

Abstract
The human ANKS1B gene encodes an activity-dependent effector of post-synaptic signaling. It was recently associated with neuropsychiatric phenotypes in genome-wide studies. While the biological function of ANKS1B has been partly elucidated, its role in behavior is poorly understood. Here, we breed and characterize a full knockout (KO) for murine Anks1b. We found that the homozygous KO genotype was partially lethal, showing significant deviation from expected segregation ratios at weaning. Behaviorally, KOs exhibited no difference in baseline acoustic startle response, but showed deficits in prepulse inhibition (PPI). KOs also exhibited locomotor hyperactivity and increased stereotypy at baseline. Administration of ketamine, a noncompetitive NMDA-receptor antagonist, greatly exacerbated locomotor activity in the KOs at lower doses, but genotype groups were almost indistinguishable as dose increased. Stereotypy showed a complex response to ketamine in the KOs, with elevated stereotypy at lower doses and markedly less at high doses, compared to wild type. Our study is the first to probe the behavioral phenotypes associated with ablation of Anks1b. Deficits in PPI, locomotor hyperactivity, elevated stereotypy and altered response to NMDA receptor antagonism are murine behavioral outcomes with translational relevance for psychiatric disorders. These findings are also consistent with the role of Anks1b as an effector of glutamatergic signaling. As an intermediary between postsynaptic receptor stimulation and long-term changes to neuronal protein expression, further investigation of Anks1b is warranted.
Keywords: Glutamatergic signaling, NMDA receptors, Sensorimotor gating, Locomotor activity, Post-synaptic density
1. Introduction
The Ankyrin Repeat and Sterile Alpha Motif Domain-containing 1 B (ANKS1B) gene is highly expressed in the CNS [1]. ANKS1B protein, also known as EB-1 and AIDA-1, plays a role in tyrosine kinase signal transduction [2] and is a scaffold of the post-synaptic density (PSD) [3], where it interacts with post-synaptic density protein 95 (PSD-95) and several classes of glutamate receptor. Stimulation of NMDA receptors (NMDAR) leads to translocation of the ANKS1B protein to the nucleus where it regulates neuronal protein expression [4]. This places ANKS1B at the interface between post-synaptic receptor stimulation and downstream changes to neuronal biology.
Interest in ANKS1B has developed following its association with psychiatric phenotypes in human studies. Genetic variants at ANKS1B were among the top findings in two genome-wide association studies (GWAS) of antipsychotic response in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) [5,6]. ANKS1B was also among the top findings in a GWAS of citalopram response [7] and a GWAS of monoamine metabolite levels in cerebrospinal fluid [8]. These findings suggest further characterization of ANKS1B is warranted.
The biological function of ANKS1B has been partly elucidated, but little is known about its effects on behavior. Here we present an initial behavioral characterization of a mouse knockout (KO) for the murine ortholog, Anks1b. We focus on the paradigms of prepulse inhibition (PPI) and locomotor activity (LA). PPI of the startle response is a widely-studied sensorimotor gating measure and pharmacological modulation of PPI in rodents is considered a translational model for deficits observed in schizophrenia [9]. LA is considered to tap neurological substrates such as dopaminergic neurotransmission in the limbic system and NMDAR function [10]. Disruptions to both PPI and LA have been previously observed in mouse knockouts for scaffolding proteins of the PSD [11]. Our study shows that elimination of Anks1b leads to effects on these behaviors and altered response the NMDAR antagonist, ketamine.
2. Methods
2.1 Ethics statement
All procedures were conducted in accordance with the “Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Academy Press, 2011) and were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
2.2 Development of the Anks1b knockout mouse and breeding program
Progenitor mice heterozygous for the Anks1btm1a(KOMP)Wtsi allele on a C57BL/6N-Atm1Brd background were generated by Knockout Mouse Project (KOMP) CSD 79332, and obtained from the Wellcome Trust Sanger Institute (Cambridge, UK). Anks1btm1a(KOMP)Wtsi is a “knockout-first” allele [12,13] in which an En2:IRES:lacZ reporter cassette was inserted into intron 16, and efficient splicing to the En2 splice acceptor site results in the truncation of the Anks1b transcript. Exon 17 (Ensembl accession number: ENSMUSE00001274863), which encodes amino acid residues 871-921, is also floxed in the Anks1btm1a(KOMP)Wtsi allele, and was targeted for deletion because it overlaps with the second Sterile Alpha Motif (SAM) domain (residues 877-938). Helix 5 of this domain contains the Anks1b nuclear localization signal [14]. Anks1btm1a mice were bred to a transgenic line expressing Cre recombinase in the germline (B6.FVB-Tg(EIIa-cre)C5379Lmgd/J - Jackson Laboratory stock #003724), generating the Anks1btm1b KO line in which the En2:IRES:lacZ cassette is retained, but in which exon 17 is deleted (Fig. 1). The Ella-cre mice used donor strain FVB/N bred to C57BL/6N for 11 generations. Thus both the KOMP Anks1btm1a(KOMP)Wtsi allele and the Ella-cre gene were on C57BL/6N backgrounds. Genotyping methods to screen for conversion of the Anks1btm1a allele to the Anks1btm1b allele via Cre-mediated deletion of exon 17 (Supplementary Fig. S1) and to screen for the EIIa-cre transgene are provided in the Supplementary Material.
