

Abstract
Aims
Oxidative bioactivation of amodiaquine (AQ) by cytochrome P450s to a reactive quinoneimine is considered as an important mechanism underlying its idiosyncratic hepatotoxicity. However, because internal exposure to its major metabolite N‐desethylamodiaquine (DEAQ) is up to 240‐fold higher than AQ, bioactivation of DEAQ might significantly contribute to covalent binding. The aim of the present study was to compare the kinetics of bioactivation of AQ and DEAQ by human liver microsomes (HLM) and to characterize the CYPs involved in bioactivation of AQ and DEAQ.
Methods
Glutathione was used to trap reactive metabolites formed in incubations of AQ and DEAQ with HLM and recombinant human cytochrome P450s (hCYPs). Kinetics of bioactivation of AQ and DEAQ in HLM and involvement of hCYPs were characterized by measuring corresponding glutathione conjugates (AQ‐SG and DEAQ‐SG) using a high‐performance liquid chromatography method.
Results
Bioactivation of AQ and DEAQ in HLM both exhibited Michaelis–Menten kinetics. For AQ bioactivation, enzyme kinetical parameters were K m, 11.5 ± 2.0 μmol l–1, V max , 59.2 ± 3.2 pmol min−1 mg−1 and CL int, 5.15 μl min−1 mg−1. For DEAQ, parameters for bioactivation were K m, 6.1 ± 1.3 μmol l–1, V max , 5.5 ± 0.4 pmol min−1 mg−1 and CL int 0.90 μl min−1 mg−1. Recombinant hCYPs and inhibition studies with HLM showed involvement of CYP3A4, CYP2C8, CYP2C9 and CYP2D6 in bioactivation.
Conclusions
The major metabolite DEAQ is likely to be quantitatively more important than AQ with respect to hepatic exposure to reactive metabolites in vivo. High expression of CYP3A4, CYP2C8, CYP2C9, and CYP2D6 may be risk factors for hepatotoxicity caused by AQ‐therapy.
Keywords: amodiaquine, bioactivation, glutathione conjugate, human cytochrome P450s, N‐desethylamodiaquine
What is Already Known about this Subject
Oxidative bioactivation of amodiaquine by cytochrome P450s is considered as an important mechanism underlying its idiosyncratic hepatotoxicity.
N‐desethylamodiaquine, the principal metabolite of amodiaquine, shows 100‐ to 240‐fold higher internal exposure than amodiaquine and is considered to determine pharmacological activity of amodiaquine‐treatment.
The contribution of individual human cytochrome P450s to hepatic bioactivation of amodiaquine and N‐desethylamodiaquine has not yet been characterized.
What this Study Adds
Enzyme kinetic parameters of microsomal bioactivation of amodiaquine and N‐desethylamodiaquine were determined.
Based on differences in internal exposure and intrinsic clearances, bioactivation of N‐desethylamodiaquine is expected to contribute more to hepatic exposure to reactive intermediates than amodiaquine.
Multiple human cytochrome contributed to amodiaquine and N‐desethylamodiaquine bioactivation; CYP3 A4, CYP2C8, CYP2C9 and CYP2D6 are the most important isoforms.
Tables of Links
These Tables lists key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2.
Introduction
Amodiaquine (AQ), a 4‐aminoquinoline antimalarial drug, has been widely used in the endemic areas of Africa and Asia for more than 50 years. As an antimalarial drug, AQ has a higher efficacy compared with other 4‐aminoquinoline antimalarial drugs 3. For example, AQ shows efficacy against chloroquine‐resistant strains of Plasmodium falciparum 4 and was proposed to replace chloroquine as the first line drug in Africa. However, severe idiosyncratic agranulocytosis and hepatotoxicity occurring in 1 of 2000 patients overshadow the therapeutic advantages of AQ 5, 6, 7. This eventually led to either the withdrawal of AQ in several countries or the prohibition of usage as single administration or as prophylaxis 8. Identification of the risk factors underlying these adverse drug reactions might assist in protection of susceptible patients against AQ toxicity. Although the exact mechanisms of these idiosyncratic reactions remain to be established, oxidative bioactivation to protein‐reactive metabolites is considered to play an important role 9, 10, 11.
After oral administration, AQ is rapidly absorbed and metabolized by CYP2C8 to N‐desethylamodiaquine (DEAQ), its major stable metabolite 12. Other minor metabolites identified in the circulation include N‐bisdesethylamodiaquine (bis‐DEAQ) and 2‐hydroxyl‐desethylamodiaquine (Figure 1) 13, 14. Because of its rapid conversion to DEAQ, and the fact that DEAQ has a 20‐ to 50‐fold longer half‐life than AQ, the area‐under‐curve (AUC) of DEAQ is 100‐ to 240‐fold higher when compared to that of AQ 15, 16, 17. Therefore, although in vitro pharmacological studies showed that AQ is about three‐fold more potent than DEAQ, the antimalarial activity of AQ will be mainly determined by DEAQ 13.
Figure 1.
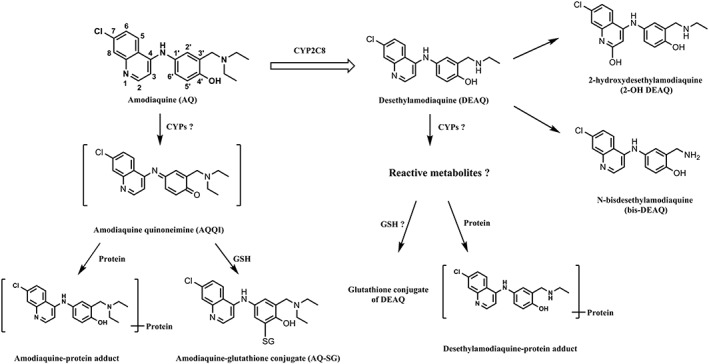
Previously identified metabolites of amodiaquine and N‐desethylamodiaquine. Question markers indicate pathways to be characterized in the present study
Besides the formation of stable metabolites, it has been demonstrated that AQ is bioactivated to reactive metabolites in vitro and in vivo 9, 18, 19. Based on the structure of a glutathione (GSH) conjugate identified in rat bile, the reactive metabolite was identified as amodiaquine quinoneimine (AQ‐QI) resulting from dehydrogenation of the 4‐aminophenol moiety (Figure 1). Covalent binding of AQ‐QI to cellular macromolecules therefore is considered to be involved in the immune‐mediated toxicity of AQ 19, 20. Although covalent binding of radiolabeled AQ to liver microsomes appeared to result mainly from autoxidation 9, 21, cytotoxicity to hepatocytes and neutrophils appeared to be strongly dependent on bioactivation to AQ‐QI by cytochrome P450 and myeloperoxidase, respectively 22, 23, 24.
Although P450‐dependent bioactivation of AQ by human liver microsomes (HLM) has been demonstrated previously 25, 26, bioactivation of DEAQ has not yet been studied. Furthermore, the individual human cytochrome P450s (hCYPs) involved in bioactivation of AQ and DEAQ have not yet been characterized. Because the expression level and activity of hepatic hCYPs can vary strongly among the population 27, 28, overexpression of bioactivating hCYPs may be important risk factors determining individual susceptibility to AQ‐induced toxicity. Recently, the extrahepatic isoforms CYP1A1, CYP1B1 and CYP2J2 were shown to bioactivate both AQ and DEAQ to their corresponding quinoneimines, as demonstrated by trapping experiments with N‐acetyl cysteine as nucleophile 29. However, the activity of the hepatic hCYPs have not been reported yet. The aims of the present study were: (1) to study and characterize the kinetics of bioactivation of AQ and DEAQ in HLM; and (2) to identify the hCYP isozymes involved in the bioactivation of AQ and DEAQ using recombinant hCYPs and by incubations of HLM in presence of specific inhibitors. Formation of reactive metabolites was determined by trapping and quantitative measurement of GSH conjugates of AQ and DEAQ from incubation samples.
