 32: 1596–1597. [DOI] [PubMed] [Google Scholar]
- Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P., Mar B., et al. (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371: 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James C. (2008) The JAK2V617F mutation in polycythemia vera and other myeloproliferative disorders: one mutation for three diseases? Hematology Am Soc Hematol Educ Program: 69–75. [DOI] [PubMed] [Google Scholar]
- Johnson S., Michalak M., Opas M., Eggleton P. (2001) The ins and outs of calreticulin: from the ERlumen to the extracellular space. Trends Cell Biol 11: 122–129. [DOI] [PubMed] [Google Scholar]
- Jones A., Chase A., Silver R., Oscier D., Zoi K., Wang Y., et al. (2009) JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet 41: 446–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keohane C., Mclornan D., Sanchez K., Connor C., Radia D., Harrison C. (2015) The effects of JAK inhibitor therapy upon novel markers of thrombosis in myeloproliferative neoplasms. Haematologica 100: e348–e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiladjian J., Cervantes F., Leebeek F., Marzac C., Cassinat B., Chevret S., et al. (2008) The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: a report on 241 cases. Blood 111: 4922–4929. [DOI] [PubMed] [Google Scholar]
- Klampfl T., Gisslinger H., Harutyunyan A., Nivarthi H., Rumi E., Milosevic J., et al. (2013) Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 369: 2379–2390. [DOI] [PubMed] [Google Scholar]
- Kouroupi E., Kiladjian J., Chomienne C., Dosquet C., Bellucci S., Valla D., et al. (2011) The JAK2 46/1 haplotype in splanchnic vein thrombosis. Blood 117: 5777–5778. [DOI] [PubMed] [Google Scholar]
- Kreher S., Ochsenreither S., Trappe R., Pabinger I., Bergmann F., Petrides P., et al. (2014) Prophylaxis and management of venous thromboembolism in patients with myeloproliferative neoplasms: consensus statement of the Haemostasis Working Party of the German Society of Hematology and Oncology (DGHO), the Austrian Society of Hematology and Oncology (OGHO) and Society of Thrombosis and Haemostasis Research (GTH e.V.). Ann Hematol 93: 1953–1963. [DOI] [PubMed] [Google Scholar]
- Lamrani L., Lacout C., Ollivier V., Denis C., Gardiner E., Ho Tin Noe B., et al. (2014) Hemostatic disorders in a JAK2V617F-driven mouse model of myeloproliferative neoplasm. Blood 124: 1136–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy T., Devillers A., Bernard M., Moisan A., Grulois I., Drenou B., et al. (1997) Inapparent polycythemia vera: an unrecognized diagnosis. Am J Med 102: 14–20. [DOI] [PubMed] [Google Scholar]
- Landolfi R., Di Gennaro L., Barbui T., De Stefano V., Finazzi G., Marfisi R., et al. (2007) Leukocytosis as a Major thrombotic risk factor in patients with polycythemia vera. Blood 109: 2446–2452. [DOI] [PubMed] [Google Scholar]
- Li S., Zhang P., Sun G., Lu Z. (2014) The JAK2 46/1 haplotype (GGCC) in myeloproliferative neoplasms and splanchnic vein thrombosis: a pooled analysis of 26 observational studies. Ann Hematol 93: 1845–1852. [DOI] [PubMed] [Google Scholar]
- Lussana F., Carobbio A., Randi M., Elena C., Rumi E., Finazzi G., et al. (2014) A lower intensity of treatment May underlie the increased risk of thrombosis in young patients with masked polycythaemia vera. Br J Haematol 167: 541–546. [DOI] [PubMed] [Google Scholar]
- Marchioli R., Finazzi G., Landolfi R., Kutti J., Gisslinger H., Patrono C., et al. (2005) Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 23: 2224–2232. [DOI] [PubMed] [Google Scholar]
- Marty C., Pecquet C., Nivarthi H., El-Khoury M., Chachoua I., Tulliez M., et al. (2016) Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 127: 1317–1324. [DOI] [PubMed] [Google Scholar]
- Marzac C., Plessier A., Kiladjian J., Andreoli A., De Raucourt E., Goria O., et al. (2015) O079: Calr somatic mutations in a prospective cohort of 308 patients with splanchnic vein thrombosis. J Hepatol 62: S230. [DOI] [PubMed] [Google Scholar]
- Nangalia J., Massie C., Baxter E., Nice F., Gundem G., Wedge D., et al. (2013) Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 369: 2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S., Gotlib J. (2010) JAK2 V617F and beyond: role of genetics and aberrant signaling in the pathogenesis of myeloproliferative neoplasms. Expert Rev Hematol 3: 323–337. [DOI] [PubMed] [Google Scholar]
- Patel R., Lea N., Heneghan M., Westwood N., Milojkovic D., Thanigaikumar M., et al. (2006) Prevalence of the activating JAK2 tyrosine kinase mutation V617F in the Budd-Chiari syndrome. Gastroenterology 130: 2031–2038. [DOI] [PubMed] [Google Scholar]
- Pikman Y., Lee B., Mercher T., McDowell E., Ebert B., Gozo M., et al. (2006) MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 3: e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plompen E., Valk P., Chu I., Darwish Murad S., Plessier A., Turon F., et al. (2015) Somatic calreticulin mutations in patients with Budd-Chiari syndrome and portal vein thrombosis. Haematologica 100: e226–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajani R., Almer S. (2009) Incidence and prevalence rates in Budd-Chiari syndrome. Gut 58: 889. [PubMed] [Google Scholar]
