

Abstract
siRNA is considered as a potent therapeutic agent because of its high specificity and efficiency in suppressing genes that are overexpressed during disease development. For nearly two decades, a significant amount of efforts has been dedicated to bringing the siRNA technology into clinical uses. However, only limited success has been achieved to date, largely due to the lack of a cell type-specific, safe, and efficient delivery technology to carry siRNA into the target cells' cytosol where RNA interference takes place. Among the emerging candidate nanocarriers for siRNA delivery, peptides have gained popularity because of their structural and functional diversity. A variety of peptides have been discovered for their ability to translocate siRNA into living cells via different mechanisms such as direct penetration through the cellular membrane, endocytosis-mediated cell entry followed by endosomolysis, and receptor-mediated uptake. This review is focused on the multiple roles played by peptides in siRNA delivery, such as membrane penetration, endosome disruption, targeting, as well as the combination of these functionalities.
Keywords: Cell penetrating peptide, endosome disruption, endosomolytic peptide, siRNA, targeting, silencing, multifunctional
Graphical abstract
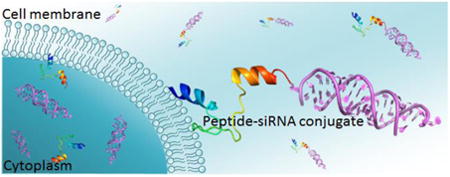
1. Introduction
Short interfering RNAs (siRNAs), also known as small interfering RNAs, are short 21-23 base-pair RNA duplexes including two 3′ 2-nucleotide overhangs. The antisense strand of siRNA with sequence complementarity to a target mRNA can induce sequence-specific gene expression suppression (silencing), which is referred to as RNA interference (RNAi) [1]. RNAi was first discovered in Caenorhabditis elegans, and later was found in plant, animal and human cells. Naturally, endogenous RNAi is a process to help regulate gene expression and function as an innate immune system to help defend viruses and other foreign genetic materials [2]. Taking advantage of the high selectivity and efficiency of this natural mechanism, RNAi has become an indispensable research tool for analysis of gene functions. The programmable degradation of target mRNA by RNAi also opens new opportunities to suppress protein expressions involved in disease pathogenesis[3]. Indeed, siRNA has been demonstrated to be a potential drug candidate for many difficult-to-treat diseases such as viral infections and cancers [4]. Many clinical trials have also been initiated over the last decade, but only very limited siRNA treatments have been approved by the FDA (e.g., topical administration of naked siRNA in ocular and retinal diseases) [5].
The primary barrier to its clinical applications is cell type-specific cytosolic delivery. siRNAs are densely negatively charged macromolecules with a molecular weight of approximately ∼ 14 kDa. They generally can't cross the cell membrane by passive diffusion like many small molecules and ions [6]. Moreover, siRNA trapped in endocytic compartments is susceptible to rapid degradation during endosome–lysosome trafficking [7], since most highly charged macromolecules are taken up by cells via endocytosis.
In this context, carriers that enable efficient siRNA delivery are required. A number of delivery technologies have been developed based on cationic lipids, polymers, dendrimers and peptides [6, 8, 9]. Among these non-viral carrier materials, peptides have gained increasing popularity because of their sequence and function diversity. Various combinations of the 20 natural amino acids result in peptides with different 3-D conformations, electric charges, polarity, hydrophobicity, and hydrophilicity. These unique sequences, within a relatively small molecule-weight range, can display various functions including siRNA binding, membrane penetration, endosome disruption, and targeting, all of which are essential for targeted siRNA delivery. Furthermore, as natural biomolecules, peptides (and their larger counterparts, proteins) with known functionalities are highly abundant in human bodies and other organisms, offering the possibility of directly picking functional peptides of interest from the enormous proteome libraries. For example, some viral coat proteins naturally carry out the biological function of shuttling exogenous biomaterials into cells, which can be useful for drug delivery. In addition to natural proteins, sequences that do not exist in current organisms can also be designed computationally or identified through panning techniques (e.g., phage and bacteria display). In this context, the aim of this review is to systematically discuss the broad functionalities of peptides and their applications in siRNA delivery. We will begin the discussion with individual functionalities such as membrane penetration, endosome disruption, and targeting, and proceed to multifunctional designs and applications.
2. Cell penetrating peptides: siRNA intracellular uptake
Cell-penetrating peptides (CPPs) are a class of short peptides that can penetrate the cell membrane and translocate into the cytoplasm. By chemical conjugation or noncovalent complex formation with siRNA, CPPs can also bring siRNA inside cells. As of now, more than 100 peptides with this cell-penetrating ability have been identified, and they exhibit a great variety in terms of amino acid sequence, length, and polarity. It is important to point out that although CPPs, as their names indicate, can penetrate through the cell membranes and directly enter the cytosol, most CPPs enter cells through multiple pathways, such as combinations of hole punching in the cell membrane and endocytic uptake. siRNA diffusion through the punched holes and entry into the cytosol lead to RNAi; whereas the endocytic uptake requires additional mechanisms to facilitate siRNA escape from the intracellular compartments. Sharing no sequence homology, CPPs can be divided into several categories (Table 1).
Table 1.
siRNA delivery by CPPs.
| Peptide | Subgroup | Sequence | Ref. |
|---|---|---|---|
| Hydrophilic CPPs | |||
| Tat | --- | RKKRRQRRR | [17, 18, 24, 25] |
| Arginine oligomer | --- | Rn; 6 < n < 16 | [22, 23, 26, 27] |
| Amphiphilic CPPs | |||
| MPGΔNLS | Bipartite peptide | GALFLGFLGAAGSTMGAWSQPKSKRKV | [30-35] |
| MPG-8 | Bipartite peptide | bAFLGWLGAWGTMGWSPKKKRK-Cya | [36] |
| MPGα | Bipartite peptide | Ac-GALFLAFLAAALSLMGLWSQPKKKRKV-Cya | [37, 38] |
| BPrPp | Bipartite peptide | MVKSKIGSWILVLFVAMWSDVGLCKKRPKP-amide | [39] |
| Pep-1 | Bipartite peptide | Ac-KETWWETWWTEWSQPKKKRKV | [40] |
| Penetratin | α-helical peptide | RQIKIWFQNRRMKWKK-amide | [18, 39, 44-46] |
| CADY | α-helical peptide | Ac-GLWRALWRLLRSLWRLLWRA-cysteamide | [36, 49, 50] |
| KALA | α-helical peptide | WEAKLAKALAKALAKHLAKALAKALKACEA | [52, 53] |
| Targeting ligand-CPP conjugates | |||
| Fab-Protamine | Antibody-based | Fab-ARYRCCRSQSRSRYYRQRQRSRRRRRRSCQTRR RAMRCCRPRYRPRCRRH | [55, 56] |
| scFv-dR9 | Antibody-based | scFv-dRdRdRdRdRdRdRdRdR | [57] |
| mAb-R9 | Antibody-based | mAb-RRRRRRRRR | [58] |
| A1-Tat | Homing peptide-based | WFLLTM-RKKRRQRRR | [68] |
| RVG-R9 | Homing peptide-based | YTIWMPENPRPGTPCDIFTNSRGKRASNGGGG-RRRRRRRRR | [69-73] |
| Activatable CPPs | |||
| ACPP | Enzyme activated | EEEEEEE-GALGLP-RRRRRRRRKKR | [78] |
2. 1. Hydrophilic cationic peptides
Hydrophilic cationic CPPs contain one or more stretches of positive charges that are critical for siRNA condensation and membrane penetration. Due to the high charge density, cationic CPPs are highly hydrophilic and often exhibit random coils in aqueous solutions. Among basic amino acids, arginine plays the key role in membrane penetration [10]. Studies indicate that the guanidine group of arginine has a strong affinity to the negatively charged phospholipids in the cell membrane. As a result, arginine-based peptides have higher cell penetrating ability than cationic peptides without arginines [11]. A minimum of eight positive charges is required for efficient penetration across the cellular membrane [12], and the charged side-chains can quickly insert into the cell lipid bilayer and nucleate a transient pore, followed by the translocation of CPPs through the pore [13]. Because the pores quickly close as the peptides cross the plasma membrane, the cytotoxicity of CPPs is low, often undetectable in their working range for siRNA delivery.
The first CPP (Tat peptide, RKKRRQRRR) was discovered from the HIV-1 Tat protein in the late 1980s [14]. The Tat protein is a transcription regulator in the human immunodeficiency virus 1(HIV-1) genome, and it can freely cross the plasma membrane [15]. Further study indicates that the transduction domain is confined in a small stretch of basic amino acids (Amino Acid 48-56) [16]. A chemically synthesized peptide of this stretch (the Tat peptide) can rapidly translocate across the plasma membrane and accumulate in the cell nucleus [16]. Since then, Tat peptide has been used as a universal carrier for intracellular delivery of a variety of cargo molecules (Fig. 1A1). Chiu et al. covalently conjugated Tat peptide to the 3′ end of siRNA antisense strand via a non-cleavable crosslinker SMPB (succinimidyl 4-(p-maleimidophenyl) butyrate) [17]. After incubation with cells, the siRNA was quickly translocated inside cells as evidenced by a punctuate distribution of Cy3 labeled siRNA-Tat in the perinuclear region, which suggested that siRNA was primarily inside endosomes. Intriguingly, the siRNA-Tat conjugate was still able to induce targeted gene silencing (exogenously transfected eGFP and endogenous CDK9) likely due to partial siRNA release. An additional detailed study by Moschos et al., however, showed that siRNA-Tat conjugate purified with High-Performance Liquid Chromatography (HPLC) was only able to induce 30% gene expression reduction at a siRNA concentration of 10 μM, which was approximately two orders of magnitude higher than the concentration used by Chiu and coworkers [18]. Taking these results together, the silencing effect of the siRNA-Tat conjugate is likely induced by siRNA complexed with Tat instead of siRNA covalently linked to the Tat peptide since the peptide may cause steric hindrance for siRNA loading into the RNA-induced silencing complexes (RISC), but systematic mechanistic studies are needed to confirm this hypothesis.
Fig. 1. CPP-mediated siRNA intracellular delivery.
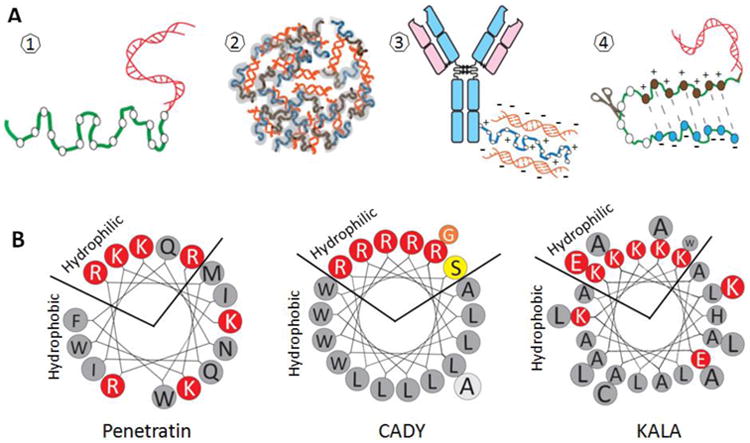
A). Schematic illustration of formulation schemes for CPP-mediated siRNA cell entry. (1) CPP-siRNA conjugate; (2) CPP-siRNA nanocomplex; (3) siRNA anchored on an antibody-CPP conjugate; (4) hairpin-structured activatable CPP conjugated with siRNA. B) The helical wheel showing the amphiphilicity of α-helical CPPs: Penetratin, CADY, and KALA. Amino acids are color coded (red: charged; orange: apolar; yellow: polar; gray: hydrophobic).