Methods
Materials
AQ dihydrochloride was obtained from INC Biomedicals (Aurora, OH, USA). DEAQ was purchased from BD Biosciences (Franklin Lakes, NJ, USA). Human liver microsomes (Lot No. 1 210 347), pooled from 200 donors, were purchased from Xenotech (Lenexa, KS, USA). Supersomes containing cDNA‐baculovirus‐insect cell‐expressed P450 enzymes were obtained from BD Biosciences (Breda, the Netherlands). The enzymes used were CYP1A1 (Lot No. 456 211), CYP1A2 (Lot No. 456 203), CYP2A6 (Lot No. 456 254), CYP2B6 (Lot No. 456 255), CYP2C8 (Lot No. 456 252), CYP2C9*1(Arg144) (Lot No. 456 258), CYP2C18 (Lot No. 456 222), CYP2C19 (Lot No. 456 259), CYP2D6*1 (Lot No. 456 217), CYP2E1 (Lot No. 456 206), CYP2J2 (Lot No. 456 264), CYP3A4 (Lot No. 456 207), CYP3A5 (Lot No. 456 256) and CYP3A7 (Lot No. 456 237). All other chemicals and reagents were of analytical grade and obtained from standard suppliers.
Identification and structural elucidation of GSH conjugates of AQ and DEAQ
The GSH conjugate of AQ was synthesized chemically as described by Harrison et al. 18. The GSH conjugate of DEAQ (DEAQ‐SG) was prepared biosynthetically by performing a large scale incubation of DEAQ, GSH and a drug metabolizing mutant of bacterial CYP102A1 30, as detailed in the Supplemental Information. After isolation of the GSH‐conjugates of AQ and DEAQ by preparative high‐performance liquid chromatography (HPLC), their purity and identity were determined by analytical HPLC‐UV and LC–time‐of‐flight (TOF)–mass spectrometry (MS; see Section 2.6), and by 1H–nuclear magnetic resonance (NMR) spectroscopy. 1H–NMR spectra were recorded on a Bruker Avance 500 (Fallanden, Switzerland) operating at 500.1 MHz to characterize the structure of the GSH conjugates. The 1H–NMR spectrum of the GSH‐conjugate of AQ was identical to that published previously 18, and corresponds to a GSH‐conjugate with the GSH‐moiety attached to the C′5‐position of AQ. The biosynthetical GSH conjugate of DEAQ showed almost identical chemical shifts and coupling patterns of aromatic protons, when compared to those of AQ‐SG, and is therefore also consistent with a GSH‐moiety attached to the C′5‐position of DEAQ (see Figure S1 and Table S1). The H‐H correlation spectroscopy spectrum confirmed the structure of DEAQ‐SG (see Figure S2).
Characterization of enzyme kinetics of oxidative metabolism of AQ and DEAQ by HLM
Incubations with HLM were performed at 37°C in 100 mmol l–1 potassium phosphate buffer (pH 7.4) containing 2 mmol l–1 ethylenediaminetetraacetic acid and 5 mmol l–1 magnesium chloride (MgCl2). Firstly, one single concentration of AQ (10 μmol l–1) and DEAQ (5 μmol l–1) were incubated with HLM in the presence of 5 mmol l–1 GSH for the identification of metabolites of AQ and DEAQ. Because a GSH dependency experiment showed maximal trapping of reactive quinonimines of AQ and DEAQ at a GSH concentration of 5 mmol l–1 (data no shown) this GSH concentration was used in all incubations. After assessing linearity of product formation with respect to incubation time and protein concentration, incubations with HLM were done at 1 mg ml−1 and with incubation times of 10 min for AQ and 45 min for DEAQ. Enzyme kinetics was determined by incubating AQ and DEAQ at seven concentrations and in a final volume of 100 μl in duplicate. Reactions were initiated by adding NADPH‐regenerating system (NRS) resulting in final concentrations of 0.1 mmol l–1 NADP+, 10 mmol l–1 glucose‐6‐phosphate, and 0.5 U ml−1 glucose‐6‐phosphate dehydrogenase. To determine involvement of autoxidation, incubations of AQ and DEAQ without NRS were performed. Reactions were terminated by the addition of ice‐cold perchloric acid, final concentration 1% (v/v), and cooled on ice for 10 min. Precipitates were removed by centrifugation for 15 min at 14 000 rpm. The supernatants were analyzed by HPLC‐UV and HPLC‐TOF‐MS, as described in Section 2.6. After correction for autoxidation, enzyme kinetical parameters were calculated by nonlinear regression using the Michaelis–Menten equation using GraphPad Prism 5 software (San Diego, CA, USA).
Incubations of AQ and DEAQ with recombinant human CYPs
Incubations of AQ and DEAQ with recombinant human CYPs were conducted in 100 mmol l–1 potassium phosphate buffer (pH 7.4) containing 5 mmol l–1 GSH, 2 mmol l–1 EDTA and 5 mmol l–1 MgCl2 at a final volume of 100 μl in duplicate. Each individual recombinant human CYP was incubated at a concentration of 100 nmol l–1 at 37°C in presence of AQ at 10 μmol l–1 and 100 μmol l–1 and DEAQ at 5 μmol l–1 and 50 μmol l–1, respectively. Incubation times for AQ and DEAQ were 10 and 60 min, respectively. The reactions were initiated and terminated as described above. The supernatants were analyzed by HPLC‐UV, as described in Analytical methods.
Effect of specific inhibitors of hCYPs on metabolite formation in incubations of AQ and DEAQ with HLM
To identify the hCYPs involved in oxidative metabolism of AQ and DEAQ, incubations were performed with HLM (1 mg ml–1) in the presence or absence of isoenzyme‐specific inhibitors. Incubations were performed in 100 mmol l–1 potassium phosphate buffer (pH 7.4, containing 5 mmol l–1 GSH, 2 mmol l–1 EDTA and 5 mmol l–1 MgCl2) containing 10 μmol l–1 AQ or 5 μmol l–1 DEAQ at a final volume of 100 μl in duplicate at 37°C. Inhibitors used were α‐naphtoflavone (α‐NF, 10 μmol l–1), quercetin (QCT, 15 μmol l–1), sulfaphenazole (SPZ, 10 μmol l–1), (+)‐N‐3‐benzyl‐nirvanol (BNV, 1 μmol l–1), quinidine (QND, 2 μmol l–1), diethylthiocarbamate (DDC, 20 μmol l–1) and ketoconazole (KTZ, 2 μmol l–1) to investigate the involvement of human P4501A2, P4502C8, P4502C9, P4502C19, P4502D6, P4502E1 and P4503A4, respectively 31. All inhibitors except DDC were dissolved in methanol. Concentrations of methanol in all incubations were kept below 0.5% (v/v). All reactions were initiated by the addition of NRS as described above, except incubations containing mechanism‐based inhibitor DDC. DDC was preincubated for 15 min in the presence of NRS before the addition of AQ or DEAQ. Reactions with AQ proceeded for 10 min and with DEAQ proceeded for 60 min regarding the product formation linearity and then terminated with ice‐cold perchloric acid at a final concentration of 1% (v/v). After centrifugation, supernatants were analyzed by HPLC‐UV as described in Analytical methods.
Analytical methods
After centrifugation of terminated incubations, the supernatants were analyzed by reverse‐phase LC using a Phenomenex Luna 5‐μm C18 column (4.6 × 150 mm) as stationary phase, protected by a 4.0 × 3.0 mm i.d. security guard (5 μm) C18 guard column from Phenomenex (Torrance, CA, USA). An isocratic method (9% acetonitrile in 5 mmol l–1 of ammonium acetate buffer, pH 2.4, adjusted by formic acid) was used for the separation of AQ and DEAQ related metabolites. The flow rate was 0.75 ml min−1 and the UV detector was set at 342 nm. To quantify metabolites, standard curves were constructed for AQ‐SG (ranging from 0.05 to 5 μmol l–1), DEAQ‐SG (ranging from 0.01 to 5 μmol l–1) and DEAQ (ranging from 0.02 to 20 μmol l–1). The lower limits of quantification of AQ‐SG, DEAQ‐SG and DEAQ were 0.05 μmol l–1, 0.01 μmol l–1 and 0.02 μmol l–1, respectively.
