

ABSTRACT
The retinal tissue of warm-blooded vertebrates performs surprisingly complex and accurate transduction of visual information. To achieve precision, a multilayered neuroglia structure is established throughout the embryonic development, and the presence of radial Müller (glial) cells ensure differentiation, growth and survival for the neuronal elements within retinal environment. It is assumed that Müller cells serve as a dynamic reservoir of progenitors, capable of expressing transcription factors, differentiating and proliferating as either neuronal or glial cells depending on extrinsic cues. In the postnatal period, Müller glia may re-enter cell cycle and produce new retinal neurons in response to acute damage. In this context, glutathione (GSH), a virtually ubiquitous tripeptide antioxidant, which is found at milimolar concentrations in central glial cells, plays a vital role as a reducing agent, buffering radical oxygen species (ROS) and preventing cell death in severely injured retinal tissues. Despite its antioxidant role, data also point to GSH as a signaling agent, suggesting that GABA release and P2X7R-mediated calcium inwards occur in Müller cells in a GSH-enriched environment. These phenomena indicate a novel mechanistic response to damage in the vertebrate retinal tissue, particularly in neuron-glia networks.
KEYWORDS: calcium, GABA, Glutathione, Müller glia, P2X7, redox signaling
Introduction
Water-soluble antioxidants are defined as low molecular weight compounds, usually found intracellularly at millimolar levels, that protect the organism against reactive oxygen (ROS) and nitrogen (RNS) species. The 2 main antioxidants found in the nervous system are ascorbic acid (AA) and glutathione (GSH), known to be compartmentalized (Fig. 1) in a way that AA is preferentially found in neurons (∼10mM, 10:1 neuron: glia), while GSH is mainly present in glial cells (∼4mM, 2:1 glia: neuron).1,2
Figure 1.
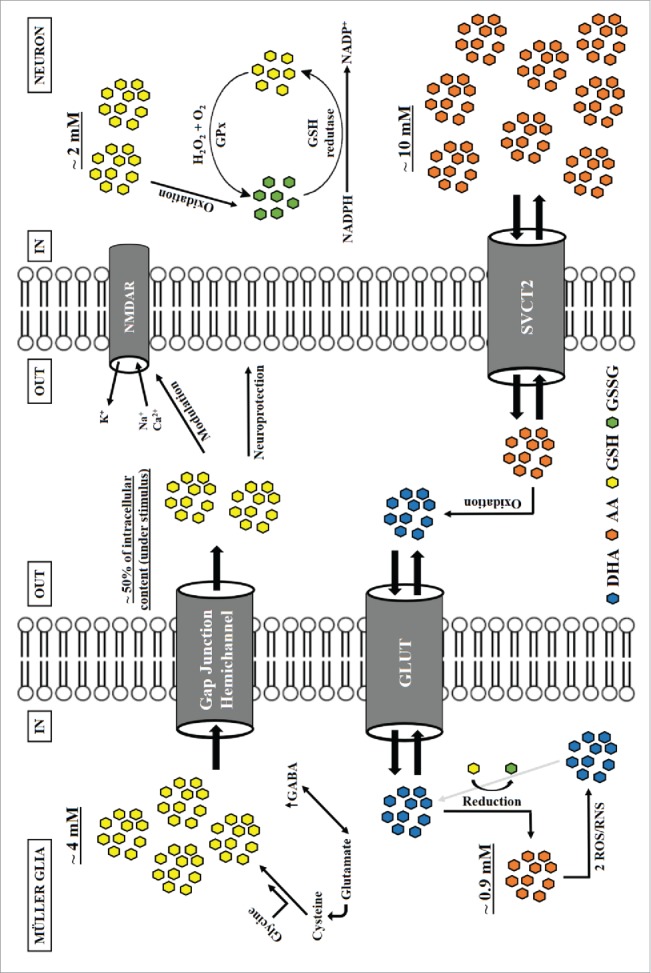
Antioxidant compartmentalization in the neuroglial environment. Neurons and glia (e.g. Müller glia) accumulate different levels of low molecular weight antioxidants within the intracellular space. While ascorbic acid (AA) is preferentially found in neurons (∼10mM, 10:1 neuron: glia), GSH is mainly present in glial cells (∼4mM, 2:1 glia: neuron).1,2 When stimulated, glia cells release nearly half of its GSH content to the extracellular compartment, possibly through formation of a gap junction hemichannel.24 When released, GSH may provide neuroprotection and modulate the activity of several membrane receptors, including NMDAR.21 In this context, neuronal ascorbate has been shown to serve as a compensation for loss or disruption of components from the antioxidant network (e.g., GSH, GSH peroxidase, catalase), providing basal antioxidant stability.35 While GSH recycling (GSSG reduction) depends on cell energy status (i.e., NADPH to NADP+ ratio), AA is ultimately recovered from and enzymatic reaction of GSH with DHA.36 Also, both GABA and GSH synthesis depend on plasma glutamate, which indicates interplay between antioxidant and neurotransmitter production in glial cells.37 Finally, release of both GSH and AA act as major components of extracellular protection and communication in neuroglia systems.35