Figure 1.
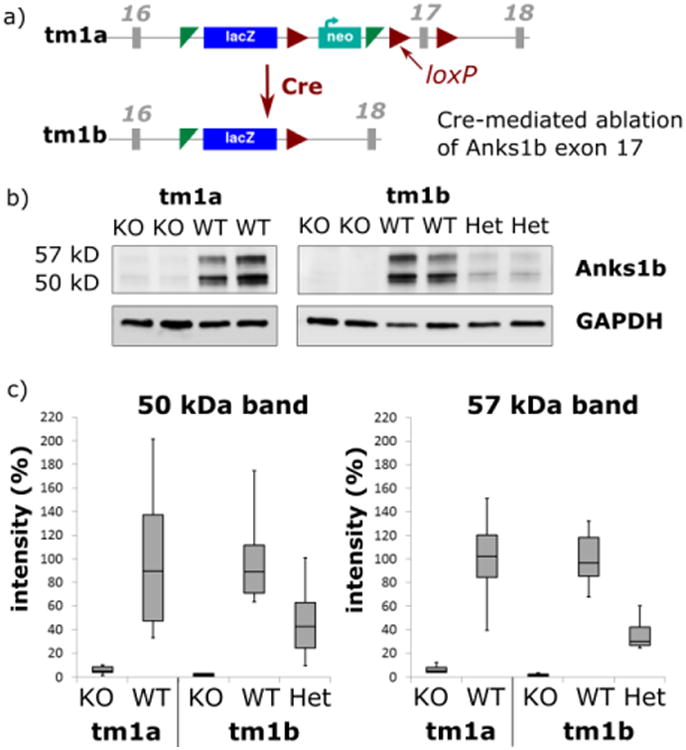
a) Schematic of the Anks1b KOMP project allele variants. The ‘knockout-first’ allele (tm1a) contains an En2:IRES:lacZ reporter cassette inserted into intron 16, disrupting gene function. Cre deletes the promoter driven selection cassette and floxed (loxP-flanked) exon 17 to generate a lacZ tagged allele (tm1b) [12]. b) Western blots showing the approximately 50 and 57 kD isoforms of Anks1b (AIDA-1e and AIDA-1d respectively) in mice of different genotypes for the tm1a and tm1b alleles. WT = wild type, KO = homozygous tm1a or tm1b, Het = heterozygote. d) Quantitative analysis of 3 technical replicates of Western blots showing results grouped by genotype, where quantities are normalized relative to WT, where mean (WT) = 100%. Box and whisker plots show median and interquartile ranges.
Subjects for behavioral analysis were generated by matings of EIIa-cre negative Anks1btm1b heterozygous pairs. Pups were genotyped at three weeks of age using DNA extracted from tail snips and a three-primer PCR protocol (Supplementary Figure S2). We aimed for approximately equal numbers of wild-type (WT), heterozygous (Het), and KO mice of both sexes in our experiments. Mice were tested in cohorts as they became available, with testing staggered so that each cohort received tests around the same age. Baseline behavioral testing was conducted at approximately 10 weeks of age with inter-test intervals ranging from 1-5 days. PPI ketamine testing was completed at approximately 15 weeks, while ketamine LA assays were completed at approximately 20 weeks. When not in testing, mice were allowed ad libitum access to standard diet (7012 Teklad LM-485, Harlan Labs Inc., Indianapolis, IN) and water, and were housed at a maximum of five mice per cage in a temperature-controlled (22°C) facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International. Mice were kept under a 12-h/12-h light/dark cycle (lights on from 0600-h to 1800-h), with all testing occurring during the light segment.