The LC–MS system used for metabolite identification consisted of an Agilent 1200 Series Rapid resolution LC system connecting to a TOF Agilent 6230 mass spectrometer, equipped with electrospray ionization source, and operating in the positive mode. Metabolites were separated using a Phenomenex Luna 5 μm C18 column and a gradient constructed of eluent A (5 mmol l–1 ammonium acetate buffer, pH adjusted to 2.4 with formic acid) and eluent B (acetonitrile) according to the following program: 0–5 min, isocratic at 10% eluent B; 5–30 min, linear increase to 15% eluent B; 30–35 min, linear increase to 40% B; decrease to 10% B in 0.5 min, and reequilibration at 10% solvent B until 45.5 min. The MS ion source parameters were set as follows: capillary voltage, 3500 V; nitrogen gas temperature, 350°C; nitrogen drying gas rate, 10 l min−1; nitrogen nebulizing gas pressure, 344738 Pa. Data analysis was performed using the Agilent MassHunter Qualitative Analysis software, Version B. 05. 00.
Results
Identification and structural elucidation of AQ and DEAQ metabolites from HLM incubations
To identify all stable metabolites and GSH conjugates, AQ and DEAQ were first incubated with HLM in the presence of 5 mmol l–1 GSH. As shown in Figure 2A, DEAQ was the major metabolite formed in incubations of AQ with HLM, consistent with previous studies. In addition a minor metabolite eluted at 17.7 min, which was identified as the GSH‐conjugate of AQ, based on its identical retention time and mass spectrum ([M + H]+ at 661.21, see supplemental Figure S3) when compared to synthetic AQ‐SG. In addition, a minor metabolite was eluting at 12.6 min and with m/z of 300.09 ([M + H]+), which corresponds to bis‐DEAQ formed by deethylation of DEAQ.
Figure 2.
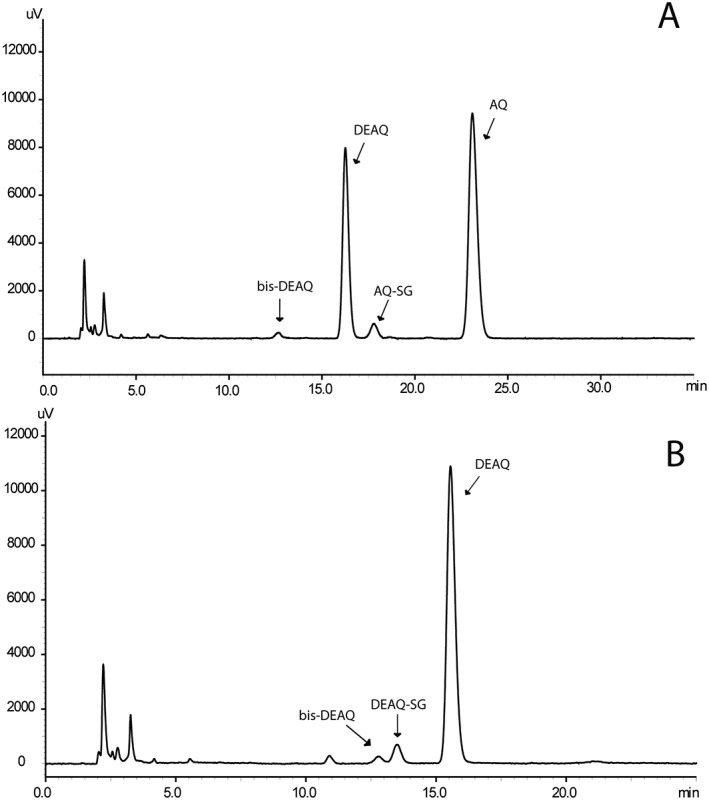
High‐performance liquid chromatography–UV chromatograms of incubations of human liver microsomes (1 mg ml−1) in presence of 5 mmol l–1 glutathione and 10 μmol l–1 amodiaquine (AQ; A) or 5 μmol l–1 N‐desethylamodiaquine (DEAQ; B). AQ was incubated for 10 minutes; DEAQ was incubated for 45 min
In incubations of HLM with DEAQ, one GSH conjugate was found eluting at 13.5 min, Figure 2B, and with m/z of 633.19 ([M + H]+), Figure S3. The retention time and mass spectrum was identical to that of the biosynthetic GSH‐conjugate of DEAQ, with the GSH‐moiety attached to the C′5‐position. Next to DEAQ‐SG, only a minor amount of bis‐DEAQ was found, indicating that, in contrast to AQ, N‐deethylation is a minor pathway compared to the bioactivation pathway.
Characterization of enzyme kinetics of AQ and DEAQ bioactivation by HLM
Figure 3A shows the concentration dependency of formation of DEAQ with AQ concentrations varying from 1 to 100 μmol l–1. As expected, the corresponding Eadie–Hofstee plot (insert Figure 3A) exhibits monophasic behavior, since N‐deethylation of AQ is considered CYP2C8‐specific 12. By applying nonlinear regression according to the Michaelis–Menten equation, the enzyme kinetical parameters found were: K m, 13.3 ± 1.9 μmol l–1, V max, 1609 ± 73 pmol min−1 mg−1 and CL int, 120.6 μl min−1 mg−1 (Table 1).
Figure 3.
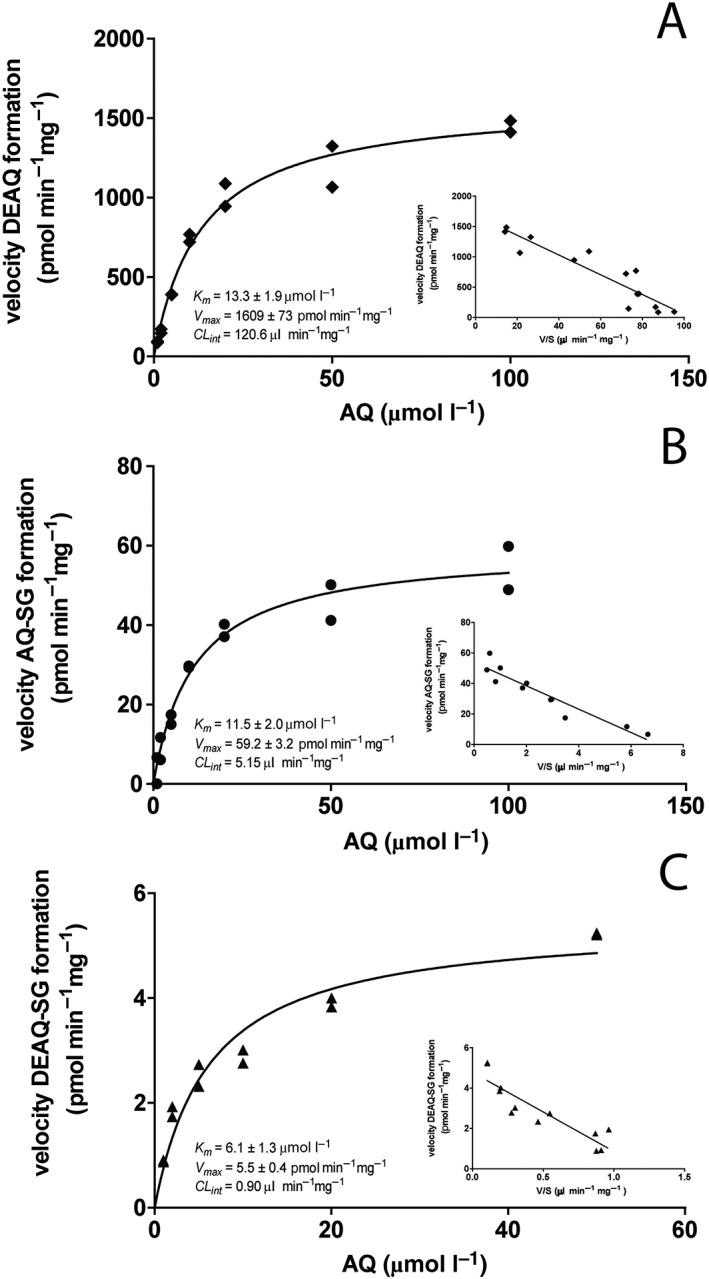
Enzyme kinetic analysis of oxidative metabolism of amodiaquine (AQ) and N‐desethylamodiaquine (DEAQ) by pooled human liver microsomes. (A) Concentration dependency curve of DEAQ formation from AQ; (B) concentration dependency curve of AQ‐SG formation from AQ; (C) concentration dependency curve of DEAQ‐SG from DEAQ. Solid lines are obtained by nonlinear regression using the Michaelis–Menten equation. Inserts show corresponding EadieHofstee plots
Table 1.