In addition to perform reduction–oxidation (redox) reactions, it has been proposed that GSH may act as a signal to activate or modulate receptors in both neurons and glial cells.3,4 For instance, GSH was shown to interact on high-affinity binding sites of pig cerebral cortical synaptic membranes,5 to communicate with the NMDA recognition domain independently of the thiol moiety,4 and to release [3H]dopamine in mouse striatal slices.6 Also, astrocytes secrete GSH into the extracellular space,7-9 which is taken up by neuronal transporters.9,10 AA uptake, however, occurs through SVCT2 transporters, which are only expressed in neurons.11 Therefore, GSH and AA could activate complementary pathways in neuron-glial circuits.
A rich neuron-glial environment, the vertebrate retina, is characterized by complexity and diversity of its multilayered structure. In the tissue, Müller glial cells enwrap most of the neuronal elements, exchanging transmitters and factors12 to maintain the balance of K+ ions, the flux of energy through neuron-glia metabolism and other vital functions (reviewed by13). The concept of gliotransmitters, however, only emerged in the last 2 decades, implying that glutamate, ATP and D-serine signal from astrocytes to neurons to support survival, development, plasticity and proliferation. Additionally, novel data indicate that disruption of gliotransmission leads to neuronal dysfunction and abnormal behavior in animal models,14 and despite most results in glial behavior refer to astrocytic cells, the intrinsic radial morphology of Müller glia in the retina clearly indicates its critical role for neuronal survival and function.12
As a transmitter, ATP activates ionotropic P2X and metabotropic P2Y receptors, and both types are found in Müller glia15 as well as on retinal neurons.16 ATP induces membrane currents at millimolar levels mainly through activation of P2X7 receptor (P2X7R), permeating Ca2+ influx through the emergence of a macro pore.17 We have recently shown that GSH selectively induces Ca2+ shifts in the avian Müller glia18 through activation of purinergic pathway, once such shifts were inhibited by brilliant blue G (BBG) antagonism of P2X7R. Also, GSH treatment induced observable GABA release from both retinal tissue and Müller cells in culture. Thus, it's tempting to elaborate on emerging communicative roles for antioxidants in the neuroglial environment.
Glutathione signaling
The idea of a tripeptide (Glu-Cys-Gly) antioxidant acting in the brain dates back to the late 80s. Initial propositions suggested cysteine (R-SH domain) and glutamate residues in the GSH molecule as potential modulators of glutamate binding and neurotransmitter release. Ogita et al. (1986) showed that 10−7–10−3 M GSH or oxidized GSH (GSSG) decreased the affinity of binding sites for [3H]glutamic acid (Glu) through either interacting with synaptic receptors or regulating Glu uptake by central neurons in the rat brain.19 Afterwards, GSSG but not GSH was shown to selectively attenuate intracellular calcium ([Ca2+]i) and whole-cell currents induced by N-methyl-D-aspartate (NMDA) in rat cortical and retinal ganglion neuron cells. This suggested a regulatory effect for GSSG over NMDA receptor-channel (NMDAR) complex, acting through oxidation of thiol groups to form peptide disulfide bonds (R-S-S-R) in the redox site of NMDAR.20,21 Also, GSSG had no impact on [Ca2+]i responses by kainate or high extracellular K+20.
