

Abstract
Aim
The aim of the present analysis was to develop a core population pharmacokinetic model for the pharmacokinetic properties of immediate‐release (IR) exenatide, which can be used in subsequent analyses of novel sustained‐release formulations.
Methods
Data from eight clinical trials, evaluating a wide range of doses and different administration routes, were available for analysis. All modelling and simulations were conducted using the nonlinear mixed‐effect modelling program NONMEM. External model validation was performed using data from the phase III clinical trials programme through standard visual predictive checks.
Results
The pharmacokinetics of IR exenatide was described by a two‐compartment model, and the absorption of subcutaneous exenatide was described with a sequential zero‐order rate constant followed by a saturable nonlinear absorption process. Drug elimination was characterized by two parallel routes (linear and nonlinear), with significant relationships between renal function and the linear elimination route, and between body weight and volume of distribution. For a subject with normal renal function, the linear clearance was estimated to be 5.06 l hr−1. The nonlinear elimination was quantified with a Michaelis–Menten constant (K m) of 567 pg ml−1 and a maximum rate of metabolism (V max) of 1.6 μg h−1. For subcutaneous administration, 37% of the subcutaneous dose is absorbed via the zero‐order process, and the remaining 63% via the nonlinear pathway.
Conclusions
The present analysis provides a comprehensive population pharmacokinetic model for exenatide, expanding the elimination process to include both linear and nonlinear components, providing a suitable platform for a broad range of concentrations and patient conditions that can be leveraged in future modelling efforts of sustained‐release exenatide formulations.
Keywords: exenatide, pharmacokinetics, renal impairment, type 2 diabetes mellitus
What is Already Known about this Subject
Exenatide, a peptide that controls glucose in patients with type 2 diabetes, is eliminated with a half‐life of ~2.4 h, primarily via renal mechanisms.
However, a nonrenal elimination pathway had been posited, based on preclinical experiments and a limited clinical study.
What this Study Adds
Extensive clinical data were used to develop a core population pharmacokinetic model for exenatide, incorporating influential covariates.
The new model demonstrated the existence of renal and nonrenal elimination pathways for exenatide and estimated percentages eliminated by each pathway.
Tables of Links
| TARGETS | |
|---|---|
| G protein‐coupled receptors 2 | Enzymes 3 |
| GLP‐1 receptor | Dipeptidyl peptidase‐IV |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3.
Introduction
Exenatide is a glucagon‐like peptide‐1 (GLP‐1) receptor agonist used to improve glycaemic control for patients with type 2 diabetes mellitus. The drug is administered subcutaneously and is given either twice daily as an immediate‐release (IR) formulation (Byetta®, AstraZeneca, Wilmington, DE, USA) 4 or once weekly as an extended‐release (ER) preparation (Bydureon®, AstraZeneca, Wilmington, DE, USA) 5. Exenatide binds to the GLP‐1 receptor and decreases glucose concentrations through multiple mechanisms of action, including slowing gastric emptying, suppressing glucagon concentrations, increasing satiety, and increasing the glucose‐dependent stimulation of insulin release 4, 6. The exenatide peptide that circulates systemically following the release and absorption of each of the various formulations of exenatide (IR, ER, and an experimental once‐monthly preparation7) is identical 8, allowing for the bridging of the relevant pharmacokinetic (PK) properties across the formulations.
Exenatide has approximately 53% sequence homology with native GLP‐1. Although it binds to and activates the mammalian GLP‐1 receptor to allow for similar glycaemic benefits, exenatide is a poor substrate of the dipeptidyl peptidase‐IV enzyme, resulting in a relatively longer half‐life (2.4 h) in comparison with the native GLP‐1 peptide (<2 min) 9, 10. Preclinical studies show that exenatide is primarily eliminated by the kidney via renal filtration and enzymatic degradation in the tubules, with little of the intact peptide excreted into the urine 11. Clinically, the IR formulation of exenatide is absorbed relatively quickly, achieving peak concentrations (C max) of approximately 211 pg ml−1 around 2 h after subcutaneous injection 4. The apparent clearance is 9.1 l h−1, with detectable serum concentrations for about 10 h after a 10 μg subcutaneous dose 4.
Consistent with the mechanisms of exenatide excretion, subjects with renal impairment are likely to exhibit increased exenatide concentrations. The PK and tolerability of the IR formulation of exenatide have been assessed in subjects with renal impairment 12. The elimination rate of exenatide is reduced in subjects with renal impairment, with reported mean half‐lives of 2.1, 3.2, and 6.0 h for subjects with mild and moderate renal impairment and end‐stage renal disease, respectively 12. The IR and ER formulations of exenatide are approved in the United States for use in patients with normal renal function [estimated glomerular filtration rate (eGFR) ≥90 ml min−1 1.73 m− 2] and mild renal impairment (eGFR 60–90 ml min−1 1.73 m− 2) and with caution in patients with moderate renal impairment (eGFR ≥30–59 ml min−1 1.73 m− 2) 4, 5, 13. The elimination of exenatide is not affected by hepatic dysfunction 11.
A minor saturable route of elimination has been identified for exenatide 14, 15, 16 and is hypothesized to result from target‐mediated drug disposition (TMDD), in which binding to the pharmacological target influences the PK of the drug 17, 18. In rats with functional nephrectomy, exenatide is cleared more slowly (five‐fold increase in half‐life), suggesting a minor nonrenal elimination pathway 19. Furthermore, an increase in exposure was observed in GLP‐1 receptor knockout mice, suggesting that the GLP‐1 receptor may play a role in the clearance of exenatide 20. Concentration‐dependent nonlinear elimination was also demonstrated at supratherapeutic doses that were achieved in a recent thorough QT study 21.
