

Abstract
Cotinine is the major metabolite of nicotine and has displayed some capacity for improving cognition in mouse models following chronic administration. We tested if acute cotinine treatment is capable of improving cognition in the mouse model of Fragile X syndrome, Fmr1−/− knockout mice, and if this is related to inhibition by cotinine treatment of glycogen synthase kinase-3β (GSK3β), which is abnormally active in Fmr1−/− mice. Acute cotinine treatment increased the inhibitory serine-phosphorylation of GSK3β and the activating phosphorylation of AKT, which can mediate serine-phosphorylation of GSK3β, in both wild-type and Fmr1−/− mouse hippocampus. Acute cotinine treatment improved cognitive functions of Fmr1−/− mice in coordinate and categorical spatial processing, novel object recognition, and temporal ordering. However, cotinine failed to restore impaired cognition in GSK3β knockin mice, in which a serine9-to-alanine9 mutation blocks the inhibitory serine phosphorylation of GSK3β, causing GSK3β to be hyperactive. These results indicate that acute cotinine treatment effectively repairs impairments of these four cognitive tasks in Fmr1−/− mice, and suggest that this cognition-enhancing effect of cotinine is linked to its induction of inhibitory serine-phosphorylation of GSK3. Taken together, these results show nicotinic receptor agonists can act as cognitive enhancers in a mouse model of Fragile X syndrome and highlight the potential role of inhibiting GSK3β in mediating the effects of cotinine on memory.
Keywords: cotinine, glycogen synthase kinase-3, fragile X syndrome, novel object recognition, spatial memory
Graphical Abstract
Introduction
Eighty percent of nicotine is metabolized to cotinine, [(5S)-1-methyl-5-(3-pyridyl)-pyrrolidin-2-one], which is under investigation as a new agent to improve cognitive functions in animal models of a variety of psychiatric and neurological diseases (Moran 2012; Terry et al., 2005). Importantly for its potential clinical use, the pharmacological half-life of cotinine is 15–24 hr (Buccafusco et al., 2007; Terry et al., 2005), much longer than the half-life of nicotine (2–3 hr), cotinine has low toxicity (Li et al., 2015), and cotinine crosses the blood brain barrier (Riah et al., 1998). The actions of cotinine are thought to be mediated by its effect as a weak agonist with lower affinity than nicotine of nicotinic acetylcholine receptors (nAChRs) (Vainio and Tuominen, 2001). Activation of nAChRs promotes the expression of synaptic proteins in the brain and enhances cognition in animal models (Buccafusco and Terry, 2003; McKay et al., 2007; Grizzell et al., 2014; Terry et al., 2005, 2012). For example, cotinine treatment restored impaired memory in mouse and monkey models of impaired hippocampal function (Echeverria et al., 2011; Gao et al., 2014; Grizzell et al., 2014; Patel et al., 2014; Terry et al., 2012), ameliorated impaired cognition in mouse models of Alzheimer’s disease, including working memory (Echeverria et al., 2011; Patel et al., 2014), memory deficits in animal models of schizophrenia (Buccafusco and Terry, 2009; Buccafusco et al., 2009; Terry et al., 2012) and stress-related memory impairments (Grizzell et al., 2014; Zeitlin et al., 2012).
Cotinine has a regulatory effect on glycogen synthase kinase-3 (GSK3), which is involved in regulating cognition and mood (Pardo et al., 2016; Beurel et al., 2015; King et al., 2014). GSK3 is constitutively active and is regulated by inhibitory phosphorylation on serine-21 of the GSK3α isoform and on serine-9 of the GSK3β isoform (Jope and Johnson, 2004). This inhibitory serine-phosphorylation of GSK3 is often mediated by Akt, which itself is activated by phosphorylation on threonine-308 and serine-473. The functional effects of impaired inhibitory serine-phosphorylation of GSK3β can be studied with GSK3β9A/9A knockin mice (McManus et al 2005; Polter et al., 2010) that have a serine-to-alanine mutation that prevents the inhibition by serine-phosphorylation of GSK3β. This mutation maintains GSK3β maximally active, but importantly within the physiological range since GSK3β is expressed at normal levels. Abnormally active GSK3 contributes to several pathological conditions that involve learning and memory impairments (Pardo et al., 2015, 2016; King et al., 2014), such as Alzheimer’s disease (Avila et al., 2010), mood disorders (Jope, 2011), and Fragile X syndrome (Mines and Jope, 2011). Furthermore, numerous studies have reported beneficial effects of GSK3 inhibition for improving cognitive deficits in many diverse conditions (King et al., 2014). The link between cotinine and GSK3 was first described in an in vitro study in cultured cells showing that cotinine treatment increased the phosphorylation of Akt and the inhibitory serine-phosphorylation of GSK3β (Rehani et al., 2008). Further studies demonstrated in vivo increases in GSK3β serine-phosphorylation (Grizzell et al., 2014; Echeverria et al., 2011) and in phosphorylated Akt on serine 473 (Echeverria et al., 2011) after chronic oral cotinine treatment in mouse hippocampus. Chronic cotinine treatment increased GSK3β phosphorylation in the brains of healthy mice as well as in two psychopathological mouse models, chronic stress (Grizzell et al., 2014) and a mouse model of Alzheimer’s disease (Echeverria et al., 2011). It was suggested that cotinine’s actions to promote neuronal survival and synaptic plasticity could be mediated in part by its action on GSK3β (Echeverria et al., 2011).