GSH and GSSG were shown to negatively modulate dithiothreitol (DTT) effects and to prolong glutamate-induced [3H]GABA release from rat hippocampal slices. Redox properties of GSH and GSSG were proposed as regulators of NMDAR.20 However, it's plausible that GSSG, not GSH, is responsible for counteracting (through oxidation) DTT-related effects on thiol residues in the NMDAR complex, as previously proposed by.21 On the other hand, GSH may act competitively with DTT to modulate Glu-NMDAR binding, sustained activation, and [3H]GABA release.
Reticular ([Ca2+]r) storage may also be affected by GSH, specifically by the intracellular GSH/GSSG ratio, which is highly dependent on the amount of substrate for reduction reactions with GSH. At the milimolar range, GSH, as well as DTT and βmercaptoethanol, inhibited high affinity [3H]ryanodine binding to sarcoplasmic reticulum in skeletal muscle cells. Oppositely, GSSG acted as a potent stimulator of [3H]ryanodine binding and subsequent Ca2+ release from the reticular compartment.22 Redox dynamics of GSH play an important role in regulating bidirectional influx of Ca2+ into the cytoplasm, and may participate in global responses to variations in the extracellular GSH content, which is referred as both directly and indirectly involved in the mammalian synaptic transmission.4 Ultimately, GSH oxidation may lead to oxidative interactions with several intra and extracellular targets, such as surface receptors and signal transduction chains.23
Beyond behaving as a neuromodulator, mainly through interference with glutamatergic signaling, a population of specific binding sites for GSH was identified in the pig cerebral cortical membranes. GSH, GSSG, S-nitrosoglutathione, gamma-L-glutamylcysteine, cysteinylglycine, cysteine and cysteamine were shown to displace [3H]glutathione binding from the cortical membrane when at the micromollar range. Also, oxidation or alkylation in the cysteinyl moiety of GSH attenuated binding affinity, suggesting that receptor-agonist interactions may be stabilized through the presence of a reduced cysteine thiol group.5 In fact, GSH and cysteine were shown to enhance glutamatergic-induced (NMDA and non-NMDA agonists) release of [3H]dopamine in mouse striatal slices. Differently, the K+-evoked release was inhibited by GSH, while enhanced by cysteine. Authors suggested a role for GSH in the cysteine (R-SH)/cysteine (R-S-S-R) redox modulation in voltage-gated Ca2+ channels, attenuating the depolarization effect on [3H]dopamine release.6
As previously discussed, glial cells are the main GSH compartment in the central nervous system (∼4mM, 2:1 glia: neuron), and considering its relevance in both inside and outside the cell membrane, a mechanism for abundant GSH release is necessary. Rana and Dringen (2007) showed that astrocyte-rich cultures from neonatal rat brain release GSH through the formation of a gap junction hemichannel, as indicated by blockage with the gap junction inhibitors carbenoxolone, flufenamic acid and lanthanum chloride. Also, when stimulated, cells released about 50% of the total intracellular GSH content, which creates a large increase in the reducing potential of the brain environment.24
Finally, GSH appears to be closely related to the glutamatergic signaling, and a recent work presented evidence for the coupling of NMDAR activation to the transcriptional control of GSH metabolism (i.e. synthesis, recycling and effects). This mechanism may represent an important strategy for astrocytes to provide neurons with large amounts of GSH in face of excitotoxicity and oxidative insults, thus preventing associated neurodevelopmental disorders.25
Glutathione in retinal Müller glia
Despite the flourishing literature on antioxidant systems in the retinal environment, our group was the first to examine the signaling role of GSH on both Müller glia and retinal neurons from the vertebrate (avian) eye.18 Initial characterization of the [Ca2+]i dynamics showed that, while cultured neurons selectively respond to KCl (labeled as βIII tubulin positive cells), large Ca2+ inwards were observed in GSH-stimulated Müller cells (labeled and identified as 2M6 positive cells). We have also observed that GSH selectively evokes abundant [Ca2+]i variations in Müller cells at the milimolar range, and that purified retinal neurons do not respond to GSH in a broad concentration gradient (10 µM – 10 mM).
