

Abstract
Aims
The main objectives of these two phase I studies were to investigate safety and tolerability as well as the pharmacokinetic/pharmacodynamic profile of the novel potent and selective formyl peptide receptor type 2 (FPR2)/Lipoxin A4 receptor (ALX) agonist ACT‐389949. A challenge model was used to assess the drug's anti‐inflammatory potential, with the aim of selecting a dosing regimen for future patient studies.
Methods
Two double‐blind, randomized phase I studies investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of ACT‐389949 at different doses and dosing regimens. Drug exposure was correlated with target engagement markers such as receptor internalization and cytokine measurements. The effect of FPR2/ALX agonism on neutrophil migration was studied in a lipopolysaccharide (LPS) inhalation model.
Results
ACT‐389949 was well tolerated. Maximum concentrations were reached around 2 h after dosing, with a mean terminal half‐life of 29.3 h [95% confidence interval (CI) 25.5, 33.7]. After multiple‐dose administration, exposure increased by 111% (95% CI 89, 136), indicating drug accumulation.
Administration of ACT‐389949 resulted in a dose‐dependent, long‐lasting internalization of FPR2/ALX into leukocytes. Pro‐ and anti‐inflammatory cytokines were dose‐dependently but transiently upregulated only after the first dose. No pharmacological effect on neutrophil count was observed in the LPS challenge test performed at steady state.
Conclusions
FPR2/ALX agonism with ACT‐389949 was shown to be safe and well tolerated in healthy subjects. Receptor internalization and downstream mediators pointed towards a desensitization of the system, which may explain the lack of effect on neutrophil recruitment in the LPS challenge model.
Keywords: biomarker, cytokines, dose finding, FACS, first in class, flow cytometry, FPR2/ALX, inflammation, LPS challenge, sputum
What is Already Known about this Subject
Formyl Peptide Receptor Type 2 (FPR2/ALX) receptor agonism is a new therapeutic concept to treat inflammatory conditions.
In vivo activation of FPR2/ALX is dependent on the nature of the ligand and leads to either pro‐ or anti inflammatory activity.
Currently, there is no published clinical data on targeting FPR2/ALX receptors.
What this Study Adds
Two Phase 1 studies have shown that the first‐in‐class FPR2/ALX agonist ACT‐389949 was safe and well tolerated in healthy subjects.
Biomarkers revealed a dose‐dependent pharmacological response on Day 1 that was not retained at steady state.
The biomarker evaluation suggests a transient pro‐inflammatory rather than an anti‐inflammatory profile for this compound.
ACT‐389949 may not be a viable drug due to rapid receptor desensitisation.
Tables of Links
| TARGETS | |
|---|---|
| G protein‐coupled receptors 2 | |
| ADRB2 http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=249 | |
| FPR2/ALX |
| LIGANDS | |
|---|---|
| Annexin I | LPS |
| CCL2 | LXA4 |
| Chemerin | SAA |
| CXCL8 | salbutamol |
| Formoterol | salmeterol |
| IL‐6 | TNF‐α |
| IL‐10 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2.
Introduction
Inflammation is a protective response of the immune system, triggered by injury or infection. The inflammatory response is tightly regulated and chronic inflammation can be due to a sustained proinflammatory response or to a lack of resolution 3, 4, 5. The absence or dysregulation of resolution of inflammation leads to chronic inflammation, a hallmark of many inflammatory medical conditions 4. Many lipid ligands, such as lipoxin, resolvins and maresins, as well as peptidic ligands such as annexin 1 and chemerin, are associated with anti‐inflammatory activities, including the suppression of proinflammatory cytokine secretion and inhibition of polymorphonuclear leukocyte (neutrophil) recruitment 3, 6, 7, 8. Bioactive lipids exert their effects through binding to specific G protein‐coupled receptors (GPCRs) 9, 10.
The formyl peptide receptor type 2 (FPR2 or FPR2/ALX), formerly known as FPRL1, is a GPCR and is expressed on monocytes and neutrophils 11. Activation of FPR2/ALX is mediated by a number of lipid as well as protein ligands, and is functionally implicated in anti‐inflammatory processes. In particular, activation by lipoxin A4 and annexin 1 has been linked to resolution of inflammation, including upregulation of anti‐inflammatory cytokines such as IL‐10 12, 13. Limited preclinical evidence of FPR2/ALX modulation suggests an exaggerated inflammatory response in a mouse model in which Fpr2 was deleted 14. However, some protein ligands of FPR2/ALX [e.g. serum amyloid A (SAA)] are associated with proinflammatory responses such as secretion of CXCL8 [interleukin (IL) 8] by human neutrophils 15, 16. Although there is strong interest in the concept 4, 5, thus far, the clinical benefits of mediators of resolution of inflammation have not been demonstrated.
ACT‐389949 (2‐methyl‐5‐m‐tolyl‐oxazole‐4‐carboxylic acid {2‐[4‐(1,1‐difluoro‐ethyl)‐oxazol‐2‐ylmethyl]‐2H‐[1,2,3]triazol‐4‐yl}‐amide) is a new potent [half maximal effective concentration (EC50) 3 nM = 1.3 ng ml−1 for FPR2/ALX internalization into monocytes] and selective FPR2/ALX agonist that has potential for the treatment of inflammatory disorders.
The safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of single‐ and multiple‐ascending doses of ACT‐389949 over a wide dose range (1–1000 mg) were investigated in healthy subjects, including dosing once daily (o.d.), every other day (q2d) and every 3 days (q3d). Biomarkers were included to investigate the relationship between exposure to and receptor activation (PD) of ACT‐389949. In addition, a challenge model of lipopolysaccharide (LPS) inhalation was used to assess the potential of this novel compound in resolving inflammation.
Methods
Study design and subjects
ACT‐389949 was investigated in a single‐ascending dose (SAD) as well as a multiple‐ascending dose (MAD) study. The SAD study was a prospective, single‐centre, double‐blind, parallel‐group, randomized, placebo‐controlled study. Sixty‐four healthy males aged 18–45 years were included. In each of the eight dose groups (1, 5, 20, 50, 100, 200, 500 and 1000 mg), eight subjects were randomized to receive a single oral dose of either ACT‐389949 (six subjects) or matching placebo (two subjects) in the fasted state (Table S1). In the MAD study, a total of 65 healthy subjects (62 male and 3 female, aged 18–58 years), 13 in each group, were enrolled and received ACT‐389949 (n = 10 per group) or placebo (n = 3 per group). Multiple oral doses of 40, 200 or 800 mg ACT‐389949 were administered, either o.d., q2d or q3d in the morning in the fasted state. In the o.d. groups, nine doses, and in the q2d/q3d groups, five doses were administered (Table S1).
