

Abstract
Aims
Netazepide, a gastrin/cholecystokinin 2 receptor antagonist, once daily for 12 weeks reduced the number of tumours and size of the largest one in 16 patients with autoimmune chronic atrophic gastritis (CAG), achlorhydria, hypergastrinaemia and multiple gastric neuroendocrine tumours (type 1 gastric NETs), and normalized circulating chromogranin A (CgA) produced by enterochromaffin‐like cells, the source of the tumours. The aim was to assess whether longer‐term netazepide treatment can eradicate type 1 gastric NETs.
Methods
After a mean 14 months off netazepide, 13 of the 16 patients took it for another 52 weeks. Assessments were: gastroscopy; gene‐transcript expression in corpus biopsies using quantitative polymerase chain reaction; blood CgA and gastrin concentrations; and safety assessments.
Results
While off‐treatment, the number of tumours, the size of the largest one, and CgA all increased again. Netazepide for 52 weeks: cleared all tumours in 5 patients; cleared all but one tumour in one patient; reduced the number of tumours and size of the largest one in the other patients; normalized CgA in all patients; and reduced mRNA abundances of CgA and histidine decarboxylase in biopsies. Gastrin did not increase further, confirming that the patients had achlorhydria. Netazepide was safe and well tolerated.
Conclusions
A gastrin/cholecystokinin 2 receptor antagonist is a potential medical and targeted treatment for type 1 gastric NETs, and an alternative to regular gastroscopy or surgery. Treatment should be continuous because the tumours will regrow if it is stopped. Progress can be monitored by CgA in blood or biomarkers in mucosal biopsies.
Keywords: chromogranin A, chronic atrophic gastritis, gastric neuroendocrine tumours, gastrin, gastrin/cholecystokinin‐2 receptor antagonist, netazepide
What is Already Known about this Subject
Patients with autoimmune CAG develop multiple type 1 gastric NETs as result of hypergastrinaemia secondary to achlorhydria.
Short‐term treatment with netazepide, a gastrin/CCK2 receptor antagonist, reduces the number of tumours, but does not eradicate them.
What this Study Adds
The tumours regrow if netazepide treatment is stopped.
Long‐term, continuous netazepide treatment has the potential to eradicate them.
Table of Links
| TARGETS | |
|---|---|
| G protein‐coupled receptors 2 | SST2 receptor |
| CCK2 receptor | SST5 receptors |
Introduction
Gastric neuroendocrine tumours (NETs), previously called carcinoids 3, 4, arise from enterochromaffin‐like (ECL) cells in the gastric mucosa, which express gastrin/cholecystokinin 2 (CCK2) receptors 5. Gastric NETs can be separated into three types 6, 7. Type 1 occur in patients with autoimmune chronic atrophic gastritis (CAG), are usually multiple and <1–2 cm in diameter, and comprise about 80% of gastric NETs. An estimated 5% of type 1 gastric NETs metastasize 7. The features of CAG are: chronic inflammation and atrophy of the gastric corpus; achlorhydria and secondary hypergastrinaemia; ECL‐cell growth; and malabsorption of vitamin B12, which leads to pernicious anaemia and neurological signs in some patients 8, 9.
The management of type 1 gastric NETs is controversial. The European Neuroendocrine Tumour Society (ENETS) recommends that patients undergo yearly gastroscopy, during which the tumours are mapped and biopsied 10. If there are concerns about the morphology and histology of the tumours, they can be treated surgically by endoscopic polypectomy or gastric antrectomy. Polypectomy does not remove the source of the hypergastrinaemia, and the tumours can recur 11. Antrectomy leads to a reduction in circulating gastrin by removing the source of the hypergastrinaemia, but it is not effective in all patients, and it carries the risk of morbidity and mortality 12, 13. Somatostatin (SST) analogues are sometimes used off‐label to treat patients with type 1 gastric NETs; they reduce gastrin by negative feedback on the gastrin‐secreting G cells in the gastric antrum, and by a direct effect on ECL cells 14, 15, which possess SST2 and SST5 receptors. Several studies have shown that SST analogues can cause regression of type 1 NETs, and are generally well‐tolerated 14, 15, 16, 17, 18, 19. However, SST analogues inhibit release of various hormones, such as growth hormone, insulin, glucagon, thyroid stimulating hormone and cholecystokinin, and neither ENETS nor the North American NETS recommends them for treating type 1 gastric NETs 10, 20.
A gastrin/CCK2 receptor antagonist is a more logical treatment of type 1 gastric NETs, because they are gastrin driven. Many have been described, but to date none has been developed into a medicine 21. In nonclinical studies, netazepide (YF476) is a potent, highly‐selective, competitive gastrin/CCK2 receptor antagonist with good oral bioavailability 22. Netazepide prevented hypergastrinaemia‐induced increases in ECL‐cell activity, density and oxyntic mucosal thickness in rats 23, and reduced substantially the incidence of ECL‐cell carcinomas in a strain of female cotton rats that develop such tumours spontaneously as a result of hypergastrinaemia secondary to gastric hypoacidity 24. Furthermore, netazepide not only prevented formation of gastric NETs accelerated by hypergastrinaemia induced by loxtidine, an insurmountable histamine H2‐receptor antagonist, in Mastomys rodents – which have a genetic predisposition to gastric NETs – but also caused shrinkage of formed lesions 25.
Netazepide is also an orally‐active gastrin/CCK2 receptor antagonist in healthy subjects. It caused dose‐dependent, persistent inhibition of pentagastrin‐induced gastric acid secretion 26 and prevented the increase in plasma CgA – a biomarker of ECL‐cell hyperactivity – resulting from proton pump inhibitor‐induced hypergastrinaemia. Furthermore, netazepide reduced baseline plasma CgA – a sign of ECL‐cell hypoactivity 27.