In general, conjugation of bulky chemical groups to siRNA can adversely affect siRNA incorporation into the RISC complex and consequently its gene silencing activity. In the RNAi pathway, internalized siRNA must be loaded into RISC, where the sense strand is cleaved during RISC activation and the antisense strand binds with the argonaute protein and directs gene silencing. Due to the compact space of siRNA binding pocket in RISC, terminal modification of siRNA often is intolerable, with the exception of some small chemical groups or cleavable bulky groups [19, 20]. Considering the bulky structure of Tat peptide, peptide modification of siRNA terminus could dramatically limit RNAi activity.
Nonaarginine (R9) is an arginine oligomer and exhibits a higher arginine density than the Tat peptide [21]. Similarly, it can interact with siRNA through electrostatic interactions. In the presence of excess R9 peptides, the negative charges of siRNA backbone are neutralized and condensed by R9. Their final complexes are still highly positively charged due to the excess amount of R9, enabling membrane penetration. The first study in cell lines stably transfected with GFP demonstrated that the R9/siRNA complexes can efficiently reach the perinuclear regions and reduce eGFP expression [22]. In vivo application has also been explored by using a longer arginine oligomer R12 for better siRNA condensation. Treatment of a mouse tumor xenograft model with anti-Her2 siRNA/R12 complexes resulted in a marked reduction of tumor growth [23].
The delivery of siRNAs by noncovalent condensation with hydrophilic cationic CPPs is a simple and effective strategy. However, excess CPP (regarding N/P molar ratio) is required for efficient siRNA condensation and delivery. For example, in the case of the R9/siRNA complex, the peptides and siRNA were mixed at a 12:1 N/P ratio [22]. Although the N/P ratio can be decreased to 3:1 by elongating the arginine oligomer to 15 mer, the number of positive changes is still overwhelmingly high [23]. The excessive cationic CPP is efficient in siRNA condensation and cell entry, but at the same time promotes nonspecific interactions with other anionic molecules and cells, thereby affecting the colloidal stability of the CPP/siRNA complexes and targeting during in vitro transfection and in vivo circulation. A common strategy to address this issue is to conjugate the CPP to polymers, which not only enhance the CPP's condensation ability through multivalency, but also reduce nonspecific binding with serum proteins. For example, block copolymers anchored with Tat peptides (MPEG-PCL-Tat) can form stable nanoparticles (60 to 200 nm) with siRNA and efficiently deliver siRNA to brain tissues via intranasal administration [24]. Similarly, treatment with an anti-Ataxin siRNA and Tat-tagged PEG-chitosan successfully suppressed Ataxin-1 gene expression in an established model of ND Spinocerebellar ataxia (SCA1) [25]. Increasing the hydrophobicity of cationic CPPs has also been proposed to overcome the inherent instability of CPP/siRNA complex. It was demonstrated that simple modification of octaarginine (R8) with a long chain fatty acid promotes siRNA condensation, and the resulting highly condensed nanoparticle shows improved stability against particle disassembly and enzymatic degradation [26]. Furthermore, the complex using modified R8 also exhibits 40–50 times higher cell uptake than the unmodified R8 [27].
2. 2. Amphiphilic CPPs
The common feature of CPPs is that they are able to effectively cross the cellular membrane while carrying cargoes. In this process, the phospholipid bilayer in cell membrane prevents transportation of cargoes in and out of cells. Amphiphilic peptides, which share similar amphiphilic properties with phospholipids, can insert into the lipid bilayer and cross the cell membrane by formation of lipid rafts or transient channels. Many amphiphilic CPPs have been studied for intracellular delivery of siRNA. These amphiphilic CPPs are often classified into bipartite CPPs (based on their primary sequences) and α-helical CPPs (based on surfaces or domains formed in the secondary structures).
2.2.1. Amphiphilic bipartite peptides
Amphiphilic bipartite CPPs are linear amphiphilic peptides with a hydrophobic domain at one end and a cationic domain at the other. The cationic end plays a major role in siRNA condensation, whereas the hydrophobic domain helps stabilize the CPP/siRNA complex through intermolecular hydrophobic interactions. Besides stabilizing the complex, the hydrophobic domain also tends to interact with the lipid bilayer, facilitating membrane fusion during cell entry. MPG (GLAFLGFLGAAGSTMGAWSQPKKKRKV) and Pep-1 (KETWWETWWTEWSQPKKRKV) represent two excellent examples of this type of peptide. The hydrophobic domain of MPG (underlined) is derived from the fusogenic sequence of the HIV glycoprotein 41 (gp 41), while that of Pep-1 (underlined) is a tryptophan-rich cluster that has high affinity to the lipid membrane [28]. Both MPG and Pep-1 contain a cationic nuclear localization signal (NLS) sequence (italic) that can bind with siRNA. A short peptide linker (bold) is inserted between the hydrophobic and hydrophilic domains to improve the flexibility of the bipartite peptides.
Among all the bipartite CPPs, MPG and its derivatives have been the major focus for siRNA delivery. MPG is originally designed for rapid and efficient delivery of plasmid DNA into the nucleus [29]. It was later optimized for siRNA delivery by a single mutation on the second lysine residue in NLS to serine (KKKRKV to KSKRKV, annotated as ΔNLS), which abolishes the nuclear translocation function and enables rapid release of siRNA in the cytosol [30]. MPGΔNLS and siRNA form complexes via electrostatic interactions, which are further stabilized by the gp41 hydrophobic domain, resisting enzymatic degradation (Fig. 1A2). siRNA intracellular delivery by MPGΔNLS is enabled by direct membrane penetration, and the endocytic pathway only plays a minor role here [31]. Studies have shown that MPGΔNLS interacts strongly with the phospholipid cell membrane through its hydrophobic gp41 sequence, which can adopt a transient β-sheet structure and insert into the plasma membrane. The β-sheet insertion temporarily changes cell membrane organization and creates a transient channel, allowing the siRNA/MPGΔNLS complex to enter the cytoplasm [31-33]. MPGΔNLS has been applied for siRNA transfection in a large panel of cells including primary cells and embryonic stem cells [34, 35].
A 6-residue shorter version of the MPGΔNLS, MPG-8, has also been developed to improve siRNA delivery efficiency [36]. Two hydrophobic residues (Phe7 and Ala11) are mutated to Trp to promote membrane fusion. The siRNA delivery efficiency of MPG-8 has been compared with that of the parent MPGΔNLS peptide by delivering anti-cyclin B1 (Cyc-B1) siRNA to HeLa and HS68 cells. At a similar peptide/siRNA molar ratio (20/1), MPG-8 condenses siRNA into nanocomplex with a diameter of 120 ± 50 nm, significantly smaller than complexes formed with MPGΔNLS (diameter: 260 ± 50nm), while its RNAi effect is 2-fold more potent than the original MPGΔNLS. Further modification of this peptide with cholesterol enhances its hydrophobicity, making the siRNA nanoparticles sufficiently stable in blood circulation. More importantly, the Chol-MPG-8 improves siRNA in vivo biodistribution under intravenous administration without activating the innate immune response [36].
Another derivative of the MPGΔNLS peptide is MPGα, which differs from the parent peptide by six amino acids in the hydrophobic domain. The mutations in the gp41 domain allow this domain to adopt a helical conformation rather than a β-sheet structure when it approaches the phospholipid membrane [37]. Penetration experiments show that MPGα undergoes a conformational transition into α-helix and can spontaneously insert into the phospholipid membranes. Further studies indicate that endocytosis is the main pathway for the uptake of MPGα/siRNA complexes, different from MPGΔNLS. Despite the difference in cellular uptake mechanism, MPGα is equally effective in siRNA delivery when compared to MPGΔNLS. Under optimal conditions, a model gene (luciferase) was inhibited by up to 90% at a siRNA concentration of sub-nanomolar range [38]. A few other amphiphilic CPPs (such as BPrPp and Pep-1) have been reported, but only limited silencing has been achieved, likely due to their low binding affinity to siRNA [39, 40].
2.2.2. Amphiphilic α-helical peptides
Besides the primary sequences (hydrophobic block), the amphiphilic property of a CPP can also be enabled by its secondary structure, such as the α-helix structure. The amphiphilicity of α-helix can be visualized through the helical wheel projection, in which hydrophilic and hydrophobic amino acids are showed in separate faces (Fig 1B). As membranolytic peptides, amphiphilic α-helical CPPs generally have a cationic patch on one face for siRNA binding, whereas the other face is hydrophobic to embed into the lipid membrane. It is worth noting that most amphiphilic α-helical CPPs are unstructured in aqueous solution, but spontaneously adopt the α-helix configuration driven by interactions with anionic siRNA molecules or hydrophobic membrane lipids.
Penetratin is the first amphiphilic α-helical CPP discovered. The sequence of penetratin is derived from the third helix of the Drosophila Antennapedia protein homeodomain [41]. Penetratin adopts various secondary structures in different environments. It has been shown to adopt a α-helix when in contact with a model membrane, become a random coil and β-strand in the cytoplasm, and switch to a β-sheet in the nucleus [42, 43]. Penetratin binds with siRNA tightly at a relatively low peptide/siRNA molar ratio (10/1), resulting in high propensity of siRNA condensation and cell uptake. Despite the high uptake efficiency, penetratin/siRNA complex is mainly trapped inside the endosome/lysosome, resulting in no obvious gene silencing effect in vitro [39]. Surprisingly, penetratin has found to be able to transport exogenous siRNA into cytoplasm when it is covalently conjugated with siRNA [44, 45]. Muratovska et al. constructed a penetratin-siRNA conjugate via a disulfide bond formation (labile in the reducing environment of the cytoplasm). Using this approach, they were able to down-regulate luciferase reporter gene expression to a level equivalent or better than using cationic liposomes [44]. A similar result has also been reported by Davidson et al. [45]. In these studies, however, the penetratin-siRNA conjugates were not purified before testing on cells. Similar to the discussions above, there is a possibility that the RNAi effect is induced by siRNA complexed with penetratin (instead of conjugated). Indeed, when Moschos et al. used formamide as a disaggregant and purified the penetratin-siRNA conjugate by HPLC, only moderate suppression of p38 (20%) was observed using penetratin-siRNA conjugate at high concentration (10 μM) [18]. Interestingly, the RNAi efficiency can be dramatically improved if the conjugation site to siRNA is switched from the 5′-end to 3′-end of the sense strand. 50% knockdown was obtained with a concentration of 1 μM [46].
CADY is another amphiphilic α-helical peptide designed for siRNA delivery. This 20-amino acid peptide can bind siRNA with high affinity through electrostatic and hydrophobic interactions. It is derived from two amphiphilic peptides: JST1 and PPTG1, but some residues are mutated into Arg and Trp to improve siRNA binding ability and membrane fusion activity (Fig. 1B) [47, 48]. The N- and C-termini of CADY are modified by acetyl and cysteamide groups to improve peptide stability and its cell membrane crossing efficiency, similar to the MPG peptides discussed above. Atomic Force Microscopy (AFM) studies have revealed that CADY molecules can self-assemble around siRNA and form a “raspberry”-like nanoparticle, in which each single complex constitutes a “drupelet” [49]. The penetrating mechanism of CADY/siRNA complex is not entirely clear yet, but a popular hypothesis is that the single complexes insert into the cell membrane through peptide conformational transition and induce membrane pinching, and translocate across the membrane through a transient channel [50]. The CADY/siRNA complexes can enter a wide variety of cell lines independent of the major endosomal pathway, leading to sustained knockdown of target genes at low siRNA concentrations (nanomolar range) with negligible toxicity and off-target effects [48].