Population PK/pharmacodynamic modelling was instrumental in the selection of the fixed, nonweight‐based dose levels of 5 μg and 10 μg recommended for the exenatide IR formulation 22, 23. The PK of the IR formulation is typically described with a one‐compartment model with dual linear and saturable absorption and linear elimination. Covariate selection procedures identified body weight, gender, and antiexenatide antibodies as factors influencing exenatide PK. Although these models were pivotal to the development of the IR formulation, they need to be expanded for the quantitative assessment of the PK of novel ER formulations of exenatide. The present retrospective population analysis aimed to combine data from diverse studies evaluating multiple routes of administration (subcutaneous, intravenous infusion, and intravenous bolus), from a wide range of doses and concentrations, allowing for the quantification of absorption, linear and nonlinear elimination, key influential factors (e.g. body weight and renal function), and the magnitude of intersubject and residual variability (RV). Our final model will serve as a core population PK system for the PK properties of IR exenatide that can be used in subsequent analyses for novel ER formulations.
Methods
Data collection
Data from eight clinical trials evaluating IR exenatide in subjects with type 2 diabetes mellitus and healthy volunteers were obtained for analysis (Table 1). In order to provide a robust assessment, pooled studies included a range of doses and infusion rates to provide a range of concentrations associated with intravenous (bolus and infusion) and subcutaneous administration. This dataset also included plasma drug concentrations collected from a renal impairment study, enriching the range of renal function to study its influence on the overall elimination of exenatide. In addition, the intravenous PK profiles allowed for an estimation of bioavailability and the linear and concentration‐dependent elimination parameters. All studies contributed PK profiles with at least six samples, collected between 10 min and 18 h after drug administration.
Table 1.
Clinical studies included in analysis
| Study | Administration route | Dose level | Population | Number of subjects | Number of observations | Reference |
|---|---|---|---|---|---|---|
| 1 | SC | 0.1 μg kg−1, 0.2 μg kg−1, 0.3 μg kg−1, 0.4 μg kg−1 | T2DM | 6 | 403 | 37 |
| 2 | SC | 0.1 μg kg−1 | T2DM | 23 | 666 | 38 |
| 3 | SC | 0.02 μg kg−1, 0.05 μg kg−1, 0.1 μg kg−1 | T2DM | 8 | 215 | 37 |
| 4 | SC | 0.05 μg kg−1, 0.1 μg kg−1, 0.2 μg kg−1 | T2DM | 12 | 444 | 38 |
| 5 & 6 a | IV infusion | Continuous IV infusion for 5 days | Healthy | 92 | 2235 | 21 |
| 7 | SC, IV bolus | 10 μg SC, 1 μg IV | T2DM | 24 | 1132 | 39 |
| 8 | SC | 5 μg and 10 μg SC | Renal impairment | 30 | 223 | 12 |
IV, intravenous; SC, subcutaneous; T2DM, type 2 diabetes mellitus
This reflects two studies with similar IV infusion designs [study 5 21 (n = 73) and a pilot study (study 6; n = 19)]
All studies were conducted in accordance with the principles described in the Declaration of Helsinki (1946) up to and including the Seoul revision (1997). A common clinical protocol was approved for each site by an appropriate institutional review board, and all subjects provided written informed consent prior to participation.
Analytical assay
Exenatide concentrations were measured from plasma samples using a validated two‐site sandwich enzyme‐linked immunosorbent assay. Specificity for exenatide was conferred by the monoclonal capture antibody which recognizes a C‐terminal epitope on exenatide and does not cross‐react with native GLP‐1(7–36), or glucagon. The monoclonal detection antibody recognizes an N‐terminal epitope on exenatide, GLP‐1(7–36), and glucagon. The requirement that both capture and detection antibodies recognize the peptide in order to generate a signal in this assay minimizes cross‐reactivity with other peptides or metabolites. The assay showed no cross‐reactivity with 1 ng ml−1 exendin‐4(2–39), exendin‐4(3–39), GLP‐1(7–36), glucagon, or insulin. Assay performance met accuracy and precision specifications published for method validation of ligand‐binding assays 24.
Pharmacokinetic modelling
All modelling and simulations were conducted using the first‐order conditional estimation method with interaction in the nonlinear mixed‐effects modelling (NONMEM) software VI (ICON Development Solutions, Ellicott City, MD, USA), and all graphical displays were created using SAS 9.2 (SAS Institute Inc., Cary, NC, USA). One‐ and two‐compartment models with a linear and/or nonlinear elimination were evaluated. First‐order, saturable, zero‐order, and combination models describing absorption from the subcutaneous space were also examined. The final population PK model for IR exenatide is a two‐compartment model [i.e. distribution clearance (Cl d), volume of central compartment (Vc), and volume of peripheral compartment (Vp)] with parallel linear clearance (Cl) and nonlinear elimination characterized with Michaelis–Menten kinetics [maximum rate of metabolism (V max), concentration required for half of the nonlinear elimination rate (K m)]. The absorption following subcutaneous administration was best described by a sequential process, with zero‐order absorption for a fraction (1–fr) of the dose, followed by a nonlinear process for the remainder of the available dose (fr, fraction absorbed through first‐order process; k a_max, maximum absorption rate constant for the nonlinear absorption process; K m_ka, amount of drug required for 50% of k a_max) (Figure 1; Table 2). This PK system is defined by the following differential equations:
| (1) |
| (2) |
| (3) |
with K 0 as the zero‐order absorption rate constant for the sequential absorption process:
| (4) |
and k 1 is a saturable absorption process:
| (5) |
Figure 1.
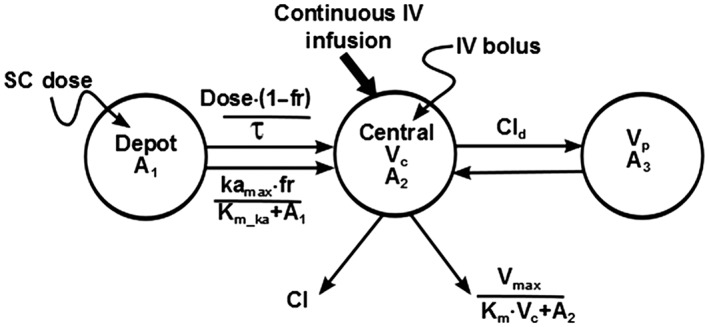
Structural pharmacokinetic model diagram. Exenatide pharmacokinetics were characterized using a two‐compartment model with sequential zero‐order and nonlinear absorption and parallel linear and nonlinear elimination (see Table 2 for parameter definitions)
Table 2.