Enzyme kinetic parameters of formation of N‐desethylamodiaquine (DEAQ) and C′5‐glutathionly‐amodiaquine (AQ‐SG) from amodiaquine and of C′5‐glutathionly‐N‐desethylamodiaquine (DEAQ‐SG) formation from DEAQ in human liver microsomes
| K m (μmol l –1 ) | V max ( pmol min −1 mg −1 ) | CL int b (μl min −1 mg −1 ) | |
|---|---|---|---|
| DEAQ Formation | 13.3 ± 1.9 | 1609 ± 73 | 120.6 |
| AQ‐SG Formation | 11.5 ± 2.0 | 59.2 ± 3.2 | 5.15 |
| DEAQ‐SG Formation | 6.1 ± 1.3 | 5.5 ± 0.4 | 0.90 |
Figure 3B shows the concentration dependent formation of AQ‐SG in HLM after correction for the contribution of autoxidation of AQ. Between 10 and 100 μmol l–1 AQ, autoxidation of AQ accounted for approximately 20–30% of AQ‐SG formation. Below 10 μmol l–1 AQ, formation of AQ‐SG was below the limit of detection in absence of NRS. The enzyme‐dependent formation of AQ‐SG obeyed Michaelis–Menten kinetics with kinetical K m, 11.5 ± 2.0 μmol l–1; V max, 59.2 ± 3.2 pmol min−1 mg−1 and CL int, 5.15 μl min−1 mg−1.
Autoxidation was also observed for DEAQ and accounted for approximately 10–20% of formation of DEAQ‐SG. Figure 3C shows concentration dependency of DEAQ‐SG formation in incubations of HLM with DEAQ ranging from 1 to 50 μmol l–1, after correction for the contribution of autoxidation. The enzyme kinetic parameters calculated using nonlinear regression were: K m, 6.1 ± 1.3 μmol l–1, V max, 5.5 ± 0.4 pmol min−1 mg−1 and CL int, 0.90 μl min−1 mg−1.
Activity of recombinant hCYPs in the bioactivation of AQ and DEAQ
To identify the individual hCYPs involved in the bioactivation of AQ and DEAQ, incubations were conducted with recombinant hCYP isozymes at AQ concentrations of 10 and 100 μmol l–1, and at DEAQ concentrations of 5 and 50 μmol l–1 in order to study the contributions of P450 isozymes under non‐saturated and saturated conditions.
Consistent with previous studies 12, the major metabolite in AQ incubations, DEAQ, was produced at highest activity by CYP2C8 and to a lesser extent by CYP2D6 at both AQ concentrations, Figure 4A. As shown in Figure 4B, multiple recombinant CYPs were active in the formation of AQ‐SG at both 10 and 100 μmol l–1. Among them, CYP2J2 and CYP2D6 appeared the most active isoforms, followed by CYP2C8 and CYP3A4. All other recombinant hCYPs show <10% activity under 10 μmol l–1 AQ and <19% under 100 μmol l–1 AQ, when compared to the most active enzyme CYP2J2. The formed AQ‐SG in control samples at both 10 and 100 μmol l–1 AQ were below the detection limit, indicating that the autoxidation of AQ under these conditions was negligible.
Figure 4.
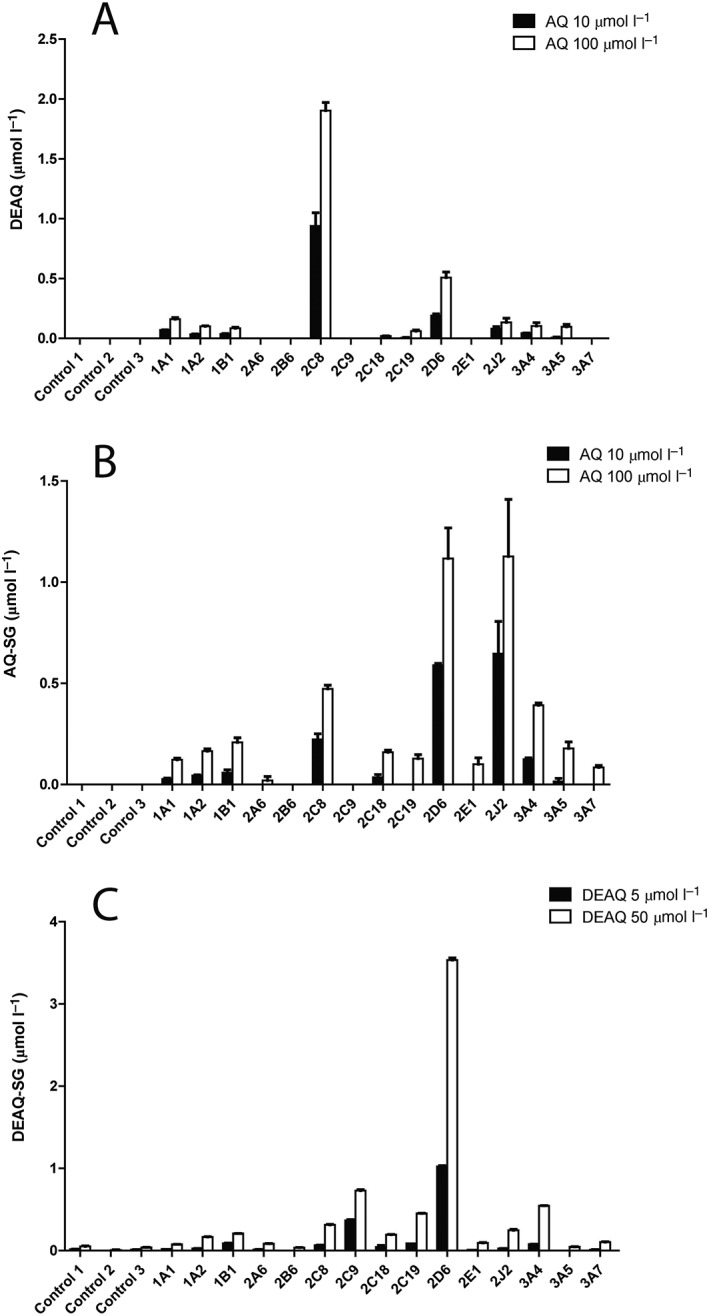
Oxidative metabolism of amodiaquine (AQ) and N‐desethylamodiaquine (DEAQ) by recombinant human P450 (supersomes). (A) Formation of DEAQ from AQ (A); (B) formation of AQ‐SG from AQ; (C) formation of DEAQ‐SG from DEAQ. AQ and DEAQ were incubated at two concentrations in presence of 100 nmol l–1 of each recombinant hCYPs. Control 1, empty baculosomes; Control 2, human CYP reductase; Control 3, baculosomes expressing only human CYP reductase
Similar to AQ, multiple CYPs also showed activity in the bioactivation of DEAQ, Figure 4C. Recombinant CYP2D6 appeared to be the most active enzyme at both substrate concentrations. At 5 μmol l–1 DEAQ, CYP2C9 also showed significant activity, being approximately 40% of that of CYP2D6. At the high DEAQ concentration, CYP2C8, CYP2C19 and CYP3A4 showed activities of approximately 10% of that of CYP2D6.