Similarly, KALA is a cationic amphiphilic peptide that undergoes a pH-dependent conformational change from a random coil to amphiphilic α-helical as the pH increases from 5.0 to 7.5 [51]. At physiological pH, one face of the α-helix displays hydrophobic leucine residues, whereas the opposite face displays hydrophilic lysine residues (Fig. 1B). The cationic and amphiphilic properties facilitate siRNA condensation and cellular membrane penetration, which is useful for siRNA transfection. It has successfully transfected anti-GFP and -VEGF siRNAs into tumor cells and suppressed the target gene expressions by 80% [52, 53]. Since KALA adopts a α-helical structure at physiological pH, its toxicity can be a potential concern. It has been reported that KALA peptide exhibits strong hemolytic activity due to the nonspecific membrane disruption [54].
2. 3. Targetable CPPs and activatable CPPs
The unique property of shuttling siRNA into virtually any cell makes CPPs potential carriers in RNAi therapy. However, this property is double-edged because CPPs enter cells without selectivity. For cell-type specific delivery, further developments are needed. A couple of strategies have been demonstrated recently by linking CPPs with other targeting ligands or making CPPs' cell entry capability activatable (initially masked, then activated by disease-associated enzymes).
2.3.1. CPP-targeting ligand bioconjugates
The targeting ability of CPPs can be enhanced by linking them to a wide variety of ligands such as antibodies, aptamers, homing peptides, and small molecules. Monoclonal antibodies and their fragments are a popular choice for targeted drug delivery in general because of their high specificity, strong binding avidity, and broad availability. Genetic fusion or chemical conjugation with cationic CPPs makes antibodies positively charged for electrostatic interaction with siRNA (Fig. 1A3). Song et al. used an antibody-protamine fusion protein to deliver non-covalently bound siRNA into target cells to treat HIV and breast cancer [55, 56]. Shankar's group conjugated oligo-9-D-arginine (DR9) peptide with a single-chain fragment variable (scFv) to deliver siRNA for suppressing HIV-1 infection in humanized mice [57]. Ma et al. reported a siRNA delivery platform based on a Lewis-Y specific antibody conjugated to a nine-arginine peptide (R9) [58]. Although the results are highly encouraging, the binding between antibody-CPP and siRNA relies on electrostatic interactions, which are prone to aggregation, and the excess positive charge of the final complexes may also induce non-specific binding during systemic delivery. To assess and improve this delivery strategy, Schneider et al. constructed a bispecific antibody that binds to cell-surface antigens and digoxigenin (Dig). In parallel, siRNA was digoxigeninylated at its 3′ end. When mixed together, the bispecific antibody and siRNA self-assembled at the single molecule level and entered cells efficiently, avoiding the aggregation and non-specific binding problems discussed above. However, RNAi effect was not observed, because the antibody-siRNA complex was not able to escape from the endosomal compartments [59]. A similar result was also reported by Cuellar et al. [60]. Overall, such coupling methodology is capable of creating monomeric antibody–siRNA conjugates with retained antibody and siRNA bioactivities, but additional endosomoytic functionality is needed for efficient RNAi [60].
Homing peptides are another category of popular ligands that can be linked with CPPs. Compared to antibody-CPP fusion proteins, homing peptide-CPP conjugates are much smaller (generally < 50 amino acids) and can be chemically synthesized by automated solid-phase reactions. Panning technologies such as phage display have led to the discovery of a large number of homing peptides targeting a wide variety of target molecules [61-63]. Some of them have been linked to CPPs to enable targeted delivery of various cargos, including siRNA [64-67]. For example, Tat peptide has been conjugated to a homing peptide A1, a 6-mer peptide showing high affinity to vascular endothelial growth factor receptor-1 (VEGFR1), for selective delivery of siRNA into tumor cells in the presence of co-cultured normal cells [68]. Successful in vivo delivery of siRNA was also reported by using a chimeric peptide consisting of an RVG peptide and an R9 CPP. The 29-aa short RVG peptide is derived from the rabies virus glycoprotein (RVG). It specifically binds to its natural target acetylcholine receptor (AchR) that has been reported to be expressed in dendritic cells, macrophages, and neuronal cells. The RVG-R9 chimera peptide was shown to bind and deliver siRNA selectively, resulting in efficient gene silencing in AchR expressing cells. Post intravenous injection, RVG-R9 was able to deliver HMGB1 siRNA to macrophages and dendritic cells and enable specific gene knockdown in these immune cells, and help suppress inflammation in brain and blood [69, 70]. Another application of RVG-R9 for antiviral siRNA delivery led to robust protection against fatal viral encephalitis in mice [71, 72]. It is worth mentioning that the chirality of the arginines (L and D isomers) significantly affected the potency and duration of RNAi [73]. Microscopy study revealed that siRNA trapped in endosome can be slowly released when the arginines are the L-isomer due to enzymatic degradation, but not the protease-resistant D-isomer [73].
2.3.2. Activatable CPPs
The first activatable CPP is a hairpin-structure peptide comprising a polycationic domain and a polyanionic domain connected by a cleavable peptide loop [74, 75]. The positive charges of the cationic domain are neutralized by the intramolecular electrostatic interactions with the polyanionic domain, reducing cell uptake [76]. Cleavage of the linker loop by a disease-associated protease activates the cell penetrating function of the cationic domain (Fig. 1A4). Using far-red fluorescent dye as a model cargo, activatable hairpin CPP has shown a 10-fold increase of cargo uptake in cultured cells and 3.1-fold increase in xenograft tumors once it was activated by proteases [77]. For selective delivery of siRNA, Li et al. constructed a matrix metalloproteinase-2 (MMP2) activatable CPP and delivered hTERT siRNA into hepatocellular carcinoma cells (SMMC-7721) that over-express MMP2 [78]. For in vivo studies, it has been found that activatable CPPs can accumulate in cartilage non-specifically. To address this problem, Olson and coworkers appended CPPs with a long PEG chain to the polyanionic domain to improve blood circulation time and biodistribution in mice [74].
Another type of activatable CPPs is made by chemical modification of the key residues in CPP via cleavable bonds [79, 80]. Most frequent modifications were made on lysines, while arginine's cationic charges were unmasked, making them currently not suitable for cell type-specific delivery of siRNA.
3. Endosome disrupting peptides facilitating siRNA endosomal escape
As aforementioned, most CPPs enter cells through more than one pathway. For example, they can directly penetrate through cell membranes and carry siRNA molecules into the cytosol where RNAi takes place. At the same time, majority of CPPs enter cells through the endocytic pathway. Taking cationic CPP R9 as an example, R9/siRNA complexes can enter the cytosol directly, but majority tend to rapidly accumulate inside endosomes [73]. In general, the endosomolytic activity of CPPs is poor [81, 82], showing the need of other peptide sequences for endosome/lysosome disruption.
3. 1. Conformation-changing fusogenic peptides
Fusogenic peptides are typically dormant at neutral pH, but its fusogenic functionality can be activated by lower pH. When the pH of surrounding environment drops (e.g., in acidic endosome and lysosome), they undergo a conformational change, such as forming an amphiphilic helix. The transformed peptides often self-associate into multimeric clusters and fuse with and disrupt the endosome membranes to aid in cargo release [83]. In contrast to the amphiphilic α-helical CPPs, fusogenic peptides often have glutamic acid or histidine, because their side chains can be de-protonated or protonated in response to small changes in pH.
HA2 (GLFGAIAGFIENGWEGMIDGWYG) derived from hemagglutinin (HA) is a potent pH-responsive fusogenic peptide. The side chains of the two Glu and Asp residues are anionic, rendering the peptide to extend to a random coil structure at physiological pH. Inside the acidic endosome, however, the carboxyl groups of those side chains became protonated, reducing the negative charges. As a result, the peptide adopts a α-helical conformation (Fig. 2A) [84], and fuses with and destabilizes the endosome membrane [85]. Because it is anionic, HA2 is often conjugated to cationic peptides for siRNA condensation. For example, HA2 has been linked with penetratin, nona(D-arginine), or cationic GFP to deliver siRNA to a broad range of cancer cells [39, 86, 87]. To enhance its fusogenic activity, HA2 has also been mutated into several derivatives. Common strategies include adding more glutamic acid to increase the pH sensitivity and incorporating hydrophobic amino acids such as Trp or Leu to improve the membrane fusion ability. The HA2 derivatives such as INF7, E5, E5WYG and C6M1 have been applied for plasmid DNA and siRNA delivery (Table 2) [83, 88-93].
Fig. 2.
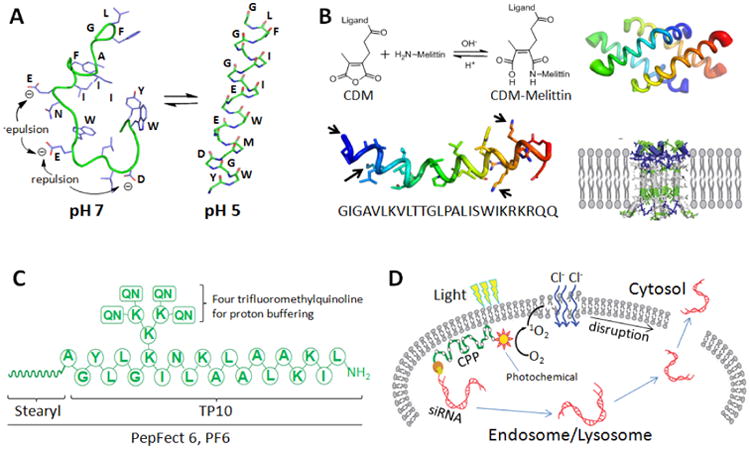
Endosome disrupting peptides. A) Conformation-changing fusogenic peptide HA2 exhibits a random coil at neutral pH and adopts a α-helical conformation at lower pH. Adapted with permission from ref. [84]. Copyright 1994 National Academy of Sciences. B) A chemical structure-changing peptide made by CDM modification of the NH2 groups in melittin (the N-terminus and the Lys side chains indicated by arrows). The membrane activity of melittin is abolished after the modification, but can be recovered in a weakly acidic environment of endosome where it refolds into a homotetramer and disrupts the endosome membrane by pore formation. C) The chemical structure PF6 with a stearyl modified N-terminus and four trifluoromethyl quinoline moieties added onto the TP10 backbone. D) Photo-induced endosomal escape by modifying CPPs with photosensitizers. Light excitation of the sensitizer triggers the generation of singlet oxygen (1O2), which lyses the endosome membrane for siRNA escape.
Table 2.
Endosome disrupting peptides for siRNA delivery.
| Peptide | Mechanism of action | Sequence | Ref. |
|---|---|---|---|
| HA2 | Conformation changing | GLFGAIAGFIENGWEGMIDGWYG | [39, 86, 87] |
| INF7 | Conformation changing | GLFEAIEGFIENGWEGMIWDYG | [88] |
| E5 | Conformation changing | GLFEAIAEFIEGGWEGLIEG | [92] |
| E5WYG | Conformation changing | GLFEAIAEFIEGGWEGLIEGWYG | [92] |
| C6M1 | Conformation changing | Ac-RLWRLLWRLWRRLWRLLR-amide | [93] |
| GALA | Conformation changing | WEAALAEALAEALAEHLAEALAEALEALAA | [96-100] |
| EB1 | Conformation changing | LIRLWSHLIHIWFQNRRLKWKKK-amide | [39] |
| H5WYG | Conformation changing | GLFHAIAHFIHGGWHGLIHGWYG | [105, 106] |
| Endoporter | Conformation changing | LHKLLHHLLHHLHKLLHHLHHLLHKL | [40, 102, 107] |
| Melittin | Chemical structure changing | GIGAVLKVLTTGLPALISWIKRKRQQ-amide (CDM modified) | [109] |
| H3K8b | Proton buffering | K3(HHHHNHHHH(HHHKHHHKHHHKHHH)2)4 | [115, 116] |
| PepFect 6 | Proton buffering | Stearyl-AGYLLGK(K3QN4)INLKALAALAKKIL-amide | [118] |
| TatU1A-Alexa | Photo induced endosomolysis | RKKRRQRRR-U1A-Alexa Fluor 546 | [125] |
Inspired by the natural HA2 fusogenic peptide, Francis C. Szoka's group has designed an artificial peptide with a glutamic acid-alanine-leucine-alanine (EALA) repeat. In this repeated sequence, the amino acid E is pH-responsive whereas ALA provides hydrophobicity. This peptide was named “GALA”. In the endosome, the helical GALA folds into a transmembrane peptide pore comprising an array of approximately 10 GALA α-helical monomers [94]. As an anionic endosomolytic peptide, GALA is a popular choice to aid in endosomal escape of DNA/cationic polymer complexes (by simple mixing) [84, 95]. Covalent conjugation is another common strategy, although it is known that modification of the peptide at the terminus compromises its pH sensitivity and endosomolytic activity. Nevertheless, it has been shown in several independent reports that GALA modified liposomes are significantly more efficient than liposomes alone in siRNA in vitro delivery [96-100].