The present study examined if acute cotinine treatment also was able to inhibit GSK3, and if there were differential effects of cotinine administration on the two isoforms of GSK3, GSK3α and GSK3β, in mouse hippocampus. We also tested if cotinine administration improved performance of Fmr1−/− mice in coordinate and categorical spatial processing, novel object recognition, and temporal ordering, each of which was recently found to be impaired in these mice and to be rescued by administration of GSK3 inhibitors (King and Jope, 2013; Franklin et al., 2014). Since cotinine treatment improved cognitive parameters in Fmr1−/− mice that exhibit hyperactive GSK3 (Min et al., 2009), we tested if blocked inhibitory serine-phosphorylation in GSK3β knockin mice interfered with the ability of cotinine treatment to improve impaired performances in these cognitive tasks.
Materials and methods
Mice and behavior measurements
Male adult (8–10 weeks old) C57BL/6 wild-type mice (N=45), C57BL/6 mice with a disruption of the Fmr1 gene (N=46) (originally kindly provided with matched controls by Dr. W. Greenough, University of Illinois), and GSK3β9A/9A knockin C57BL/6 mice (N=19) (originally kindly provided with matched controls by Dr. D. Alessi, University of Dundee), were used. This mutation disables the inhibitory serine phosphorylation of GSK3β, but both GSK3 isoforms are expressed at normal levels so GSK3 retains maximal activities within the normal physiological range (McManus et al., 2005). Mice were housed in groups of 3–5 in standard cages in light and temperature controlled rooms and were treated in accordance with NIH and the University of Miami Institutional Animal Care and Use Committee regulations. Cotinine (#C5923; Sigma Aldrich) was administered intraperitoneally (i.p.) at the dose of 3 mg/kg where indicated, 30 min before behavioral testing, which lasted 60 min, or 30 or 90 min before sample collection.
Four cognitive tasks were tested: coordinate and categorical spatial processing, novel object recognition, and temporal order memory, carried out as previously described (King and Jope, 2013; Pardo et al., 2015). Mice were habituated to the testing room with a white noise generator (55 dB) for 1 hr. Behavioral tests were conducted during 2 consecutive days, two tests every 24 hr. 70% ethanol was used to clean each apparatus and object used between each test session, test sessions were filmed, and films were scored by an investigator blind to the genotype and treatment.
For the coordinate spatial processing task, each mouse was allowed to explore two novel objects that were 45 cm apart for 15 min. After 5 min in an opaque chamber, each mouse was allowed to explore the same two objects that had been moved closer together (30 cm) for 5 min. Increased exploration of the objects during the test session compared with the last 5 min of the habituation phase is considered a measure of memory of the distance between objects. The exploration ratio was calculated as time (exploring during the 5 min test session)/(exploring during the 5 min test session plus the last 5 min of the habituation session).
For the categorical spatial processing task, each mouse was allowed to explore two novel objects that were 45 cm apart for 15 min. After 5 min in an opaque chamber, each mouse was allowed to explore the same two objects that had been transposed for 5 min. Increased exploration of the objects during the test session compared with the last 5 min of the habituation phase is considered a measure of memory of the position of objects. The exploration ratio was calculated as time (exploring during the 5 min test session)/(exploring during the 5 min test session plus the last 5 min of the habituation session).
For the novel object recognition task, time spent exploring an object only included the mouse sniffing or touching the object with its nose, vibrissa, mouth, or forepaws. As described previously (Pardo et al., 2015), novel object recognition was measured by allowing each mouse to explore two identical objects for 5 min, and after a 5 min period in an opaque chamber, mice were allowed to explore an unused familiar object and a novel object for 5 min. The discrimination index was calculated as the times ((exploring Object 2 − exploring Object 1)/total time of object exploration) × 100). To measure temporal order memory, each mouse underwent three sessions exploring three new sets of objects (Objects 1, 2, 3). The duration of the sessions and the interval between sessions were 5 min. During the test session, the mouse was allowed to explore an unused copy of Object 1 and an unused copy of Object 3 for 5 min. Intact temporal order memory is evident when mice spend more time exploring the first object presented (Object 1) than the most recent object presented (Object 3). A discrimination index was calculated as the times ((exploring Object 3 − exploring Object 1)/total time of object exploration) × 100).