Preincubation with DNQX (70 μM) and MK-801 (20μM), non-NMDA and NMDA receptor blockers, did not influence the Ca2+ behavior in Müller cells, however, when cultures were treated with a P2X7R antagonist, BBG (100 nM), there was full inhibition of Ca2+ responses, which were only recovered after complete washing out the BBG solution from the cells. Also, GSSG did not alter [Ca2+]i levels in purified Müller cultures, indicating that GSH acts through redox mechanisms to regulate the activation of P2X7R receptors. This proposition was later confirmed by incorporation of propidium iodide (PI) by Müller cells when stimulated with either GSH or ATP. In both cases, the P2X7R inhibitor, BBG (500 nM), a selective P2X7R blocker, A740003 (1 µM), and an intracellular Ca2+ chelator, BAPTA-AM (5 µM), significantly reduced PI incorporation by these cells.
We have shown that intracellular GSH levels are rapidly increased in Müller cells after 60s incubation, which is sufficient to promote cell survival and resilience to highly oxidant (H2O2 0.1%) environments, as seen through Live/Dead and dichlorofluorescein oxidation assays. Posteriorly, through a dUTP nick end labeling (TUNEL) assay, GSH showed no influence in the number of TUNEL+ cells in culture, as seen by comparison with negative control and thapsigargin (30 nM) treatments.
Finally, GSH induced GABA release from the embryonic retinal tissue, mixed neuron-glia and from Müller cell cultures. Retinal tissue release of GABA, as observed through high performance liquid chromatography (HPLC) analysis, occurred at GSH milimolar levels, corresponding to previous data on Ca2+ transients and PI uptake. In the tissue, a GSH synthesis inhibitor, buthionine sulphoximine (BSO 50 µM), blocked the basal GABA release. Posterior quantification of GSH-induced [3H]GABA release showed that mixed neuron-glial cell cultures are more susceptible to GSH stimulus for [3H]GABA than purified Müller cells only, indicating that the Müller glia, when stimulated with GSH, provoke [3H]GABA release from retinal neurons in culture. Also, treatment with BBG (100 nM) or absence of sodium caused impairment of [3H]GABA mobilization after GSH treatment. This points to a P2X7R-mediated mechanism, in which [Ca2+]i levels may promote GABA release through reversal of the GAT transporter activity (Fig. 2).
Figure 2.
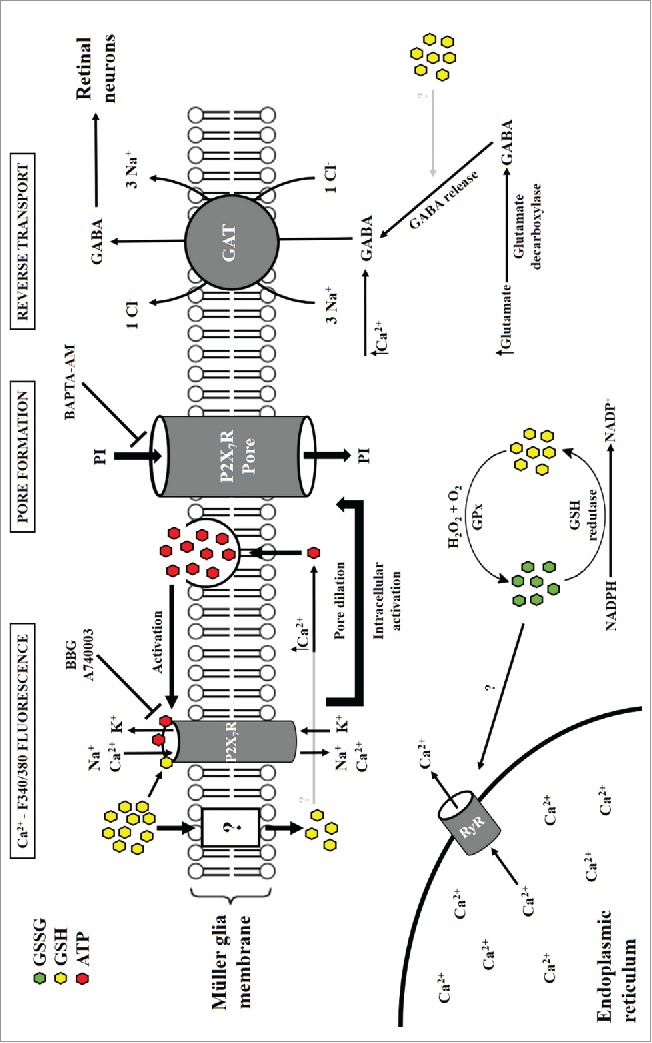
Glutathione signals through P2X7R on Müller glia and releases GABA. Müller cells rapidly uptake glutathione (GSH), which regulates intracellular calcium ([Ca2+]i) levels by P2X7R-mediated activation and/or release from an endoplasmic reticulum storage by stimulus with newly formed GSSG molecules. Increased [Ca2+]i, in turn, modulates ATP and GABA release, with a possible mechanistic role for GSH in the process of GABA formation and/or release. Either by GSH or ATP interaction (both inhibited by BBG and A740003), P2X7R promotes the formation of a P2X7R-associated large pore, as seen by incorporation of propidium iodide (PI), which was blocked by the Ca2+ chelator BAPTA-AM. GABA released from Müller glia under stress or excitotoxic conditions may act as an important neuroprotectant gliotransmitter.