Thus, there is evidence from animal models and healthy subjects to justify developing a gastrin/CCK2 receptor antagonist as a medical treatment for patients with gastric NETs type 1, which are gastrin‐driven and comprise the majority of all gastric NETs 6, 7. Indeed, in two separate studies in Trondheim, Norway, and Liverpool, UK, in patients with type 1 gastric NETs, netazepide 50 mg once daily for 12 weeks, reduced the number of tumours and the size of the largest one 28, 29. Current toxicology studies of netazepide allow treatment for only 13 weeks. However, because of those favourable results, the UK Medicines and Healthcare products Regulatory Agency and the Norwegian Regulatory Agency (NoMA) allowed netazepide treatment for longer without the extended toxicology studies normally required 30.
Aim
Our aim was to treat patients with type 1 gastric NETs from the aforementioned 12‐week studies with netazepide for another 52 weeks, and pool the results: (1) to assess if netazepide for longer can eradicate the tumours; (2) to identify tumour biomarkers; and (3) to continue to assess the safety and tolerability of netazepide.
Methods
Study design
The two centres in Trondheim and Liverpool first did an open study in which Helicobacter pylori negative patients with CAG, hypergastrinaemia, multiple type 1 gastric NETs and raised circulating CgA were treated with netazepide 50 mg once daily for 12 weeks, with 12‐week follow‐up. The same protocol was used for both studies. There were seven outpatient visits. Visits 1 and 2 were to assess eligibility and to obtain consent. Visits 3, 4, 5 and 6 were at 3, 6, 9 and 12 weeks after starting treatment, respectively. Visit 7 was at 12 weeks after stopping treatment. Gastroscopy was done at visits 2, 4, 6 and 7, during which the number of tumours was counted, the diameter of the largest tumour was measured against the open biopsy forceps, and the tumours and flat corpus mucosa were biopsied. Blood was collected at visits 2, 3, 4, 5, 6 and 7 for assay of fasting serum gastrin, plasma or serum CgA, and plasma netazepide, as described previously 28, 29. Safety and tolerability were assessed by: medical examination; echocardiography; blood and urine tests; and adverse events, which were recorded by the patient on a diary card and transferred to the patient's case report form by the investigator. Trio Medicines (London, UK) supplied netazepide 25 mg capsules.
The Medicines and Healthcare products Regulatory Agency approved a protocol amendment for the Liverpool patients to receive netazepide 50 mg once daily for another 52 weeks. The frequency of clinic visits and gastroscopy was reduced to 3‐ and 6‐monthly, respectively, to make the extension to the study less demanding. NoMA would not agree to a similar protocol amendment for the Trondheim study. However, they did agree to long‐term treatment with netazepide 25 mg once daily under the Norwegian ‘named patient’ scheme. Cambridgeshire 1 Research Ethics Committee (REC) and East of England – Cambridge East approved the Liverpool 12‐ and 52‐week studies, respectively (reference 10/H0304/51). The Regional REC approved the Trondheim studies (reference 2010/1617). Subjects gave written, informed consent. The 12‐week studies were completed during January 2011 to July 2012, and the 52‐week studies during October 2012 to April 2014. There was a mean interval of 14 (range 8–19) months between the end of dosing patients in the 12‐week studies and the start of dosing them in the 52‐week studies. The interval off‐treatment was required to obtain regulatory and REC approvals. The studies complied with the ICH Guideline for Good Clinical Practice and the EU Clinical Trial Directive. The Liverpool study was registered as ClinicalTrials.gov NCT01339169, and the Trondheim study as ClinicalTrials.gov NCT01444014.
Biopsies
The Liverpool centre took biopsies of the tumours and flat corpus mucosa at each endoscopy during both the 12‐ and 52‐week studies and assessed them in detail, so only those procedures are described here. Results of biopsies taken during the 12‐week study in Trondheim are published elsewhere 28.
Histology
The extent of ECL‐cell proliferation in tumour biopsies was classified as linear ECL‐cell hyperplasia, micronodular ECL‐cell hyperplasia, ECL‐cell dysplasia or NET, as described previously 29. The histopathologist was blind to the clinical and endoscopic findings.
Biomarkers
RNA was extracted from corpus mucosal biopsies by Tri‐Reagent and reverse transcribed before assessment of real‐time polymerase chain reaction (qPCR) abundances of CgA, histidine decarboxylase (HDC) and matrix metalloproteinase‐7 (MMP‐7). qPCR used the primers, probes, master mix, standards, and 7500 real‐time PCR machine (Applied Biosystems, Warrington, UK), as described previously 29. Absolute abundances relative to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) were calculated.
Statistics
Wilcoxon signed rank test was used to test for significant differences with respect to pooled data from the two centres for the number of tumours, size of the largest tumour, and circulating gastrin and CgA for the 12‐week and 52‐week studies, and for any changes during the period off‐treatment. Data were illustrated by box–whisker plots for median, interquartile range and range of results. Because the two centres used different assays with different units for CgA 28, 29, we adjusted measurements to the upper limit of normal (Liverpool 22 U l−1; Trondheim 6 nmol l−1). Data for qPCR were analysed by SPSS v20 after testing for normality using the Kolmogorov–Smirnov test, and then analysed using a Wilcoxon signed rank test. Differences were deemed significant when P < 0.05.
Results
12 weeks' netazepide treatment
Sixteen patients, eight per centre, mean age 61 (range 50–76) years, entered and completed the study. Eleven were receiving vitamin B12 for treatment of pernicious anaemia.