- Rajani R., Bjornsson E., Bergquist A., Danielsson A., Gustavsson A., Grip O., et al. (2010) The epidemiology and clinical features of portal vein thrombosis: a multicentre study. Aliment Pharmacol Ther 32: 1154–1162. [DOI] [PubMed] [Google Scholar]
- Robertson B., Urquhart C., Ford I., Townend J., Watson H., Vickers M., et al. (2007) Platelet and coagulation activation markers in myeloproliferative diseases: relationships with JAK2 V6I7 F status, clonality, and antiphospholipid antibodies. J Thromb Haemost 5: 1679–1685. [DOI] [PubMed] [Google Scholar]
- Roques M., Park J., Minello A., Bastie J., Girodon F. (2015) Detection of the CALR mutation in the diagnosis of splanchnic vein thrombosis. Br J Haematol 169: 601–603. [DOI] [PubMed] [Google Scholar]
- Rosenberg R., Aird W. (1999) Vascular-bed-specific hemostasis and hypercoagulable states. N Engl J Med 340: 1555–1564. [DOI] [PubMed] [Google Scholar]
- Rosti V., Villani L., Riboni R., Poletto V., Bonetti E., Tozzi L., et al. (2013) Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood 121: 360–368. [DOI] [PubMed] [Google Scholar]
- Rumi E., Pietra D., Ferretti V., Klampfl T., Harutyunyan A., Milosevic J., et al. (2014) JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 123: 1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumi E., Pietra D., Guglielmelli P., Bordoni R., Casetti I., Milanesi C., et al. (2013) Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 121: 4388–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhar M., McVinnie K., Burroughs A. (2013) Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 162: 730–747. [DOI] [PubMed] [Google Scholar]
- Smalberg J., Arends L., Valla D., Kiladjian J., Janssen H., Leebeek F. (2012) Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 120: 4921–4928. [DOI] [PubMed] [Google Scholar]
- Smalberg J., Darwish Murad S., Braakman E., Valk P., Janssen H., Leebeek F. (2006) Myeloproliferative disease in the pathogenesis and survival of Budd-Chiari syndrome. Haematologica 91: 1712–1713. [PubMed] [Google Scholar]
- Smalberg J., Koehler E., Darwish Murad S., Plessier A., Seijo S., Trebicka J., et al. (2011) The JAK2 46/1 haplotype in Budd-Chiari Syndrome and portal vein thrombosis. Blood 117: 3968–3973. [DOI] [PubMed] [Google Scholar]
- Sozer S., Fiel M., Schiano T., Xu M., Mascarenhas J., Hoffman R. (2009) The presence of JAK2 V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood 113: 5246–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam J. (2005) Thrombosis of the cerebral veins and sinuses. N Engl J Med 352: 1791–1798. [DOI] [PubMed] [Google Scholar]
- Stein B., Saraf S., Sobol U., Halpern A., Shammo J., Rondelli D., et al. (2013) Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymphoma 54: 1989–1995. [DOI] [PubMed] [Google Scholar]
- Stein B., Williams D., Wang N., Rogers O., Isaacs M., Pemmaraju N., et al. (2010) Sex differences in the JAK2 V617F allele burden in chronic myeloproliferative disorders. Haematologica 95: 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A., Barbui T. (2015) Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk-stratification and management. Am J Hematol 90: 162–173. [DOI] [PubMed] [Google Scholar]
- Tefferi A., Vardiman J. (2008) classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 22: 14–22. [DOI] [PubMed] [Google Scholar]
- Tefferi A., Guglielmelli P., Larson D., Finke C., Wassie E., Pieri L., et al. (2014) Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 124: 2507–2513; quiz 2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teofili L., Martini M., Iachininoto M., Capodimonti S., Nuzzolo E., Torti L., et al. (2011) Endothelial progenitor cells are clonal and exhibit the JAK2(V617F) mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms. Blood 117: 2700–2707. [DOI] [PubMed] [Google Scholar]
- Turon F., Cervantes F., Colomer D., Baiges A., Hernandez-Gea V., Garcia-Pagan J. (2015) Role of calreticulin mutations in the aetiological diagnosis of splanchnic vein thrombosis. J Hepatol 62: 72–74. [DOI] [PubMed] [Google Scholar]
- Valla D. (2015) Splanchnic vein thrombosis. Semin Thromb Hemost 41: 494–502. [DOI] [PubMed] [Google Scholar]
- Van Bijnen S., van Heerde W., Muus P. (2012) Mechanisms and clinical implications of thrombosis in paroxysmal nocturnal hemoglobinuria. J Thromb Haemost 10: 1–10. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke J., Koster T., Briet E., Reitsma P., Bertina R., Rosendaal F. (1994) increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet 344: 1453–1457. [DOI] [PubMed] [Google Scholar]
- Wautier J., Wautier M. (2013) Molecular basis of erythrocyte adhesion to endothelial cells in diseases. Clin Hemorheol Microcirc 53: 11–21. [DOI] [PubMed] [Google Scholar]
- Westbrook R., Lea N., Mohamedali A., Smith A., Orr D., Roberts L., et al. (2012) Prevalence and clinical outcomes of the 46/1 haplotype, Janus kinase 2 mutations, and ten-eleven translocation 2 mutations in Budd-Chiari syndrome and their impact on thrombotic complications post liver transplantation. Liver Transpl 18: 819–827. [DOI] [PubMed] [Google Scholar]
- White R. (2003) The epidemiology of venous thromboembolism. Circulation 107(23 Suppl. 1): I4–8. [DOI] [PubMed] [Google Scholar]
- Xie M., Lu C., Wang J., McLellan M., Johnson K., Wendl M., et al. (2014) Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 20: 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]