2.3 Validation of the knockout allele via Western blotting
Drug-naive mice of each genotype were euthanized by CO2 asphyxiation, the whole brain excised and immediately frozen. Cortical tissue samples were lysed in Pierce® RIPA buffer (Thermo Scientific, Rockford, IL) and homogenized. Samples were resolved on polyacrylamide gels and transferred to a PVDF membrane (Bio-Rad, Hercules, CA), which was then treated with either mouse anti-AIDA-1/Anks1b antibody (C-10, Santa Cruz Biotechnology, Dallas, TX), or mouse anti-GAPDH antibody (Thermo Scientific), followed by anti-mouse IgG secondary antibody (Cell Signaling Technology, Danvers, MA). A ChemiDoc system (Bio-Rad) was used to capture and process blot images. Protein band intensity was quantified by normalizing the chemiluminescent image to the stain-free blot image captured before antibody treatment. All experiments were repeated at least three times.
2.4 Drugs
Ketamine (KetaVed, Vedco, Inc., St. Joseph, MO) was prepared in 0.9% saline. Intraperitoneal (i.p.) injections were given in a volume equivalent to 10 ml/kg body weight.
2.5 Prepulse Inhibition of the startle reflex (PPI)
Commercially-supplied startle chambers (San Diego Instruments, San Diego, CA) were used to record startle following acoustic stimuli. Beginning with the onset of the startle pulse (STIM) for each trial, 1000 readings were taken at 1-ms intervals. A test session consisted of 75, 200-ms trials, with five trial types: STIM alone, 73pp (i.e., “73dB prepulse”)+STIM, 77pp+STIM, 85pp+STIM, and NO STIM. Both prepulse and pulse stimuli were 20-ms in duration, with an interstimulus interval of 100-ms between the onset of their presentations, and an intertrial interval average of ∼15-s across the session (range: 10-20-s). STIM intensity was set at 119 dB, while prepulses were set at 4, 8, and 16 dB above the background level of 69 dB (i.e., 73, 77, and 85 dB). A test session was initiated with a 10-min acclimation period to the background level of noise before presentation of five STIM alone trials. Following this, 12-14 replicates of each trial type were presented in a mixed sequence to prevent habituation. Test sessions ended with five STIM alone trials. Total session time was approximately 30 min.
2.6 Locomotor activity (LA)
For LA studies, mice were placed in automated activity monitoring chambers that were sound- and light-attenuating (AccuScan Instruments, Columbus OH). Each arena was 20×20×30 cm, with photobeam sensors spaced 2.5 cm apart along each axis to detect movement. For baseline measurements, mice were placed in chambers for a 2-h test session with distance traveled recorded in 10-min bins. For the LA ketamine challenge, each subject received a cumulative dosing regimen. In this procedure, mice were habituated to chambers for 30-min before the trial, then removed and administered an i.p. injection of saline before being returned to the chamber for a 6-min recording period. Mice were then removed, administered an i.p. injection of ketamine, and placed back into the chamber for the next 6-min recording period. This process was repeated three more times to obtain the complete dose-effect curve. Subjects were administered acute injections of 0, 10, 20, 26, and 44 mg/kg ketamine, to generate cumulative doses of 0, 10, 30, 56, and 100 mg/kg.
2.7 Behavioral data analysis
For both PPI and LA, electronically captured data files from test days were combined and processed using scripts written in R (www.r-project.org). Percentage PPI (% PPI) was calculated using the mean peak startle amplitude (vmax) of STIM alone trials (vmaxSTIM alone) or prepulse+STIM trials (vmaxprepulse), according to standard methods [15].
LA used the “total distance traveled” variable. For stereotypy, we used the “stereotypy count” variable as defined by the software that records the number of times the mouse breaks the same beam in succession without breaking an adjacent beam [16].
Analysis of behavioral outcomes by genotype used linear regression implemented via the linear model (lm) function in R. Genotypes were coded 0 = KO homozygote; 1 = heterozygote and 2 = wild type. Degrees of freedom and P values were output for regression coefficients. Data were visualized using the ggplot2 package.
3. Results
3.1 Biological validation of the KO and lethality
The knockout-first (KO-first) targeting strategy used by KOMP [12,13] produces a KO at the transcript level due to a splice acceptor in the cassette. KO-first (tm1a) homozygotes are typically null, but hypomorphic expression may occur if the RNA processing module is bypassed [17]. Western blotting of cortical tissue lysate (Fig. 1) revealed 5-6% Anks1b protein expression in Anks1btm1a (KO-first) animals compared to WT for both the ∼50 and ∼57 kD isoforms (AIDA-1e and AIDA-1d respectively). In contrast, ablation of exon 17 (Anks1btm1b) resulted in no detectable protein above background (Fig. 1) confirming the validity of this KO. Protein levels in Anks1btm1b heterozygotes were intermediate between KOs and WT (approximately 40-50%), indicating the use of an additive statistical model would be valid in our phenotypic analysis.