Effect of isozyme‐selective inhibitors on the bioactivation of AQ and DEAQ by HLM
Based on the enzyme kinetic parameters, inhibition experiments with HLM were performed at 10 μmol l–1 of AQ and 5 μmol l–1 of DEAQ, respectively, to get substrate concentrations in the range of the K m values. Figure 5 shows the percentage of inhibition by specific CYP inhibitors on the bioactivation of AQ and DEAQ and the detailed values of the inhibitory effects are shown in Table 2. As expected, in incubations of HLM with 10 μmol l–1 AQ, formation of DEAQ was most strongly inhibited by the selective CYP2C8 inhibitor QCT. Formation of AQ‐SG was strongly reduced by 53.1% and 40.1%, by QCT and the CYP3A4 inhibitor KTZ, respectively. Although recombinant CYP2D6 showed activity in AQ‐SG formation, the CYP2D6 inhibitor QND did not significantly reduce AQ‐SG formation in HLM incubations. Combining the inhibitors of CYP2C8, CYP2D6 and CYP3A4 resulted in 84.2% reduced DEAQ formation and 79.8% reduced AQ‐SG formation, respectively (Figure 5A).
Figure 5.

Effect of specific CYP450 inhibitors on metabolism of amodiaquine (AQ) at 10 μmol l–1 (A) and N‐desethylamodiaquine (DEAQ) at 5 μmol l–1 (B) by human liver microsomes. Data are expressed as % of the control experiments where inhibitors were omitted and represented as mean ± standard deviation of duplicate determinations
Table 2.
Inhibitory effects of specific P450‐inhibitors on the formation of N‐desethylamodiaquine (DEAQ), C′5‐glutathionly‐amodiaquine (AQ‐SG) and C′5‐glutathionly‐N‐desethylamodiaquine (DEAQ‐SG) in human liver microsomes incubations
| Specific P450 inhibitor | DEAQ formation | AQ‐SG formation | DEAQ‐SG formation |
|---|---|---|---|
| α‐Naphtoflavone (1 A2) | 86.4 ± 8.7 | 106.3 ± 5.4 | 104.1 ± 5.8 |
| Quercetin (2C8) | 22.9 ± 0.4 | 46.9 ± 1.5 | 80.3 ± 9.6 |
| Sulfaphenazole (2C9) | 113.1 ± 3.5 | 106.5 ± 3.5 | 89.3 ± 1.3 |
| (+)‐N‐3‐Benzyl‐nirvanol (2C19) | 101.1 ± 5.1 | 101.3 ± 0.9 | 96.5 ± 0.6 |
| Quinidine (2D6) | 111.0 ± 5.1 | 113.5 ± 14.5 | 70.9 ± 2.6 |
| Diethylthiocarbamate (2E1) | 122.0 ± 5.4 | 115.1 ± 8.6 | 96.4 ± 5.6 |
| Ketoconazole (3 A4) | 98.3 ± 1.1 | 59.9 ± 1.8 | 76.4 ± 2.7 |
| Combinantion inhibition a | 15.8 ± 1.3 | 20.2 ± 0.2 | 30.6 ± 0.3 |
Values are the expressed as mean ± standard deviation from duplicated experiments and as percentage (%) relative to control incubations without inhibitors
Combination inhibition for AQ‐SG and DEAQ‐SG formation: quercetin (CYP2C8) + quinidine (CYP2D6) + ketoconazole (CYP3A4)
In incubations of HLM with DEAQ, the formation of DEAQ‐SG was moderately inhibited by selective CYP2C8, CYP2D6 and CYP3A4 inhibitors by 19.7%, 29.1%, and 23.6%, respectively (Figure 5B). The combined inhibition of CYP2C8, CYP2D6 and CYP3A4 showed the strongest inhibition of 69.4% of DEAQ‐SG formation compared to corresponding control incubations. Although the recombinant CYP2C9 showed the second highest activity, inhibition of CYP2C9 only gave a minor decrease of 10.7% regarding the formation of DEAQ‐SG.
Discussion
The aim of present study was to characterize the kinetics of bioactivation of AQ and DEAQ by HLM in order to evaluate which compound will have the highest contribution to covalent binding to hepatic tissue. In addition, which hCYP isozymes contribute to bioactivation of AQ and DEAQ was investigated.
Previous studies showed that AQ is prone to autoxidation, leading to covalent binding to HLM even in absence of the cofactor NADPH 9, 21. In the present study, we confirmed this finding by observation of AQ‐SG formation in incubations of HLM in absence of NRS. In addition, DEAQ also showed the potency of autoxidation in incubations without NRS. However, addition of NRS resulted in a three‐ to nine‐fold strong increase in formation of AQ‐SG and DEAQ‐SG, dependent on substrate concentration (Figure S4). Also, the fact that addition of a mixture of P450‐inhibitors to HLM could decrease formation of AQ‐SG by more than 80% (Figure 4A) only can be explained by inhibition of P450‐dependent bioactivation, since these inhibitors lack antioxidant properties (data not shown). Similarly, in incubations of AQ with recombinant hCYPs, formation of AQ‐SG showed large differences in formation of GSH‐conjugates, which would not be expected if bioactivation would mainly occur by autoxidation. Although the contribution of autoxidation in our in vitro experiments ranged to 30% in incubations with 1 mg ml−1 of HLM, the average concentration of microsomal proteins of 128 human livers was reported to be 39.46 mg g−1 liver 32. Therefore, because of this almost 40‐fold higher enzyme concentration the bioactivation of AQ and DEAQ will be mainly P450‐dependent in vivo.
Since the peak concentration of DEAQ is more than 20‐fold higher than that of AQ itself after oral administration of AQ, whereas the half‐life of DEAQ is much longer, the internal exposure to DEAQ is up to 240‐fold higher than that of AQ, when considering the ratio AUCDEAQ/AUCAQ 16. The much shorter half‐life of AQ is explained by the more rapid oxidative metabolism by hepatic enzymes. As shown in Figure 2, AQ was metabolized for 50% by HLM within 10 min, whereas DEAQ was metabolized for only 5% after 45 min. Because of the much higher internal exposure to DEAQ, and the fact that bioactivation appears to be the major pathway of oxidative metabolism, an enzyme kinetical analysis of the bioactivation pathways was performed to estimate difference in internal exposure to the quinonimines of AQ and DEAQ. Since the maximal plasma concentrations of AQ and DEAQ found clinically are in the range of 50 nmol l–1 (AQ) and 1.5–2 μmol l–1 (DEAQ), the intrinsic clearances CL int and the local substrate concentration will determine the rate by which their corresponding quinonimines will be produced. Although the CL int of DEAQ‐SG formation appeared to be 5.7‐fold lower compared to that of AQ‐SG formation, the fact that the DEAQ concentration is over 40 times higher, and also present for a longer time because of it long half‐life, implies that the overall internal exposure to chemically reactive quinonimine of DEAQ will be much higher compared to that of AQ. Because the body burden of covalent binding is considered an important risk factor for idiosyncratic drug reactions 33, DEAQ probably not only determines the pharmacological effect of AQ‐treatment, but also may determine its toxic side effects.