GSH induces [Ca2+]i transients and GABA release through glial P2X7R
Previous data have shown that activation of P2X7R in rat hippocampal slices leads to GABA release. In this model, ATP and the selective P2X7 agonist N6-Benzyl-ATP (BzATP) induced GABA release through neuronal P2X7R located in CA1 and CA3 neurons, as revealed by in situ hybridization.26 Other groups demonstrated neuronal or astrocytic P2X7R coupled to glutamate release in the spinal cord and hippocampus.27 In the spinal cord, astrocytic glutamate release through a P2X7R-dependent manner induces a gliogenic long-term potentiation involved in nociceptive pathways that could explain pain hypersensitivity.28
GSH induces a dose-dependent GABA release in embryonic avian retinal tissue (stage E14, embryonic day 14), and both neurons and Müller cells are known to release GABA. [3H] GABA release induced by GSH was also observed in cultured Müller cells or in dense mixed neuron-glia cultures, but not in purified neuronal cells.18 GSH might be interacting at the P2X7 receptor, as the effect was blocked by BBG or A74003. Addition of BAPTA-AM, a calcium chelator, also blocked propidium iodine uptake induced by GSH (or ATP) in Müller glial cells.18
It is known that a subset of amacrine neurons29 releases GABA in the retina. In addition, extracellular GABA levels are regulated by activation of glial P2X receptors.30 The mechanism of GABA release is known to be Ca2+-dependent and independent, or to involve Na+-dependent and independent mechanisms.31,32 Based in what is known in the literature and in our results, it is possible that both neurons and Müller glia might be involved in GABA release induced by GSH. Calcium increase evoked by GSH in Müller cells might secrete a signal that could mediate GABA release from amacrine neurons in the mixed culture. This could be dependent on Ca2+, as in exocytosis, or through reversal of GABA transporters (GAT), that are known to be Na+ dependent. However, it cannot be discarded a Ca2+-independent mechanism for GABA release in retinal cells that produce/accumulate GABA. Indeed, at least 80% of GABA secretion from chick retinal cell occurs through GAT-1/3-like transporters.31 Also, data from our group show that GAT-3 transporter is regulated by glutamate in Müller glia.33 In summary, ATP induces Ca2+ increase that might lead to neurotoxicity in retinal neuronal-glial cell cultures,34 but GABA released by GSH through a P2X7 glial receptor could represent a mechanism of protection in the avian retina, where Müller cells have a pivotal protective role.
Concluding remarks and perspectives
Despite the simple molecular structure, amino acid moieties (Glu-Cys-Gly) from GSH may interact with neuronal and glial elements in the whole brain, depending on the density of receptor expression and susceptibility to membrane redox modulation. Surprisingly, GSH played no role in altering neuronal [Ca2+]i levels in the avian retina, which was largely reported by other studies with cortical and striatal membranes. It's possible, however, that GSH may only modulate glutamatergic binding in retinal neurons, as shown in previous studies with cortical slices. Our data point to novel roles for the neuroglial compartmentalization of low molecular weight antioxidants (GSH and AA) and intra/extracellular signaling mechanisms, presenting a provocative view of redox control in neuroprotection and neurodevelopment.