We established heterozygous breeding pairs for the Anks1btm1b line and noticed that many pups did not survive to maturity. Over the 9 month breeding period, we recorded 221 live births, of which 202 survived to weaning. We genotyped some early losses and found them to all be homozygous KOs. Genotyping of all surviving mice at weaning revealed a significant deviation from expected segregation ratios (Chi square = 8.257, 2df, P = 0.016) due to mortality in the Anks1btm1b KO homozygotes. Therefore, the Anks1btm1b allele is partially lethal. Conversely, neither we nor KOMP observed any deviation from expected ratios in the Anks1btm1a KO-first line (www.mousephenotype.org/data/genes/MGI:1924781).
3.2 Baseline PPI in the Anks1b KO
Baseline acoustic startle responses revealed no significant differences across Anks1btm1b genotypes, indicating that these mice startled normally. Furthermore, startle diminished in proportion to increasing prepulse intensities (Supplementary Figure S3). Together, these data indicated that hearing was normal in the KOs. Male mice (all genotypes combined) exhibited a greater startle amplitude than females (t(87) = 3.51, P = 0.0007), but this sex difference did not extend to PPI and was not observed at any of the prepulse intensities used. This pattern of sexual dimorphism in startle but not PPI is in agreement with previous studies of C57s [18]. Therefore, we present baseline PPI for both sexes combined in Fig. 2, with results for each sex provided separately in Supplementary Figures S4-5. Our analysis showed that mice carrying the Anks1btm1b allele had significant PPI deficits using a 73 dB prepulse (t(87) = 3.46, P = 0.0008), with nonsignificant deficits at higher prepulse intensities. Global PPI, or the average effect across all prepulse intensities, was significant (t(87) = 2.28, P = 0.025).
Figure 2.
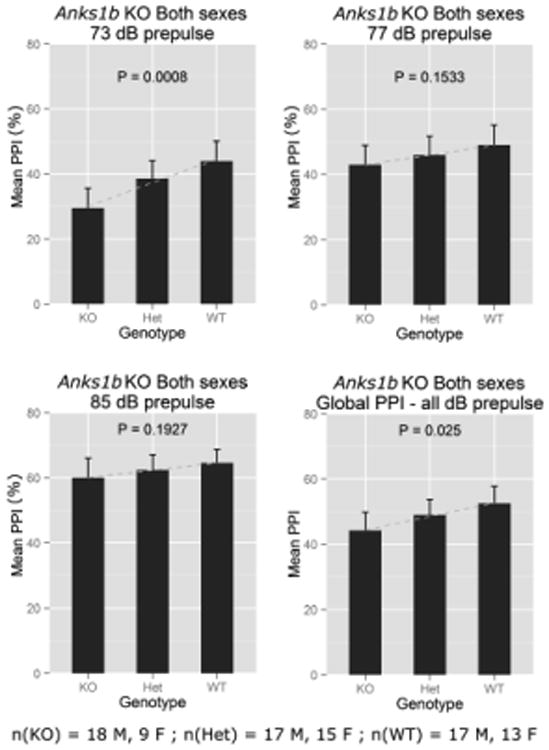
Baseline percentage (%) PPI at three different prepulse intensities and global PPI by genotype for the Anks1btm1b allele. Means are plotted for each group and error bars show 95% confidence intervals.
3.3 Baseline LA and stereotypy
Female mice (all genotypes combined) exhibited elevated baseline LA (Fig. 3) relative to males (t(87) = -2.56, P = 0.012). Therefore, in Fig. 3, we show results separately by sex. A test of the summed total distance traveled across all time periods showed significant differences between genotypes for males (t(50) = -3.54, P = 0.0009) and females (t(35) = -3.08, P = 0.004), with KOs showing the highest activity levels.
Figure 3.
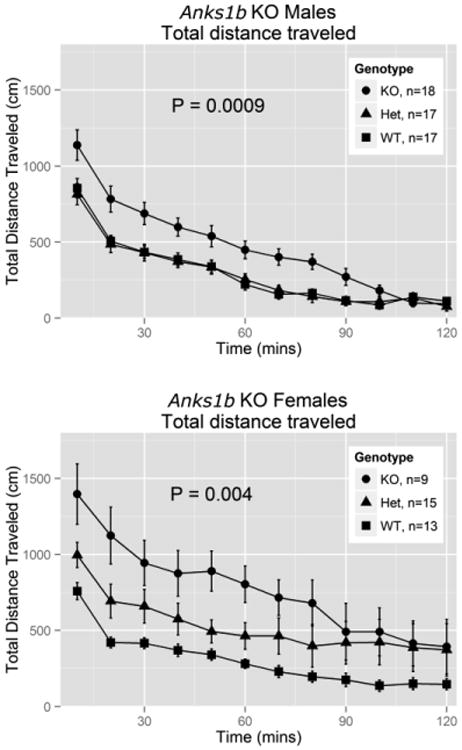
Baseline locomotor activity (total distance traveled in cm) in Anks1btm1b mice over a 2-h observation period, displayed in 12, consecutive 10 min periods. Means are plotted for each group for each 10 min period and error bars show 95% confidence intervals. We observed a significant sex difference and so have plotted both sexes independently. The reported P-values are for tests of total distance traveled by genotype over the entire 2-h period.