At baseline, the median number of tumours was 10 (range 4–30) and the median size of the largest one was 6 (range 3–15) mm (Table S1; Figure 1). Netazepide 50 mg once daily for 12 weeks reduced significantly the number of tumours (P < 0.001) and the size of the largest tumour (P < 0.001), and reduced plasma or serum CgA in all patients to within normal limits (P < 0.001). The reduction in CgA was evident at the first assessment, at 3 weeks.
Figure 1.
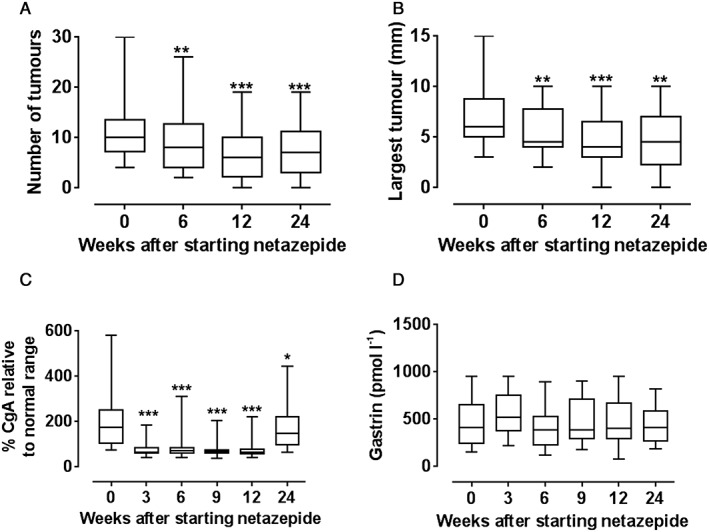
Effect (n = 16 patients) of netazepide 50 mg once daily for 12 weeks, and 12 weeks off treatment, on: (A) number of tumours; (B) size of largest tumour; (C) chromogranin A (adjusted); and (D) gastrin. *P < 0.05; **P < 0.01; ***P < 0.001. (Normal ranges. chromogranin A: Trondheim ≤6 nmol l−1; Liverpool ≤22 U l−1. Gastrin: <40 pmol l−1)
Netazepide did not affect (P < 0.1) the high serum gastrin observed at baseline (median 415 pmol l−1; normal ≤40 pmol l–1 for both centres). At 24 weeks, when patients had been off‐treatment for 12 weeks, the number of tumours, the size of the largest tumour, and circulating CgA had all increased again, but the effect of netazepide was still significant, albeit less so.
Plasma netazepide concentrations before and 1 h after dosing ranged from 4.6 to 7.0 ng ml−1 and 87 to 220 ng ml−1, respectively, at 3, 6, 9 and 12 weeks. There were no clinically relevant changes in safety tests, and 10 mild‐to‐moderate adverse events, none of which was deemed related to treatment by the investigator. No patient had their treatment stopped.
Interval off‐treatment
All eight patients from the Liverpool study and six from the Trondheim study consented to a further 52 weeks' treatment with netazepide. One Trondheim patient withdrew soon after, for personal reasons. The mean interval between the end of 12 weeks' netazepide treatment and the start of 52 weeks' treatment in 13 patients was 14 (range 8–19) months. During the period off‐treatment, the number of tumours (P < 0.01), the size of the largest tumour (P < 0.05), and plasma or serum CgA (P < 0.001) all increased. Serum gastrin was unaffected (Figure 2).
Figure 2.
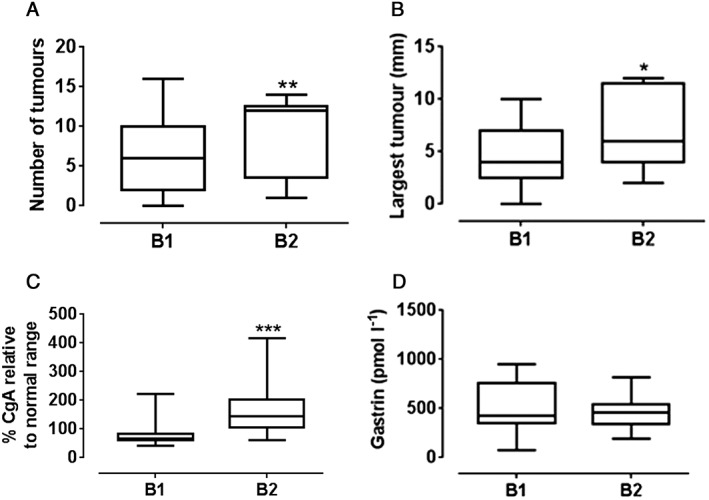
Changes (n = 13 patients) during the period off netazepide treatment in: (A). number of tumours; (B). size of the largest tumour; (C). chromogranin A (adjusted); and (D). gastrin. (Normal ranges. chromogranin A: Trondheim ≤6 nmol l−1; Liverpool ≤22 U l−1; Gastrin: ≤40 pmol l−1). *P < 0.05; **P < 0.01; ***P < 0.001. B1 = end of 12‐weeks' netazepide. B2 = start of 52 weeks' netazepide
52 weeks' netazepide treatment
Thirteen patients from the two centres entered and completed another 52 weeks' treatment with netazepide 50 mg (Liverpool) or 25 mg (Trondheim) once daily for 52 weeks. Netazepide cleared all tumours in five of the 13 patients and reduced the number of tumours (P < 0.01) and the size of the largest one (P < 0.001) in the other patients, and reduced plasma or serum CgA to within normal limits in all patients (P < 0.001; Table S2; Figure 3). One patient was left with only one tumour. Serum gastrin was unaffected. There were no clinically relevant changes in safety tests and 28 adverse events. One adverse event was considered severe and the others mild‐to‐moderate. None of them was deemed related to treatment by the investigator. No patient had their treatment stopped.