Final pharmacokinetic model parameter estimates for exenatide
| Symbol (unit) | Parameter estimate | %RSE | IIV(%) | %RSE | |
|---|---|---|---|---|---|
| Cl_int (l h −1 ) | 4.58 | 5.68 | 33.9 | 19.1 | |
| Cl_eGFR | 0.838 | 12.3 | |||
| Cl = Cl_int Cl_eGFR | |||||
| Cl d (l h −1 ) | 3.72 | 21.7 | – | – | |
| K m (pg ml −1 ) | 567 | 20.6 | 95.7 | 21.9 | |
| V max (μg h −1 ) | 1.55 | 22.1 | – | – | |
| V p (l) | 7.04 | 9.49 | – | – | |
| Vc_int (l) | 7.03 | 10.1 | 80.5 | 13.2 | |
| Vc_wtkg | 2.67 | 13.3 | |||
|
| |||||
| V p (l) | 7.04 | 9.49 | – | – | |
| k a_max (μg h −1 ) | 12.8 | 42.5 | |||
| K m_ka (μg) | 16.9 | 54.9 | – | – | |
| τ (h) | 1.35 | NE | – | – | |
| F | 1 | NE | – | – | |
| fr | 0.628 | 3.5 | – | – | |
| RV (log units) SD | Y = LOG(F) + EPS(1) | ||||
| Study 1 | 0.39 | 11 | – | – | |
| Study 5 | 0.08 | 11.9 | – | – | |
| All other studies | 0.373 | 21.7 | – | – | |
Cl d, distribution clearance; Cl_eGFR, power for estimated glomerular filtration rate (eGFR) effect on linear clearance; Cl_int, linear elimination rate for subject with eGFR of 80 ml min−1 1.73 m− 2; F, bioavailability; fr, fraction absorbed during the first‐order process; IIV, interindividual variability; k a_max, maximum rate of absorption for the nonlinear absorption process; K m, concentration required for half of the nonlinear elimination rate (V max); K m_ka, amount required for 50% of the maximum rate (k a_max); NE, not estimated; RSE, relative standard error of the mean; RV, residual variability; SD, standard deviation; Vc_int, volume of the central compartment for an 84.8 kg person; Vc_wtkg, power for weight effect on volume of the central compartment; V max, maximum nonlinear elimination rate; V p, volume of the peripheral component; τ, duration of the zero‐order process
The duration of the initial zero‐order process (τ) was fixed to 1.35 h based on the results from a sensitivity analysis of an initial model run (data not shown). Data from all studies and all routes of administration were fit simultaneously. The absolute bioavailability of exenatide was estimated to be approximately 100%, and F (absolute bioavailability) was fixed to 1 in all subsequent modelling. During model development, approximately 4% of the measured concentrations were determined to be outliers and removed from the analysis. These outliers were identified through a combination of visual inspection of the concentration–time profile as well as minimization difficulties with NONMEM. Outliers were associated with weighted residuals greater than 7 and resulted in minimization difficulties in NONMEM. The final model was re‐evaluated by including the outlier concentrations. This resulted in minimization difficulties or poor precision of parameter estimates; however, the parameter estimates remained consistent with those obtained for the final model. Therefore, it was deemed appropriate to exclude outlier concentrations from the final model. Concentrations identified as below the limit of quantification (3% of total observations) were also excluded.
Interindividual variability and residual error
Interindividual variability (IIV) was evaluated using a log‐normal distribution model, with the random variability distributed with a mean of zero and a variance of ω2. The RV was described by a log error model, owing to the range of doses evaluated, with natural log transformed data and an additive model. Alternative RV structures were evaluated but did not improve model performance.
Covariate evaluation
Subject characteristics were evaluated for their influence on exenatide PK parameters, including age (years), gender, race, ideal body weight (kg), total body weight (kg), body mass index (kg m−2), and renal function (expressed as eGFR in ml min−1 1.73 m− 2 using the equation developed from the Modification of Diet in Renal Disease Study 13):
| (6) |
where Scr is serum creatinine concentration.
A univariate analysis of each covariate, with an observable trend in the graphical analyses, was performed using NONMEM. Covariates contributing at least a 10.83 change in the minimum value of the objective function (MVOF; α = 0.001, one degree of freedom for χ2 distribution) were retained in the model.
Model assessment
Model adequacy, at multiple stages of development, was assessed by the precision of the parameter estimates and shrinkage 25, assessment of the MVOF, and graphical assessments and evaluation of predictive performance through standard diagnostic plots and visual predictive checks (VPCs) 26. The fixed and random‐effect parameters from the final model were used to simulate 500 replicates of the observed dataset. The 5th, 50th (median), and 95th percentiles of the distributions of the simulated concentration values at each sampling time were calculated. Graphical displays of the simulated percentiles overlaid on the observed data were assessed. For external model validation, PK samples (n = 5) up to 3 h after drug administration were obtained from three phase III clinical trials. Prior to the external validation, phase III data were excluded for several anomalies, including: missing values, concentrations assayed as below the limit of quantitation, and concentration–time profiles that significantly deviated from anticipated curves. This resulted in a total of 669 concentrations from 80 subjects available for this assessment. Subjects received twice‐daily subcutaneous exenatide doses of 5 μg for 4 weeks, followed by 26 weeks of 5 μg or 10 μg twice‐daily 27, 28, 29. Given the relatively sparse sampling strategies in these studies, the data were reserved for an external predictive check. The adequacy of the model to predict the external data was evaluated through standard VPCs.