Immunoblot analysis
Mice were sacrificed by decapitation 30 or 90 min, where indicated, after i.p. cotinine treatment. Mouse hippocampi were rapidly dissected in ice-cold phosphate-buffered saline and homogenized in ice-cold lysis buffer containing 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol, 1 µg/ml leupeptin, 1 µg/ml aprotinin, 1 µg/ml pepstatin, 1 mM phenylmethanesulfonyl fluoride, 50 mM NaF, 1 mM sodium orthovanadate, and 100 nM okadaic acid. The lysates were centrifuged at 20,800xg for 10 min. Protein concentrations in the supernatants were determined using the Bradford protein assay (Bradford, 1976). Lysates were mixed with Laemmli sample buffer (2% SDS) and placed in a boiling water bath for 5 min. Proteins (10 µg) were resolved in SDS-polyacrylamide gels, transferred to nitrocellulose, and incubated with primary antibodies to phospho-Ser21-GSK3α (1:2000; #9316L; Cell Signaling Technology), phospho-Ser9-GSK3β (1:2000; #9336; Cell Signaling Technology), total GSK3α/β (1:2000; #05-412; Millipore), phospho-Ser473-Akt (1:2000; #9271; Cell Signaling Technology), phospho-Thr308-Akt (1:1000; #9275; Cell Signaling Technology) and total Akt (1:1000; #9272; Cell Signaling Technology). Separate blots were used to measure the ratio of phosphorylation/total levels for each protein. Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse (1:2000), or goat anti-rabbit IgG (1:2000), followed by detection with enhanced chemiluminescence. Images were acquired and quantitated with an Amersham Imager 600.
Statistical analyses
Results were analyzed by one-way ANOVA followed by Dunnett’s post-hoc analyses to determine statistical significance or by Student’s t-test where indicated. P values less than 0.05 were considered statistically significant.
Results
Acute cotinine treatment increases phosphorylation of GSK3 and Akt in the hippocampus of wild-type mice
Previous reports that chronic cotinine treatment increased the serine phosphorylation of GSK3β in mouse brain regions (Echeverria et al., 2011; Grizzell et al., 2014) were extended to test if acute cotinine treatment altered the serine-phosphorylation of either GSK3 isoform in mouse hippocampus.
Acute cotinine treatment, (3 mg/kg), was used to study the effects and the duration of the effects on the phosphorylation of GSK3 and Akt for a time period necessary to perform the memory tasks. At both 30 and 90 min after treatment, cotinine administration significantly increased the phosphorylation of Ser-9-GSK3β (30 min: t(12)=3.15, p<0.01; 90 min: t(13)=2.48, p<0.05; Fig. 1A), Ser-21-GSK3α (30 min: t(12)=2.75, p<0.05; 90 min: t(13)=3.49, p<0.01; Fig. 1B), Thr-308-Akt (30 min: t(9)=2.18, p<0.05; 90 min: t(13)=5.07, p<0.05; Fig. 1C) and Ser-473-Akt (30 min: t(10)=2.40, p<0.05; 90 min: t(11)=4.24, p<0.01; Fig. 1D).
Figure 1.

Acute cotinine treatment increases in phosphorylation of GSK3 and AKT in the hippocampus of wild-type mice. Immunoblots from representative mice in each group, at 30 and 90 min after treatment, showing the increase in the hippocampus of phosphorylation of (A) Ser-9-GSK3β, (B) Ser-21-GSK3α, (C) Thr-308-Akt and (D) Ser-473-Akt. Data was normalized relative to control (vehicle (0 mg/kg)). Values are means ±SEM. *p<0.05 **p<0.01 compared to vehicle-treated mice. (n=4–8 per group).
Cotinine treatment increases phosphorylation of GSK3 and Akt in the hippocampus of Fmr1−/− mice
We also tested if acute cotinine treatment increased the inhibitory serine-phosphorylation of GSK3 and the activating phosphorylations of Akt in the hippocampus of Fmr1−/− mice because GSK3 was previously reported to be abnormally active via reduced serine-phosphorylation in the hippocampus of these mice (Yuskaitis et al., 2010). Cotinine produced similar effects in Fmr1−/− mice as in wild—type mice at both 30 and 90 min (Fig. 2). Treatment with cotinine increased hippocampal phosphorylation of Ser-9-GK3β (30 min: t(10)=2.20, p<0.05; 90 min: t(11)= 3.53, p<0.01; Fig. 2A), Ser-21-GSK3α (30 min: t(14)=2.28, p<0.05; 90 min: t(13)=3.36, p<0.01; Fig. 2B), Thr-308-Akt (30 min: t(12)=2.60, p<0.05; 90 min: t(11)=3.43, p<0.01; Fig. 2C) and Ser-473-Akt (30 min: t(11)=2.19, p<0.05; 90 min: t(11)=2.47, p<0.05; Fig. 2D) in Fmr1−/− mice.
Figure 2.

Cotinine treatment increases phosphorylation of GSK3 and AKT in the hippocampus of Fmr1−/− mice. Immunoblots from representative mice in each group, at 30 and 90 min after treatment, showing the increase in the hippocampus of phosphorylation of (A) Ser-9-GSK3β, (B) Ser-21-GSK3α, (C) Thr-308-Akt and (D) Ser-473-Akt. Data was normalized relative to control (vehicle (0 mg/kg)). Values are means ±SEM. *p<0.05 **p<0.01 compared to vehicle treated mice. (n=5–8 per group).