For both studies, healthy Caucasian subjects with no clinically significant findings on physical examination were selected, using the following inclusion criteria: body mass index (BMI) between 18.0 kg m−2 and 30.0 kg m−2, tympanic temperature (T°) 35.5–37.5°C at screening and prior to (first) dosing, total and differential white blood cell (WBC) count strictly within normal ranges at screening and on Day −1, and C‐reactive protein (CRP) levels below 5 mg l−1. Subjects were excluded if they had treatment with any prescribed or over‐the‐counter medications (e.g. vaccines, acetylsalicylic acid, steroidal or nonsteroidal anti‐inflammatory drugs, and herbal medicines such as St John's Wort) during the study and 2 weeks prior to (first) study drug administration or five half‐lives of the medication (whichever was longer), or signs of infection (viral, systemic fungal, bacterial or protozoal) within 4 weeks prior to first study drug administration.
The studies were conducted in accordance with the Declaration of Helsinki, performed according to good clinical practice guidelines and registered with ClinicalTrials.gov (NCT02099071, NCT02099201). The subjects consented in writing to the study after receiving a full explanation of the trial; the subjects all signed an Informed Consent. The SAD study was approved by the ethics committee METC Assen, the Netherlands, study ID: 115 358‐CS0171, on 18 November 2011. The MAD study was approved by the Office for Research Ethics Committees Northern Ireland (ORECNI), study ID 12/NI/0145, on 18 October 2012.
Rationale for dose selection
The dose range (1–1000 mg) for the SAD study was based on safety margin calculations vs. the most sensitive preclinical species (rat). The predicted safety margin based on human‐equivalent doses was in the range 3429–39 714‐fold at the starting dose of 1 mg, and 3.4–38.9‐fold at a dose of 1000 mg. MAD doses were based on the prediction that the highest dose of 800 mg o.d. would achieve exposure at steady state similar to that following a single dose of 1000 mg. The SAD data showed pharmacological activity starting at 20 mg (Figure 1A,B). Therefore, 40 mg was chosen as the starting dose in the MAD study. Based on the less than dose‐proportional increase in ACT‐389949 exposure in the SAD study, an intermediate dose of 200 mg was studied as a bridge between the 40 mg and 800 mg doses. The safety data from the SAD study supported the selected dose range.
Figure 1.
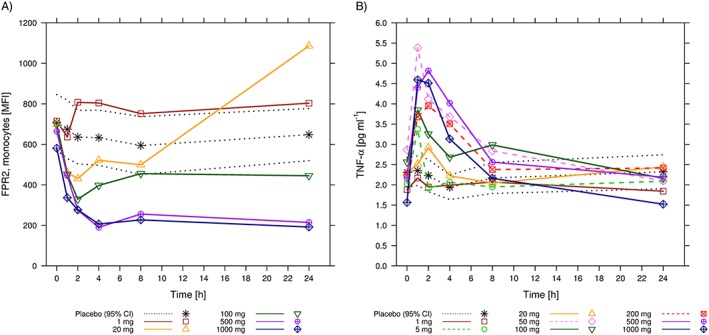
Pharmacodynamic markers over time following single doses of ACT‐389949. (A) Arithmetic mean [and 95% confidence interval (CI) for placebo] formyl peptide receptor type 2 (FPR2)/ surface expression on monocytes measured as mean fluorescence intensity (MFI). (B) Arithmetic mean (and 95% CI for placebo) tumour necrosis factor α (TNF‐α) levels in plasma by dose group
Safety and tolerability
In both studies, vital signs, physical examination, spirometry and an electrocardiogram (ECG) were assessed and adverse events (AEs) were recorded. For each assessment of routine laboratory variables, blood was taken for haematology (including WBC), clinical chemistry, coagulation tests and erythrocyte sedimentation rate.
PK
At each PK time point, blood was collected into an ethylenediaminetetraacetic acid (EDTA) tube. Concentrations of ACT‐389949 in plasma were determined using a validated liquid chromatography with tandem mass spectrometry (LC–MS/MS) assay. The lower limit of quantification (LLOQ) was 0.50 ng ml−1. In the SAD study, samples were collected prior to the first dose on Day 1 and at 20 min and 40 min, and 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48 and 60 h after drug intake. In the MAD study, samples were taken prior to the first dose and 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 18 and 24 h postdose on Day 1; predose and 2 h postdose (q2d dosing only) from Day 1 to Day 7/10; and 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 18, 24, 48 and 72 h postdose on Day 7/10.
PD
Target engagement biomarker
Flow cytometry analysis of cell surface levels of FPR2/ALX was performed in selected dose groups. In the SAD study, the time profile was analysed for Day 1 (0, 1, 2, 4, 8, and 24 h) and in the MAD study for Day 1 and at steady state (i.e., Day 7 or 10; 0, 2, 6, 12 and 24 h).
Blood specimens collected in heparin tubes were stored at 4°C until processing following established protocols. All samples were acquired using a BD fluorescence‐activated cell sorting (FACS) Canto II flow cytometer using the FACSDiva software (BD Biosciences, St Jose, CA, USA). Gating of the cell population was established with neutrophils (CD16+ CD14–) and monocytes (CD16– CD14+). Processed samples were acquired until 2000 monocytes had been collected. Placebo data from all dose groups (SAD: 1, 20, 100, 500 and 800 mg) or MAD (40 mg and 200 mg) were pooled (SAD: n = 10, MAD n = 6). Mean and 95% confidence interval (CI) were shown for placebo and active treatment (SAD and MAD, n = 10). In the overview analysis, only the 2 h postdose time point on Day 1 and at steady state (Day 7 for o.d. and q2d dosing and Day 10 for q3d dosing) were compared.
Plasma cytokine measurements
Cytokine measurements were performed in all treatment groups at predose, and 1, 2, 6 and 24 h postdose on Day 1 and at steady state (Day 7/10).