To better understand the interindividual variability in susceptibility to AQ‐induced hepatotoxicity, identification of the hCYPs involved in bioactivation of AQ and DEAQ is important. As shown in Figure 4, multiple recombinant hCYPs appeared to be active in bioactivation of AQ and DEAQ. For AQ, highest activity in formation of AQ‐SG was observed with recombinant CYP2D6 and CYP2J2, while significant activity was also found with CYP2C8 and CYP3A4. When considering the different expression levels of hCYPs in human liver (Table 3), the largest contribution to bioactivation of AQ in the average liver is expected from CYP3A4, CYP2D6 and CYP2C8, in decreasing order. The strong inhibition observed by the inhibitors of CYP2C8 (QCT) and CYP3A4 (KTZ) in incubations with HLM confirmed their contribution to bioactivation of AQ. However, the contribution of CYP2D6 in hepatic bioactivation appears to be overpredicted when based on supersomes expressing CYP2D6, since no significant inhibition of HLM‐catalyzed bioactivation of AQ was observed with the CYP2D6‐specific inhibitor QND.
Table 3.
Specific activities of recombinant human cytochrome P450s (hCYPs) in the formation of glutathione conjugates of amodiaquine (AQ‐SG) and desethylamodiaquine (DEAQ‐SG) and predicted contribution of each hCYP isozyme in the average human liver
| AQ 10 μmol l –1 | DEAQ 5 μmol l –1 | ||||
|---|---|---|---|---|---|
| Average abundance a (range) | AQ‐SG formation b ( pmol min −1 nmol −1 P450) | Relative contribution c (%) | DEAQ‐SG formation b ( pmol min −1 nmol −1 P450) | Relative contribution c (%) | |
| CYP1A1 | 1.8 | 27.1 ± 6.2 | 0.4 | 3.3 ± 0.1 | 0.2 |
| CYP1A2 | 39 (1–263) | 44.4 ± 5.5 | 15.0 | 4.3 ± 0.2 | 4.5 |
| CYP2A6 | 27 (0–191) | ND | 0 | 2.2 ± 0.3 | 1.6 |
| CYP2B6 | 16 (0–180) | ND | 0 | ND | 0 |
| CYP2C8 | 22.4 (0–85) | 223.8 ± 29.0 | 43.3 | 10.7 ± 0.4 | 6.4 |
| CYP2C9 | 61 (0–277) | ND | 0 | 61.2 ± 2.1 | 100.0 |
| CYP2C18 | 0.4 (0.2–0.7) | 35.9 ± 17.9 | 0.1 | 7.2 ± 4.7 | 0.1 |
| CYP2C19 | 11 (0–67) | ND | 0 | 14.8 ± 0.1 | 4.4 |
| CYP2D6 | 12.6 (0–75) | 589.6 ± 13.6 | 64.1 | 170.8 ± 2.0 | 57.7 |
| CYP2E1 | 64.5 (2–201) | ND | 0 | 1.7 ± 0.1 | 3.0 |
| CYP2J2 | 1.2 (0–3) | 646.8 ± 224.6 | 6.7 | 4.2 ± 0.1 | 0.1 |
| CYP3A4 | 93 (0–601) | 124.6 ± 10.5 | 100.0 | 13.1 ± 0.2 | 32.6 |
| CYP3A5 | 17 (0–291) | 15.1 ± 21.4 | 2.2 | ND | 0 |
| CYP3A7 | 9 (0–90) | ND | 0 | 1.8 ± 0.2 | 0.4 |
Values in pmol mg−1, adapted from Reference 28
Specific activities were determined from duplicated experiments. Values were presented as mean ± standard deviation (n = 2)
Values were calculated by multiplying the mean specific activity of each hCYP isozyme with its average abundance in liver
ND, not detectable
For DEAQ, which overall may cause a higher degree of covalent binding to that of AQ, as described above, also several recombinant hCYPs showed activity in bioactivation to its quinonimine (Figure 4C). At the low concentration of DEAQ, the highest amount of DEAQ‐SG was found with recombinant CYP2D6 and CYP2C9, whereas CYP3A4, CYP2C19 and CYP2C8 also showed significant activity at 50 μmol l–1 DEAQ. Considering the different expression levels of these CYPs in the average human liver, CYP2C9, CYP2D6 and CYP3A4 were predicted to contribute significantly to DEAQ bioactivation, which appear to be confirmed by the inhibition study with P450‐specific inhibitors. Although so‐called “intersystem extrapolation factors” 34 have been used to improve extrapolation of activities of recombinant enzymes to that of HLM, it was decided not to include this in the present study because prediction of relative contributions of individual hCYPs in the average human liver, might not be representative for the relative contribution of hCYPs in the liver of susceptible individuals.
As shown by the meta‐analysis of Achour et al. 28, extremely large variabilities occur in the level of hepatic CYPs, which may be due to genetic factors, enzyme induction and epigenetic factors. For almost each CYP individuals have been identified with up to 10‐fold higher hepatic levels than present in the average liver, Table 3. The very large variability in CYP2C8 expression, the main enzyme involved in oxidative metabolism of AQ, probably underlies the variability in pharmacokinetics of AQ. Individuals with high levels of CYP3A4, CYP2C8, CYP2C9 and CYP2D6 will be exposed to higher levels of quinonimines of AQ and DEAQ. The meta‐analysis of Achour et al. 28, showed significant correlations between abundance of CYP3A4 and CYP2C8 and between CYP3A4 and CYP2C9. This may be partially explained by the fact that these enzymes are inducible, mediated by the same transcription factors. This implicates that individuals with high expression levels of all three CYP isoforms might be exposed to an increased risk of AQ‐induced toxicity.
Identification of CYPs involved in oxidative metabolism of AQ and DEAQ may not only explain interindividual differences in pharmacokinetics, but also explain the consequences of drug–drug interactions caused by drugs administered simultaneously. AQ is often administered together with other antimalarial drugs, such as artesunate, and antiretroviral drugs, such as nevirapine 17 and efavirenz 35. A pharmacokinetic interaction study between efavirenz and amodiaquine/artesunate, was prematurely discontinued because the first two of the five healthy volunteers developed strong increases in plasma transaminase levels 35. Efavirenz is known to cause enzyme induction of CYP3A4 and CYP2B6 in humans 36, 37. A strong increase in CYP3A4‐dependent bioactivation of AQ and DEAQ therefore may explain the hepatic damage observed in these volunteers. Since artesunate is mainly metabolized by CYP2A6, drug–drug interaction between artesunate and AQ or DEAQ is unlikely to occur 38. However, both artemisinin and dehydroartemesinin have been shown to induce CYP3A4 in human hepatocytes, which may lead to increased bioactivation of AQ and DEAQ when coadministered 39.
In conclusion, based on the present study covalent binding by the quinoneimine of both AQ and DEAQ might significantly contribute to the hepatotoxicity observed in a small subgroup of AQ‐treated patients, several human CYPs, such as CYP3A4, CYP2D6, CYP2C8 and CYP2C9 appear to be involved in bioactivation of AQ and DEAQ to their respective quinoneimines, as depicted in Figure 6. Therefore, genetic and nongenetic factors influencing interindividual variability of these isozymes in the liver are likely to be important factors determining the susceptibility of patients to AQ‐induced toxicity. Future investigations focusing on the genotyping of relevant hCYPs of patients encountered AQ‐induced toxicity would be of clinical importance.
Figure 6.