Neuron-glia circuits in the retina are activated by several types of transmitters, of which glutamate and ATP are among the most abundant driving neuronal excitability and synaptic efficacy; glutamate is released by photoreceptors, bipolar and retinal ganglion cells, while ATP is secreted by both neurons and Müller glia. They increase ROS and RNS particularly in neurons, which are highly susceptible to attack if not involved by Müller glia in a symbiotic manner. The antioxidant GSH provides neuroprotection through a thiol moiety, and evidence in the literature show that this agent might operate as a neuromodulator or as a neurotransmitter on classical receptors, such as NMDAR.4 Here we discussed the potential of GSH to behave as a signal to induce Ca2+ transients through P2X7R in Müller cells, releasing GABA to ensure protection for retinal neurons (Fig. 2). Further work is necessary to elucidate this phenomenon as present in other systems and in mammal circuitry.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998; 82:1213-23; PMID:9466441; http://dx.doi.org/ 10.1016/S0306-4522(97)00347-3 [DOI] [PubMed] [Google Scholar]
- [2].Huster D, Reichenbach A, Reichelt W. The glutathione content of retinal Muller (glial) cells: effect of pathological conditions. Neurochem Int 2000; 36:461-9; PMID:10733014; http://dx.doi.org/ 10.1016/S0197-0186(99)00149-7 [DOI] [PubMed] [Google Scholar]
- [3].Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem 2009; 390:191-214; PMID:19166318; http://dx.doi.org/ 10.1515/BC.2009.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Janaky R, Ogita K, Pasqualotto BA, Bains JS, Oja SS, Yoneda Y, Shaw CA. Glutathione and signal transduction in the mammalian CNS. J Neurochem 1999; 73:889-902; PMID:10461878; http://dx.doi.org/ 10.1046/j.1471-4159.1999.0730889.x [DOI] [PubMed] [Google Scholar]
- [5].Janaky R, Shaw CA, Varga V, Hermann A, Dohovics R, Saransaari P, Oja SS. Specific glutathione binding sites in pig cerebral cortical synaptic membranes. Neuroscience 2000; 95:617-24; PMID:10658641; http://dx.doi.org/ 10.1016/S0306-4522(99)00442-X [DOI] [PubMed] [Google Scholar]
- [6].Janaky R, Dohovics R, Saransaari P, Oja SS. Modulation of [3H]dopamine release by glutathione in mouse striatal slices. Neurochem Res 2007; 32:1357-64; PMID:17401648; http://dx.doi.org/ 10.1007/s11064-007-9315-z [DOI] [PubMed] [Google Scholar]
- [7].Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol 2000; 62:649-71; PMID:10880854; http://dx.doi.org/ 10.1016/S0301-0082(99)00060-X [DOI] [PubMed] [Google Scholar]
- [8].Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem 2003; 384:505-16; PMID:12751781; http://dx.doi.org/ 10.1515/BC.2003.059 [DOI] [PubMed] [Google Scholar]
- [9].Johnson WM, Wilson-Delfosse AL, Mieyal JJ. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012; 4:1399-440; PMID:23201762; http://dx.doi.org/ 10.3390/nu4101399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Keelan J, Allen NJ, Antcliffe D, Pal S, Duchen MR. Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J Neurosci Res 2001; 66:873-84; PMID:11746414; http://dx.doi.org/ 10.1002/jnr.10085 [DOI] [PubMed] [Google Scholar]
- [11].Portugal CC, Miya VS, Calaza Kda C, Santos RA, Paes-de-Carvalho R. Glutamate receptors modulate sodium-dependent and calcium-independent vitamin C bidirectional transport in cultured avian retinal cells. J Neurochem 2009; 108:507-20; PMID:19054286; http://dx.doi.org/ 10.1111/j.1471-4159.2008.05786.x [DOI] [PubMed] [Google Scholar]
- [12].de Melo Reis RA, Ventura AL, Schitine CS, de Mello MC, de Mello FG. Muller glia as an active compartment modulating nervous activity in the vertebrate retina: neurotransmitters and trophic factors. Neurochem Res 2008; 33:1466-74; PMID:18273703; http://dx.doi.org/ 10.1007/s11064-008-9604-1 [DOI] [PubMed] [Google Scholar]