Elevated stereotypy is characteristic of mice with disrupted NMDAR signaling [19,20], so we hypothesized that stereotypy would differ by Anks1b genotype in our LA data. We observed no significant sex difference in stereotypy and present the results for both sexes combined in Fig. 4, with results for each sex separately in Supplementary Figures S6-7. As predicted, stereotypy count was significantly elevated in Anks1btm1b mice (t(87) = -3.22, P = 0.0018).
Figure 4.
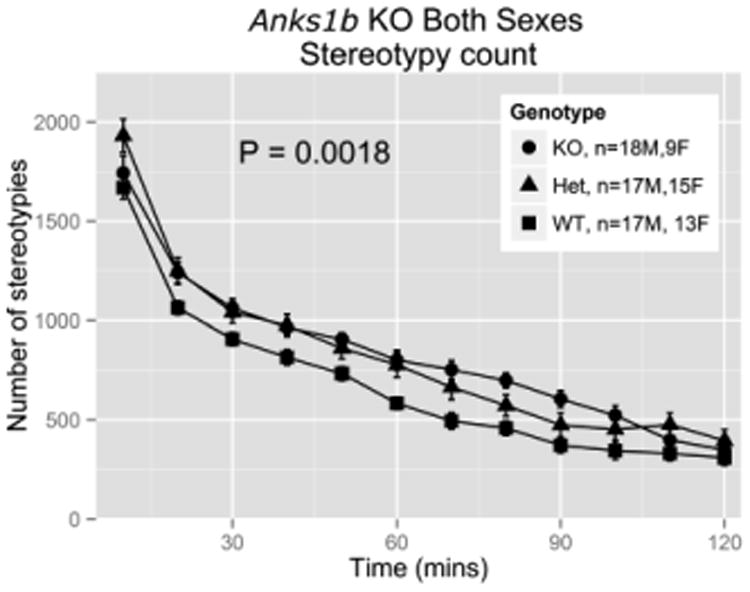
Baseline stereotypies in Anks1btm1b mice over a 2-h observation period, displayed as 12, consecutive 10 min periods. Means are plotted for each group for each 10 min period and error bars show 95% confidence intervals. The reported P-value is for total stereotypies by genotype over the entire 2-h period.
3.4 Response to ketamine in Anks1b KO mice
Altered response to glutamatergic psychotomimetic drugs is a psychosis-related endophenotype and a correlate of glutamatergic dysfunction [21]. We hypothesized that Anks1btm1b mice would exhibit altered behavioral response to the NMDAR antagonist ketamine. Our ketamine challenge experiments were conducted after baseline testing and by then too few female KOs (n=3) survived to analyze. Hence the following results include males only.
We first tested ketamine's effects on PPI. Ketamine typically induces PPI deficits, so our choice of dose (56 mg/kg) was based on PPI disruption in a preliminary study of male C57BL/6Js (Supplementary Figure S8). Analysis of all genotypes combined confirmed that 56 mg/kg ketamine had a significant effect (t(42) = -4.16 = 1.55×10-4). However, a test of interaction between ketamine exposure and genotype was not significant (t(40) = -0.58, P = 0.564), indicating no detectable difference in response across genotypes.
We evaluated the LA effects of ketamine at several doses, to characterize its dose-response curve. Subjects were administered acute injections of ketamine prior to each of five, 6-min recording periods to generate cumulative doses of 0 (vehicle), 10, 30, 56, and 100 mg/kg (Fig.5A). Over the 30-min trial, mice carrying the Anks1btm1b allele showed elevated LA (t(42) = -4.28, P = 0.0001). In particular, they showed extreme sensitivity to ketamine's locomotor activating effects at lower doses. At 10 mg/kg ketamine, the lowest dose tested, KOs showed significant increases in total distance traveled relative to their own scores at baseline (t(28) = 6.95, P = 1.5 × 10-7) and relative to the other genotype groups (t(42) = -6.94, P = 1.78 × 10-8). However, at rate-decreasing doses of ketamine (i.e. ≥ 56 mg/kg) those differences diminished, such that the LA levels of the KOs were indistinguishable from the other subjects.