Model simulations
To evaluate the influence of renal function on the PK of IR exenatide, 7500 subjects weighing 90 kg with normal (eGFR 90 ml min−1 1.73 m− 2), mild (eGFR 75 ml min−1 1.73 m− 2), and moderate (eGFR 45 ml min−1 1.73 m− 2) renal impairment were simulated. In addition, 7500 subjects with normal renal function (eGFR 90 ml min−1 1.73 m− 2) weighing 75, 100, and 125 kg were simulated to evaluate the impact of body weight independently of renal function. Forest plots of the simulated data were created to evaluate the change in expected drug exposure [area under the curve from time zero to infinity (AUC(0–∞)) and C max] from subjects with normal renal function and weighing 75 kg.
Results
Subject pharmacodynamics and demographics
A total of 195 subjects and 5318 exenatide observations were available for analysis. The population was 22% female and 67% Caucasian. The mean [standard deviation (SD)] age was 48 (12.9) years (range, 18–76 years). Body weight ranged from 52.6 kg to 162 kg, with a mean (SD) of 86 (16.2) kg. The range of renal function was relatively wide, with eGFR values as low as 4.1 ml min−1 1.73 m− 2 and a mean of 88.7 (31.4) ml min−1 1.73 m− 2. Using the guidance for industry criteria for renal impairment classification 13, the number of subjects with normal renal function, mild, moderate, and severe renal impairment, and end‐stage renal disease in the interdialytic period was 129, 47, 10, 1, and 8, respectively.
Exenatide pharmacokinetics
The final model characterized the absorption process of subcutaneous exenatide with sequential zero‐order absorption followed by a saturable nonlinear absorption process. Elimination of exenatide was characterized by two parallel routes (linear and nonlinear). Individual profiles from representative subjects illustrating the types of profiles (subcutaneous, intravenous, and continuous infusion) are shown in Figure 2. These profiles show the time to peak concentration after subcutaneous administration at approximately 2 h, and a subsequent decrease in concentrations over 10–15 h postdosing, depending on renal function. The relatively rapid decrease suggests no anticipated accumulation on multiple dosing. The VPCs stratified by route of administration (Figure 3), and individual profiles for representative subjects (Figure 2), suggest that the model adequately describes the PK data.
Figure 2.
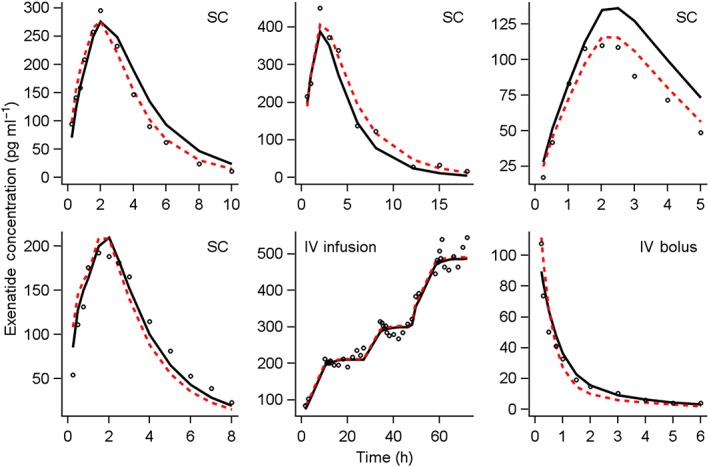
Observed, population‐ and individual‐predicted concentration–time profiles for select representative subjects. Panel labels (SC, IV infusion, IV bolus) indicate the route of exenatide administration. Open circles represent the observed data points. Solid black lines represent the population mean predicted concentrations. Dashed red lines represent the individual predicted concentrations. IV, intravenous; SC, subcutaneous
Figure 3.
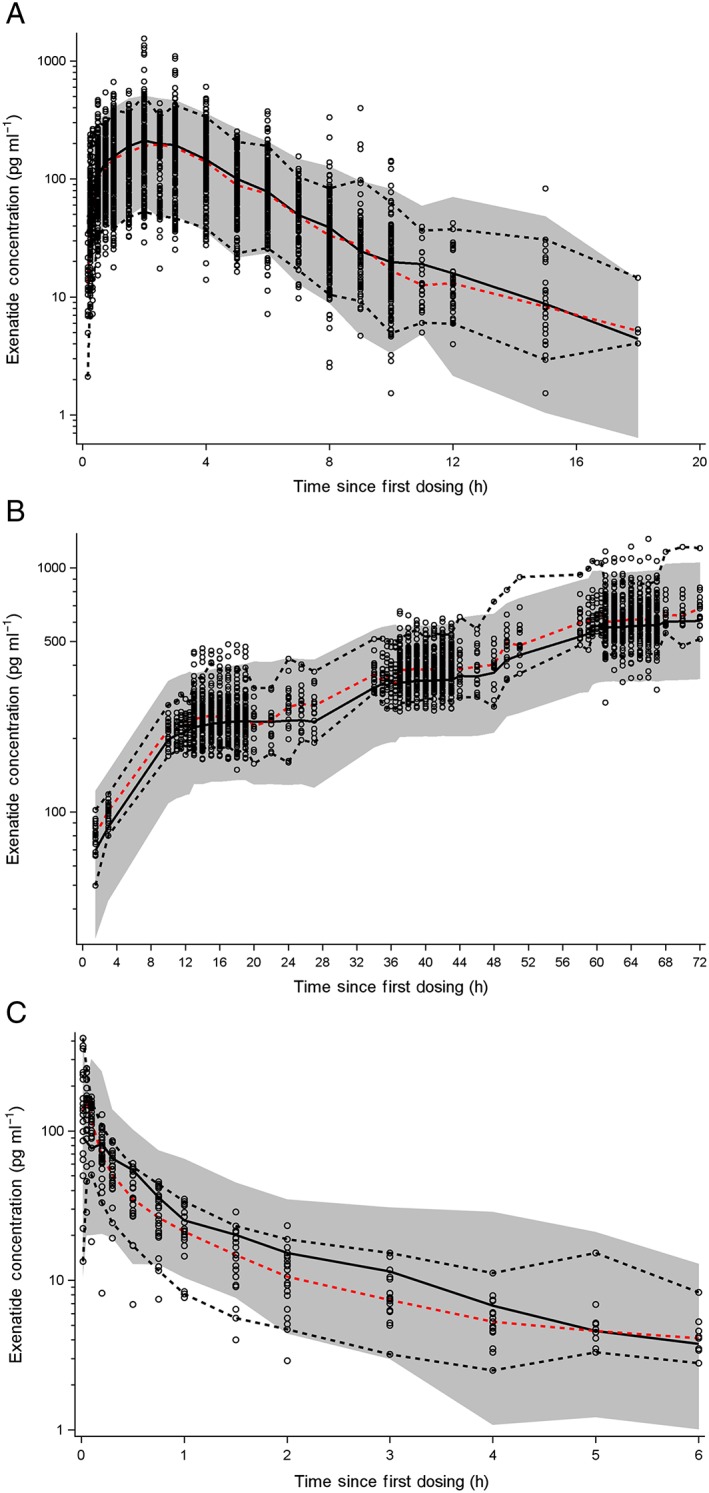