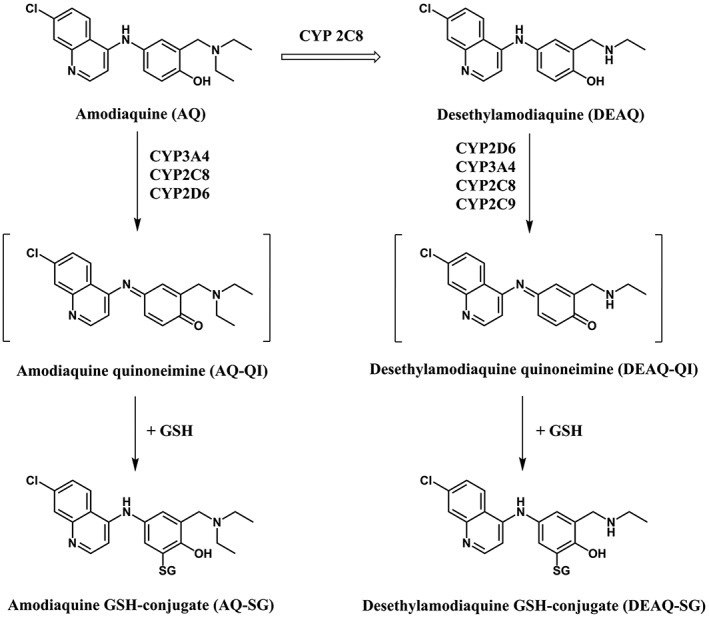
Bioactivation of amodiaquine and N‐desethylamodiaquine by human CYPs
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
Y.Z. is funded by the China Scholarship Council Scholarship . No funding bodies had any role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributors
Y.Z. conducted the study and obtained the data. Y.Z. and J.C. designed the study and interpreted the results. J.C. and N.V. supervised the work. All authors contributed to the writing of the manuscript and approved the final draft.
Supporting information
Figure S1 Comparison of aromatic regions of 1H–nuclear magnetic resonance spectra of (A) C′5‐glutathionyl‐amodiaquine (AQ‐SG) and (B) C′5‐glutathionly‐N‐desethylamodiaquine (DEAQ‐SG)
Figure S2 H‐H correlation spectroscopy spectrum of C′5‐glutathionly‐N‐desethylamodiaquine
Figure S3 Mass spectra of C′5‐glutathionyl‐amodiaquine (A) and C′5‐glutathionly‐N‐desethylamodiaquine (B) extracted from human liver microsome incubations with amodiaquine and N‐desethylamodiaquine, respectively
Figure S4 Concentration dependency of C′5‐glutathionyl‐amodiaquine formation (A) in the presence (closed circle) and absence (open circle) of NADPH‐regenerating system when incubating amodiaquine varying from 1 to 100 μmol l–1 with 1 mg ml−1 human liver microsomes and C′5‐glutathionly‐N‐desethylamodiaquine formation (B) in the presence (closed triangle) and absence (open triangle) of NADPH‐regenerating system when incubating N‐desethylamodiaquine varying from 1 to 50 μmol l–1 with 1 mg ml−1 human liver microsomes. Lines are only intended to guide the eye. Detailed incubation conditions can be found in the text
Table S1 1H‐nuclear magnetic resonance spectra of aromatic protons of glutathione conjugates of amodiaquine and N‐desethylamodiaquine
Supporting info item
Zhang, Y. , Vermeulen, N. P. E. , and Commandeur, J. N. M. (2017) Characterization of human cytochrome P450 mediated bioactivation of amodiaquine and its major metabolite N‐desethylamodiaquine. Br J Clin Pharmacol, 83: 572–583. doi: 10.1111/bcp.13148.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hawley SR, Bray PG, Park BK, Ward SA. Amodiaquine accumulation in Plasmodium falciparum as a possible explanation for its superior antimalarial activity over chloroquine. Mol Biochem Parasitol 1996; 80: 15–25. [DOI] [PubMed] [Google Scholar]
- 4. Watkins WM, Sixsmith DG, Spencer HC, Boriga DA, Kariuki DM, Kipingor T, et al. Effectiveness of amodiaquine as treatment for chloroquine‐resistant Plasmodium falciparum infections in Kenya. Lancet 1984; 1: 357–359. [DOI] [PubMed] [Google Scholar]
- 5. Larrey D, Castot A, Pessayre D, Merigot P, Machayekhy JP, Feldmann G, et al. Amodiaquine‐induced hepatitis: a report of seven cases. Ann Intern Med 1986; 104: 801–803. [DOI] [PubMed] [Google Scholar]
- 6. Neftel KA, Woodtly W, Schmid M, Frick PG, Fehr J. Amodiaquine induced agranulocytosis and liver damage. Br Med J 1986; 292: 721–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Phillips‐Howard PA, West LJ. Serious adverse drug reactions to pyrimethamine–sulphadoxine, pyrimethamine–dapsone and to amodiaquine in Britain. J R Soc Med 1990; 83: 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olliaro P, Nevill C, LeBras J, Ringwald P, Mussano P, Garner P, et al. Systematic review of amodiaquine treatment in uncomplicated malaria. Lancet 1996; 348: 1196–1201. [DOI] [PubMed] [Google Scholar]
- 9. Jewell H, Maggs JL, Harrison AC, O'Neill PM, Ruscoe JE, Park BK. Role of hepatic metabolism in the bioactivation and detoxication of amodiaquine. Xenobiotica 1995; 25: 199–217. [DOI] [PubMed] [Google Scholar]
- 10. Shimizu S, Atsumi R, Itokawa K, Iwasaki M, Aoki T, Ono C, et al. Metabolism‐dependent hepatotoxicity of amodiaquine in glutathione‐depleted mice. Arch Toxicol 2009; 83: 701–707. [DOI] [PubMed] [Google Scholar]
- 11. Srivastava A, Maggs JL, Antoine DJ, Williams DP, Smith DA, Park BK. Role of reactive metabolites in drug‐induced hepatotoxicity. Handb Exp Pharmacol 2010; 196: 165–194. [DOI] [PubMed] [Google Scholar]
- 12. Li XQ, Björkman A, Andersson TB, Ridderström M, Masimirembwa CM. Amodiaquine clearance and its metabolism to N‐desethylamodiaquine is mediated by CYP2C8: a new high affinity and turnover enzyme‐specific probe substrate. J Pharmacol Exp Ther 2002; 300: 399–407. [DOI] [PubMed] [Google Scholar]
- 13. Churchill FC, Patchen LC, Campbell CC, Schwartz IK, Nguyen‐Dinh P, Dickinson CM. Amodiaquine as a prodrug: importance of metabolite(s) in the antimalarial effect of amodiaquine in humans. Life Sci 1985; 36: 53–62. [DOI] [PubMed] [Google Scholar]
- 14. Mount DL, Patchen LC, Nguyen‐Dinh P, Barber AM, Schwartz IK, Churchill FC. Sensitive analysis of blood for amodiaquine and three metabolites by high‐performance liquid chromatography with electrochemical detection. J Chromatogr 1986; 383: 375–386. [DOI] [PubMed] [Google Scholar]
- 15. Laurent F, Saivin S, Chretien P, Magnaval JF, Peyron F, Sqalli A, et al. Pharmacokinetic and pharmacodynamic study of amodiaquine and its two metabolites after a single oral dose in human volunteers. Arzneimittelforschung 1993; 43: 612–616. [PubMed] [Google Scholar]
- 16. Lai CS, Nair NK, Muniandy A, Mansor SM, Olliaro PL, Navaratnam V. Validation of high performance liquid chromatography‐electrochemical detection methods with simultaneous extraction procedure for the determination of artesunate, dihydroartemisinin, amodiaquine and desethylamodiaquine in human plasma for application in clinical pharmacological studies of artesunate‐amodiaquine drug combination. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877: 558–562. [DOI] [PubMed] [Google Scholar]
- 17. Scarsi KK, Fehintola FA, Ma Q, Aweeka FT, Darin KM, Morse GD, et al. Disposition of amodiaquine and desethylamodiaquine in HIV‐infected Nigerian subjects on nevirapine‐containing antiretroviral therapy. J Antimicrob Chemother 2014; 69: 1370–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harrison AC, Kitteringham NR, Clarke JB, Park BK. The mechanism of bioactivation and antigen formation of amodiaquine in the rat. Biochem Pharmacol 1992; 43: 1421–1430. [DOI] [PubMed] [Google Scholar]
- 19. Lobach AR, Uetrecht J. Involvement of myeloperoxidase and NADPH oxidase in the covalent binding of amodiaquine and clozapine to neutrophils: implications for drug‐induced agranulocytosis. Chem Res Toxicol 2014; 27: 699–709. [DOI] [PubMed] [Google Scholar]