Similar to glutamic acid, histidine is another pH sensitive amino acid commonly used in destabilizing endosomes. The imidazole group of histidine has a pKa around 6.0, falling in the range of pH transition in the endocytic pathway (from neutral to pH 6.0–6.5 in early endosomes and pH 4.5–5.5 in late endosomes). Naturally, histidine is a common component in modulating pH-dependent functions of proteins. For example, the pH-dependent binding activity of human prolactin and its receptor is believed to be linked to the presence of several histidines within the binding interface [101]. Taking advantage of histidine's natural pH sensitivity, Lundberg et al. replaced the two amino acids in penetratin by histidine and created a new pH-sensitive fusogenic peptide, EB1 (LIRLWSHLIHIWFQNRRLKWKKK-amide). This peptide does not disrupt the cell membrane (because outside cells the pH is neutral), but it adopts a membranolytic α-helix upon protonation in the acidic endosome [39]. Similarly, histidine has also been utilized to control the α-helix adoption in response to a pH change in many other fusogenic peptides such as LAH4, H5WYG, and Endoporter [102-105]. These peptides are widely used to facilitate endosome escape in DNA and siRNA delivery [40, 105, 106]. For example, Endoporter appears to be a highly potent fusogenic peptide. Effective delivery of morpholino RNA (synthetic molecules with nucleic acid bases bound to morpholine rings instead of deoxyribose rings) into the cytoplasm has been demonstrated at a peptide concentration of 1 μM in the presence of 10% FBS [107]. Similarly, peptides, proteins, siRNAs, antisense oligos and other high-molecular-weight cargos have been delivered into cells using the Endoporter peptide via the endocytic pathway [107].
3. 2. Chemical structure-changing peptides
In addition to fusogenic peptides switched on and off by pH-triggered confirmation change, peptides have also been developed by transiently masking the key residues critical for endosome disruption. Upon cell entry, the acidic environment of endosomes helps remove the masking moieties through chemical reactions, activating the membrane destabilization function. Carboxydimethylmaleic (CDM) modified melittin is an outstanding representative of this strategy. CDM is a maleic anhydride that reacts with nucleophiles such as primary amines in peptides, and this modification can be removed in acidic environments in as soon as 5 minutes [108]. With CDM modification, a carboxylic acid group is added to melittin. The anionic charge inhibits the membrane disruption activity of melittin (Fig. 2B). Microscopy studies show that unmodified melittin at a concentration of 10 μg/mL can completely destroy cells in less than 10 minutes, whereas CDM-modified melittin has no cytotoxicity at concentration as high as 400 μg/mL. Co-incubation of DNA plasmid and CDM-melittin results in simultaneous accumulation of both agents in the endosome, followed by their escape in approximately 35 minutes, and consequently a 10-fold enhancement in gene expression[108]. For in vivo targeted uses, a hepatocyte-targeted N-acetylgalactosamine ligand has been linked to CDM-melittin through the free carboxylic acid group of CDM. A simple co-injection of this pH-activatable peptide (named NAG-MLP) and a liver-tropic cholesterol-conjugated siRNA (chol-siRNA) targeting coagulation factor VII (F7) resulted in an efficient F7 knockdown in mice and nonhuman primates without changes in clinical chemistry biomarkers or induction of cytokines [109].
ARC-520 is also an NAG-MLP based RNAi therapeutic in development for the treatment of chronic hepatitis B virus (HBV) infection. It consists of a cocktail of two cholesterol-conjugated siRNAs against the highly conserved sequences of HBV genomes, and NAG-MLP that promotes endosomal escape of the HBV chol-siRNAs. A phase I study consisting of 36 subjects showed that ARC-520 produced no detectable side effects, except urticarial rash developed in one subject at the highest dose. Comparable to placebo, ARC-520 appears to be safe and well-tolerated even at a dose as high as 2 mg/kg [110]. A phase 2a clinical trial revealed that ARC-520 can dramatically reduce s-antigen, e-antigen, and core-related antigen in humans after a single dose, strong evidence that ARC-520 can enable reconstitution of the immune system (a pharmacological outcome of HBV protein suppression by siRNA) [111]. A Phase 2b clinical study is ongoing to evaluate its therapeutic consistency.
3. 3. Proton buffering peptides
Accumulation of acid-absorbing peptides inside the endosome helps cargo molecules to escape the endosome through a process known as the proton sponge effect, which arises from a large number of weak bases with buffering capabilities at pH 5-6. Proton absorption in acidic organelles leads to osmotic pressure buildup across the organelle membrane and subsequent swelling and/or rupture of the acidic endosomes.
Histidine, with a pKa value of 6.0, has excellent proton absorbing capability. Histidine-rich polypeptides have been shown to enhance DNA transfection efficiency of liposomes by 100-folds more than using liposome alone [112]. Shorter linear peptides containing multiple histidines were also reported to increase DNA transfection efficiency [113], which can be further improved by using branched peptides where histidines are incorporated at higher density [114]. For siRNA delivery, an 8-arm peptide, H3K8b, was complexed with siRNA targeting β-galactosidase and applied to mouse endothelial cells that were stably expressing β-galactosidase. The branched peptide was capable of reducing β-galactosidase expression by 80% at a siRNA concentration of 300 nM. Combination of H3K8b with an integrin-targeting ligand (RGD peptide) further enhanced siRNA delivery efficiency [115]. Systemic delivery of anti-raf1 siRNA to tumor sites by the branched peptide has been tested in mice bearing MDA-MB-435 tumor cells. Despite significant accumulation in the kidney, siRNA tumor uptake was evident, and significant tumor shrinkage was observed after multiple tail-vain injections [116].
In addition to natural amino acid histidine, a proton-buffering agent, chloroquine, can be covalently conjugated to peptides for promoting cargo endosomal escape as well. PepFect6 (PF6) is designed this way (Fig. 2C), by modifying a CPP (stearyl-TP10) with four trifluoromethyl quinolines (a chloroquine analog) [117]. Unlike the parental TP-10, PF6 does not generate pores in the cell membrane. Instead, it enters cells via the endocytotic pathway. Trifluoromethylquinoline molecules neutralize excess protons and eventually break the endosomal compartment by osmotic swelling. Due to this endosome disruption capability, robust RNAi in various cell types, including primary cells, has been achieved. The stable PF6/siRNA nanoparticles are also suitable for systemic siRNA delivery. RNAi in mice following systemic injections was achieved without detectable toxicities [118].
3. 4. Photo-induced endosomolysis
Besides chemical-triggered endosomolysis, endosome escape also can be achieved using external stimuli such as lights. When photosensitizers are included in the delivery systems, they can produce reactive oxygen species (ROS) upon light excitation. Common photosensitizers, such as sulfonated tetraphenyl porphyrins (TPPS2a or TPPS4a), sulfonated aluminum phthalocyanines (AlPcS), and 5-(4-carboxyphenyl)-10,15,20-triphenyl-2,3-dihydroxychlorin (TPC) generate highly reactive singlet oxygen (1O2), which damages endosomal membranes by reacting with membrane lipids, resulting in release of siRNA into the cytosol (Fig. 2D) [119]. For example, cationic CPPs Tat and hepta-arginine (R7) have been conjugated with photosensitizer AlPcS and TPC to promote endosome escape [120, 121]. Although the mechanism remains unclear, it has been speculated that the cationic CPPs play an important role in this process by bringing the conjugated photosensitizers close to the endosome membrane through electrostatic interactions [122].
Some conventional fluorescent dyes have also been explored following a similar mechanism. For example, fluorescein-labeled arginine-rich CPPs can be released from endosomes upon irradiation with a laser at 488 nm [123]. The similar endosomal release was also observed using another organic fluorophore, Alexa Fluor 633 [124]. Ohtsuki and co-workers explored siRNA delivery using Alexa Fluor 546-labeled TatU1A peptide (TatU1A-Alexa) [125]. The endosomal release of TatU1A-Alexa and its cargo U1A-siRNA was observed 10 seconds post photo-irradiation. Once in the cytoplasm, U1A-siRNA induced gene silencing of eGFP in CHO cells and the endogenous epidermal growth factor receptor (EGFR) gene in A431 cells. In contrast, no silencing effect was observed in areas where cells were not irradiated by laser.
Despite the fast response time of photochemical-induced endosomolysis, the experimental conditions must be precisely controlled because photostimulation of photosensitizers and fluorophores can cause cell death for light overdose [126]. Apoptosis and necrotic cell damage have been observed after laser excitation of R7-TPC and Tat-TMR [120, 127]. Cytoplasm membrane permeabilization and blebbing, and cell shrinkage were seen shortly after photo-illumination of TMR-TAT at 560 nm [127]. Another potential limitation of photochemically induced endosomolysis is the difficulty of in vivo light delivery. Similar to optical-based imaging technologies, light penetration depth is limited to millimeters, including near-infrared (NIR) light [128].
4. Targeting peptide-siRNA bioconjugates
A large number of peptides with specific affinity to disease-associated biomolecules have been identified through biopanning, site-specific mutagenesis of natural peptide sequences, and computer-aided design. Some peptides, such as BBN [7-14], exhibit affinity and specificity to antigens comparable to therapeutic antibodies [129], while being much more compact in size and cheaper to make. Because extensive reviews exist in the literature, here, we will only highlight a few that have been directly conjugated to siRNAs.
An IGF1-targeting cyclic peptide D-(Cys-Ser-Lys-Cys) has been conjugated to the sense strand of a siRNA at its 5′ end by an ester bond. Without the use of cationic lipids or electroporation, the peptide-siRNA conjugate was shown to knock down the target gene by 35-55% in MCF-7 cells that overexpress IGF1 receptor at a siRNA concentration of 100 nM [130]. Similarly, an integrin-binding peptide cRGD was conjugated to anti-VEGFR2 siRNA and applied to integrin-positive Human Umbilical Vein Endothelial Cells (HUVEC). The cRGD-siRNA conjugate can enter HUVEC cells selectively and silence targeted genes both in vitro and in vivo. Systemic delivery of cRGD-siRNA through multiple administrations resulted in down-regulation of the corresponding mRNA by 45 to 55% and of corresponding protein by 45 to 65% in tumor tissues. Impressively, the overall tumor volume was also reduced by 70-90% [131]. The integrin targeting efficiency of RGD peptide can be further enhanced by engineering a multivalent version. Alam et al. constructed a multivalent cRGD (mcRGD) with higher affinity and specificity than the monomeric peptide, and conjugated it to an anti-luciferase siRNA. Integrin-positive human melanoma cells M21+ showed strong receptor-mediated uptake of the mcRGD-siRNA conjugate, but the cargo siRNA was primarily accumulated in the endosome. Despite the endosome trapping problem, the mcRGD-siRNA conjugate produced dose-dependent luciferase reduction markedly, whereas the monovalent version had little effect [132]. A similar endosome trapping problem has also been observed by Park's group when they tested a luteinizing hormone-releasing hormone (LHRH) peptide-siRNA conjugate (siRNA-PEG-LHRH) on an ovarian cancer cell line, A2780. Despite strong cellular uptake, siRNA-PEG-LHRH exhibited limited gene silencing compared to siRNA-PEG-LHRH delivered by polyethyleneimine (PEI) [133]. All of these studies point to the significance of combining targeting ligands with peptides that can get siRNA out of intracellular compartments.