Comparison of observed and predicted exenatide pharmacokinetics. Visual predictive check (VPC) plots for subcutaneous (SC) and intravenous (IV) infusion are prediction‐corrected VPCs in order to represent multiple levels of dosing. Dashed red lines represent the median of the observed data. Dashed black lines represent the 5th and 95th percentiles for the observed data. Black solid lines represent the median of the predicted data. Shaded grey regions represent the 90% prediction interval. (A) SC injection, (B) IV infusion, and (C) IV bolus
All parameters were well estimated [% relative standard error (RSE) < 23%; Table 2], with the exception of the parameters related to the nonlinear absorption process (K m_ka; %RSE 54.9, k a_max; 42.5%). Estimates of the magnitude of IIV were included in Cl, Vc, and the K m of elimination with good precision and low shrinkage (<22% on all parameters), with the largest IIV of 96% associated with the K m of elimination. The inclusion of renal function in the linear elimination estimate, and weight in the volume estimate reduced the IIV in Cl and Vc by 19% and 17%, respectively. For a subject with normal renal function (eGFR 90 ml min−1 1.73 m− 2), the linear clearance was estimated to be 5.06 l h−1. The nonlinear elimination was quantified with a K m of 567 pg ml−1 and V max of 1.6 μg h−1. Figure 4A shows the decrease in total and nonlinear clearance with increasing concentration and illustrates that, for a subject with normal renal function, plasma concentrations well beyond 1000 pg ml−1 would be required to saturate fully the concentration‐dependent elimination pathway. The linear elimination route decreases with increasing renal impairment (Figure 4B). Within the peak plasma concentration range expected for the IR formulation of 200–300 pg ml−1, for subjects with normal renal function, approximately 75% of the drug is cleared via the linear route, and total exenatide clearance at the concentration of 210 pg ml−1 is predicted to be 5.1, 6.3, and 7.5 l h−1 for subjects with eGFR estimates of 50, 75, and 100 ml min−1 1.73 m− 2, respectively. Figure 5 shows the overall impact of renal function on the secondary parameters of C max (pg ml−1) and AUC(0–∞) (pg h ml−1) from 7500 simulated subjects, showing a 7% (273 pg ml−1) and 24% (321 pg ml−1) increase in C max and a 12% and 48% increase in AUC for subjects with mild and moderate renal impairment, relative to normal renal function (C max = 256 pg ml−1), with a large overlap across renal impairment levels. Total body weight was identified as a significant predictor of the variability in V c. Compared with the peak concentration for the typical 75 kg subject (C max 287 pg ml−1), an 18% (C max 237 pg ml−1) and 34% (C max 189 pg ml−1) decrease in C max is expected with body weights of 100 kg and 125 kg (Figure 5).
Figure 4.
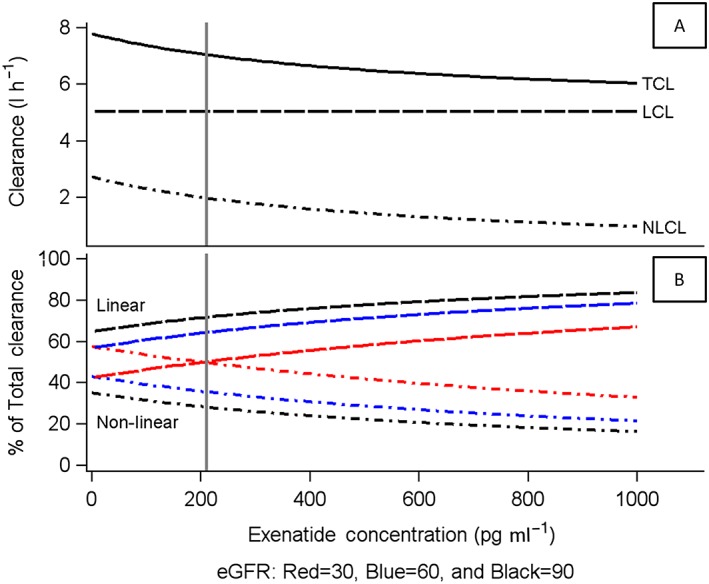
Relationship between exenatide clearance and plasma concentration. Profiles are stratified by relative contribution (A) and renal function (B). Vertical reference line at 210 pg ml−1 represents the mean peak concentration for subcutaneous exenatide. Units of eGFR are ml min−1 1.72 m− 2. eGFR, estimated glomerular filtration rate; LCL, linear clearance; NLCL, nonlinear clearance; TCL, total clearance
Figure 5.