Figure 3.
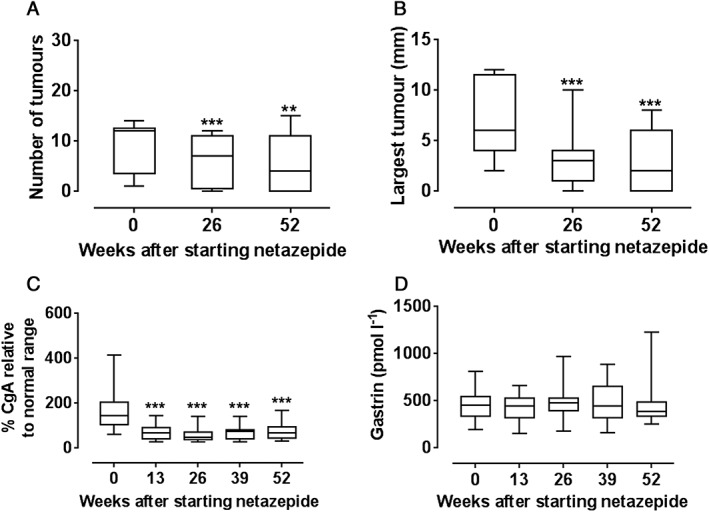
Effect (n = 13 patients) of netazepide 25 mg (n = 5) or 50 mg (n = 8) once daily for 52 weeks on: (A) number of tumours; (B) size of largest tumour; (C) chromogranin A (adjusted); and (D) gastrin. **P < 0.01; ***P < 0.001. (Normal ranges. chromogranin A: Trondheim ≤6 nmol l−1; Liverpool ≤22 U l−1; Gastrin: ≤40 pmol l−1)
Biopsies
Histology
When Liverpool patients were screened for the study, biopsies of their tumours and flat mucosa showed that all eight patients had NETs and ECL‐cell hyperplasia, respectively (Table 1). After 12 weeks' treatment, four patients had NETs and four had micronodular ECL‐cell hyperplasia 29. After 52 weeks' treatment, only three patients had NETs.
Table 1.
Histology of tumour biopsies (n = 8 patients)
| 12‐week study | 52‐week study | |||||||
|---|---|---|---|---|---|---|---|---|
| Patient | Screen | Week 0 | Week 6 | Week 12 | Week 24 | Week 0 | Week 24 | Week 52 |
| 1 | NET | NET | ECL‐M | ECL‐M | ECL‐M | NET | ECL‐M | ECL‐M |
| 2 | NET | NET | NET | NET | ECL‐M | ECL‐D | ECL‐M | ECL‐M |
| 3 | NET | ECL‐M | ECL‐M | ECL‐M | ECL‐M | ECL‐M | ECL‐M | ECL‐L |
| 4 | NET | NET | ECL‐M | ECL‐M | NET | ECL‐M | ECL‐M | ECL‐M |
| 5 | NET | NET | NET | NET | NET | NET | ECL‐M | NET |
| 6 | NET | NET | NET | ECL‐M | NET | NET | NET | NET |
| 7 | NET | NET | NET | NET | NET | NET | NET | NET |
| 8 | NET | NET | NET | NET | ECL‐M | nb | ECL‐M | ECL‐M |
ECL‐D, ECL‐cell dysplasia; ECL‐L, linear ECL cell hyperplasia; ECL‐M, micronodular ECL‐cell hyperplasia; nb, no biopsy; NET, neuroendocrine tumour
Biomarkers
After 26 and 52 weeks' netazepide treatment, there were significant reductions in qPCR abundances (normalized to GAPDH) of the biomarkers CgA and HDC, but not MMP‐7, relative to baseline of the 12‐week study and to the baseline after the period off‐treatment (Figure 4).
Figure 4.
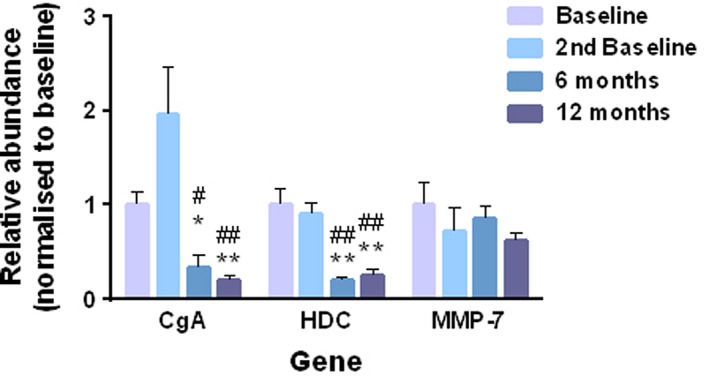
Absolute quantities of abundances of biomarkers in gastric mucosal biopsies (n = 8) relative to GAPDH during 52 weeks of netazepide treatment. * P < 0.05 and ** P < 0.01 compared to baseline. # P < 0.05 and ## P < 0.01 compared to second baseline
Discussion
The limitations of the studies were: the open design; the subjective counting of the tumours and measurement of the diameter of the largest one; tumour but not mucosal biopsies may have contributed to tumour clearance in some patients; and the small numbers of patients. Placebo controls seemed unreasonable given that: the studies were exploratory or proof‐of‐concept; gastric NETs are rare and recognized as an orphan disease in Europe and the USA 31, so the number of eligible patients per centre was limited; and the studies were very demanding for the patient. Also, the gastroscopies were done by only two experienced physicians per centre, and they counted and measured the tumours carefully. Furthermore, the histology of the gastric biopsies was assessed blind, and the biomarker measurements were objective.