- [13].Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC. Glia-neuron interactions in the mammalian retina. Prog Retin Eye Res 2016; 51:1-40; PMID:26113209; http://dx.doi.org/ 10.1016/j.preteyeres.2015.06.003 [DOI] [PubMed] [Google Scholar]
- [14].Harada K, Kamiya T, Tsuboi T. Gliotransmitter Release from Astrocytes: Functional, Developmental, and Pathological Implications in the Brain. Frontiers in neuroscience 2015; 9:499; PMID:26793048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reichenbach A, Bringmann A. Role of Purines in Muller Glia. J Ocul Pharmacol Ther 2016; 32:518-33. [DOI] [PubMed] [Google Scholar]
- [16].Sanderson J, Dartt DA, Trinkaus-Randall V, Pintor J, Civan MM, Delamere NA, Fletcher EL, Salt TE, Grosche A, Mitchell CH. Purines in the eye: recent evidence for the physiological and pathological role of purines in the RPE, retinal neurons, astrocytes, Muller cells, lens, trabecular meshwork, cornea and lacrimal gland. Exp Eye Res 2014; 127:270-9; PMID:25151301; http://dx.doi.org/ 10.1016/j.exer.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Alves LA, de Melo Reis RA, de Souza CA, de Freitas MS, Teixeira PC, Neto Moreira Ferreira D, Xavier RF. The P2X7 receptor: shifting from a low- to a high-conductance channel - an enigmatic phenomenon? Biochim Biophys Acta 2014; 1838:2578-87; PMID:24857862; http://dx.doi.org/ 10.1016/j.bbamem.2014.05.015 [DOI] [PubMed] [Google Scholar]
- [18].Freitas HR, Ferraz G, Ferreira GC, Ribeiro-Resende VT, Chiarini LB, do Nascimento JL, Matos Oliveira KR, Pereira TL, Ferreira LG, Kubrusly RC, et al.. Glutathione-induced calcium shifts in chick retinal glial cells. PLoS One 2016; 11:e0153677; PMID:27078878; http://dx.doi.org/ 10.1371/journal.pone.0153677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ogita K, Kitago T, Nakamuta H, Fukuda Y, Koida M, Ogawa Y, Yoneda Y. Glutathione-induced inhibition of Na+-independent and -dependent bindings of L-[3H]glutamate in rat brain. Life Sci 1986; 39:2411-8; PMID:3796201; http://dx.doi.org/ 10.1016/0024-3205(86)90482-0 [DOI] [PubMed] [Google Scholar]
- [20].Janaky R, Varga V, Oja SS, Saransaari P. Release of [3H]GABA evoked by glutamate agonists from hippocampal slices: effects of dithiothreitol and glutathione. Neurochem Int 1994; 24:575-82; PMID:7981640; http://dx.doi.org/ 10.1016/0197-0186(94)90010-8 [DOI] [PubMed] [Google Scholar]
- [21].Sucher NJ, Lipton SA. Redox modulatory site of the NMDA receptor-channel complex: regulation by oxidized glutathione. J Neurosci Res 1991; 30:582-91; PMID:1666131; http://dx.doi.org/ 10.1002/jnr.490300316 [DOI] [PubMed] [Google Scholar]
- [22].Zable AC, Favero TG, Abramson JJ. Glutathione modulates ryanodine receptor from skeletal muscle sarcoplasmic reticulum. Evidence for redox regulation of the Ca2+ release mechanism. J Biol Chem 1997; 272:7069-77; PMID:9054399; http://dx.doi.org/ 10.1074/jbc.272.11.7069 [DOI] [PubMed] [Google Scholar]
- [23].Paolicchi A, Dominici S, Pieri L, Maellaro E, Pompella A. Glutathione catabolism as a signaling mechanism. Biochem Pharmacol 2002; 64:1027-35; PMID:12213602; http://dx.doi.org/ 10.1016/S0006-2952(02)01173-5 [DOI] [PubMed] [Google Scholar]
- [24].Rana S, Dringen R. Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci Lett 2007; 415:45-8; PMID:17222973; http://dx.doi.org/ 10.1016/j.neulet.2006.12.043 [DOI] [PubMed] [Google Scholar]