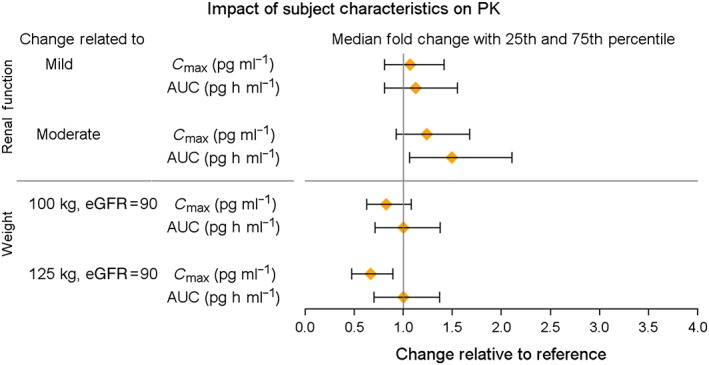
Forest plot of the impact of renal function and body weight on exenatide pharmacokinetics (PK). Renal function is relative to the normal condition (eGFR 90 ml min−1 1.73 m− 2). Mild renal impairment reflects an eGFR of 75 ml min−1 1.73 m− 2, and moderate renal impairment with an eGFR of 45 ml min−1 1.73 m− 2. Weight is relative to a reference of 75 kg. AUC, area under the concentration–time curve; C max, maximum plasma concentration; eGFR, estimated glomerular filtration rate
For subcutaneous administration, 37% of the subcutaneous dose is absorbed via the zero‐order process, and the remaining 63% via the nonlinear pathway. The nonlinear absorption had a K m of 16.9 μg and a maximum capacity of 12.8 μg h−1. As the maximum approved dose of the IR formulation is 10 μg twice‐daily, saturation of the absorption process is not expected.
External validation
The final model was used to predict the sparse sampling PK profiles from phase III trials. Whereas the final model generally described the central tendency of the 3‐h profile, there was a slight overprediction of the phase III data (Figure 6). The observed profiles exhibited a relatively large degree of variability that was not anticipated by the final model and parameters.
Figure 6.
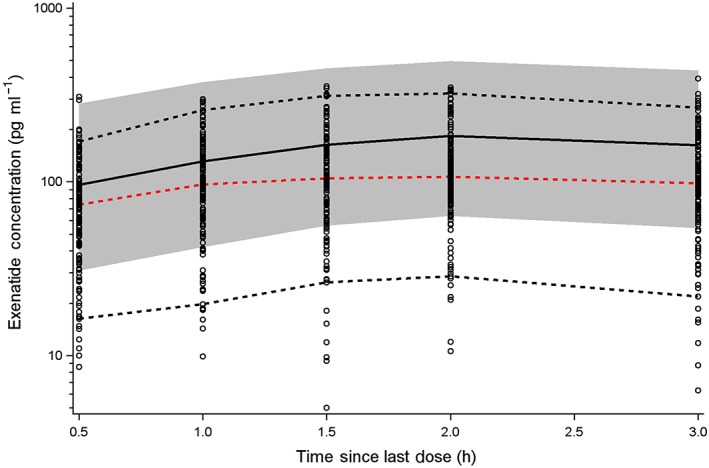
External model validation. The final model was used to predict the sparse sampling pharmacokinetic profiles from a phase III trial. Dashed red lines represent the median of the observed data. Dashed black lines represent the 5th and 95th percentiles for the observed data. Black solid lines represent the median of the predicted data. Shaded grey regions represent the 90% prediction interval
Discussion
The refinement of the population PK model for exenatide described herein provides a foundation for use in modelling efforts for novel formulations and their potential pharmacodynamic effects. It incorporates the important covariates of renal function on clearance and body weight on the volume of distribution, as well as expands the range of concentrations that this model can accommodate by incorporating several nonlinear processes.
In contrast to prior population models of exenatide based on simple linear kinetics, the elimination of exenatide was described by linear and nonlinear routes of elimination. The estimated K m from this model (567 pg ml−1) is greater than the C max achieved with the largest dose of the IR formulation of exenatide (211 pg ml−1 for a 10 μg dose), placing the therapeutically relevant concentrations well within the linear region of the elimination process characterized by first‐order kinetics. The supratherapeutic dose levels included in a thorough QT study resulted in the evaluation of concentrations greater than typical clinical profiles, providing the opportunity to quantify the concentration ranges in humans in which saturable elimination might be observed. There is prior evidence of saturable exenatide elimination, and preclinical and clinical data for the IR formulation have been characterized by TMDD 14, 15, 16. Attempts to use the general TMDD model 17, 18 resulted in poor precision and convergence difficulties. Therefore, a linear plus Michaelis–Menten elimination function for clearance was used, which has been shown to approximate target‐mediated systems under certain conditions 30.
Exenatide is predominately eliminated by the kidney, and increased exposures are observed in subjects with decreased renal function. The IR formulation is not indicated for use in subjects with severe or end‐stage renal impairment in the United States, and the drug label indicates that caution should be taken when initiating or escalating doses from 5 μg to 10 μg in patients with moderate renal impairment 4. The 12% and 48% increases in AUC predicted for subjects with mild and moderate renal impairment are similar to the values reported by Linnebjerg et al. 12, confirming the adequacy of the model to predict the impact of renal function on exenatide exposure. At the clinically relevant doses of 5 μg and 10 μg, the safety and tolerability were acceptable for subjects with either mild or moderate renal impairment 12. In addition, the results of a clinical trial evaluating exenatide PK in elderly subjects concluded that, after accounting for renal function, no additional effects of age on exenatide exposure were apparent 31.
Despite the influence of body weight on the volume of distribution, population PK/pharmacodynamic analysis supports a fixed dosing algorithm 22, 23. Our final model is also consistent with these findings. Notwithstanding the identification of body weight as a significant predictor of exenatide volume, with greater body weight associated with increased volume (and lower exenatide concentrations), the observed and simulated concentrations support that even in heavier subjects (weight of about 125 kg), the range of C max values is well above the minimum effective concentration of 50 pg ml−1 32. Thus, despite the lower concentrations with increased body weight, the concentrations achieved with the approved dose in heavier subjects exceed the effective concentration, and thus no dose adjustments are warranted.