The clearance of tumours in five of 13 patients during 52 weeks' treatment, and the reduction in circulating CgA in all patients, are consistent with a treatment effect. One patient was left with only one tumour. The reduction in the number of tumours and the size of the largest one in the other patients are supportive evidence of a treatment effect. The reduction in mRNA abundances of the biomarkers CgA and HDC and the changes in the histology in the biopsies of the tumours and flat mucosa, respectively, during both the 12‐week 29 and 52‐week studies are further evidence of a treatment effect.
While patients were off‐treatment for a mean of 14 (range 8–19) months, the number of tumours, size of the largest tumour, circulating CgA and tumour biomarkers all increased again. Circulating CgA is a valid biomarker of ECL‐cell activity in patients with type 1 gastric NETs 32, 33, 34 and in healthy subjects 27. Type 1 gastric NETs are derived from ECL cells, which are also the source of the increased circulating CgA in CAG patients 33. The normalisation of circulating CgA during both the initial and extended studies is consistent with netazepide inhibiting ECL‐cell growth via antagonism of gastrin/CCK2 receptors on the ECL cells. Circulating CgA increased again during the interval off‐treatment, when gastrin/CCK2 receptors would not have been blocked.
Apart from its well‐known effect on gastric acid secretion and its effect on ECL‐cell growth via gastrin/CCK2 receptors, gastrin also has an effect on various cellular mechanisms, including proliferation, apoptosis, migration, differentiation and angiogenesis 35, 36, 37, 38, 39. Proteins such as Reg, MMP‐7, MMP‐1, and plasminogen activator inhibitor (PAI)‐1 and PAI‐2 are overexpressed in the stomach of patients with hypergastrinaemia, and may contribute to the formation of gastric tumours 40, 41, 42, 43. Therefore, abundances of MMP‐7, PAI‐1 and PAI‐2, as well as CgA and HDC, were studied in gastric mucosal biopsies as potential biomarkers for type 1 gastric NETs before and during netazepide treatment. In the 12‐week study, CgA, HDC and MMP‐7 were reduced relative to baseline 29. They had almost returned to pretreatment levels 12 weeks after the end of treatment. PAI‐1 and PAI‐2 did not change significantly. In the 52‐week study, CDA and HDC were again reduced significantly, at 26 and 52 weeks, but at neither time was there an effect on MMP‐7.
The results are strengthened further by a recent report of the effect of netazepide on microRNA‐222 (miR‐222) in corpus mucosal biopsies and serum during the 12‐week study in Liverpool patients 44. miRNAs are nonprotein coding short RNAs that regulate ~30% of the human genome, and inhibit the translation, increase cleavage or induce degradation of target mRNAs. One gene can be regulated by several miRNAs, including tumour suppressor genes and oncogenes. There was a small but significant increase in miR‐222 expression in mucosal biopsies before netazepide compared to subjects with normal serum gastrin. Expression decreased significantly during netazepide treatment, and returned to baseline after treatment cessation. Serum miR‐222 expression was also increased significantly, by 5.7‐fold, before netazepide treatment compared to controls. As with mucosal biopsies, expression decreased significantly during netazepide treatment and returned towards baseline after treatment cessation. Gastrin‐induced miR‐222 overexpression in a human gastric adenocarcinoma cell line transfected with the CCK2 receptor resulted in suppression of the oncogene p27kip1, which was reversed by pretreatment with netazepide 44. miR‐222 is also dysregulated in gastric adenocarcinoma and in the stomach infected with H. pylori 45, and may be a useful biomarker for monitoring gastrin‐induced premalignant changes in the stomach 46.
Our rationale for pooling the results is as follows. The protocol for the 12‐week and 52‐week studies was the same for both centres, apart from the reduction in netazepide dose from 50 mg to 25 mg daily in the 52‐week study at the Trondheim centre, for reasons explained earlier, and the two centres used different assays for circulating CgA. Also, the Liverpool centre did a more detailed assessment of mucosal and blood biomarkers in the 52‐week study. The results of the 12‐week studies, which are published elsewhere 28, 29, were remarkably similar for the two centres. Each centre was independent, and there was no contact between them during either of the studies. The effects of netazepide for 52 weeks on tumour number and size and circulating CgA and gastrin were also similar for the two centres, despite the difference in doses. Circulating CgA was reduced by netazepide to within the normal range in both centres and in both studies, so it seemed reasonable to adjust measurements to the upper limit of normal. Patients in the Trondheim centre were given the lower dose of netazepide because NoMA was more receptive to allowing a lower dose for long‐term treatment, and netazepide 25 mg of a similar formulation was top of the dose–response curve for increasing gastric pH in healthy subjects 47. Netazepide is a competitive antagonist at CCK2 receptors 22, 26, so patients with hypergastrinaemia may require adjustment of netazepide dose according to their serum gastrin concentration. Finding the dose for phase 3 studies is the most challenging aspect of proof‐of‐concept studies: 25 mg once daily was enough for patients in one centre, and might be enough for all patients with type 1 gastric NETs.
In animal models 24 and healthy subjects 27, acid suppression by netazepide leads to a secondary increase in serum gastrin, which is ‘harmless’ because the gastrin/CCK2 receptors are blocked. Netazepide did not increase serum gastrin further in the CAG patients, confirming that they had achlorhydria as a result of parietal‐cell atrophy. In other words, their serum gastrin was already maximally increased.