- 20. Christie G, Breckenridge AM, Park BK. Drug‐protein conjugates‐XVIII. Detection of antibodies towards the antimalarial amodiaquine and its quinone imine metabolite in man and the rat. Biochem Pharmacol 1989; 38: 1451–1458. [DOI] [PubMed] [Google Scholar]
- 21. Masubuchi N, Makino C, Murayama N. Prediction of in vivo potential for metabolic activation of drugs into chemically reactive metabolite: correlation of in vitro and in vivo generation of reactive intermediates and in vitro glutathione conjugate formation in rats and humans. Chem Res Toxicol 2007; 20: 455–464. [DOI] [PubMed] [Google Scholar]
- 22. Tafazoli S, O'Brien PJ. Amodiaquine‐induced oxidative stress in a hepatocyte inflammation model. Toxicology 2009; 256: 101–109. [DOI] [PubMed] [Google Scholar]
- 23. Naisbitt DJ, Williams DP, O'Neill PM, Maggs JL, Willock DJ, Pirmohamed M, et al. Metabolism‐dependent neutrophil cytotoxicity of amodiaquine: a comparison with pyronaridine and related antimalarial drugs. Chem Res Toxicol 1998; 11: 1586–1595. [DOI] [PubMed] [Google Scholar]
- 24. Tingle MD, Jewell H, Maggs JL, O'Neill PM, Park BK. The bioactivation of amodiaquine by human polymorphonuclear leucocytes in vitro: chemical mechanisms and the effects of fluorine substitution. Biochem Pharmacol 1995; 50: 1113–1119. [DOI] [PubMed] [Google Scholar]
- 25. Madsen KG, Olsen J, Skonberg C, Hansen SH, Jurva U. Development and evaluation of an electrochemical method for studying reactive phase I metabolites: correlation to in vitro drug metabolism. Chem Res Toxicol 2007; 20: 821–831. [DOI] [PubMed] [Google Scholar]
- 26. Jurva U, Holmén A, Grönberg G, Masimirembwa C, Weidolf L. Electrochemical generation of amodiaquine quinoneimine and cysteinyl conjugates by MS, IR and NMR. Chem Res Toxicol 2008; 21: 928–935. [DOI] [PubMed] [Google Scholar]
- 27. Zanger UM, Klein K, Thomas M, Rieger JK, Tremmel R, Kandel BA, et al. Genetics, epigenetics, and regulation of drug‐metabolizing cytochrome P450 enzymes. Clin Pharmacol Ther 2014; 95: 258–261. [DOI] [PubMed] [Google Scholar]
- 28. Achour B, Barber J, Rostami‐Hodjegan A. Expression of hepatic drug‐metabolizing cytochrome P450 enzymes and their intercorrelations: a meta‐analysis. Drug Metab Dispos 2014; 42: 1349–1356. [DOI] [PubMed] [Google Scholar]
- 29. Johansson T, Jurva U, Grönberg G, Weidolf L, Masimirembwa C. Novel metabolites of amodiaquine formed by CYP1A1 and CYP1B1: structure elucidation using electrochemistry, mass spectrometry, and NMR. Drug Metab Dispos 2009; 37: 571–579. [DOI] [PubMed] [Google Scholar]
- 30. Damsten MC, van Vugt‐Lussenburg BM, Zeldenthuis T, de Vlieger JS, Commandeur JN, Vermeulen NP. Application of drug metabolising mutants of cytochrome P450 BM3 (CYP102A1) as biocatalysts for the generation of reactive metabolites. Chem Biol Interact 2008; 171: 96–107. [DOI] [PubMed] [Google Scholar]
- 31. Khojasteh SC, Prabhu S, Kenny JR, Halladay JS, Lu AY. Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: a re‐evaluation of P450 isoform selectivity. Eur J Drug Metab Pharmacokinet 2011; 36: 1–16. [DOI] [PubMed] [Google Scholar]
- 32. Zhang H, Gao N, Tian X, Liu T, Fang Y, Zhou J, et al. Content and activity of human liver microsomal protein and prediction of individual hepatic clearance in vivo . Sci Rep 2015. doi:10.1038/srep17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thompson RA, Isin EM, Li Y, Weidolf L, Page K, Wilson I, et al. In vitro approach to assess the potential for risk of idiosyncratic adverse reactions caused by candidate drugs. Chem Res Toxicol 2012; 25: 1616–1632. [DOI] [PubMed] [Google Scholar]
- 34. Crewe HK, Barter ZE, Yeo KR, Rostami‐Hodjegan A. Are there differences in the catalytic activity per unit enzyme of recombinantly expressed and human liver microsomal cytochrome P450 2C9? A systematic investigation into inter‐system extrapolation factors. Biopharm Drug Dispos 2011; 32: 303–318. [DOI] [PubMed] [Google Scholar]
- 35. German P, Greenhouse B, Coates C, Dorsey G, Rosenthal PJ, Charlebois E, et al. Hepatotoxicity due to a drug interaction between amodiaquine plus artesunate and efavirenz. Clin Infect Dis 2007; 44: 889–891. [DOI] [PubMed] [Google Scholar]
- 36. Mouly S, Lown KS, Kornhauser D, Joseph JL, Fiske WD, Benedek IH, et al. Hepatic but not intestinal CYP3A4 displays dose‐dependent induction by efavirenz in humans. Clin Pharmacol Ther 2002; 72: 1–9. [DOI] [PubMed] [Google Scholar]
- 37. Kharasch ED, Whittington D, Ensign D, Hoffer C, Bedynek PS, Campbell S, et al. Mechanism of efavirenz influence on methadone pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 2012; 91: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li XQ, Björkman A, Andersson TB, Gustafsson LL, Masimirembwa CM. Identification of human cytochrome P450s that metabolise anti‐parasitic drugs and predictions of in vivo drug hepatic clearance from in vitro data. Eur J Clin Pharmacol 2003; 59: 429–442. [DOI] [PubMed] [Google Scholar]
- 39. Xing J, Kirby BJ, Whittington D, Wan Y, Goodlett DR. Evaluation of CYPs inhibition and induction by artemisinin antimalarials in human liver microsomes and primary human hepatocytes. Drug Metab Dispos 2012; 40: 1757–1764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Comparison of aromatic regions of 1H–nuclear magnetic resonance spectra of (A) C′5‐glutathionyl‐amodiaquine (AQ‐SG) and (B) C′5‐glutathionly‐N‐desethylamodiaquine (DEAQ‐SG)
Figure S2 H‐H correlation spectroscopy spectrum of C′5‐glutathionly‐N‐desethylamodiaquine
Figure S3 Mass spectra of C′5‐glutathionyl‐amodiaquine (A) and C′5‐glutathionly‐N‐desethylamodiaquine (B) extracted from human liver microsome incubations with amodiaquine and N‐desethylamodiaquine, respectively
Figure S4 Concentration dependency of C′5‐glutathionyl‐amodiaquine formation (A) in the presence (closed circle) and absence (open circle) of NADPH‐regenerating system when incubating amodiaquine varying from 1 to 100 μmol l–1 with 1 mg ml−1 human liver microsomes and C′5‐glutathionly‐N‐desethylamodiaquine formation (B) in the presence (closed triangle) and absence (open triangle) of NADPH‐regenerating system when incubating N‐desethylamodiaquine varying from 1 to 50 μmol l–1 with 1 mg ml−1 human liver microsomes. Lines are only intended to guide the eye. Detailed incubation conditions can be found in the text
Table S1 1H‐nuclear magnetic resonance spectra of aromatic protons of glutathione conjugates of amodiaquine and N‐desethylamodiaquine
Supporting info item