Detzer et al. covalently attached a localization signal peptide to facilitate intracellular delivery [134]. Although phosphorothioate (PS) modified siRNA can stimulate cellular uptake via the caveosomal uptake pathway [135], most of the siRNA is trapped in the ER-specific perinuclear sites. A localization signal peptide TQIENLKEKG has been shown to promote siRNA release because it is recognized by the trans-membrane transporter and transferred into the cytoplasm from the perinuclear sites [134].
5. Multifunctional peptides
Besides the two strategies discussed above where siRNAs are either covalently linked to peptides or complexed with cationic peptides through electrostatic interactions, another emerging approach is to utilize double-strand RNA (dsRNA) binding peptides. Many dsRNA binding peptides exist in nature. For example, the first 172 amino acids in human protein kinase R (PKR) is a double-stranded RNA binding domain (dsRBD). It is composed of a tandem repeat of double-stranded RNA binding motif 1 and 2 (dsRBM1 and dsRBM2) linked by a flexible unstructured peptide linker (33 amino acids). Once mixed with siRNA, dsRBM1 and dsRBM2 wrap around the siRNA backbone with an affinity as low as 50 nM (Kd) [136]. Therefore, it can be used as an adaptor to link siRNA molecules with other functional peptides. Compared with cationic peptides and polymers, dsRBD does not introduce excessive positive charges, avoiding non-specific binding and aggregation. Compared with covalent linkage, the plug-and-play preparation is much simpler than running chemical reactions.
5. 1. DNA binding peptides for gene delivery
The strategy was first demonstrated for gene delivery by using a DNA-binding peptide. Histone H1 is a natural protein that can bind to DNA and fold linear DNA into a higher-order structure. By using a histone H1-based recombinant fusion peptide, Hatefi's group constructed a multi-domain peptide for targeted delivery of genes to ZR-75-1 breast cancer cells (Fig. 3A) [137]. The multifunctional peptide was made with two tandem repeat units of truncated histone H1 for DNA condensation, a short peptide KALA for promotion of endosome escape, a cyclic peptide TP1 for ZR-75-1 cell targeting, and an NLS peptide (from human immunodeficiency virus) for enhancement of DNA translocation toward the cell nucleus. A gel shift assay proved that the multifunctional peptide efficiently binds to DNA and protects it from nuclease degradation, and a cell transfection study indicated that both the KALA and NLS motifs maintain their functions after being chained together with the other peptide domains, as evidenced by efficient luciferase transfection. His lab further developed the gene delivery system by inserting a cathepsin D substrate linker between the targeting motif and histone H1 peptide [138]. The protease substrate facilitates the dissociation of the targeting motif in endosome where cathepsin D is abundant.
Fig. 3.
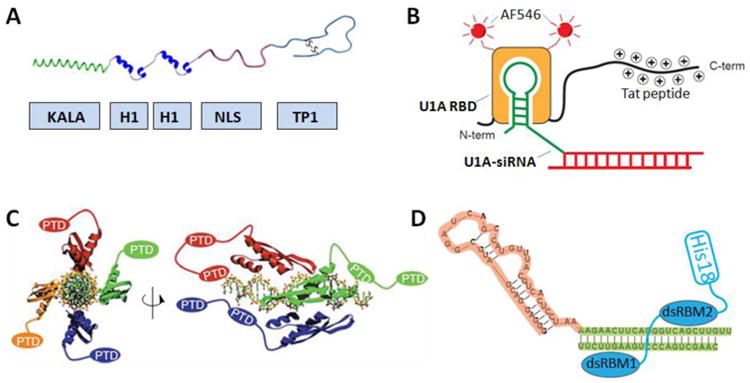
Gene and siRNA delivery by multifunctional peptides. A) Schematic of the trifunctional peptide for targeted gene delivery. The KALA domain promotes endosome escape; the tandem H1 repeats enable DNA condensation; the NLS sequence enhances nucleus translocation; and the TP1 block serves as a ligand for cell-specific homing. B) Sequence-specific RNA binding peptide U1A-RBD-Tat forms a complex with U1A modified siRNA, which can escape endosome aided by photo-induced endosomolysis. AF546: Alexa Fluor 546. C) Schematic drawing of a siRNA associated with PTD modified dsRBD. Adapted with permission from ref. [139]. Copyright 2009 Nature Publishing Group. D) dsRBD-His18 chimeric peptide as a general platform for targeted delivery of aptamer-siRNA chimeras. Orange: PSAM targeting aptamer; green: siRNA.
5. 2. dsRNA binding peptides for siRNA delivery
Endoh and coworkers constructed the first sequence-specific siRNA binding system using a U1A RNA-binding peptide. siRNAs containing a short 5′-extension of the U1A (U1A-siRNA) can be specifically recognized by the U1A RNA-binding peptide (U1A RBD) and form a biomolecular complex (Fig. 3B). In their experiments, the U1A peptide was labeled with a Tat peptide and a fluorophore [125]. When the peptide-siRNA complex enters cells through endocytosis, the fluorophore was excited with UV light for photo-induced endosome destabilization. Similarly, Eguchi et al. reported a siRNA delivery approach that uses dsRBD, which binds with siRNA in a sequence-independent fashion (Fig. 3C). The recombinant dsRBD was fused with several cationic peptides (Tat peptide) to mask the negative charges of siRNA and promote cell uptake. Rapid gene knockout was observed without significant toxicity, off-target effect, or immune response [139]. This approach, although effective in the cultured cells, is likely to have problems under in vivo conditions, because the cationic peptides could induce interactions with serum proteins and cells non-specifically. Gopal group has extended this approach by fusing the dsRNA binding peptide with target homing peptides such as BTP (binding to GM1 and GT1b) and Her2 affibody (binding to Her2 receptor). By replacing the cationic peptides with homing ligands, the new constructs have a better chance to reach the target cells [140, 141]. Similarly, Geoghegan et al. constructed a tandem repeat of dsRBD for higher binding affinity to siRNA. However, without an endosome destabilization mechanism, the majority of the delivery siRNA was trapped without eliciting the desired RNAi effect [142]. Most recently, our group designed a dsRBD-polyhistidine recombinant protein. This simple dual-block protein complements the siRNA-aptamer chimera structures reported by Dassie et al. [143], because the complexes exhibit cell targeting, endosome escape, and therapeutic functionalities simultaneously, without introducing excessive positive charges (Fig. 3D) [144].
6. Conclusions
siRNA has gained considerable interest in biology and medicine because it can elicit potent, target-specific knockdown of virtually any mRNA, creating a useful tool in basic sciences to study gene functions as well as a therapeutic option in pharmaceutics. Unfortunately, as with other antisense approaches, clinical translation of the powerful siRNA technology has proven challenging. Problems include enhancing siRNA stability, minimizing off-target effects, identifying sensitive sites in the target RNAs, and particularly ensuring efficient delivery. For RNA molecule stability, they are inherently less stable. Chemical modifications have been extensively explored to improve their serum stability and in vivo pharmacokinetics. The chemical modification of siRNA is often performed at the phosphodiester backbone and ribose 2′-OH group [145]. For example, the ribose 2′-OH ground can be substituted by 2′-OMe or 2′-F group, which can help extend the half-life of siRNA in serum beyond 48 hour [146, 147]. For in vivo therapeutics uses, despite limited success in some niche applications (e.g., deliver to the liver), a general platform for in vivo cell type-specific delivery does not exist. To address the delivery problem, recent advances in bioengineering and nanotechnology have produced a number of non-viral siRNA carriers including liposomes, inorganic nanoparticles, cationic polymers, and the focus of this review, peptides. However, the in vivo delivery problem remains because these current delivery technologies all suffer from one or another shortcoming, preventing efficient, specific delivery of siRNA, and consequently calling for truly innovative solutions to this difficult problem. A ‘perfect’ delivery platform should simultaneously achieve a compact size, low immunogenicity, low cytotoxicity, selective targeting (free of excessive positive charges), in vivo stability, high payload, efficient endosomolysis, ease of production, flexibility in siRNA and target ligand selection, and affordability. In this regard, peptides are of great promise to achieve this goal because of their diverse functionalities, biocompatibility, and the recent development and maturation of the recombinant protein technique that allows various peptides and proteins to be assembled into precisely controlled nanostructures. Thus, multifunctional peptides are of great excitement and promise to fill in the long-standing gap of RNAi clinical translation.