Netazepide has so far been safe and well tolerated in clinical trials. To date, about 220 healthy subjects have taken netazepide by mouth for up to 6 weeks, and 27 patients have taken it for 3–36 months. Adverse events have been minor, transient, independent of netazepide dose, and as common in placebo or comparator groups. Plasma netazepide levels at 1 h after 25 or 50 mg doses daily for 12 weeks in CAG patients were within the range seen with those doses at that time in studies of healthy subjects that used a similar formulation 47. Thus, achlorhydria does not appear to affect the bioavailability of netazepide in CAG patients.
Patients with pernicious anaemia, one of the possible presentations of type 1 gastric NETs, have a nearly seven‐fold increased risk of gastric adenocarcinoma 48. Children with hypergastrinaemia caused by genetic mutations of KCNQ1 or KCNE1 49, 50, 51, 52 or ATP4A 53, genes that control acid secretion by the parietal cell, not only develop gastric NETs, but also have a high risk of gastric adenocarcinoma. Some patients with those genetic mutations have needed gastrectomy. These reports all support the concept that hypergastrinaemia has malignant potential. Indeed, all gastric NETs have the potential to metastasize, especially ones that are >2 cm in size, infiltrate the muscularis propria, are angioinvasive and/or are G2 grade 10. The percentage of type 1 gastric NETs that metastasize may be an underestimate 31.
A survey of cancer registries and a PubMed search yielded a prevalence rate for gastric NETs of 0.32 (range 0.09–0.92), 0.17 and 0.05 per 10 000 population of 10 European countries, USA and Japan, respectively 31. There has been a 10–20 fold increase in the prevalence of gastric NETs in the USA, Japan and the countries of Europe in recent decades. The reasons for the increases are unknown, but they may include improvements in clinical practice, such as diagnostic gastroscopy and biopsies, and greater awareness of the condition 54, 55.
In the absence of a licensed medical treatment for type 1 gastric NETs, SST analogues, such as octreotide and lanreotide, are sometimes used off‐label 14, 15, 16, 17, 18, 19, despite neither ENETS nor NANETS recommending them for that purpose 10, 20. SST (somatotropin release inhibiting factor) is an abundant neuropeptide, which acts on five subtypes of SST receptor (SST1–SST5). Activation of those receptors produces a wide range of physiological effects throughout the body, including inhibition of secretion of many hormones, modulation of neurotransmission, inhibition of cell proliferation and smooth muscle contractility 5. SST analogues are licensed only for treatment of patients with: acromegaly; symptoms associated with functional gastroentero‐pancreatic NETs, such as carcinoid tumours, insulinomas and glucagonomas; and progression of well‐differentiated advanced NETs 56, 57. An estimated 80% of those tumours express SST receptors. In clinical trials of octreotide, very common (≥1/10) adverse drug reactions (ADR), based on the CIOMS III convention, included: diarrhoea, headache, gall stones and hyperglycaemia, and common ADR (≥1/100 to <1/10) included: steatorrhoea, dizziness, hypothyroidism, cholecystitis, dyspnoea, hypoglycaemia and bradycardia 56. Glucose and thyroid function monitoring, and ultrasonic examination of the gallbladder for gall stones before and every 6 months after the start of treatment, are recommended. However, SST analogues have been generally well tolerated when used off‐label in patients with type 1 gastric NETs to inhibit gastrin release, reduce ECL‐cell hyperplasia and to reduce the tumour load 14, 15, 16, 17, 18, 19. The tumours regrow when treatment is stopped.
Of 254 consecutive patients with type 1 gastric NETs from five countries, 20 (7.9%) had metastases to lymph nodes or liver at presentation 58. Of those 20 patients, 10 underwent gastrectomy and lymph node dissection, four underwent antrectomy and wedge resection, and one underwent only primary tumour biopsy. Five patients were treated with SST analogues; one had a complete response, and the disease stabilized in another. The other three patients had liver metastases, which progressed. All 20 patients were alive after follow‐up for a mean 83 (range 12–360) months. SST analogues were generally well tolerated, apart from deterioration in control of one patient's diabetes.
Thus, there is evidence that SST analogues are an effective medical treatment for patients with type 1 gastric NETs, perhaps even ones that have metastasized. However, they are not licensed for that purpose and there have been no controlled trials. Furthermore, their ADR profile is unfavourable 56, they must be given by injection, and they are expensive 19. An orally active gastrin/CCK2 receptor antagonist may offer advantages over an SST analogue in the treatment of type 1 gastric NETs. Further studies are required to confirm whether that is so.
Conclusions
This study confirms that type 1 gastric NETs are gastrin driven. A gastrin/CCK2 receptor antagonist such as netazepide is a potential targeted treatment for type 1 gastric NETs, and an alternative to regular gastroscopy or surgery. But that would need confirmation in a placebo‐controlled study in a larger number of patients. Given the rarity of the tumours, the trial would need to be multicentre and international.
Treatment of type 1 gastric NETs with a gastrin/CCK2 antagonist should be continuous, because they will eventually regrow if treatment is stopped. Measuring circulating biomarkers CgA or miR‐222 is a simple way to monitor treatment. Measuring gastric mucosal biomarkers CgA, HDC or miR‐222 is an alternative method.
A longer trial is required to assess whether a gastrin/CCK2 receptor antagonist can clear type 1 gastric NETs in all patients. The capacity of atrophic gastric mucosa to remodel itself may be limited, and a gastrin/CCK2 receptor antagonist may not be capable of causing complete regression of large type 1 gastric NETs that have been present for many years. However, it might reduce the risk of invasion/metastasis.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). None of the authors has any conflict of interest. The Liverpool centre was funded by Trio, a subsidiary of Hammersmith Medicines Research (HMR), a contract research organisation. The biopsy work was part‐funded by a grant from UKINETS (TRANSNETS). Trio monitored the study and measured serum gastrin, plasma CgA for the Liverpool centre and plasma netazepide for both centres. M.B. is an employee of HMR, and owns both companies. Trio holds the patent for netazepide.