Figure 5.
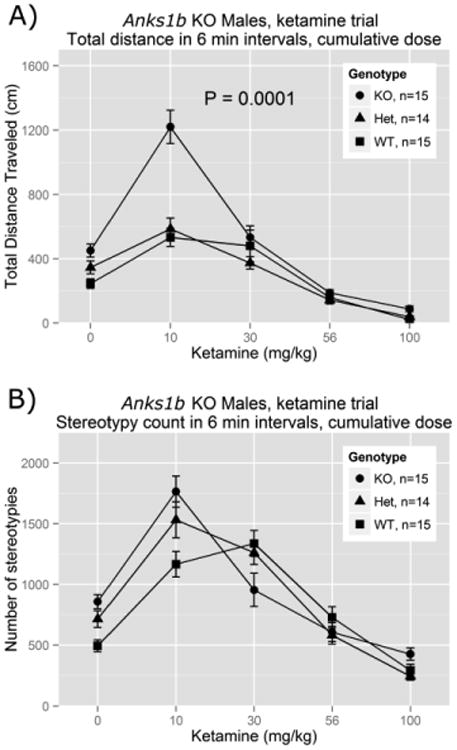
Total distance traveled in cm (panel A) and stereotypy counts (panel B) as a function of cumulative ketamine dose. Dependent measures were summed within 6-min intervals following each ketamine (cumulative) dose. Reported P-values are for differences between genotype groups considered cumulatively over the entire 30-min test period.
In Fig. 5B, we show stereotypy findings for the same experiment. For all groups, stereotypy counts first increased and then decreased as ketamine dose increased. KOs exhibited significant increases in stereotypy induced by low dose (10 mg/kg) ketamine relative to their baseline levels (t(28) = 6.46, P = 5.33 × 10-7), as well as relative to the other genotype groups (t(42) = -2.62, P = 0.012). KOs also showed the greatest sensitivity to ketamine's suppressive effects on stereotypy at high doses, which were decreased relative to other genotype groups (t(42) = 2.48, P = 0.018) and were close to vehicle levels by 30 mg/kg. Because of these dose-induced, bimodal effects there was a net nonsignificant effect on cumulative stereotypy counts across the entire 30-min period (t(42) = -1.39, P = 0.171).
Discussion
4.1 Overview and the lethal phenotype
We bred, validated and characterized a KO for murine Anks1b, achieved through ablation of exon 17 (Anks1btm1b). We observed that the homozygous Anks1btm1b genotype was partially lethal. However, hypomorphic expression in the KO-first (Anks1btm1a) homozygotes appeared sufficient to rescue the phenotype. A previous study of a forebrain-specific (CaMKIIα) conditional KO for Anks1b did not report lethality [22], suggesting that the lethality is caused by deletion of Anks1b in another tissue.
4.2 Behavioral comparison to other KOs
Anks1b protein is activated upon stimulation of NMDARs [4] and regulates NMDAR subunit composition at synapses [22]. NMDARs are heterodimers of two obligatory NR1 subunits and two variable NR2 subunits (NR2A-D). NMDAR subunit KOs exhibit specific behavioral anomalies [23]. Hypomorphic NR1 mice exhibit increased LA and deficits in PPI [19,24], mirroring our findings here. NR2A mutant mice exhibit increased LA only, while NR2B and NR3C mutants exhibit no changes to sensory or motor behavior and NR2D exhibit reduced LA only [23]. These observations suggest that, behaviorally, ablation of Anks1b shows some similarities to reduced NR1 expression. However, NR1 mutant mice exhibit a broad spectrum of behavioral abnormalities and the extent to which Anks1b KOs would align on these additional domains remains untested.
Scaffolding proteins of the PSD, including Anks1b [3,25], are principal organizers of glutamatergic neurotransmission. KOs for PSD scaffolds such as homer, shank, etc, exhibit specific behavioral phenotypes [11]. However, insofar as characterizations have been carried out, no other PSD scaffold deletion exhibits diminished PPI while enhancing LA (see [11], Table 1 for review). Therefore, deletion of Anks1b has a different effect on behavior than other PSD scaffolds.