Acknowledgments
This work was supported in part by NIH (R01CA140295), and the Department of Bioengineering at the University of Washington. We also thank Drew Matsuura for manuscript proof reading.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 2.Obbard DJ, Gordon KHJ, Buck AH, Jiggins FM. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc Lond, B, Biol Sci. 2009;364:99–115. doi: 10.1098/rstb.2008.0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Fougerolles A, Vornlocher HP, Maraganore J, Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov. 2007;6:443–453. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ozcan G, Ozpolat B, Coleman RL, Sood AK, Lopez-Berestein G. Preclinical and clinical development of siRNA-based therapeutics. Adv Drug Deliv Rev. 2015;87:108–119. doi: 10.1016/j.addr.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Lu Z, Wientjes MG, Au JL. Delivery of siRNA therapeutics: barriers and carriers. AAPS J. 2010;12:492–503. doi: 10.1208/s12248-010-9210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. J Cell Sci. 2010;123:1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 8.Endoh T, Ohtsuki T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv Drug Delivery Rev. 2009;61:704–709. doi: 10.1016/j.addr.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 10.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 11.Tunnemann G, Ter-Avetisyan G, Martin RM, Stockl M, Herrmann A, Cardoso MC. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J Pept Sci. 2008;14:469–476. doi: 10.1002/psc.968. [DOI] [PubMed] [Google Scholar]
- 12.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 13.Herce HD, Garcia AE, Litt J, Kane RS, Martin P, Enrique N, Rebolledo A, Milesi V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys J. 2009;97:1917–1925. doi: 10.1016/j.bpj.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green M, Ishino M, Loewenstein PM. Mutational analysis of HIV-1 Tat minimal domain peptides: identification of trans-dominant mutants that suppress HIV-LTR-driven gene expression. Cell. 1989;58:215–223. doi: 10.1016/0092-8674(89)90417-0. [DOI] [PubMed] [Google Scholar]
- 15.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 16.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 17.Chiu YL, Ali A, Chu CY, Cao H, Rana TM. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem Biol. 2004;11:1165–1175. doi: 10.1016/j.chembiol.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Moschos SA, Jones SW, Perry MM, Williams AE, Erjefalt JS, Turner JJ, Barnes PJ, Sproat BS, Gait MJ, Lindsay MA. Lung Delivery Studies Using siRNA Conjugated to TAT(48-60) and Penetratin Reveal Peptide Induced Reduction in Gene Expression and Induction of Innate Immunity. Bioconjug Chem. 2007;18:1450–1459. doi: 10.1021/bc070077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh N, Agrawal A, Leung AK, Sharp PA, Bhatia SN. Effect of nanoparticle conjugation on gene silencing by RNA interference. J Am Chem Soc. 132:8241–8243. doi: 10.1021/ja102132e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain PK, Shah S, Friedman SH. Patterning of Gene Expression Using New Photolabile Groups Applied to Light Activated RNAi. J Am Chem Soc. 133:440–446. doi: 10.1021/ja107226e. [DOI] [PubMed] [Google Scholar]
- 21.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang YH, Hou YW, Lee HJ. An intracellular delivery method for siRNA by an arginine-rich peptide. J Biochem Biophys Methods. 2007;70:579–586. doi: 10.1016/j.jbbm.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 23.Kim SW, Kim NY, Choi YB, Park SH, Yang JM, Shin S. RNA interference in vitro and in vivo using an arginine peptide/siRNA complex system. J Control Release. 2010;143:335–343. doi: 10.1016/j.jconrel.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 24.Kanazawa T, Morisaki K, Suzuki S, Takashima Y. Prolongation of life in rats with malignant glioma by intranasal siRNA/drug codelivery to the brain with cell-penetrating peptide-modified micelles. Mol Pharm. 2014;11:1471–1478. doi: 10.1021/mp400644e. [DOI] [PubMed] [Google Scholar]
- 25.Malhotra M, Tomaro-Duchesneau C, Prakash S. Synthesis of TAT peptide-tagged PEGylated chitosan nanoparticles for siRNA delivery targeting neurodegenerative diseases. Biomaterials. 2013;34:1270–1280. doi: 10.1016/j.biomaterials.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 26.Tonges L, Lingor P, Egle R, Dietz GP, Fahr A, Bahr M. Stearylated octaarginine and artificial virus-like particles for transfection of siRNA into primary rat neurons. RNA. 2006;12:1431–1438. doi: 10.1261/rna.2252206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Li Y, Wang X, Lee RJ, Teng L. Fatty acid modified octa-arginine for delivery of siRNA. Int J Pharm. 2015;495:527–535. doi: 10.1016/j.ijpharm.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 28.Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today. 2012;17:850–860. doi: 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Morris MC, Vidal P, Chaloin L, Heitz F, Divita G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997;25:2730–2736. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simeoni F, Morris MC, Heitz F, Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31:2717–2724. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crombez L, Charnet A, Morris MC, Aldrian-Herrada G, Heitz F, Divita G. A non-covalent peptide-based strategy for siRNA delivery. Biochem Soc Trans. 2007;35:44–46. doi: 10.1042/BST0350044. [DOI] [PubMed] [Google Scholar]
- 32.Deshayes S, Gerbal-Chaloin S, Morris MC, Aldrian-Herrada G, Charnet P, Divita G, Heitz F. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochimica et Biophysica Acta. 2004;1667:141–147. doi: 10.1016/j.bbamem.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 33.Deshayes S, Morris MC, Divita G, Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cellular and Molecular Life Sciences. 2005;62:1839–1849. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simeoni F, Morris MC, Heitz F, Divita G. Peptide-based strategy for siRNA delivery into mammalian cells. Methods Mol Biol. 2005;309:251–260. doi: 10.1385/1-59259-935-4:251. [DOI] [PubMed] [Google Scholar]
- 35.Zeineddine D, Papadimou E, Chebli K, Gineste M, Liu J, Grey C, Thurig S, Behfar A, Wallace VA, Skerjanc IS, Puceat M. Oct-3/4 dose dependently regulates specification of embryonic stem cells toward a cardiac lineage and early heart development. Developmental Cell. 2006;11:535–546. doi: 10.1016/j.devcel.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 36.Crombez L, Morris MC, Dufort S, Aldrian-Herrada G, Nguyen Q, Mc Master G, Coll JL, Heitz F, Divita G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009;37:4559–4569. doi: 10.1093/nar/gkp451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deshayes S, Plenat T, Aldrian-Herrada G, Divita G, Le Grimellec C, Heitz F. Primary amphipathic cell-penetrating peptides: structural requirements and interactions with model membranes. Biochemistry (Mosc) 2004;43:7698–7706. doi: 10.1021/bi049298m. [DOI] [PubMed] [Google Scholar]
- 38.Veldhoen S, Laufer SD, Trampe A, Restle T. Cellular delivery of small interfering RNA by a non-covalently attached cell-penetrating peptide: quantitative analysis of uptake and biological effect. Nucleic Acids Res. 2006;34:6561–6573. doi: 10.1093/nar/gkl941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lundberg P, El-Andaloussi S, Sütlü T, Johansson H, Langel Ü. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB Journal. 2007;21:2664–2671. doi: 10.1096/fj.06-6502com. [DOI] [PubMed] [Google Scholar]
- 40.Bartz R, Fan H, Zhang J, Innocent N, Cherrin C, Beck Stephen C, Pei Y, Momose A, Jadhav V, Tellers David M, Meng F, Crocker Louis S, Sepp-Lorenzino L, Barnett Stanley F. Effective siRNA delivery and target mRNA degradation using an amphipathic peptide to facilitate pH-dependent endosomal escape. Biochemical Journal. 2011;435:475–487. doi: 10.1042/BJ20101021. [DOI] [PubMed] [Google Scholar]
- 41.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 42.Magzoub M, Eriksson LEG, Gräslund A. Conformational states of the cell-penetrating peptide penetratin when interacting with phospholipid vesicles: effects of surface charge and peptide concentration. Biochim Biophys Acta, Biomembr. 2002;1563:53–63. doi: 10.1016/s0005-2736(02)00373-5. [DOI] [PubMed] [Google Scholar]
- 43.Ye J, Fox SA, Cudic M, Rezler EM, Lauer JL, Fields GB, Terentis AC. Determination of penetratin secondary structure in live cells with Raman microscopy. J Am Chem Soc. 2010;132:980–988. doi: 10.1021/ja9043196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muratovska A, Eccles MR. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004;558:63–68. doi: 10.1016/S0014-5793(03)01505-9. [DOI] [PubMed] [Google Scholar]
- 45.Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, Troy CM. Highly efficient small interfering RNA delivery to primary mammalian neurons induces MicroRNA-like effects before mRNA degradation. J Neurosci. 2004;24:10040–10046. doi: 10.1523/JNEUROSCI.3643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turner JJ, Jones S, Fabani MM, Ivanova G, Arzumanov AA, Gait MJ. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Mol Dis. 2007;38:1–7. doi: 10.1016/j.bcmd.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 47.Gottschalk S, Sparrow JT, Hauer J, Mims MP, Leland FE, Woo SL, Smith LC. A novel DNA-peptide complex for efficient gene transfer and expression in mammalian cells. Gene Ther. 1996;3:448–457. [PubMed] [Google Scholar]
- 48.Crombez L, Aldrian-Herrada G, Konate K, Nguyen QN, McMaster GK, Brasseur R, Heitz F, Divita G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol Ther. 2009;17:95–103. doi: 10.1038/mt.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konate K, Rydstrom A, Divita G, Deshayes S. Everything you always wanted to know about CADY-mediated siRNA delivery* (* but afraid to ask) Current Pharmaceutical Design. 2013;19:2869–2877. doi: 10.2174/1381612811319160004. [DOI] [PubMed] [Google Scholar]
- 50.Rydström A, Deshayes S, Konate K, Crombez L, Padari K, Boukhaddaoui H, Aldrian G, Pooga M, Divita G. Direct Translocation as Major Cellular Uptake for CADY Self-Assembling Peptide-Based Nanoparticles. PLoS One. 2011;6:e25924. doi: 10.1371/journal.pone.0025924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wyman TB, Nicol F, Zelphati O, Scaria PV, Plank C, Szoka FC. Design, Synthesis, and Characterization of a Cationic Peptide That Binds to Nucleic Acids and Permeabilizes Bilayers. Biochemistry (Mosc) 1997;36:3008–3017. doi: 10.1021/bi9618474. [DOI] [PubMed] [Google Scholar]
- 52.Mok H, Park TG. Self-crosslinked and reducible fusogenic peptides for intracellular delivery of siRNA. Biopolymers. 2008;89:881–888. doi: 10.1002/bip.21032. [DOI] [PubMed] [Google Scholar]
- 53.Lee SH, Kim SH, Park TG. Intracellular siRNA delivery system using polyelectrolyte complex micelles prepared from VEGF siRNA-PEG conjugate and cationic fusogenic peptide. Biochemical and Biophysical Research Communications. 2007;357:511–516. doi: 10.1016/j.bbrc.2007.03.185. [DOI] [PubMed] [Google Scholar]
- 54.Lee H, Jeong JH, Park TG. A new gene delivery formulation of polyethylenimine/DNA complexes coated with PEG conjugated fusogenic peptide. J Control Release. 2001;76:183–192. doi: 10.1016/s0168-3659(01)00426-6. [DOI] [PubMed] [Google Scholar]
- 55.Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, Marasco WA, Lieberman J. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 56.Yao YD, Sun TM, Huang SY, Dou S, Lin L, Chen JN, Ruan JB, Mao CQ, Yu FY, Zeng MS, Zang JY, Liu Q, Su FX, Zhang P, Lieberman J, Wang J, Song E. Targeted delivery of PLK1-siRNA by ScFv suppresses Her2+ breast cancer growth and metastasis. Sci Transl Med. 2012;4:130ra148. doi: 10.1126/scitranslmed.3003601. [DOI] [PubMed] [Google Scholar]