Contributors
M.B. designed the studies and wrote the draft manuscript. D.M.P., A.R.M., L.S. and R.F. did the gastroscopies. F.C. did the histopathology. B.N.P. and A.V. did the qPCR. All authors contributed to and approved the final version of the manuscript.
Supporting information
Table S1 12‐week studies (n = 16 patients): A. number of tumours; B. size of largest tumour (mm); C. CgA (adjusted); and D. gastrin (pmol l−1)
Table S2 52‐week studies (n = 13 patients): A. number of tumours; B. size of largest tumour (mm); C. CgA (adjusted); and D. gastrin (pmol l−1)
Supporting info item
Supporting info item
Boyce, M. , Moore, A. R. , Sagatun, L. , Parsons, B. N. , Varro, A. , Campbell, F. , Fossmark, R. , Waldum, H. L. , and Pritchard, D. M. (2017) Netazepide, a gastrin/cholecystokinin‐2 receptor antagonist, can eradicate gastric neuroendocrine tumours in patients with autoimmune chronic atrophic gastritis. Br J Clin Pharmacol, 83: 466–475. doi: 10.1111/bcp.13146.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Davenport AP, Kelly E, Marrion N, Peters JA, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rindi G, Arnold R, Bosman FT. Nomenclature and classification of neuroendocrine neoplasms of the digestive system In: WHO classification of tumors of the digestive system, eds Bosman FT, Carneiro F, Hruban RH, Theise ND. Lyon: IARC, 2010; 13–14. [Google Scholar]
- 4. Klöppel G. Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer 2011; 18 (Suppl. 1): S1–16. [DOI] [PubMed] [Google Scholar]
- 5. Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rindi G, Luinetti O, Cornaggia M, Capella C, Solcia E. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma: a clinicopathologic study. Gastroenterology 1993; 104: 994–1006. [DOI] [PubMed] [Google Scholar]
- 7. Burkitt MD, Pritchard DM. Pathogenesis and management of gastric carcinoid tumours. Aliment Pharmacol Ther 2006; 24: 1305–1320. [DOI] [PubMed] [Google Scholar]
- 8. Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis – pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol 2013; 10: 529–541. [DOI] [PubMed] [Google Scholar]
- 9. Annibale B, Lahner E, Fave GD. Diagnosis and management of pernicious anemia. Curr Gastroenterol Rep 2011; 13: 518–524. [DOI] [PubMed] [Google Scholar]
- 10. Delle Fave G, Kwekkeboom DJ, Van Cutsem E, Rindi G, Kos‐Kudla B, Knigge U, et al. ENETS Consensus Guidelines for the management of patients with gastroduodenal neoplasms. Neuroendocrinology 2012; 95: 74–87. [DOI] [PubMed] [Google Scholar]
- 11. Merola E, Sbrozzi‐Vanni A, Panzuto F, D'Ambra G, Di Giulio E, Pilozzi E, et al. Type I gastric carcinoids: a prospective study on endoscopic management and recurrence rate. Neuroendocrinology 2012; 95: 207–213. [DOI] [PubMed] [Google Scholar]
- 12. Hirschowitz B, Griffith J, Pellegrin D, Cummings O. Rapid regression of ECL‐cell gastric carcinoids in pernicious anemia after antrectomy. Gastroenterology 1992; 102: 1409–1418. [PubMed] [Google Scholar]
- 13. Ozao‐Choy J, Buch K, Strauchen JA, Warner RR, Divino CM. Laparoscopic antrectomy for the treatment of type I gastric carcinoid tumors. J Surg Res 2010; 162: 22–25. [DOI] [PubMed] [Google Scholar]
- 14. Fykse V, Sandvik A, Qvigstad G, Falkmer S, Syversen U, Waldum H. Treatment of ECL cell carcinoids with octreotide LAR. Scand J Gastroenterol 2004; 39: 621–628. [DOI] [PubMed] [Google Scholar]
- 15. Grozinsky‐Glasberg S, Kaltsas G, Gur C, Gal E, Thomas D, Fichman S, et al. Long‐acting somatostatin analogues are an effective treatment for type 1 gastric carcinoid tumours. Eur J Endocrinol 2008; 159: 475–482. [DOI] [PubMed] [Google Scholar]
- 16. Campana D, Nori F, Pezzilli R, Piscitelli L, Santini D, Brocchi E, et al. Gastric endocrine tumors type I: treatment with long‐acting somatostatin analogs. Endocr Relat Cancer 2008; 15: 337–342. [DOI] [PubMed] [Google Scholar]
- 17. Li TT, Qui F, Qian Z, Wan J, Qi XK, Wu BY. Classification, clinicopathologic features and treatment of gastric neuroendocrine tumors. World J Gastroenterol 2014; 20: 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas D, Tsolakis AV, Grozinsky‐Glasberg S, Fraenkel M, Alexandraki K, Sougioultzis S, et al. Long‐term follow‐up of a large series of patients with type 1 gastric carcinoid tumors: data from a multicenter study. Eur J Endocrinol 2013; 168: 185–193. [DOI] [PubMed] [Google Scholar]
- 19. Massironi S, Zilli A, Fanetti I, Ciafardini C, Conte D, Peracchi M. Intermittent treatment of recurrent type‐1 gastric carcinoids with somatostatin analogues in patients with chronic autoimmune atrophic gastritis. Dig Liver Dis 2015; 47: 978–983. [DOI] [PubMed] [Google Scholar]