IL‐8 (CXCL8) and monocyte chemoattractant protein 1 [CCL2 (MCP‐1)] were quantified in serum using a custom duplex immunoassay (N45IA‐1) from Mesoscale (MSD, Rockville, MD, USA). The assay was performed according to the manufacturer's instructions. The assay range for CCL2 (MCP‐1) was 2.4–2500 pg ml−1, with a coefficient of variation (CV) <20%, and for IL‐8 0.61–2500 pg ml−1, CV < 20%.
Tumour necrosis factor α (TNF‐α), IL‐6 and IL‐10 were quantified in serum using Singulex kits following the manufacturer's instructions (Singulex Inc., Alameda, CA, USA). The assay range for TNF‐α was 0.075–50 pg ml−1, CV < 15%; for IL‐6 was 0.035–50 pg ml−1, CV13%; and for IL‐10 was 0.6–50 pg ml−1, CV < 12%.
Data are represented as mean (SAD: n = 16 placebo, n = 6/dosing group; MAD: n = 15 placebo, n = 10 per dosing group) and 95% CI. No further statistical analysis was performed.
Cytokine levels were compared at the time of maximum induction (2 h postdose) on Day 1 and at steady state. Steady state was reached on Day 7 for 40 mg and 200 mg o.d., and on Day 10 for 200 mg q3d.
LPS challenge test
On Day 8 (o.d. and q2d) or Day 12 (q3d), the sponsor and investigator jointly reviewed all safety and tolerability data. A decision was subsequently made as to whether the subject should receive an inhaled dose of 50 μg LPS (Sigma‐Aldrich, Dorset, UK, Lot No.091M4054) on Day 9 (o.d. and q2d) or Day 13 (q3d). LPS was inhaled 2 h postdose, followed by sputum induction at 8 h postdose.
Not all subjects were able to produce amounts of sputum sufficient for analysis on Days 9/13, and two subjects did not receive inhaled LPS; therefore, the number of subjects pooled for analysis was lower than the total number of subjects in the dataset.
The absolute counts and percentages (of total cell counts) for neutrophils, monocytes, eosinophils, endothelial cells and lymphocytes were calculated and presented as a box‐whisker plot.
Data analysis
Tolerability parameters were analysed descriptively. The maximum plasma concentration (Cmax), time to reach maximum plasma concentration (tmax) and terminal half‐life (t1/2) were derived by noncompartmental analysis using WinNonLin Version 5.2 Software (Pharsight, Mountain View, CA, USA). The area under the plasma concentration–time curve from time zero to time t of the last measured concentration above the LLOQ (AUC0–t), as well as over one dosing interval (AUCτ), were calculated using the linear trapezoidal rule. The t1/2 was calculated from t1/2 = ln(2/λz) where λz represents the terminal elimination rate constant determined by log linear regression of the measured plasma concentrations in the terminal elimination phase. The accumulation index (AI) was calculated by dividing AUCτ on Day 7/10 by AUCτ on Day 1. AUC and Cmax values were assumed to be log‐normally distributed.
Statistical analysis
Dose proportionality in the PK of ACT‐389949 was analysed in the SAD study by comparing dose‐normalized Cmax and AUC0–t values using a power model 17.
The nomenclature of receptors and ligands was used in accordance with International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification (IUPHAR) classification and nomenclature 2.
Results
Safety and tolerability
ACT‐389949 was well tolerated in healthy subjects with daily doses of up to 800 mg for 9 days. All reported AEs were of mild to moderate intensity and resolved without sequelae (Table S2).
No clinically significant findings were observed in vital signs, ECGs, spirometry, haematology (except WBC counts), coagulation tests, clinical chemistry or urinalysis (Table S2).
Doses from 40 mg to 800 mg on Day 1 resulted in a dose‐dependent systemic WBC decrease (in particular, monocytes and neutrophils) 1–2 h postdose compared with placebo (Figure 2). Following this decrease, WBC counts rapidly increased again, returning to predose baseline values by 2 h postdose for some subjects and by 6 h postdose for all subjects. Drug‐related decreases in WBC on Day 1 were reported as an AE in 7/65 (11%) subjects, observed in all groups above 40 mg (three subjects on 200 mg, four subjects on 800 mg). No effect was observed on Day 7 following multiple oral dosing for any dose or dosing regimen. Other reported AEs are summarized in Table S2.
Figure 2.
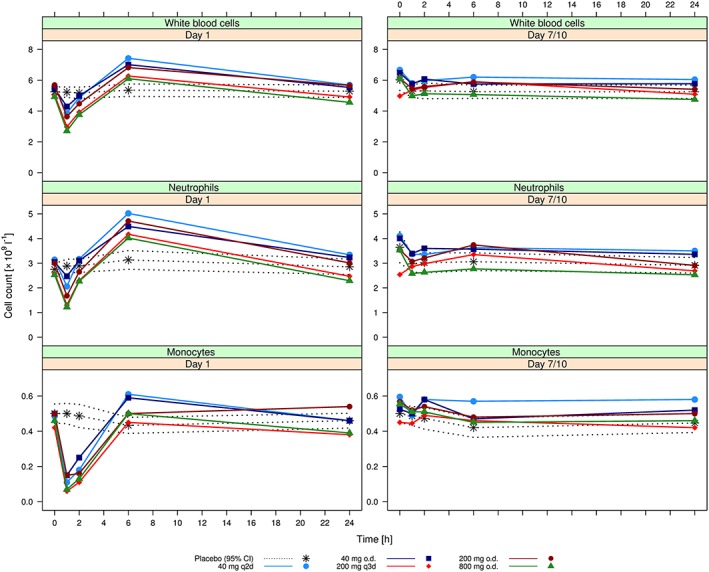
Arithmetic mean [and 95% confidence interval (CI) for placebo] cell counts for white blood cells (top), neutrophils (middle) and monocytes (bottom) following administration of the first dose of ACT‐389949 on Day 1 (left) and at steady state on Day 7/10 (right) by dose group
PK
Following both single and repeat oral dosing, ACT‐389949 was rapidly absorbed, with a median tmax between 1.0 h and 4.0 h. After attainment of Cmax, plasma concentrations decreased in a multiphasic manner, with a mean terminal t1/2 of 29.3 h (95% CI 25.5, 33.7) (Figure 3). A second peak was observed at 12–24 h postdose following single‐dose administration, more prominently at higher doses. A summary of ACT‐389949 PK parameters is shown in Table 1.