We also compared the basal and cotinine-stimulated phosphorylation levels of GSK3 and Akt in the hippocampus of wild-type mice and Fmr1−/− mice. Compared to untreated wild-type mice, the hippocampus of untreated Fmr1−/− mice had lower phosphorylation of Ser-9-GSK3β (Fig. 3A; one-way ANOVA, F(3,13)=28.43, p<0.01), and Ser-21-GSK3α (Fig. 3B; one-way ANOVA, F(3,13)=21.31, p<0.01), as reported previously (Yuskaitis et al., 2010), and greater phosphorylation of Thr-308-Akt (Fig. 3C; one-way ANOVA, F(3,12)=13.27), and Ser-473-Akt (Fig. 3D; one-way ANOVA, F(3,12)=16.57, p<0.01), as reported previously (Liu et al., 2012). Despite these differences in basal phosphorylation levels, cotinine treatment increased the phosphorylation of GSK3 and Akt similarly in wild-type and Fmr1−/− mice.
Figure 3.

Comparisons of the phosphorylation of GSK3 and AKT in the hippocampus of wild-type (WT) mice and Fmr1−/− mice. Immunoblots from representative mice in each group, at 30 min after treatment, showing the basal levels and cotinine-induced increase in the hippocampus of phosphorylation of (A) Ser-9-GSK3β, (B) Ser-21-GSK3α, (C) Thr-308-Akt and (D) Ser-473-Akt. Data was normalized relative to vehicle treated wild-type mice. Values are means ±SEM. *p<0.05 **p<0.01 compared to wild-type vehicle-treated mice; #p<0.05 ##p<0.01 compared to Fmr1−/− vehicle (0 mg/kg) mice. (n=4–5 per group).
Cotinine effects on performance of Fmr1−/− mice in four cognitive tasks
Since acute cotinine treatment effectively increased the inhibitory serine-phosphorylation of GSK3 in Fmr1−/− mice, we tested if acute cotinine treatment (3 mg/kg) was able to ameliorate cognitive impairments displayed by Fmr1−/− mice that were previously shown to be due to hyperactive GSK3, including novel object recognition, temporal ordering, and coordinate and categorical spatial processing (King and Jope, 2013, Franklin et al., 2014). The performance of wild-type mice was unaltered by acute administration of cotinine in all tasks tested. However, cotinine treatment reversed impairments in spatial memory of the Fmr1−/− mice (Fig. 4A, B), normalizing performance in the coordinate spatial processing task (one-way ANOVA, F(3,29)=14.87, p<0.01; Fig. 4A) and in the categorical spatial processing task (one-way ANOVA, F(3,30)=8.39, p<0.01; Fig. 4B). Post-hoc analyses for both tasks revealed Fmr1−/− impairments (p<0.05 compared to wild type vehicle-treated mice) but no significant differences between wild-type and Fmr1−/− mice after cotinine treatment. In the novel object recognition task, cotinine treatment significantly increased the preference of Fmr1−/− mice for the novel object (% time exploring the objects: Fmr1−/− mice vehicle: familiar object 65.0 ± 4.9 vs novel object 35.0 ± 4.9, t(16)= 4.34, p<0.05; Fmr1−/− mice cotinine: familiar object 39.3 ± 3.2 vs novel object 60.71 ± 3.23, t(10)=4.66, p<0.05; Fig. 4C) and normalized the discrimination index (one-way ANOVA, F(3,30)=10.47, p<0.01; Fig. 4D). Post-hoc analyses yielded significant differences between vehicle-treated wild-type and Fmr1−/− mice (p<0.05), but no differences between wild-type and Fmr1−/− mice were present after cotinine treatment. Administration of cotinine to Fmr1−/− mice rescued the impairment in temporal order memory by reallocating the % of time spent exploring the objects (Fmr1−/− mice vehicle: first object 45.3 ± 5.3 vs third object 54.9 ± 5.3, t(16)= 1.26, p=0.23; Fmr1−/− mice cotinine: first object 67.2 ± 43.7 vs third object 32.8 ± 43.7, t(10)=6.68, p<0.05; Fig. 4E) and the discrimination index (one-way ANOVA, F(3,30)=5.30, p<0.01; Fig. 4F). Post-hoc analyses showed significant differences between vehicle-treated wild-type and Fmr1−/− mice (p<0.05), an impairment that was normalized after cotinine treatment.
Figure 4.

Cotinine effects on performance of Fmr1−/− mice in four cognitive tasks.
(A) Performance of wild-type (WT) mice (n=15) and Fmr1−/− mice (n=14) in the coordinate spatial processing task (Means±SEM; *p<0.05 compared to vehicle-treated WT mice). (B) Performance of WT mice (n=16) and Fmr1−/− mice (n=15) in the categorical spatial processing task (Means±SEM; *p<0.05 compared to vehicle-treated WT mice). (C,D) Performance of wild-type (WT) mice (n=16), and Fmr1−/− mice (n=15) on novel object recognition. (C) Percent time spent exploring the novel (N) and familiar (F) object. (Means±SEM; *p<0.05 compared to time spent with familiar object). (D) Discrimination index (Means±SEM; *p<0.05 compared to vehicle-treated WT mice). (E,F) Performance of WT mice (n=16) and Fmr1−/− mice (n=15) on the temporal order task. (E) Percent time spent exploring the first (1) and last (3) object presented. (Means±SEM; *p<0.05 compared to time spent with object 1). (F) Discrimination index (Means±SEM; *p<0.05 compared to vehicle-treated WT mice).