Complex and rate‐limiting processes, such as interstitial transport and flow rates, control the absorption of macromolecules from the subcutaneous space, and the source of the nonlinear absorption of exenatide is unknown. A review of the PK modelling of subcutaneous absorption of protein therapeutics provides a detailed description of several factors that influence absorption and the types of available integrated PK models of protein absorption 33. During the model development process for exenatide, several combinations of zero‐order, first‐order, and saturable absorption models were evaluated. The absorption of exenatide from the subcutaneous space was best described by sequential absorption, with a zero‐order process followed by a nonlinear process, possibly reflecting absorption through both blood and lymph. Although the reason for the saturable exenatide absorption after subcutaneous injection is still unknown, similar absorption models have been reported for exenatide and other injectable proteins 14, 23, 33, 34. The relevance of this nonlinear absorption is minimal with the currently approved doses of 5 μg and 10 μg, as the amount of the dose predicted to be absorbed via the saturable pathway is below the estimated K m of the nonlinear absorption process (16.9 μg) and bioavailability is complete (100%).
The influence of antibody titre on the exposure of exenatide has previously been reported 35. Greater exposure of exenatide is expected with increasing antibody titres owing to inhibition of renal elimination. Similar effects of antibody titre on elimination have been reported for other therapeutic proteins 36. Within the current analysis, the influence of antidrug antibody was not incorporated as longer durations of drug exposure would be needed to form antibody titres. Long‐term exenatide exposure would be required to incorporate the effect of antibody titre on its PK. An external model validation was performed as part of this analysis, using sparse PK data from the phase III development programme. The results of this evaluation showed a slight overprediction of the external data (Figure 6). The phase III data are associated with increased variability across patients, and the exact reason for this overprediction is unclear.
In summary, this study focuses on the development of a comprehensive population PK model for exenatide, expanding the elimination process to include both linear and nonlinear components. Furthermore, the analysis incorporated the influence of renal function on the linear elimination pathway. A sequential dual absorption model was used to characterize the absorption of exenatide from the subcutaneous space in order to quantify exposure profiles adequately. Overall, this expanded model will have greater utility in exploring new formulations, routes of administration, and a greater range of resulting plasma concentrations than prior exenatide models. Within the exenatide development programme, formulations have progressed from IR to weekly and monthly administration regimens, but all deliver the identical underlying circulating peptide. Thus, our final model should provide a suitable platform for a broad range of concentrations and patient conditions that can be leveraged in future modelling efforts for ER formulations.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare BC is an employee of Bristol‐Myers Squibb and was an employee of Amylin Pharmaceuticals, and DEM has received personal fees from Bristol‐Myers Squibb.
This analysis was supported by Amylin Pharmaceuticals, Inc., Bristol‐Myers Squibb Company, and AstraZeneca.
The authors thank the exenatide development team for their support of this analysis. They also thank David Clawson, Dr Jeffrey Edwards, Dr Demiana Faltaos and Carl LaCerte for their SAS programming, consultation, and quality control support. The authors are also grateful to Dr Frank LaCreta for his review and clinical perspectives in support of this manuscript. In addition, we thank inScience Communications, Springer Healthcare (Philadelphia, PA, USA), for providing copyright editorial support for submission and AstraZeneca for funding support.
Cirincione, B. , and Mager, D. E. (2017) Population pharmacokinetics of exenatide. Br J Clin Pharmacol, 83: 517–526. doi: 10.1111/bcp.13135.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byetta (exenatide) injection: US prescribing information. Wilmington, DE: AstraZeneca LP; 2014.
- 5. Bydureon (exenatide extended‐release for injectable suspension): US prescribing information. Wilmington, DE: AstraZeneca LP; 2014.
- 6. Cervera A, Wajcberg E, Sriwijitkamol A, Fernandez M, Zuo P, Triplitt C, et al. Mechanism of action of exenatide to reduce postprandial hyperglycemia in type 2 diabetes. Am J Physiol Endocrinol Metab 2008; 294: E846–E852. [DOI] [PubMed] [Google Scholar]
- 7. MacConell L, Malloy J, Huang W, Cirincione B, Shen L, Porter L. Safety and efficacy of once‐monthly exenatide administration over 20 weeks in patients with type 2 diabetes. Diabetes 2011; 60: LB13–LB14. [Google Scholar]
- 8. DeYoung MB, MacConell L, Sarin V, Trautmann M, Herbert P. Encapsulation of exenatide in poly‐(D,L‐lactide‐co‐glycolide) microspheres produced an investigational long‐acting once‐weekly formulation for type 2 diabetes. Diabetes Technol Ther 2011; 13: 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nielsen LL, Young AA, Parkes DG. Pharmacology of exenatide (synthetic exendin‐4): a potential therapeutic for improved glycemic control of type 2 diabetes. Regul Pept 2004; 117: 77–88. [DOI] [PubMed] [Google Scholar]
- 10. Vilsbøll T, Agerso H, Krarup T, Holst JJ. Similar elimination rates of glucagon‐like peptide‐1 in obese type 2 diabetic patients and healthy subjects. J Clin Endocrinol Metab 2003; 88: 220–224. [DOI] [PubMed] [Google Scholar]
- 11. Copley K, McCowen K, Hiles R, Nielsen LL, Young A, Parkes DG. Investigation of exenatide elimination and its in vivo and in vitro degradation. Curr Drug Metab 2006; 7: 367–374. [DOI] [PubMed] [Google Scholar]
- 12. Linnebjerg H, Kothare PA, Park S, Mace K, Reddy S, Mitchell M, et al. Effect of renal impairment on the pharmacokinetics of exenatide. Br J Clin Pharmacol 2007; 64: 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. US Food and Drug Administration . Guidance for industry: pharmacokinetics in patients with impaired renal function – study design, data analysis, and impact on dosing and labeling (draft guidance ‐ revision 1); 2010. [online]. Available at http://www.fda.gov/downloads/Drugs/Guidances/UCM204959.pdf (last accessed 8 October 2014).