Figure 3.
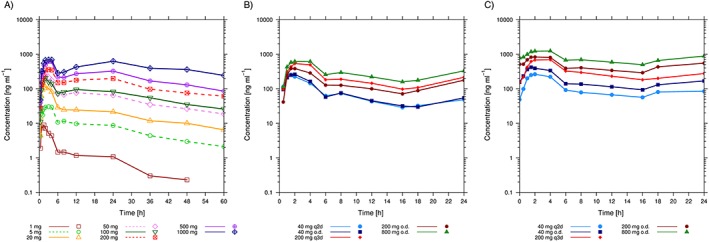
Arithmetic mean plasma concentration–time profiles of ACT‐389949 following single doses (single‐ascending dose study) (A) and multiple doses (multiple‐ascending dose study) on Day 1 (B) and at steady state on Day 7/10 (C) by dose group
Table 1.
Plasma pharmacokinetic variables of ACT‐389949 after administration of single doses of 1, 5, 20, 50, 100, 200, 500 and 1000 mg or multiple doses of 40, 200 and 800 mg of ACT‐389949 once daily (o.d.), every other day (q2d) and every 3 days (q3d)
| Dose | Period | N | C max (ng ml −1 ) | t max (h) | AUC a (ng*h ml −1 ) | t 1/2 (h) | AI AUC τ b | AI C max b |
|---|---|---|---|---|---|---|---|---|
| Single‐ascending dose study (SAD) | ||||||||
| 1 mg | ‐ | 6 | 10.2 (6.7, 15.6) | 1.3 (0.7–3.0) | 57.4 (44.4, 74.1) | 33.9 (21.2, 54.1) | – | – |
| 5 mg | ‐ | 6 | 39.6 (30.3, 51.8) | 2.0 (0.5–4.0) | 446 (310, 641) | 22.6 (12.5, 41.2) | – | – |
| 20 mg | ‐ | 6 | 146 (117, 183) | 1.5 (1.5–3.0) | 1295 (1039, 1614) | 30.4 (16.2, 56.9) | – | – |
| 50 mg | ‐ | 6 | 304 (230, 402) | 1.5 (0.7–2.0) | 3479 (2664, 4545) | 24.2 (15.4, 38.2) | – | – |
| 100 mg | ‐ | 6 | 223 (158, 315) | 1.8 (1.0–6.0) | 4059 (3581, 4602) | 21.3 (16.5, 27.5) | – | – |
| 200 mg | ‐ | 7 | 393 (264, 585) | 3.0 (1.5–4.0) | 7940 (5499, 11 464) | 38.8 (21.6, 69.9) | – | – |
| 500 mg | ‐ | 6 | 656 (477, 901) | 3.5 (1.5–4.0) | 13 137 (9750, 17 699) | 28.0 (17.0, 46.1) | – | – |
| 1000 mg | ‐ | 6 | 843 (619, 1147) | 2.5 (1.5–24.0) | 24 850 (18 460, 33 454) | 33.4 (14.6, 76.4) | – | – |
| Multiple‐ascending dose study (MAD) | ||||||||
| 40 mg o.d. | Day 1 | 10 | 281 (229, 345) | 1.75 (1.0–4.0) | 1795 (1547, 2081) | – | – | – |
| Day 7 | 10 | 417 (348, 498) | 1.5 (1.0–4.0) | 4013 (3269, 4926) | – | 2.2 (1.9, 2.6) | 1.5 (1.2, 1.8) | |
| 40 mg q2d | Day 1 | 10 | 275 (200, 379) | 1.0 (1.0–2.0) | 2652 (2262, 3109) | – | – | – |
| Day 7 | 10 | 280 (228, 345) | 2.0 (1.0–4.0) | 3950 (3350, 4657) | – | 1.5 (1.3, 1.7) | 1.0 (0.7, 1.4) | |
| 200 mg o.d. | Day 1 | 10 | 401 (310, 544) | 1.75 (1.5–4.0) | 3276 (2476, 4334) | – | – | – |
| Day 7 | 10 | 912 (750, 1108) | 2.0 (1.5–4.0) | 10 759 (8250, 14 032) | – | 3.3 (2.7, 4.0) | 2.2 (1.7, 2.9) | |
| 200 mg q3d | Day 1 | 10 | 551 (408, 745) | 2.0 (1.5–4.0) | 11 355 (8692, 14 834) | – | – | – |
| Day 10 | 10 | 736 (580, 934) | 2.0 (1.5–4.0) | 16 651 (12 503, 22 175) | – | 1.5 (1.2, 1.8) | 1.3 (1.1, 1.6) | |
| 800 mg o.d. | Day 1 | 10 | 639 (469, 870) | 2.0 (1.5–24.0) | 6633 (5114, 8605) | – | – | – |
| Day 7 | 10 | 1281 (1019, 1610) | 4.0 (1.5–4.0) | 17 488 (13 933, 21 950) | – | 2.6 (2.2, 3.1) | 2.0 (1.6, 2.5) | |
| Overall (geometric means and 95 % CI) | – | – | – | 29.3 (25.5, 33.7) | 2.1 (1.9, 2.4) | 1.6 (1.4, 1.8) | ||
Data are expressed as geometric means (and 95% CI) or for tmax as median (and range). AI, accumulation index; AUCτ, area under the plasma concentration–time curve over one dosing interval; CI, confidence interval; Cmax, maximum plasma concentration; t1/2, ; tmax, maximum plasma concentration
Area under the plasma concentration–time curve from zero to time t of the last measured concentration above the limit of quantification (24 h or 48 h postdose for 1 mg and 60 h postdose for 5–1000 mg) for single doses (SAD) and during the dosing interval τ for multiple dosing (MAD); τ is 24 h for all o.d. dosing regimens, 48 h for q2d dosing and 72 h for q3d dosing
Accumulation index = AUCτ Day 7(10)/AUCτ Day 1, Cmax Day 7(10)/Cmax Day 1
Exposure to ACT‐389949 increased in a less than dose‐proportional manner following single doses from 1 mg to 1000 mg and multiple doses from 40 mg to 800 mg. The dose‐proportionality coefficient for the power test based on the SAD data was 0.62 (90% CI 0.58, 0.67) for Cmax and 0.84 (90% CI 0.80, 0.88) for AUC.