Cotinine effects on performance of GSK3β knockin (KI) mice in two cognitive tasks
Considering that acute cotinine treatment increased the inhibitory serine-9-phosphorylation of GSK3β in wild-type and Fmr1−/− mice, and restored impaired memory in Fmr1−/− mice, we tested if GSK3β serine-phosphorylation was necessary for the cognition enhancing effects of cotinine. GSK3β knockin (KI) mice, with serine9-to-alanine9 mutations in GSK3β preventing the inhibition of GSK3β by serine-phosphorylation, were used to examine cotinine effects on their previously reported impairments in coordinate spatial processing and novel object recognition (Pardo et al., 2016). Figure 5A shows the lack of serine-9-phosphorylation in the GSK3β KI mice. GSK3β KI mice displayed impaired coordinate spatial processing memory and cotinine treatment did not modify this impairment (one-way ANOVA, F(3,32)=14.55, p<0.01; Fig. 5B). Post-hoc analyses showed a significantly lower exploration ratio in vehicle-treated GSK3β KI mice compared to vehicle-treated wild-type mice (p<0.05), and there were no effects of cotinine in either of the groups. In the novel object recognition task, cotinine treatment had no significant effect in GSK3β KI mice on % time exploring the objects (GSK3β KI mice vehicle: familiar object 49.7 ± 3.1 vs novel object 50.3 ± 3.1, t(18)= 0.14, p=0.89; GSK3β KI mice cotinine: familiar object 51.7 ± 32.6 vs novel object 48.4 ± 2.6, t(16)=0.91, p=0.38; Fig. 5C) or in the discrimination index (one-way ANOVA, F(3,34)=5.38, p<0.01; Fig. 5D). Post-hoc analyses showed a significant impairment in GSK3β KI mice compared to wild-type mice (p<0.05), an effect that persisted after cotinine treatment. These findings show that cotinine was unable to improve performances in several cognitive tasks when the inhibitory serine-phosphorylation of GSK3β was blocked.
Figure 5.

Cotinine effects on performance of GSK3β KI mice in two cognitive tasks.
A) Immunoblots from 3 mice in each group showing the absence of serine-9 phosphorylation of GSK3β in the hippocampus of male GSK3β KI mice. Total levels of GSK3β are equal in GSK3β KI and wild-type (WT) mice. (B) Performance of wild-type (WT) mice (n=15) and GSK3β KI (GSK3β KI) mice (n=18) in the coordinate spatial processing task (Means±SEM; *p<0.05 compared to vehicle-treated WT mice). (C,D) Performance of WT mice (n=16) and GSK3β KI mice (n=19) on novel object recognition. (C) Percent time spent exploring the novel (N) and familiar (F) object. (Means±SEM; *p<0.05 compared to time spent with familiar object). (D) Discrimination index (one-way ANOVA, F(3,34)=5.38, p<0.01; Means±SEM; *p<0.05 compared to vehicle-treated WT mice).
Discussion
We tested the effects of acute cotinine treatment as a cognitive enhancer in a mouse model of Fragile X syndrome, and we further examined if GSK3 may be involved in the effects of cotinine on learning and memory. Acute cotinine treatment increased phosphorylation levels of Akt and GSK3 in the hippocampus of both wild-type mice and Fmr1−/− mice. Furthermore, acute cotinine treatment restored learning and memory impairments in Fmr1−/− mice but failed to restore cognitive impairments in GSK3β knockin mice where a serine-to-alanine mutation blocks the inhibitory serine-phosphorylation of GSK3β.
Acute cotinine treatment increases phosphorylation of GSK3 and AKT in the hippocampus
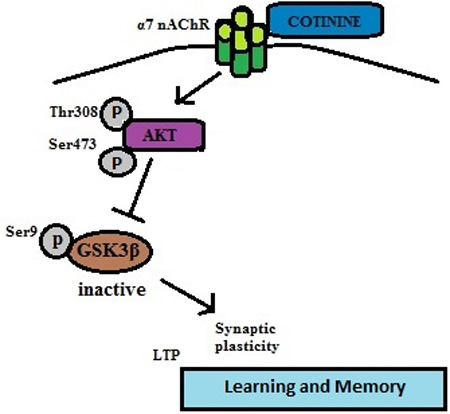
GSK3 is involved in regulating a variety of cellular functions, and abnormally active GSK3 has previously been linked to learning and memory impairments in the mouse model of Fragile X syndrome, as well as other disorders (King et al., 2014; Beurel et al., 2015). GSK3 has two isoforms, GSK3α and GSK3β, which are mainly regulated by inhibitory phosphorylation of serine-21 in GSK3α and serine-9 in GSK3β that are often mediated by Akt. Recent reports demonstrated increased brain phosphorylation of Ser-473-Akt and Ser-9-GSK3β after chronic oral cotinine treatment in the hippocampus of healthy mice, stressed mice, and a mouse model of Alzheimer’s disease (Echeverria et al., 2011; Grizzell et al., 2014). The present results show that acute cotinine treatment is sufficient to induce increased serine phosphorylation in both GSK3 isoforms and Akt serine and threonine phosphorylation in the hippocampus of wild-type mice and in Fmr1−/− mice, in which the phosphorylation levels of GSK3 on Serine 9 and 21 were previously reported to be lower than in wild-type mice (Yuskaitis et al., 2010). These results demonstrate that cotinine treatment rapidly activates Akt and inhibits GSK3. Figure 6 shows a signaling diagram based on these findings and reports that PI3K is activated following stimulation of α7 nicotinic receptors, which can promote Akt phosphorylation (Kihara et al., 2001; Zhang et al., 2014) and the subsequent inhibitory serine-phosphorylation of GSK3β.