- 57.Kumar P, Ban HS, Kim SS, Wu H, Pearson T, Greiner DL, Laouar A, Yao J, Haridas V, Habiro K, Yang YG, Jeong JH, Lee KY, Kim YH, Kim SW, Peipp M, Fey GH, Manjunath N, Shultz LD, Lee SK, Shankar P. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell. 2008;134:577–586. doi: 10.1016/j.cell.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma Y, Kowolik CM, Swiderski PM, Kortylewski M, Yu H, Horne DA, Jove R, Caballero OL, Simpson AJ, Lee FT, Pillay V, Scott AM. Humanized Lewis-Y specific antibody based delivery of STAT3 siRNA. ACS Chem Biol. 2011;6:962–970. doi: 10.1021/cb200176v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schneider B, Grote M, John M, Haas A, Bramlage B, Ickenstein LM, Jahn-Hofmann K, Bauss F, Cheng W, Croasdale R, Daub K, Dill S, Hoffmann E, Lau W, Burtscher H, Ludtke JL, Metz S, Mundigl O, Neal ZC, Scheuer W, Stracke J, Herweijer H, Brinkmann U. Targeted siRNA Delivery and mRNA Knockdown Mediated by Bispecific Digoxigenin-binding Antibodies. Mol Ther Nucleic Acids. 2012;1:e46. doi: 10.1038/mtna.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cuellar TL, Barnes D, Nelson C, Tanguay J, Yu SF, Wen X, Scales SJ, Gesch J, Davis D, van Brabant Smith A, Leake D, Vandlen R, Siebel CW. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 2015;43:1189–1203. doi: 10.1093/nar/gku1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Molek P, Strukelj B, Bratkovic T. Peptide Phage Display as a Tool for Drug Discovery: Targeting Membrane Receptors. Molecules. 2011;16:857. doi: 10.3390/molecules16010857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qin B, Tai W, Shukla R, Cheng K. Identification of a LNCaP-Specific Binding Peptide Using Phage Display. Pharm Res. 2011;28:2422–2434. doi: 10.1007/s11095-011-0469-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Z, Jin W, Liu H, Zhao Z, Cheng K. Discovery of Peptide Ligands for Hepatic Stellate Cells Using Phage Display. Mol Pharm. 2015;12:2180–2188. doi: 10.1021/acs.molpharmaceut.5b00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Myrberg H, Zhang L, Mae M, Langel U. Design of a tumor-homing cell-penetrating peptide. Bioconjug Chem. 2008;19:70–75. doi: 10.1021/bc0701139. [DOI] [PubMed] [Google Scholar]
- 65.Mäe M, Myrberg H, El-Andaloussi S, Langel Ü. Design of a Tumor Homing Cell-Penetrating Peptide for Drug Delivery. Int J Peptide Res Therapeut. 2009;15:11–15. [Google Scholar]
- 66.Snyder EL, Saenz CC, Denicourt C, Meade BR, Cui XS, Kaplan IM, Dowdy SF. Enhanced targeting and killing of tumor cells expressing the CXC chemokine receptor 4 by transducible anticancer peptides. Cancer Res. 2005;65:10646–10650. doi: 10.1158/0008-5472.CAN-05-0118. [DOI] [PubMed] [Google Scholar]
- 67.Mäe M, Rautsi O, Enbäck J, Hällbrink M, Aizman K, Lindgren M, Laakkonen P, Langel Ü. Tumour Targeting with Rationally Modified Cell-Penetrating Peptides. Int J Peptide Res Therapeut. 2012;18:361–371. [Google Scholar]
- 68.Fang B, Jiang L, Zhang M, Ren FZ. A novel cell-penetrating peptide TAT-A1 delivers siRNA into tumor cells selectively. Biochimie. 2013;95:251–257. doi: 10.1016/j.biochi.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 69.Kim SS, Ye C, Kumar P, Chiu I, Subramanya S, Wu H, Shankar P, Manjunath N. Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Molecular Therapy. 2010;18:993–1001. doi: 10.1038/mt.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ye C, Choi JG, Abraham S, Wu H, Diaz D, Terreros D, Shankar P, Manjunath N. Human macrophage and dendritic cell-specific silencing of high-mobility group protein B1 ameliorates sepsis in a humanized mouse model. Proc Natl Acad Sci U S A. 2012;109:21052–21057. doi: 10.1073/pnas.1216195109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 72.Ye C, Abraham S, Wu H, Shankar P, Manjunath N. Silencing early viral replication in macrophages and dendritic cells effectively suppresses flavivirus encephalitis. PLoS One. 2011;6:e17889. doi: 10.1371/journal.pone.0017889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zeller S, Choi CS, Uchil PD, Ban HS, Siefert A, Fahmy TM, Mothes W, Lee SK, Kumar P. Attachment of cell-binding ligands to arginine-rich cell-penetrating peptides enables cytosolic translocation of complexed siRNA. Chem Biol. 2015;22:50–62. doi: 10.1016/j.chembiol.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Olson ES, Aguilera TA, Jiang T, Ellies LG, Nguyen QT, Wong EH, Gross LA, Tsien RY. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr Biol. 2009;1:382–393. doi: 10.1039/b904890a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shi NQ, Gao W, Xiang B, Qi XR. Enhancing cellular uptake of activable cell-penetrating peptide-doxorubicin conjugate by enzymatic cleavage. Int J Nanomedicine. 2012;7:1613–1621. doi: 10.2147/IJN.S30104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martín I, Teixidó M, Giralt E. Building Cell Selectivity into CPP-Mediated Strategies. Pharmaceuticals. 2010;3:1456. doi: 10.3390/ph3051456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101:17867–17872. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H, He J, Yi H, Xiang G, Chen K, Fu B, Yang Y, Chen G. siRNA suppression of hTERT using activatable cell-penetrating peptides in hepatoma cells. Bioscience Reports. 2015;35 doi: 10.1042/BSR20140145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bode SA, Hansen MB, Oerlemans RAJF, van Hest JCM, Löwik DWPM. Enzyme-Activatable Cell-Penetrating Peptides through a Minimal Side Chain Modification. Bioconjug Chem. 2015;26:850–856. doi: 10.1021/acs.bioconjchem.5b00066. [DOI] [PubMed] [Google Scholar]
- 80.Lee SH, Moroz E, Castagner B, Leroux JC. Activatable cell penetrating peptide-peptide nucleic acid conjugate via reduction of azobenzene PEG chains. J Am Chem Soc. 2014;136:12868–12871. doi: 10.1021/ja507547w. [DOI] [PubMed] [Google Scholar]
- 81.Schwarze SR, Dowdy SF. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci. 2000;21:45–48. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]
- 82.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 83.Van Rossenberg SM, Sliedregt-Bol KM, Meeuwenoord NJ, Van Berkel TJ, Van Boom JH, Van Der Marel GA, Biessen EA. Targeted lysosome disruptive elements for improvement of parenchymal liver cell-specific gene delivery. Journal of Biological Chemistry. 2002;277:45803–45810. doi: 10.1074/jbc.M203510200. [DOI] [PubMed] [Google Scholar]
- 84.Plank C, Oberhauser B, Mechtler K, Koch C, Wagner E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J Biol Chem. 1994;269:12918–12924. [PubMed] [Google Scholar]
- 85.Lüneberg J, Martin I, Nüßler F, Ruysschaert JM, Herrmann A. Structure and Topology of the Influenza Virus Fusion Peptide in Lipid Bilayers. J Biol Chem. 1995;270:27606–27614. doi: 10.1074/jbc.270.46.27606. [DOI] [PubMed] [Google Scholar]
- 86.Cantini L, Attaway CC, Butler B, Andino LM, Sokolosky ML, Jakymiw A. Fusogenic-oligoarginine peptide-mediated delivery of siRNAs targeting the CIP2A oncogene into oral cancer cells. PLoS One. 2013;8:e73348. doi: 10.1371/journal.pone.0073348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McNaughton BR, Cronican JJ, Thompson DB, Liu DR. Mammalian cell penetration, siRNA transfection, and DNA transfection by supercharged proteins. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6111–6116. doi: 10.1073/pnas.0807883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mykhaylyk O, Sanchez-Antequera Y, Vlaskou D, Cerda M, Bokharaei M, Hammerschmid E, Anton M, Plank C. Magnetic Nanoparticle and Magnetic Field Assisted siRNA Delivery In Vitro. In: Sioud M, editor. RNA Interference. Springer; New York: 2015. pp. 53–106. [DOI] [PubMed] [Google Scholar]
- 89.Klink DT, Chao S, Glick MC, Scanlin TF. Nuclear Translocation of Lactosylated Poly-L-lysine/cDNA Complex in Cystic Fibrosis Airway Epithelial Cells. Molecular Therapy. 2001;3:831–841. doi: 10.1006/mthe.2001.0332. [DOI] [PubMed] [Google Scholar]
- 90.Midoux P, Mendes C, Legrand A, Raimond J, Mayer R, Monsigny M, Roche AC. Specific gene transfer mediated by lactosylated poly-L-lysine into hepatoma cells. Nucleic Acids Research. 1993;21:871–878. doi: 10.1093/nar/21.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Neuner-Jehle M, Berghe LVD, Bonnel S, Uteza Y, Benmeziane F, Rouillot JS, Marchant D, Kobetz A, Dufier JL, Menasche M, Abitbol M. Ocular Cell Transfection with the Human Basic Fibroblast Growth Factor Gene Delays Photoreceptor Cell Degeneration in RCS Rats. Human Gene Therapy. 2000;11:1875–1890. doi: 10.1089/10430340050129495. [DOI] [PubMed] [Google Scholar]
- 92.Freulon I, Roche AC, Monsigny M, Mayer R. Delivery of oligonucleotides into mammalian cells by anionic peptides: comparison between monomeric and dimeric peptides. Biochemical Journal. 2001;354:671–679. doi: 10.1042/0264-6021:3540671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu W, Jafari M, Yuan F, Pan R, Chen B, Ding Y, Sheinin T, Chu D, Lu S, Yuan Y, Chen P. In vitro and in vivo therapeutic siRNA delivery induced by a tryptophan-rich endosomolytic peptide. Journal of Materials Chemistry B. 2014;2:6010–6019. doi: 10.1039/c4tb00629a. [DOI] [PubMed] [Google Scholar]
- 94.Li W, Nicol F, Szoka FC., Jr GALA: a designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv Drug Deliv Rev. 2004;56:967–985. doi: 10.1016/j.addr.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 95.Simoes S, Slepushkin V, Gaspar R, de Lima MC, Duzgunes N. Gene delivery by negatively charged ternary complexes of DNA, cationic liposomes and transferrin or fusigenic peptides. Gene Ther. 1998;5:955–964. doi: 10.1038/sj.gt.3300674. [DOI] [PubMed] [Google Scholar]
- 96.Kusumoto K, Akita H, Santiwarangkool S, Harashima H. Advantages of ethanol dilution method for preparing GALA-modified liposomal siRNA carriers on the in vivo gene knockdown efficiency in pulmonary endothelium. Int J Pharm. 2014;473:144–147. doi: 10.1016/j.ijpharm.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 97.Sakurai Y, Hatakeyama H, Akita H, Oishi M, Nagasaki Y, Futaki S, Harashima H. Efficient Short Interference RNA Delivery to Tumor Cells Using a Combination of Octaarginine, GALA and Tumor-Specific, Cleavable Polyethylene Glycol System. Biol Pharm Bull. 2009;32:928–932. doi: 10.1248/bpb.32.928. [DOI] [PubMed] [Google Scholar]
- 98.Kusumoto K, Akita H, Ishitsuka T, Matsumoto Y, Nomoto T, Furukawa R, El-Sayed A, Hatakeyama H, Kajimoto K, Yamada Y, Kataoka K, Harashima H. Lipid envelope-type nanoparticle incorporating a multifunctional peptide for systemic siRNA delivery to the pulmonary endothelium. ACS Nano. 2013;7:7534–7541. doi: 10.1021/nn401317t. [DOI] [PubMed] [Google Scholar]
- 99.Toriyabe N, Hayashi Y, Harashima H. The transfection activity of R8-modified nanoparticles and siRNA condensation using pH sensitive stearylated-octahistidine. Biomaterials. 2013;34:1337–1343. doi: 10.1016/j.biomaterials.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 100.Ali HM, Maksimenko A, Urbinati G, Chapuis H, Raouane M, Desmaele D, Yasuhiro H, Harashima H, Couvreur P, Massaad-Massade L. Effects of silencing the RET/PTC1 oncogene in papillary thyroid carcinoma by siRNA-squalene nanoparticles with and without fusogenic companion GALA-cholesterol. Thyroid. 2014;24:327–338. doi: 10.1089/thy.2012.0544. [DOI] [PubMed] [Google Scholar]
- 101.Kulkarni MV, Tettamanzi MC, Murphy JW, Keeler C, Myszka DG, Chayen NE, Lolis EJ, Hodsdon ME. Two independent histidines, one in human prolactin and one in its receptor, are critical for pH-dependent receptor recognition and activation. J Biol Chem. 2010;285:38524–38533. doi: 10.1074/jbc.M110.172072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Summerton JE. Endo-Porter: A Novel Reagent for Safe, Effective Delivery of Substances into Cells. Annals of the New York Academy of Sciences. 2005;1058:62–75. doi: 10.1196/annals.1359.012. [DOI] [PubMed] [Google Scholar]