Following multiple dosing, steady‐state conditions were reached at the fourth dose. At steady state, mean AUCτ and Cmax increased by 111% (95% CI 89, 136) and 55% (95% CI 37, 75), respectively, indicating low to moderate accumulation.
PD
Target engagement biomarker
Flow cytometry was used to study receptor internalization as a characterized measure of target engagement. In the SAD study, FPR2/ALX receptor internalization into monocytes was observed at doses of 20 mg and higher following single oral doses of ACT‐389949 (Figure 4A). Minimum levels in mean fluorescence intensity (MFI) were observed at 2–4 h postdose. Following doses of 100 mg and higher, FPR2/ALX internalization was sustained from 2 h postdose up to the assessment at 24 h postdose. Receptor activity returned to baseline levels at 24 h only following the 20 mg dose. Maximum FPR2/ALX internalization was observed with doses of 500 mg and higher.
Figure 4.
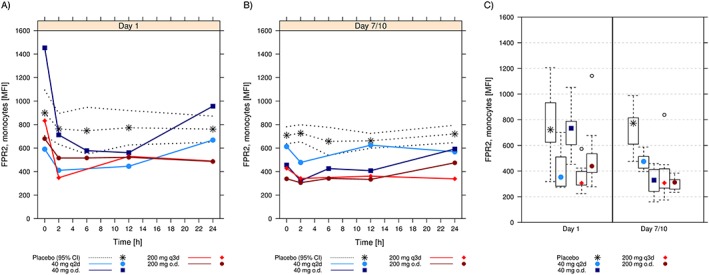
Fluorescence‐activated cell sorting (FACS) analysis of formyl peptide receptor type 2 (FPR2)/ surface expression on monocytes. Arithmetic mean [and 95% confidence interval (CI) for placebo] fluorescence intensity (MFI) over time following administration of the first dose of ACT‐389949 on Day 1 (A) and at steady‐state on Day 7/10 (B) by dose group. (C) Comparison of the surface expression at 2 h postdose on Day 1 and Day 7/10. Boxes indicate the range of the middle 50% of the data. Medians are shown as symbols in the boxes. Whiskers indicate the 95% range. Extreme values are shown individually. o.d; once daily; q2d, every other day; q3d, every 3 days
In the MAD study, FPR2/ALX surface expression levels on monocytes after the first dose were similar to those obtained in the SAD study (Figure 4A). Under steady‐state conditions at Day 7/10, the FPR2/ALX expression levels on monocytes did not change after dosing and were consistently below the levels of the placebo group (Figure 4B). Mean values showed that, while placebo values remained stable over time, FPR2/ALX surface levels were reduced and did not change (except with 40 mg o.d.) after further dosing, for any dose or dosing regimen investigated (Figure 4C).
Plasma cytokine measurements
Pro‐ and anti‐inflammatory cytokine induction was measured as downstream markers of FPR2/ALX receptor activation and as guidance to the drug's pharmacological profile.
In the SAD study, transient increases in serum levels of TNF‐α were generally observed at doses of 5 mg and higher, with the largest effect observed following 500–1000 mg (Figure 1B). This cytokine regulation was transient following Cmax and returned to baseline at 8 h postdose.
In the MAD study, serum levels of IL‐10 (Figure 5) and TNF‐α (Figure 6), as well as IL‐6, IL‐8 and MCP‐1, were assessed to investigate the change in cytokine markers following repeated dosing of ACT‐389949 (Figure S1).
Figure 5.
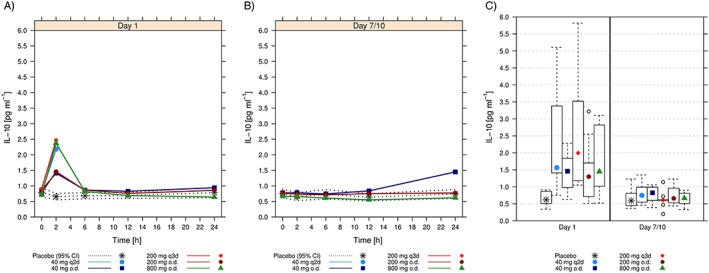
Arithmetic mean [and 95% confidence interval (CI) for placebo] interleukin (IL) 10 plasma concentrations over time following administration of the first dose of ACT‐389949 on Day 1 (A) and at steady state on Day 7/10 (B) by dose group. (C) Comparison of plasma levels 2 h postdose on Day 1 and Day 7/10. Boxes indicate the range of the middle 50% of the data. Medians are shown as symbols in the boxes. Whiskers indicate the 95% range. Extreme values are shown individually. o.d; once daily; q2d, every other day; q3d, every 3 days
Figure 6.

Arithmetic mean [and 95% confidence interval (CI) for placebo] tumour necrosis factor α (TNF‐α) plasma concentrations over time following administration of the first dose of ACT‐389949 on Day 1 (A) and at steady state on Day 7/10 (B) by dose group. (C) Comparison of plasma levels 2 h postdose on Day 1 and Day 7/10. Boxes indicate the range of the middle 50% of the data. Medians are shown as symbols in the boxes. Whiskers indicate the 95% range. Extreme values are shown individually. o.d; once daily; q2d, every other day; q3d, every 3 days
Analysis of IL‐10 and TNF‐α over 24 h on Day 1 showed a sharp, dose‐dependent increase in serum cytokine levels at 2 h postdose, starting at 40 mg (Figure 5A, Figure 6A).
This observation was consistent with serum levels of other cytokines (Figure S1). Under steady‐state conditions (Days 7/10), TNF‐α and IL‐10 (as well as IL‐6, IL‐8 and MCP‐1) were not regulated 2 h after dosing, independent of the dosing regimen (o.d., q2d, q3d) (Figures 5 and 6 B,C, Figure S1).
LPS challenge test
Neutrophil and macrophage effects (absolute counts and percentage per gram of sputum) at screening and after the LPS challenge on Day 9 (o.d. and q2d) or Day 13 (q3d) generally showed a large intersubject variability (Figure 7). Similar analyses were performed for eosinophils, epithelial cells and lymphocytes (Figure S2).