- 14. Chen T, Mager DE, Kagan L. Interspecies modeling and prediction of human exenatide pharmacokinetics. Pharm Res 2013; 30: 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gao W, Jusko WJ. Pharmacokinetic and pharmacodynamic modeling of exendin‐4 in type 2 diabetic Goto‐Kakizaki rats. J Pharmacol Exp Ther 2011; 336: 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao W, Jusko WJ. Target‐mediated pharmacokinetic and pharmacodynamic model of exendin‐4 in rats, monkeys, and humans. Drug Metab Dispos 2012; 40: 990–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. J Pharmacokinet Pharmacodyn 2001; 28: 507–532. [DOI] [PubMed] [Google Scholar]
- 18. Levy G. Pharmacologic target‐mediated drug disposition. Clin Pharmacol Ther 1994; 56: 248–252. [DOI] [PubMed] [Google Scholar]
- 19. Parkes D, Jodka C, Smith P, Nayak S, Rinehart L, Gingerich R, et al. Pharmacokinetic actions of exendin‐4 in the rat: comparison with glucagon‐like peptide‐1. Drug Dev Res 2001; 53: 260–267. [Google Scholar]
- 20. Tatarkiewicz K, Sablan E, Polizzi C, Parkes D. Long‐term metabolic benefits of exenatide in mice are mediated solely via the known glucagon‐like peptide‐1 receptor. Diabetes 2011; 60: A480. [DOI] [PubMed] [Google Scholar]
- 21. Darpo B, Sager P, MacConell L, Cirincione B, Mitchell M, Han J, et al. Exenatide at therapeutic and supratherapeutic concentrations does not prolong the QTc interval in healthy subjects. Br J Clin Pharmacol 2013; 75: 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fineman M, Phillips L, Jaworowicz DJ, Cirincione B, Ludwig E, Taylor K, et al. Model‐based evaluations to select and confirm doses in the clinical development of exenatide. Clin Pharmacol Ther 2007; 81: S111. [Google Scholar]
- 23. Phillips L, Fineman M, Taylor K, Baron A, Ludwig E, Grasela T. Population modeling to guide phase 3 dose selection for AC2993 (synthetic exendin‐4). Clin Pharmacol Ther 2002; 71: P29. [Google Scholar]
- 24. US Food and Drug Administration . Guidance for industry: bioanalytical method validation; 2001. [online]. Available at http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf (last accessed 8 October 2014).
- 25. Xu XS, Yuan M, Karlsson MO, Dunne A, Nandy P, Vermeulen A. Shrinkage in nonlinear mixed‐effects population models: quantification, influencing factors, and impact. AAPS J 2012; 14: 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holford N. The visual predictive check – superiority to standard diagnostic (Rorschach) plots. Paper presented at: PAGE 142005.
- 27. Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD, et al. Effects of exenatide (exendin‐4) on glycemic control over 30 weeks in sulfonylurea‐treated patients with type 2 diabetes. Diabetes Care 2004; 27: 2628–2635. [DOI] [PubMed] [Google Scholar]
- 28. DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin‐4) on glycemic control and weight over 30 weeks in metformin‐treated patients with type 2 diabetes. Diabetes Care 2005; 28: 1092–1100. [DOI] [PubMed] [Google Scholar]
- 29. Kendall DM, Riddle MC, Rosenstock J, Zhuang D, Kim DD, Fineman MS, et al. Effects of exenatide (exendin‐4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care 2005; 28: 1083–1091. [DOI] [PubMed] [Google Scholar]
- 30. Yan X, Mager DE, Krzyzanski W. Selection between Michaelis–Menten and target‐mediated drug disposition pharmacokinetic models. J Pharmacokinet Pharmacodyn 2010; 37: 25–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Linnebjerg H, Kothare PA, Seger M, Wolka AM, Mitchell MI. Exenatide ‐ pharmacokinetics, pharmacodynamics, safety and tolerability in patients ≥75 years of age with type 2 diabetes. Int J Clin Pharmacol Ther 2011; 49: 99–108. [DOI] [PubMed] [Google Scholar]
- 32. Bhavsar S, Mudaliar S, Cherrington A. Evolution of exenatide as a diabetes therapeutic. Curr Diabetes Rev 2013; 9: 161–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kagan L. Pharmacokinetic modeling of the subcutaneous absorption of therapeutic proteins. Drug Metab Dispos 2014; 42: 1890–1905. [DOI] [PubMed] [Google Scholar]
- 34. Watson E, Jonker DM, Jacobsen LV, Ingwersen SH. Population pharmacokinetics of liraglutide, a once‐daily human glucagon‐like peptide‐1 analog, in healthy volunteers and subjects with type 2 diabetes, and comparison to twice‐daily exenatide. J Clin Pharmacol 2010; 50: 886–894. [DOI] [PubMed] [Google Scholar]
- 35. Fineman MS, Mace KF, Diamant M, Darsow T, Cirincione BB, Booker Porter TK, et al. Clinical relevance of anti‐exenatide antibodies: safety, efficacy and cross‐reactivity with long‐term treatment. Diabetes Obes Metab 2012; 14: 546–554. [DOI] [PubMed] [Google Scholar]
- 36. Perez Ruixo JJ, Ma P, Chow AT. The utility of modeling and simulation approaches to evaluate immunogenicity effect on the therapeutic protein pharmacokinetics. AAPS J 2013; 15: 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kolterman OG, Kim DD, Shen L, Ruggles JA, Nielsen LL, Fineman MS, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm 2005; 62: 173–181. [DOI] [PubMed] [Google Scholar]
- 38. Kolterman OG, Buse JB, Fineman MS, Gaines E, Heintz S, Bicsak TA, et al. Synthetic exendin‐4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 2003; 88: 3082–3089. [DOI] [PubMed] [Google Scholar]
- 39. Calara F, Taylor K, Han J, Zabala E, Carr EM, Wintle M, et al. A randomized, open‐label, crossover study examining the effect of injection site on bioavailability of exenatide (synthetic exendin‐4). Clin Ther 2005; 27: 210–215. [DOI] [PubMed] [Google Scholar]