Figure 6.

Signaling pathway stimulated by cotinine.
Stimulation of α7 nicotinic receptors activates PI3K leading to the activating phosphorylations on Akt, which phosphorylates the inhibitory Ser-9 of GSK3β, leading to pro-cognitive effects.
Acute cotinine treatment restores impaired cognition of Fmr1−/− mice
Several previous studies reported enhanced learning and memory after chronic cotinine administration in rodents (Buccafusco and Terry, 2009; Echeverria et al., 2011; Gao et al., 2014; Grizzell et al., 2014; Grizzell and Echeverria, 2014; Moran, 2012; Patel et al., 2014; Terry et al., 2005, 2012). Chronic subcutaneous cotinine treatment did not significantly affect memory in wild-type rats (Terry et al., 2012), but chronic oral cotinine treatment enhanced spatial working memory in wild-type mice compared to vehicle-treated mice (Grizzell et al., 2014). Cotinine treatment completely reversed the cognitive impairments induced by treatment with an NMDA antagonist in animal models of schizophrenia (Buccafusco and Terry, 2009; Terry et al., 2012), and chronic cotinine treatment improved memory in transgenic mouse models of Alzheimer’s disease (Echeverria et al., 2011; Patel et al., 2014). Chronic cotinine treatment also induced improvements in spatial memory or contextual memory after impairments induced by stress (Grizzell et al., 2014; Zeitlin et al., 2012). In the present study, in which cotinine was administered acutely, cotinine did not have significant effects on the memory of wild-type mice and reversed impairments in Fmr1−/− mice.
Inhibitory phosphorylation of GSK3β is associated with the cognitive enhancing effects of cotinine
The mechanism through which cotinine exerts its effects on cognition most often has been linked to its action on nAChRs (Grizzell and Echeverria, 2014), although it also has been reported to affect the serotonergic (Fuxe et al., 1979; Grizzell and Echeverria, 2014) and dopaminergic systems (Riah et al., 2000). The increases in GSK3 inhibitory serine-phosphorylation after chronic cotinine treatment revealed a possible important role for this kinase in mediating the cognitive improvements caused by cotinine in mice exposed to chronic stress (Grizzell et al., 2014) and in a mouse model of Alzheimer’s disease (Echeverria et al., 2011). With the aim of better understanding the mechanism underlying cotinine’s effects in restoring impaired memory, and to study the role of GSK3β in mediating cotinine’s effects, cotinine was administered to mice in which the Serine-9 phosphorylation of GSK3β is blocked. Acute cotinine administration was unable to restore cognitive impairments in GSK3β knockin mice (Pardo et al., 2016), suggesting that phosphorylation of GSK3β is required to mediate the cognitive enhancing properties of cotinine. However, these results do not rule out the possibility that other mechanisms also may contribute to the cognition-enhancing effects of cotinine. There is increasing consideration of GSK3 inhibitors as potential treatments for cognitive impairments (King et al., 2014) because GSK3 activity is increased in a variety of conditions, such as in Fragile X mice (Yuskaitis et al., 2010) and Alzheimer’s disease (Maqbool et al., 2015), where memory impairments are a main symptom. A study of GSK3 isoform specificity on memory revealed that even though both GSK3 isoforms share many functions, GSK3β has a predominant role in regulating novel object recognition (Pardo et al., 2016).
Recently, there is much interest in the potential use of nAChR agonists as cognitive enhancers, particularly related to the cholinergic deficits found in disorders such as Alzheimer’s disease (Muir, 1997; Nordberg, 2001) and schizophrenia (Kenney and Gould, 2008). nAChRs activate a number of signaling cascades in the brain that lead to cellular alterations regulating short- and long-term memory (Levin and Simon, 1998; Barros et al., 2004; Kenney and Gould, 2008). The present results add to the previous literature showing that nicotinic receptor agonists can act as cognitive enhancers in a mouse model of Fragile X syndrome and highlight the potential role of inhibiting GSK3β in mediating the effects of cotinine on memory.
Acknowledgments
This research was supported by grants from the National Institute of Mental Health (MH038752, MH090236, MH095380, MH104656). Author contributions: MP performed the experiments and wrote the manuscript. EB and RJ designed the experiments and wrote the manuscript.
Footnotes
Conflicts of interest
This authors declare that they do not have any conflict of interest regarding this research.