Figure 7.
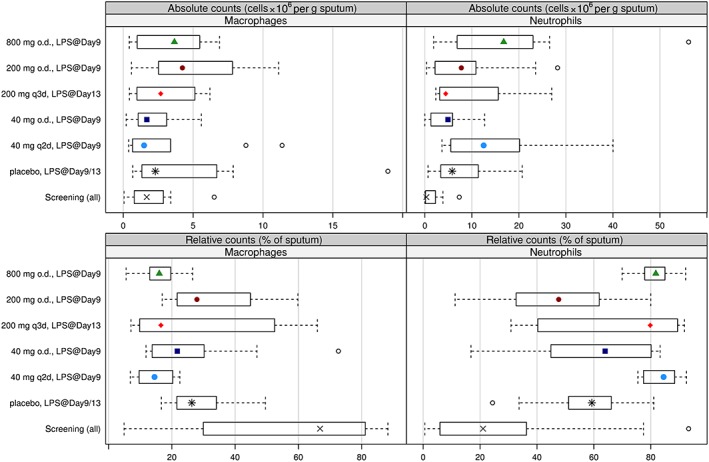
Differential cell counts in the sputum following lipopolysaccharide (LPS) challenge. Absolute (top) and relative (bottom) counts for macrophages (left) and neutrophils (right) in the sputum at 6 h post‐LPS on Day 9/13 by dose group. Boxes indicate the range of the middle 50% of the data. Medians are shown as symbols in the boxes. Whiskers indicate the 95% range. Extreme values are shown individually
Following LPS administration on Day 9 (o.d. and q2d) or Day 13 (q3d), sputum neutrophil percentages in the placebo group increased approximately threefold compared with screening values. Percentage sputum macrophages decreased from screening values following LPS administration, in line with previous observations 18. This indicates that the administration of LPS elicited the intended local inflammatory response in the lung. Sputum neutrophil and macrophage percentages of the subjects receiving ACT‐389949 were no different from those receiving placebo.
Discussion
In view of the novelty of this pharmacological class, the main aim of the SAD and MAD studies was carefully to monitor safety and tolerability. In addition, mechanistic assessments were performed to predict treatment effects better and guide dose selection in future patient studies. A biomarker‐enriched trial design was therefore applied in these entry‐into‐human studies with the first‐in‐class FPR2/ALX receptor agonist ACT‐389949.
ACT‐389949 was rapidly absorbed, reaching peak plasma concentrations 1–4 h after dosing following single‐ and multiple‐dose administration. Drug exposure increased in a less than dose‐proportional manner over the dose range investigated and showed moderate accumulation. The less than dose‐proportional increase in systemic exposure might be attributed to the low solubility of the compound. Overall, ACT‐389949 was well tolerated. Only a transient reduction in WBC count related to the drug's pharmacological effect was observed.
FPR2/ALX receptor internalization has been shown to be a valuable PD marker to demonstrate target engagement and guide dose selection 19. Recent studies demonstrated that lipoxin A4‐mediated FPR2/ALX internalization is necessary for functional activation 20, 21. In the SAD study, transient FPR2/ALX receptor internalization into monocytes (and neutrophils, data not shown) was observed following a single dose of 20 mg but there was only minimal receptor activation at this dose level, as reflected in most PD markers. Higher doses tested led to extended (>24 h) receptor internalization, with maximum but transient PD effects occurring close to tmax. Interestingly, the majority of soluble PD markers exhibited the same profile – i.e. a narrow peak around 2 h on Day 1 and a quick return to baseline levels, while FPR2/ALX expression levels remained low.
In an attempt to elicit a lower than maximum FPR2/ALX activation and avoid constant receptor internalization, the dose of 40 mg was tested in an intermittent dosing regimen (q2d). In addition, to achieve the desired pharmacological activity, 200 mg was investigated at an intermittent dosing regimen (q3d). At all doses and dosing regimens tested, FPR2/ALX showed complete desensitization within 2 days.
The fact that FPR2/ALX receptor levels did not return to baseline levels following 200 mg q3d or 40 mg q2d (at which exposure showed minimal accumulation) suggests that ACT‐389949 exposure was either not sufficiently reduced within the monitored time frame to allow the FPR2/ALX receptors to be re‐expressed on the surface of neutrophils and monocytes, or that it otherwise leads to prolonged FPR2/ALX desensitization.
In line with the dose‐dependent cell surface loss of FPR2/ALX receptors, which did not recover during the dosing regimens investigated, were the other PD findings of the transient nature of cytokine induction and the transient reduction in WBC count.
Together, these markers suggest that ACT‐389949 is able to exert a pharmacological effect on systemic neutrophils (and WBC in general) on Day 1 but not during steady‐state conditions for any dose or dosing regimen investigated in the present studies. The clinical relevance of the rapid and transient decrease in WBC count is currently unclear. However, it remains an interesting observation in the context of the mechanism of action and warrants further investigation.
Several reports suggest that lipid mediators, such as lipoxins, elicit pro‐resolution activities, while peptide ligands are pro‐inflammatory mediators when binding to FPR2/ALX 22.
In order to characterize the novel FPR2/ALX agonist ACT‐389949 clinically, pro‐ and anti‐inflammatory cytokines were measured 11. In vitro human cell stimulation experiments have shown that IL‐6, IL‐8, MCP‐1, TNF‐α and IL‐10 were dose‐dependently upregulated following stimulation with ACT‐389949 (data not shown).
While anti‐inflammatory or pro‐resolution mediators could be clinically useful under chronic inflammatory conditions, pro‐inflammatory activation is a new concept. We therefore included a set of cytokines representing pro‐inflammatory as well as anti‐inflammatory molecules. Our data from the SAD and MAD studies showed that all cytokines measured (IL‐6, IL‐8, IL‐10, MCP‐1 and TNF‐α) demonstrated the same transient, rapid activation pattern, and in the MAD study no further stimulation under steady‐state conditions was observed. A feature of a pro‐inflammatory reaction is the transient induction of neutropenia in peripheral blood following an immunological challenge 23, substantiating the observed activation pattern of FPR2/ALX by ACT‐389949.