4.3 Anks1b and response to ketamine
Pharmacological blockade of NMDARs in rodents produces PPI deficits, hyperlocomotion and stereotypies [10,26], which is exactly the pattern of effects observed with knocking out Anks1b. Therefore, we might expect that administering ketamine to the Anks1b KOs should yield larger effects in the same direction. This was the case for LA, where hyperlocomotion in the KO was potentiated further. However, KOs did not differ from WT in their PPI response to ketamine, while their stereotypy response was complex and dose-dependent. Considering all these outcomes, Anks1b KOs exhibit altered response to ketamine but this varies across behavioral domains and will require further work to fully elucidate.
4.4 Conclusion
As an effector post-synaptic NMDA signaling, Anks1b operates as an intermediary between neuronal activity and downstream changes to neuronal biology [25]. Our study shows that Anks1b is a critical in post-natal development, exhibiting partial lethality at weaning. Furthermore, ablation of Anks1b has behavioral effects consistent with disruption to NMDAR-mediated glutamatergic signaling. These findings may have implications for translational studies of psychiatric disorders.
Supplementary Material
Highlights.
Human ANKS1B was associated with psychiatric phenotypes in genome-wide studies
Here we behaviorally characterize a knockout (KO) for the murine ortholog Anks1b
Anks1b KOs exhibit PPI deficits, elevated stereotypies and hyperlocomotor activity
Anks1b KOs exhibit altered response to ketamine, an NMDA receptor antagonist
Anks1b plays a role in mediating behaviors linked to glutamatergic signaling
Acknowledgments
Funding: This study was funded through a NARSAD Young Investigator Award to JLM. RE was supported by training grant T32 DA007027 from the U.S. National Institutes of Health (NIH) and this study was completed in partial fulfillment of the doctoral requirements in Pharmacology and Toxicology at Virginia Commonwealth University (VCU). Support services were provided by the VCU Massey Cancer Center Transgenic/Knockout Mouse Shared Resource, funded in part by NIH National Cancer Institute (NIH-NCI) Cancer Center Support Grant P30 CA016059. Sponsors had no involvement in the reported research or in the writing of this manuscript.
Footnotes
Disclosure: The authors declare no financial conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ghersi E, Vito P, Lopez P, Abdallah M, D'Adamio L. The intracellular localization of amyloid beta protein precursor (AbetaPP) intracellular domain associated protein-1 (AIDA-1) is regulated by AbetaPP and alternative splicing. J Alzheimers Dis. 2004;6:67–78. doi: 10.3233/jad-2004-6108. [DOI] [PubMed] [Google Scholar]
- 2.Fu X, McGrath S, Pasillas M, Nakazawa S, Kamps MP. EB-1, a tyrosine kinase signal transduction gene, is transcriptionally activated in the t(1;19) subset of pre-B ALL, which express oncoprotein E2a-Pbx1. Oncogene. 1999;18:4920–4929. doi: 10.1038/sj.onc.1202874. [DOI] [PubMed] [Google Scholar]
- 3.Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, Neubert TA. Identification and verification of novel rodent postsynaptic density proteins. Mol Cell Proteomics MCP. 2004;3:857–871. doi: 10.1074/mcp.M400045-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Jordan BA, Fernholz BD, Khatri L, Ziff EB. Activity-dependent AIDA-1 nuclear signaling regulates nucleolar numbers and protein synthesis in neurons. Nat Neurosci. 2007;10:427–435. doi: 10.1038/nn1867. [DOI] [PubMed] [Google Scholar]
- 5.McClay JL, Adkins DE, Aberg K, Stroup S, Perkins DO, Vladimirov VI, Lieberman JA, Sullivan PF, van den Oord EJCG. Genome-wide pharmacogenomic analysis of response to treatment with antipsychotics. Mol Psychiatry. 2011;16:76–85. doi: 10.1038/mp.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McClay JL, Adkins DE, Aberg K, Bukszár J, Khachane AN, Keefe RSE, Perkins DO, McEvoy JP, Stroup TS, Vann RE, Beardsley PM, Lieberman JA, Sullivan PF, van den Oord EJCG. Genome-wide pharmacogenomic study of neurocognition as an indicator of antipsychotic treatment response in schizophrenia. Neuropsychopharmacol. 2011;36:616–626. doi: 10.1038/npp.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garriock HA, Kraft JB, Shyn SI, Peters EJ, Yokoyama JS, Jenkins GD, Reinalda MS, Slager SL, McGrath PJ, Hamilton SP. A genomewide association study of citalopram response in major depressive disorder. Biol Psychiatry. 2010;67:133–138. doi: 10.1016/j.biopsych.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luykx JJ, Bakker SC, Lentjes E, Neeleman M, Strengman E, Mentink L, DeYoung J, de Jong S, Sul JH, Eskin E, van Eijk K, van Setten J, Buizer-Voskamp JE, Cantor RM, Lu A, van Amerongen M, van Dongen EPA, Keijzers P, Kappen T, Borgdorff P, Bruins P, Derks EM, Kahn RS, Ophoff RA. Genome-wide association study of monoamine metabolite levels in human cerebrospinal fluid. Mol Psychiatry. 2014;19:228–234. doi: 10.1038/mp.2012.183. [DOI] [PubMed] [Google Scholar]