References
- Avila J, Wandosell F, Hernández F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert. Rev. Neurother. 2010;10:703–710. doi: 10.1586/ern.10.40. [DOI] [PubMed] [Google Scholar]
- Barros D, Ramirez MR, Dos Reis EA, Izquierdo I. Participation of hippocampal nicotinic receptors in acquisition, consolidation and retrieval of memory for one trial inhibitory avoidance in rats. Neuroscience. 2004;126:651–656. doi: 10.1016/j.neuroscience.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 2015;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Terry AV. The potential role of cotinine in the cognitive and neuroprotective actions of nicotine. Life Sci. 2003;72:2931–2942. doi: 10.1016/s0024-3205(03)00226-1. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Shuster LC, Terry AV. Disconnection between activation and desensitization of autonomic nicotinic receptors by nicotine and cotinine. Neurosci. Lett. 2007;413:68–71. doi: 10.1016/j.neulet.2006.11.028. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Terry AV. A reversible model of the cognitive impairment associated with schizophrenia in monkeys: potential therapeutic effects of two nicotinic acetylcholine receptor agonists. Biochem. Pharmacol. 2009;78:852–862. doi: 10.1016/j.bcp.2009.06.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buccafusco JJ, Beach JW, Terry AV., Jr Desensitization of nicotinic acetylcholine receptors as a strategy for drug development. J. Pharmacol. Exp. Ther. 2009;328:364–370. doi: 10.1124/jpet.108.145292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverria V, Zeitlin R, Burgess S, Patel S, Barman A, Thakur G, Mamcarz M, Wang L, Sattelle DB, Kirschner DA, Mori T, Leblanc RM, Prabhakar R, Arendash GW. Cotinine reduces amyloid-beta aggregation and improves memory in Alzheimer’s disease mice. J. Alzheimers Dis. 2011;24:817–835. doi: 10.3233/JAD-2011-102136. [DOI] [PubMed] [Google Scholar]
- Franklin AV, King MK, Palomo V, Martinez A, McMahon LL, Jope RS. Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol. Psychiatry. 2014;75:198–206. doi: 10.1016/j.biopsych.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxe K, Everitt BJ, Hokfelt T. On the action of nicotine and cotinine on central 5-hydroxytryptamine neurons. Pharmacol. Biochem. Behav. 1979;10:671–677. doi: 10.1016/0091-3057(79)90319-8. [DOI] [PubMed] [Google Scholar]
- Gao J, Adam BL, Terry AV. Evaluation of nicotine and cotinine analogs as potential neuroprotective agents for Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2014;24:1472–1478. doi: 10.1016/j.bmcl.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grizzell JA, Echeverria V. New Insights into the Mechanisms of Action of Cotinine and its Distinctive Effects from Nicotine. Neurochem. Res. 2014;40:2032–2046. doi: 10.1007/s11064-014-1359-2. [DOI] [PubMed] [Google Scholar]
- Grizzell JA, Iarkov A, Holmes R, Mori T, Echeverria V. Cotinine reduces depressive-like behavior, working memory deficits, and synaptic loss associated with chronic stress in mice. Behav. Brain. Res. 2014;268:55–65. doi: 10.1016/j.bbr.2014.03.047. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Jope RS. Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front. Mol. Neurosci. 2011;4:16. doi: 10.3389/fnmol.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Gould TJ. Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Mol. Neurobiol. 2008;38:101–121. doi: 10.1007/s12035-008-8037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A. alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- King MK, Jope RS. Lithium treatment alleviates impaired cognition in a mouse model of fragile X syndrome. Genes Brain Behav. 2013;12:723–731. doi: 10.1111/gbb.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MK, Pardo M, Cheng Y, Downey K, Jope RS, Beurel E. Glycogen synthase kinase-3 inhibitors: Rescuers of cognitive impairments. Pharmacol. Ther. 2014;141:1–12. doi: 10.1016/j.pharmthera.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Hall BJ, Rezvani AH. Heterogeneity across brain regions and neurotransmitter interactions with nicotinic effects on memory function. Curr. Top. Behav. Neurosci. 2015;23:87–101. doi: 10.1007/978-3-319-13665-3_4. [DOI] [PubMed] [Google Scholar]
- Levin ED, Simon BB. Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology. 1998;138:217–230. doi: 10.1007/s002130050667. [DOI] [PubMed] [Google Scholar]
- Li P, Beck WD, Callahan PM, Terry AV, Jr, Bartlett MG. Pharmacokinetics of cotinine in rats: A potential therapeutic agent for disorders of cognitive function. Pharmacol. Rep. 2015;67:494–500. doi: 10.1016/j.pharep.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZH, Huang T, Smith CB. Lithium reverses increased rates of cerebral protein synthesis in a mouse model of fragile X syndrome. Neurobiol Dis. 2012;45:1145–1152. doi: 10.1016/j.nbd.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maqbool M, Mobashir M, Hoda N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer's disease. Eur. J. Med. Chem. 2015;107:63–81. doi: 10.1016/j.ejmech.2015.10.018. [DOI] [PubMed] [Google Scholar]