Although TNF‐α and IL‐6 concentrations increased to levels measured under inflammatory conditions 24, 25, no effect on body temperature and CRP could be demonstrated and general tolerability was good.
In addition to the PD biomarkers, the study was designed to obtain proof‐of‐mechanism data following multiple doses of ACT‐389949, enabling a PK/PD dose prediction model for phase II proof‐of‐concept studies in patients.
The LPS challenge model has been proposed as one of the few available models to study the concept of anti‐inflammatory treatments and resolution of inflammation 5. To investigate whether chronic activation of FPR2/ALX modulates leukocyte recruitment, sputum neutrophils and monocytes were counted after inhalation of LPS in the MAD study as a mechanistic biomarker 26.
In the current trial, there was no pharmacological effect of ACT‐389949 on the counts of sputum neutrophils (or monocytes). This is consistent with the observation of no detectable FPR2/ALX on blood neutrophils or monocytes under repeated dosing, independent of the dosing schedule, and no measurable response in any of the investigated biomarkers further suggested a desensitization of the FPR2/ALX system.
The observed desensitization by a GPCR agonist such as ACT‐389949 and its resulting limitations for a prolonged therapeutic effect in chronic conditions shows similarities with the well‐described phenomenon for β2‐adrenoceptor (ADRB2) agonists for the treatment of asthma 27. Two different types of ADRB2 agonists activate the receptor in different ways, leading to distinct types of activation patterns 28. The less potent, long‐acting ABRB2 agonists salmeterol and formoterol do not desensitize, in contrast to the short‐acting agonist salbutamol 27, 29. Although the clinical effects of both classes are well described, the exact molecular mechanisms of desensitization only recently started to be better understood 30, 31, 32 and the clinical relevance of target modifiers has gained relevance for many drugs, particularly in the field of the central nervous system 33. Although representing a different class of GPCRs, with a different signal transduction, this shows the possibilities of different agonists having a very different therapeutic outcome.
As the FPR2/ALX system has many natural ligands that lead to a set of distinct activation patterns, it makes this receptor a likely candidate for the success of targeted molecules, when designed with a defined target profile. For this approach, a number of ways exist further to optimize molecules that make use of the concepts of functional selectivity and biased agonism 33, 34.
The data obtained by the dosing regimens assessed suggest that the therapeutic potential of this first‐in‐class molecule is limited. The consequent use of biomarkers at different levels of biological processes, and the employment of a wide variety of techniques, cumulatively draws a consistent picture. While the first activation of the receptor by ACT‐389949 led to a transient effect on all biomarkers explored, under all repeated dosing paradigms FPR2/ALX was lost from the cell surface and thus did not lead to further activation of cytokines. While ACT‐389949 may not be a suitable drug candidate owing to its desensitization, the therapeutic potential of the target warrants further investigation, to elucidate whether molecules with other physicochemical properties will have more favourable pharmacological effects under chronic dosing conditions.
Competing Interests
All authors were employees of Actelion Pharmaceuticals Ltd and received stocks or stock options.
The SAD and MAD studies were conducted by QPS Netherlands (principal investigator K. Abd‐Elaziz MD) and Celerion Belfast (principal investigator A. J. Stewart MD), respectively. We thank I. Kluge at Swiss BioAnalytics AG (Birsfelden, Switzerland) for bioanalytical determinations of ACT‐389949, and at Actelion Pharmaceuticals Ltd J. Marrie, H. Farine and J. Hoerner for technical and analytical support for the PD measurements, A. Mackie for coordination of the MAD study, and A. Klenk and C. Runser for their technical assistance.
Contributors
A.K.S, D.L., D.S.S., H.G.C., A.K., P.M.A.G. and J.D. wrote the manuscript. A.K.S. and J.D. contributed to the design of both the SAD and MAD studies, and H.G.C. contributed to the design of the SAD study. A.K.S., D.L., D.S.S., H.G.C. and A.K. analysed the data. All authors reviewed and approved the manuscript.
Supporting information
Figure S1 Arithmetic mean (and 95% confidence interval for placebo) for interleukin (IL) 6 (top), IL‐8 (middle) and monocyte chemoattractant protein 1 (bottom) following administration of the first dose of ACT‐389949 on Day 1 (left) and at steady state (Day 7/10) by dose group
Figure S2 Differential cell counts in the sputum following lipopolysaccharide (LPS) challenge. Absolute (top) and relative (bottom) counts for eosinophils (left), epithelial cells (middle) and lymphocytes (right) in the sputum at 6 h post‐LPS on Day 9/13 by dose group. Boxes indicate the range of the middle 50% of the data. Medians are shown as symbols in the boxes. Whiskers indicate the 95% range. Extreme values are shown individually
Table S1 Baseline demographic data for subjects in the single‐ascending dose and multiple‐ascending dose studies
Table S2 Most frequent (n ≥ 3 subjects) adverse events by preferred term (multiple‐ascending dose study)
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Stalder, A. K. , Lott, D. , Strasser, D. S. , Cruz, H. G. , Krause, A. , Groenen, P. M. A. , and Dingemanse, J. (2017) Biomarker‐guided clinical development of the first‐in‐class anti‐inflammatory FPR2/ALX agonist ACT‐389949. Br J Clin Pharmacol, 83: 476–486. doi: 10.1111/bcp.13149.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol 2010; 177: 1576–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perretti M, Leroy X, Bland EJ, Montero‐Melendez T. Resolution pharmacology: opportunities for therapeutic innovation in inflammation. Trends Pharmacol Sci 2015; 36: 737–755. [DOI] [PubMed] [Google Scholar]
- 5. Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov 2016; 15: 551–567. [DOI] [PubMed] [Google Scholar]
- 6. Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov 2004; 3: 401–416. [DOI] [PubMed] [Google Scholar]
- 7. Chiang N, Serhan CN. New mechanism for an old drug: aspirin triggers anti‐inflammatory lipid mediators with gender implications. Compr Ther 2006; 32: 150–157. [DOI] [PubMed] [Google Scholar]