- 9.Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- 10.Amann LC, Gandal MJ, Halene TB, Ehrlichman RS, White SL, McCarren HS, Siegel SJ. Mouse behavioral endophenotypes for schizophrenia. Brain Res Bull. 2010;83:147–161. doi: 10.1016/j.brainresbull.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Gao C, Tronson NC, Radulovic J. Modulation of behavior by scaffolding proteins of the post-synaptic density. Neurobiol Learn Mem. 2013;105:3–12. doi: 10.1016/j.nlm.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Testa G, Schaft J, van der Hoeven F, Glaser S, Anastassiadis K, Zhang Y, Hermann T, Stremmel W, Stewart AF. A reliable lacZ expression reporter cassette for multipurpose, knockout-first alleles. Genes N Y. 2004;38:151–158. doi: 10.1002/gene.20012. [DOI] [PubMed] [Google Scholar]
- 14.Kurabi A, Brener S, Mobli M, Kwan JJ, Donaldson LW. A nuclear localization signal at the SAM-SAM domain interface of AIDA-1 suggests a requirement for domain uncoupling prior to nuclear import. J Mol Biol. 2009;392:1168–1177. doi: 10.1016/j.jmb.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Geyer MA. Assessing prepulse inhibition of startle in wild-type and knockout mice. Psychopharmacology (Berl) 1999;147:11–13. doi: 10.1007/s002130051130. [DOI] [PubMed] [Google Scholar]
- 16.Tilley MR, Gu HH. Dopamine transporter inhibition is required for cocaine-induced stereotypy. Neuroreport. 2008;19:1137–1140. doi: 10.1097/WNR.0b013e3283063183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanstein R, Negoro H, Patel NK, Charollais A, Meda P, Spray DC, Suadicani SO, Scemes E. Promises and pitfalls of a Pannexin1 transgenic mouse line. Pharmacol Ion Channels Channelopathies. 2013;4:61. doi: 10.3389/fphar.2013.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plappert CF, Rodenbücher AM, Pilz PKD. Effects of sex and estrous cycle on modulation of the acoustic startle response in mice. Physiol Behav. 2005;84:585–594. doi: 10.1016/j.physbeh.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 20.Milenkovic M, Mielnik CA, Ramsey AJ. NMDA receptor-deficient mice display sexual dimorphism in the onset and severity of behavioural abnormalities. Genes Brain Behav. 2014;13:850–862. doi: 10.1111/gbb.12183. [DOI] [PubMed] [Google Scholar]
- 21.Gray L, McOmish CE, Scarr E, Dean B, Hannan AJ. Sensitivity to MK-801 in phospholipase C-β1 knockout mice reveals a specific NMDA receptor deficit. Int J Neuropsychopharmacol. 2009;12:917–928. doi: 10.1017/S1461145709009961. [DOI] [PubMed] [Google Scholar]
- 22.Tindi JO, Chávez AE, Cvejic S, Calvo-Ochoa E, Castillo PE, Jordan BA. ANKS1B Gene Product AIDA-1 Controls Hippocampal Synaptic Transmission by Regulating GluN2B Subunit Localization. J Neurosci. 2015;35:8986–8996. doi: 10.1523/JNEUROSCI.4029-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mori H, Mishina M. Roles of diverse glutamate receptors in brain functions elucidated by subunit-specific and region-specific gene targeting. Life Sci. 2003;74:329–336. doi: 10.1016/j.lfs.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 24.Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM, Lieberman JA, Snouwaert JN, Koller BH. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav Brain Res. 2004;153:507–519. doi: 10.1016/j.bbr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 25.Smirnova E, Shanbhag R, Kurabi A, Mobli M, Kwan JJ, Donaldson LW. Solution structure and peptide binding of the PTB domain from the AIDA1 postsynaptic signaling scaffolding protein. PloS One. 2013;8:e65605. doi: 10.1371/journal.pone.0065605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Razoux F, Garcia R, Léna I. Ketamine, at a dose that disrupts motor behavior and latent inhibition, enhances prefrontal cortex synaptic efficacy and glutamate release in the nucleus accumbens. Neuropsychopharmacol. 2007;32:719–727. doi: 10.1038/sj.npp.1301057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.