- McKay BE, Placzek AN, Dani JA. Regulation of synaptic trans- mission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem. Pharmacol. 2007;74:1120–1133. doi: 10.1016/j.bcp.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mines MA, Jope RS. Glycogen synthase kinase-3: a promising therapeutic target for fragile x syndrome. Front. Mol. Neurosci. 2011;4:35. doi: 10.3389/fnmol.2011.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min WW, Yuskaitis CJ, Yan Q, Sikorski C, Chen S, Jope RS, Bauchwitz RP. Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology. 2009;56:463–472. doi: 10.1016/j.neuropharm.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran VE. Cotinine: Beyond that expected, more than a biomarker of tobacco consumption. Front. Pharmacol. 2012;3:173. doi: 10.3389/fphar.2012.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir JL. Acetylcholine, aging, and Alzheimer's disease. Pharmacol. Biochem. Behav. 1997;56:687–696. doi: 10.1016/s0091-3057(96)00431-5. [DOI] [PubMed] [Google Scholar]
- Nordberg A. Nicotinic receptor abnormalities of Alzheimer's disease: therapeutic implications. Biol. Psychiatry. 2001;49:200–210. doi: 10.1016/s0006-3223(00)01125-2. [DOI] [PubMed] [Google Scholar]
- Pardo M, King MK, Perez-Costas E, Melendez-Ferro M, Martinez A, Beurel E, Jope RS. Impairments in cognition and neural precursor cell proliferation in mice expressing constitutively active glycogen synthase kinase-3. Front. Behav. Neurosci. 2015;9:55. doi: 10.3389/fnbeh.2015.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo M, Abrial E, Jope RS, Beurel E. GSK3β isoform-selective regulation of depression, memory and hippocampal cell proliferation. Genes Brain Behav. 2016;15:348–355. doi: 10.1111/gbb.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Grizzell JA, Holmes R, Zeitlin R, Solomon R, Sutton TL, Rohani A, Charry LC, Iarkov A, Mori T, Echeverria Moran V. Cotinine halts the advance of Alzheimer's disease-like pathology and associated depressive-like behavior in Tg6799 mice. Front. Aging Neurosci. 2014;6:162. doi: 10.3389/fnagi.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polter A, Beurel E, Yang S, Garner R, Song L, Miller CA, Sweatt JD, McMahon L, Bartolucci AA, Li X, Jope RS. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology. 2010;35:1761–1774. doi: 10.1038/npp.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehani K, Scott DA, Renaud D, Hamza H, Williams LR, Wang H, Martin M. Cotinine-induced convergence of the cholinergic and PI3 kinase-dependent anti-inflammatory pathways in innate immune cells. Biochim. Biophys. Acta. 2008;1783:375–382. doi: 10.1016/j.bbamcr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Riah O, Courriere P, Dousset JC, Todeschi N, Labat C. Nicotine is more efficient than cotinine at passing the blood–brain barrier in rats. Cell Mol. Neurobiol. 1998;18:311–318. doi: 10.1023/A:1022501131709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riah O, Dousset JC, Bofill-Cardona E, Courriere P. Isolation and microsequencing of a novel cotinine receptor. Cell Mol. Neurobiol. 2000;20:653–664. doi: 10.1023/A:1007094623775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry AV, Jr, Buccafusco JJ, Schade RF, Vandenhuerk L, Callahan PM, Beck WD, Hutchings EJ, Chapman JM, Li P, Bartlett MG. The nicotine metabolite, cotinine, attenuates glutamate (NMDA) antagonist-related effects on the performance of the five choice serial reaction time task (5C-SRTT) in rats. Biochem. Pharmacol. 2012;83:941–951. doi: 10.1016/j.bcp.2011.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry AV, Jr, Hernandez CM, Hohnadel EJ, Bouchard KP, Buccafusco JJ. Cotinine a neuroactive metabolite of nicotine: potential for treating disorders of impaired cognition. CNS Drug Rev. 2005;11:229–252. doi: 10.1111/j.1527-3458.2005.tb00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainio PJ, Tuominen RK. Cotinine binding to nicotinic acetylcholine receptors in bovine chromaffin cell and rat brain membranes. Nicotine Tob. Res. 2001;3:177–182. doi: 10.1080/14622200110043095. [DOI] [PubMed] [Google Scholar]
- Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem. Pharmacol. 2010;79:632–646. doi: 10.1016/j.bcp.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu M, Lu F, Luo N, He ZP, Yang H. Involvement of α7 nAChR signaling cascade in epigallocatechin gallate suppression of β-amyloid-induced apoptotic cortical neuronal insults. Mol Neurobiol. 2014;49:66–77. doi: 10.1007/s12035-013-8491-x. [DOI] [PubMed] [Google Scholar]
- Zeitlin R, Patel S, Solomon R, Tran J, Weeber EJ, Echeverria V. Cotinine enhances the extinction of contextual fear memory and reduces anxiety after fear conditioning. Behav. Brain Res. 2012;228:284–293. doi: 10.1016/j.bbr.2011.11.023. [DOI] [PubMed] [Google Scholar]