- 8. Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol 2009; 9: 62–70. [DOI] [PubMed] [Google Scholar]
- 9. Kostenis E. A glance at G‐protein‐coupled receptors for lipid mediators: a growing receptor family with remarkably diverse ligands. Pharmacol Ther 2004; 102: 243–257. [DOI] [PubMed] [Google Scholar]
- 10. Back M, Powell WS, Dahlen SE, Drazen JM, Evans JF, Serhan CN, et al. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br J Pharmacol 2014; 171: 3551–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein‐coupled receptors controlling immune responses. Cytokine Growth Factor Rev 2006; 17: 501–519. [DOI] [PubMed] [Google Scholar]
- 12. Dalli J, Consalvo AP, Ray V, Di Filippo C, D'Amico M, Mehta N, et al. Proresolving and tissue‐protective actions of annexin A1‐based cleavage‐resistant peptides are mediated by formyl peptide receptor 2/lipoxin A4 receptor. J Immunol 2013; 190: 6478–6487. [DOI] [PubMed] [Google Scholar]
- 13. Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC, et al. Annexin A1 modulates natural and glucocorticoid‐induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol 2012; 92: 249–258. [DOI] [PubMed] [Google Scholar]
- 14. Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, et al. Anti‐inflammatory role of the murine formyl‐peptide receptor 2: ligand‐specific effects on leukocyte responses and experimental inflammation. J Immunol 2010; 184: 2611–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He R, Sang H, Ye RD. Serum amyloid A induces IL‐8 secretion through a G protein‐coupled receptor, FPRL1/LXA4R. Blood 2003; 101: 1572–1581. [DOI] [PubMed] [Google Scholar]
- 16. Lee HY, Kim H, Lee SY, Jung YS, Kim SD, Bae YS. A membrane‐tethering pepducin that inhibits formyl peptide receptor 2‐induced signaling. Pharmazie 2014; 69: 293–296. [PubMed] [Google Scholar]
- 17. Gough K, Hutchison M, Keene O, Byrom B, Ellis S, Lacey L, et al. Assessment of dose proportionality: report from the statisticians in the Pharmaceutical Industry/Pharmacokinetics UK Joint Working Party. Ther Innov Regul Sci 1995; 29: 1039–1048. [Google Scholar]
- 18. Aul R, Patel S, Summerhill S, Kilty I, Plumb J, Singh D. LPS challenge in healthy subjects: an investigation of neutrophil chemotaxis mechanisms involving CXCR1 and CXCR2. Int Immunopharmacol 2012; 13: 225–231. [DOI] [PubMed] [Google Scholar]
- 19. Krause A, Zisowsky J, Strasser DS, Gehin M, Sidharta PN, Groenen PM, et al. Pharmacokinetic/pharmacodynamic modelling of receptor internalization with CRTH2 antagonists to optimize dose selection. Clin Pharmacokinet 2016; 55: 813–821. [DOI] [PubMed] [Google Scholar]
- 20. Maderna P, Cottell DC, Toivonen T, Dufton N, Dalli J, Perretti M, et al. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin‐derived peptide‐stimulated phagocytosis. FASEB J 2010; 24: 4240–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marchese A, Chen C, Kim YM, Benovic JL. The ins and outs of G protein‐coupled receptor trafficking. Trends Biochem Sci 2003; 28: 369–376. [DOI] [PubMed] [Google Scholar]
- 22. Cattaneo F, Parisi M, Ammendola R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int J Mol Sci 2013; 14: 7193–7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karavanaki K, Polychronopoulou S, Giannaki M, Haliotis F, Sider B, Brisimitzi M, et al. Transient and chronic neutropenias detected in children with different viral and bacterial infections. Acta Paediatr 2006; 95: 565–572. [DOI] [PubMed] [Google Scholar]
- 24. Zhao PW, Jiang WG, Wang L, Jiang ZY, Shan YX, Jiang YF. Plasma levels of IL‐37 and correlation with TNF‐alpha, IL‐17 A, and disease activity during DMARD treatment of rheumatoid arthritis. PLoS One 2014; 9: e95346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jin C, Roen DR, Lehmann PV, Kellermann GH. An enhanced ELISPOT assay for sensitive detection of antigen‐specific T cell responses to Borrelia burgdorferi. Cells 2013; 2: 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leaker BR, Barnes PJ, O'Connor B. Inhibition of LPS‐induced airway neutrophilic inflammation in healthy volunteers with an oral CXCR2 antagonist. Respir Res 2013; 14: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duringer C, Grundstrom G, Gurcan E, Dainty IA, Lawson M, Korn SH, et al. Agonist‐specific patterns of beta 2‐adrenoceptor responses in human airway cells during prolonged exposure. Br J Pharmacol 2009; 158: 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. Structure‐based discovery of beta2‐adrenergic receptor ligands. Proc Natl Acad Sci U S A 2009; 106: 6843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goral V, Jin Y, Sun H, Ferrie AM, Wu Q, Fang Y. Agonist‐directed desensitization of the beta2‐adrenergic receptor. PLoS One 2011; 6: e19282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther 2011; 336: 296–302. [DOI] [PubMed] [Google Scholar]
- 31. Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 2010; 62: 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G‐protein‐coupled receptors. Nature 2013; 494: 185–194. [DOI] [PubMed] [Google Scholar]
- 33. Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 2007; 320: 1–13. [DOI] [PubMed] [Google Scholar]
- 34. Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G‐protein‐coupled receptors. Nature 2009; 459: 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Arithmetic mean (and 95% confidence interval for placebo) for interleukin (IL) 6 (top), IL‐8 (middle) and monocyte chemoattractant protein 1 (bottom) following administration of the first dose of ACT‐389949 on Day 1 (left) and at steady state (Day 7/10) by dose group
Figure S2 Differential cell counts in the sputum following lipopolysaccharide (LPS) challenge. Absolute (top) and relative (bottom) counts for eosinophils (left), epithelial cells (middle) and lymphocytes (right) in the sputum at 6 h post‐LPS on Day 9/13 by dose group. Boxes indicate the range of the middle 50% of the data. Medians are shown as symbols in the boxes. Whiskers indicate the 95% range. Extreme values are shown individually
Table S1 Baseline demographic data for subjects in the single‐ascending dose and multiple‐ascending dose studies
Table S2 Most frequent (n ≥ 3 subjects) adverse events by preferred term (multiple‐ascending dose study)
Supporting info item
Supporting info item
Supporting info item
Supporting info item