- 103.Kichler A, Leborgne C, Marz J, Danos O, Bechinger B. Histidine-rich amphipathic peptide antibiotics promote efficient delivery of DNA into mammalian cells. Proc Natl Acad Sci U S A. 2003;100:1564–1568. doi: 10.1073/pnas.0337677100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Midoux P, Kichler A, Boutin V, Maurizot JC, Monsigny M. Membrane permeabilization and efficient gene transfer by a peptide containing several histidines. Bioconjug Chem. 1998;9:260–267. doi: 10.1021/bc9701611. [DOI] [PubMed] [Google Scholar]
- 105.Ashley CE, Carnes EC, Phillips GK, Durfee PN, Buley MD, Lino CA, Padilla DP, Phillips B, Carter MB, Willman CL, Brinker CJ, Caldeira Jdo C, Chackerian B, Wharton W, Peabody DS. Cell-specific delivery of diverse cargos by bacteriophage MS2 virus-like particles. ACS Nano. 2011;5:5729–5745. doi: 10.1021/nn201397z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, Carroll NJ, Jiang X, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nature Materials. 2011;10:389–397. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Summerton JE. Peptide composition and method for delivering substances into the cytosol of cells. 7084248 U.S. Patent. 2006
- 108.Rozema DB, Ekena K, Lewis DL, Loomis AG, Wolff JA. Endosomolysis by masking of a membrane-active agent (EMMA) for cytoplasmic release of macromolecules. Bioconjug Chem. 2003;14:51–57. doi: 10.1021/bc0255945. [DOI] [PubMed] [Google Scholar]
- 109.Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, Hegge JO, Klein JJ, Wakefield DH, Oropeza CE, Deckert J, Roehl I, Jahn-Hofmann K, Hadwiger P, Vornlocher HP, McLachlan A, Lewis DL. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Molecular Therapy. 2013;21:973–985. doi: 10.1038/mt.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.K L, Schluep Thomas, Wooddell Christine, Lewis David. A Phase I, first in human clinical trial of ARC-520, an siRNA-based therapeutic for the treatment of chronic hepatitis B virus infection, in normal healthy volunteers. HepDART Meeting. 2013 [Google Scholar]
- 111.C HLY, Yuen Man-Fung, Given Bruce D, Hamilton James, Schluep Thomas, Lewis David L, La Ching-Lung. Phase II, dose ranging study of ARC-520, a siRNA-based therapeutic, in patients with chronic hepatitis B virus infection; Annual Meeting of the American Association for the Study of Liver Diseases; 2014. [Google Scholar]
- 112.Chen QR, Zhang L, Stass SA, Mixson AJ. Co-polymer of histidine and lysine markedly enhances transfection efficiency of liposomes. Gene Ther. 2000;7:1698–1705. doi: 10.1038/sj.gt.3301294. [DOI] [PubMed] [Google Scholar]
- 113.Aoki Y, Hosaka S, Kawa S, Kiyosawa K. Potential tumor-targeting peptide vector of histidylated oligolysine conjugated to a tumor-homing RGD motif. Cancer Gene Ther. 2001;8:783–787. doi: 10.1038/sj.cgt.7700362. [DOI] [PubMed] [Google Scholar]
- 114.Leng Q, Mixson AJ. Modified branched peptides with a histidine-rich tail enhance in vitro gene transfection. Nucleic Acids Res. 2005;33:e40. doi: 10.1093/nar/gni040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Leng Q, Scaria P, Zhu J, Ambulos N, Campbell P, Mixson AJ. Highly branched HK peptides are effective carriers of siRNA. Journal of Gene Medicine. 2005;7:977–986. doi: 10.1002/jgm.748. [DOI] [PubMed] [Google Scholar]
- 116.Leng Q, Scaria P, Lu P, Woodle MC, Mixson AJ. Systemic delivery of HK Raf-1 siRNA polyplexes inhibits MDA-MB-435 xenografts. Cancer Gene Ther. 2008;15:485–495. doi: 10.1038/cgt.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Anko M, Majhenc J, Kogej K, Sillard R, Langel U, Anderluh G, Zorko M. Influence of stearyl and trifluoromethylquinoline modifications of the cell penetrating peptide TP10 on its interaction with a lipid membrane. Biochim Biophys Acta. 2012;1818:915–924. doi: 10.1016/j.bbamem.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 118.Andaloussi SE, Lehto T, Mager I, Rosenthal-Aizman K, Oprea II, Simonson OE, Sork H, Ezzat K, Copolovici DM, Kurrikoff K, Viola JR, Zaghloul EM, Sillard R, Johansson HJ, Said Hassane F, Guterstam P, Suhorutsenko J, Moreno PM, Oskolkov N, Halldin J, Tedebark U, Metspalu A, Lebleu B, Lehtio J, Smith CI, Langel U. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Research. 2011;39:3972–3987. doi: 10.1093/nar/gkq1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Berg K, Kristian Selbo P, Prasmickaite L, Tjelle TE, Sandvig K, Moan J, Gaudernack G, Fodstad Ø, Kjølsrud S, Anholt H, Rodal GH, Rodal SK, Høgset A. Photochemical Internalization: A Novel Technology for Delivery of Macromolecules into Cytosol. Cancer Res. 1999;59:1180–1183. [PubMed] [Google Scholar]
- 120.Choi Y, McCarthy JR, Weissleder R, Tung CH. Conjugation of a photosensitizer to an oligoarginine-based cell-penetrating peptide increases the efficacy of photodynamic therapy. ChemMedChem. 2006;1:458–463. doi: 10.1002/cmdc.200500036. [DOI] [PubMed] [Google Scholar]
- 121.Zhao JF, Chen JY, Mi L, Wang PN, Peng Q. Enhancement of intracellular delivery of anti-cancer drugs by the Tat peptide. Ultrastruct Pathol. 2011;35:119–123. doi: 10.3109/01913123.2011.557522. [DOI] [PubMed] [Google Scholar]
- 122.Erazo-Oliveras A, Muthukrishnan N, Baker R, Wang TY, Pellois JP. Improving the endosomal escape of cell-penetrating peptides and their cargos: strategies and challenges. Pharmaceuticals (Basel) 2012;5:1177–1209. doi: 10.3390/ph5111177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Matsushita M, Noguchi H, Lu YF, Tomizawa K, Michiue H, Li ST, Hirose K, Bonner-Weir S, Matsui H. Photo-acceleration of protein release from endosome in the protein transduction system. FEBS Lett. 2004;572:221–226. doi: 10.1016/j.febslet.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 124.Maiolo JR, Ottinger EA, Ferrer M. Specific Redistribution of Cell-Penetrating Peptides from Endosomes to the Cytoplasm and Nucleus upon Laser Illumination. J Am Chem Soc. 2004;126:15376–15377. doi: 10.1021/ja044867z. [DOI] [PubMed] [Google Scholar]
- 125.Endoh T, Sisido M, Ohtsuki T. Cellular siRNA delivery mediated by a cell-permeant RNA-binding protein and photoinduced RNA interference. Bioconjug Chem. 2008;19:1017–1024. doi: 10.1021/bc800020n. [DOI] [PubMed] [Google Scholar]
- 126.Oliveira S, Fretz MM, Høgset A, Storm G, Schiffelers RM. Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2007;1768:1211–1217. doi: 10.1016/j.bbamem.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 127.Srinivasan D, Muthukrishnan N, Johnson GA, Erazo-Oliveras A, Lim J, Simanek EE, Pellois JP. Conjugation to the cell-penetrating peptide TAT potentiates the photodynamic effect of carboxytetramethylrhodamine. PLoS One. 2011;6:e17732. doi: 10.1371/journal.pone.0017732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Smith AM, Mancini MC, Nie S. Bioimaging: second window for in vivo imaging. Nat Nanotechnol. 2009;4:710–711. doi: 10.1038/nnano.2009.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Parry JJ, Kelly TS, Andrews R, Rogers BE. In vitro and in vivo evaluation of 64Cu-labeled DOTA-linker-bombesin(7-14) analogues containing different amino acid linker moieties. Bioconjug Chem. 2007;18:1110–1117. doi: 10.1021/bc0603788. [DOI] [PubMed] [Google Scholar]
- 130.Cesarone G, Edupuganti OP, Chen CP, Wickstrom E. Insulin receptor substrate 1 knockdown in human MCF7 ER+ breast cancer cells by nuclease-resistant IRS1 siRNA conjugated to a disulfide-bridged D-peptide analogue of insulin-like growth factor 1. Bioconjug Chem. 2007;18:1831–1840. doi: 10.1021/bc070135v. [DOI] [PubMed] [Google Scholar]
- 131.Liu X, Wang W, Samarsky D, Liu L, Xu Q, Zhang W, Zhu G, Wu P, Zuo X, Deng H, Zhang J, Wu Z, Chen X, Zhao L, Qiu Z, Zhang Z, Zeng Q, Yang W, Zhang B, Ji A. Tumor-targeted in vivo gene silencing via systemic delivery of cRGD-conjugated siRNA. Nucleic Acids Res. 2014;42:11805–11817. doi: 10.1093/nar/gku831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Alam MR, Ming X, Fisher M, Lackey JG, Rajeev KG, Manoharan M, Juliano RL. Multivalent cyclic RGD conjugates for targeted delivery of small interfering RNA. Bioconjug Chem. 2011;22:1673–1681. doi: 10.1021/bc200235q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim SH, Jeong JH, Lee SH, Kim SW, Park TG. LHRH Receptor-Mediated Delivery of siRNA Using Polyelectrolyte Complex Micelles Self-Assembled from siRNA-PEG-LHRH Conjugate and PEI. Bioconjug Chem. 2008;19:2156–2162. doi: 10.1021/bc800249n. [DOI] [PubMed] [Google Scholar]
- 134.Detzer A, Overhoff M, Wünsche W, Rompf M, Turner JJ, Ivanova GD, Gait MJ, Sczakiel G. Increased RNAi is related to intracellular release of siRNA via a covalently attached signal peptide. RNA. 2009;15:627–636. doi: 10.1261/rna.1305209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Overhoff M, Sczakiel G. Phosphorothioate-stimulated uptake of short interfering RNA by human cells. EMBO Rep. 2005;6:1176–1181. doi: 10.1038/sj.embor.7400535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Husain B, Mukerji I, Cole JL. Analysis of high-affinity binding of protein kinase R to double-stranded RNA. Biochemistry. 51:8764–8770. doi: 10.1021/bi301226h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Soltani F, Sankian M, Hatefi A, Ramezani M. Development of a novel histone H1-based recombinant fusion peptide for targeted non-viral gene delivery. Int J Pharm. 2013;441:307–315. doi: 10.1016/j.ijpharm.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 138.Wang Y, Mangipudi SS, Canine BF, Hatefi A. A designer biomimetic vector with a chimeric architecture for targeted gene transfer. J Control Release. 2009;137:46–53. doi: 10.1016/j.jconrel.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N, Dowdy SF. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat Biotech. 2009;27:567–571. doi: 10.1038/nbt.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Haroon MM, Dar GH, Jeyalakshmi D, Venkatraman U, Saba K, Rangaraj N, Patel AB, Gopal V. A designed recombinant fusion protein for targeted delivery of siRNA to the mouse brain. J Control Release. 228:120–131. doi: 10.1016/j.jconrel.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 141.Dar GH, Gopal V, Rao M. Conformation-dependent binding and tumor-targeted delivery of siRNA by a designed TRBP2: Affibody fusion protein. Nanomed. 11:1455–1466. doi: 10.1016/j.nano.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 142.Geoghegan JC, Gilmore BL, Davidson BL. Gene Silencing Mediated by siRNA-binding Fusion Proteins Is Attenuated by Double-stranded RNA-binding Domain Structure. Mol Ther Nucleic Acids. 2012;1:e53. doi: 10.1038/mtna.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO, 2nd, Giangrande PH. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu HY, Gao X. A universal protein tag for delivery of SiRNA-aptamer chimeras. Sci Rep. 2013;3:3129. doi: 10.1038/srep03129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bramsen JB, Kjems J. Development of Therapeutic-Grade Small Interfering RNAs by Chemical Engineering. Front Genet. 3:154. doi: 10.3389/fgene.2012.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Allerson CR, Sioufi N, Jarres R, Prakash TP, Naik N, Berdeja A, Wanders L, Griffey RH, Swayze EE, Bhat B. Fully 2′-Modified Oligonucleotide Duplexes with Improved in Vitro Potency and Stability Compared to Unmodified Small Interfering RNA. J Med Chem. 2005;48:901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- 147.Choung S, Kim YJ, Kim S, Park HO, Choi YC. Chemical modification of siRNAs to improve serum stability without loss of efficacy, Biochem. Biophys Res Commun. 2006;342:919–927. doi: 10.1016/j.bbrc.2006.02.049. [DOI] [PubMed] [Google Scholar]