- [25].Baxter PS, Bell KF, Hasel P, Kaindl AM, Fricker M, Thomson D, Cregan SP, Gillingwater TH, Hardingham GE. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat Commun 2015; 6:6761; PMID:25854456; http://dx.doi.org/ 10.1038/ncomms7761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sperlagh B, Kofalvi A, Deuchars J, Atkinson L, Milligan CJ, Buckley NJ, Vizi ES. Involvement of P2X7 receptors in the regulation of neurotransmitter release in the rat hippocampus. J Neurochem 2002; 81:1196-211; PMID:12068068; http://dx.doi.org/ 10.1046/j.1471-4159.2002.00920.x [DOI] [PubMed] [Google Scholar]
- [27].Ficker C, Rozmer K, Kato E, Ando RD, Schumann L, Krugel U, Franke H, Sperlagh B, Riedel T, Illes P. Astrocyte-neuron interaction in the substantia gelatinosa of the spinal cord dorsal horn via P2X7 receptor-mediated release of glutamate and reactive oxygen species. Glia 2014; 62:1671-86; PMID:24895290; http://dx.doi.org/ 10.1002/glia.22707 [DOI] [PubMed] [Google Scholar]
- [28].Kronschlager MT, Drdla-Schutting R, Gassner M, Honsek SD, Teuchmann HL, Sandkuhler J. Gliogenic LTP spreads widely in nociceptive pathways. Science 2016; 354:1144-8; PMID:27934764; http://dx.doi.org/ 10.1126/science.aah5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hofmann HD, Mockel V. Release of gamma-amino[3H]butyric acid from cultured amacrine-like neurons mediated by different excitatory amino acid receptors. J Neurochem 1991; 56:923-32; PMID:1847190; http://dx.doi.org/ 10.1111/j.1471-4159.1991.tb02010.x [DOI] [PubMed] [Google Scholar]
- [30].Neal MJ, Cunningham JR, Dent Z. Modulation of extracellular GABA levels in the retina by activation of glial P2X-purinoceptors. Br J Pharmacol 1998; 124:317-22; PMID:9641548; http://dx.doi.org/ 10.1038/sj.bjp.0701841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].do Nascimento JL, de Mello FG. Induced release of gamma-aminobutyric acid by a carrier-mediated, high-affinity uptake of L-glutamate in cultured chick retina cells. J Neurochem 1985; 45:1820-7; PMID:2865335; http://dx.doi.org/ 10.1111/j.1471-4159.1985.tb10539.x [DOI] [PubMed] [Google Scholar]
- [32].Duarte CB, Ferreira IL, Santos PF, Oliveira CR, Carvalho AP. Ca(2+)-dependent release of [3H]GABA in cultured chick retina cells. Brain Res 1992; 591:27-32; PMID:1446230; http://dx.doi.org/ 10.1016/0006-8993(92)90974-E [DOI] [PubMed] [Google Scholar]
- [33].Schitine CS, Mendez-Flores OG, Santos LE, Ornelas I, Calaza KC, Perez-Toledo K, Lopez-Bayghen E, Ortega A, Gardino PF, de Mello FG, et al.. Functional plasticity of GAT-3 in avian Muller cells is regulated by neurons via a glutamatergic input. Neurochem Int 2015; 82:42-51; PMID:25700791; http://dx.doi.org/ 10.1016/j.neuint.2015.02.004 [DOI] [PubMed] [Google Scholar]
- [34].O.I. Anccasi RM, Cossenza M, Persechini PM, Ventura AL. ATP induces the death of developing avian retinal neurons in culture via activation of P2X7 and glutamate receptors. Purinergic Signal 2013; 9:15-29; PMID:22733428; http://dx.doi.org/ 10.1007/s11302-012-9324-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Avshalumov MV, MacGregor DG, Sehgal LM, Rice ME. The glial antioxidant network and neuronal ascorbate: protective yet permissive for H(2)O(2) signaling. Neuron Glia Biol 2004; 1:365-76; PMID:18292802; http://dx.doi.org/ 10.1017/S1740925X05000311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Covarrubias-Pinto A, Acuna AI, Beltran FA, Torres-Diaz L, Castro MA. Old things new view: Ascorbic acid protects the brain in neurodegenerative disorders. Int J Mol Sci 2015; 16:28194-217; PMID:26633354; http://dx.doi.org/ 10.3390/ijms161226095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bringmann A, Grosche A, Pannicke T, Reichenbach A. GABA and glutamate uptake and metabolism in retinal glial (Muller) cells. Front Endocrinol 2013; 4:48; PMID:23616782; http://dx.doi.org/ 10.3389/fendo.2013.00048 [DOI] [PMC free article] [PubMed] [Google Scholar]