- 20. Kulke MH, Anthony LB, Bushnell DL, de Herder WW, Goldsmith SJ, Klimstra DS, et al. North American Neuroendocrine Tumor Society (NANETS). NANETS treatment guidelines: well‐differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas 2010; 39: 735–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Herranz R. Cholecystokinin antagonists: pharmacological and therapeutic potential. Med Res Rev 2003; 23: 559–605. [DOI] [PubMed] [Google Scholar]
- 22. Takemoto Y, Yuki H, Nishida A, Ito H, Kobayashi‐Uchida A, Takinami Y, et al. Effects of YF476, a potent and selective gastrin/cholecystokinin‐B receptor antagonist, on gastric acid secretion in beagle dogs with gastric fistula. Arzneimittelforschung 1998; 48: 403–407. [PubMed] [Google Scholar]
- 23. Chen D, Zhao CM, Norlén P, Björkqvist M, Ding XQ, Kitano M, et al. Effect of cholecystokinin‐2 receptor blockade on rat stomach ECL cells. A histochemical, electron‐microscopic and chemical study. Cell Tissue Res 2000; 299: 81–95. [DOI] [PubMed] [Google Scholar]
- 24. Martinsen TC, Kawase S, Håkanson R, Torp SH, Fossmark R, Qvigstad G, et al. Spontaneous ECL cell carcinomas in cotton rats: natural course and prevention by a gastrin receptor antagonist. Carcinogenesis 2003; 24: 1887–1896. [DOI] [PubMed] [Google Scholar]
- 25. Kidd M, Siddique ZL, Drozdov I, Gustafsson BI, Camp RL, Black JW, et al. The CCK2 receptor antagonist, YF476, inhibits mastomys ECL cell hyperplasia and gastric carcinoid tumor development. Regul Pept 2010; 162: 52–60. [DOI] [PubMed] [Google Scholar]
- 26. Boyce M, Warrington S, Black J. Netazepide, a gastrin/CCK2 receptor antagonist, causes dose‐dependent, persistent inhibition of the responses to pentagastrin in healthy subjects. Br J Clin Pharmacol 2013; 76: 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boyce M, Dowen S, Turnbull G, van den Berg F, Zhao CM, Chen D, et al. Effect of netazepide, a gastrin/CCK2 receptor antagonist, on gastric acid secretion and rabeprazole‐induced hypergastrinaemia: a double‐blind trial in healthy subjects. Br J Clin Pharmacol 2015; 79: 744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fossmark R, Sørdal O, Jianu C, Qvigstad G, Nordrum I, Boyce M, et al. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment Pharmacol Ther 2012; 36: 1067–1075. [DOI] [PubMed] [Google Scholar]
- 29. Moore A, Boyce M, Steele I, Campbell F, Varro A, Pritchard DM. Netazepide, a gastrin/CCK‐2 receptor antagonist, reduces tumour biomarker gene expression and causes regression of type 1 gastric neuroendocrine tumours. PLoS One 2013; 8: e76462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. ICH M3 (R2) guidelines on non‐clinical safety studies for conduct of human clinical trials and marketing authorisation for pharmaceuticals. EMA/CPMP/ICH/286/1995. December 2009.
- 31. Boyce M, Thomsen L. Gastric neuroendocrine tumours: prevalence in Europe, USA and Japan, and rationale for treatment with a gastrin/CCK2 receptor antagonist. Scand J Gastroenterol 2015; 50: 550–559. [DOI] [PubMed] [Google Scholar]
- 32. Waldum HL, Arnestad JS, Brenna E, Syversen U, Sandvik AK. Marked increase in gastric acid secretory capacity after omeprazole treatment. Gut 1996; 39: 649–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanduleanu S, De Bruïne A, Stridsberg M, Jonkers D, Biemond I, Hameeteman W, et al. Serum chromogranin A as a screening test for gastric ECL‐cell hyperplasia during acid‐suppressive therapy. Eur J Clin Invest 2001; 31: 802–811. [DOI] [PubMed] [Google Scholar]
- 34. Peracchi M, Gebbia C, Basilisco G, Quatrini M, Tarantino C, Vescarelli C, et al. Plasma chromogranin A in patients with autoimmune chronic atrophic gastritis, enterochromaffin‐like cell lesions and gastric carcinoids. Eur J Endocrinol 2005; 152: 443–448. [DOI] [PubMed] [Google Scholar]
- 35. Dockray G, Dimaline R, Varro A. Gastrin: old hormone, new functions. Pflugers Arch 2005; 449: 344–355. [DOI] [PubMed] [Google Scholar]
- 36. Dockray G, Varro A, Dimaline R, Wang T. The gastrins: their production and biological activities. Annu Rev Physiol 2001; 63: 119–139. [DOI] [PubMed] [Google Scholar]
- 37. Dimaline R, Varro A. Novel roles of gastrin. J Physiol 2014; 592: 2951–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Watson SA, Grabowska A, El‐Zaatari M, Takhar A. Gastrin – active participant or bystander in gastric carcinogenesis? Nat Rev Cancer 2006; 6: 936–946. [DOI] [PubMed] [Google Scholar]
- 39. Cui G, Takaishi S, Ai W, Betz KS, Florholmen J, Koh TJ, et al. Gastrin‐induced apoptosis contributes to carcinogenesis in the stomach. Lab Invest 2006; 86: 1037–1051. [DOI] [PubMed] [Google Scholar]
- 40. Higham AD, Bishop LA, Dimaline R, Blackmore CG, Dobbins AC, Varro A, et al. Mutations of RegIalpha are associated with enterochromaffin‐like cell tumor development in patients with hypergastrinemia. Gastroenterology 1999; 116: 1310–1318. [DOI] [PubMed] [Google Scholar]
- 41. Varro A, Kenny S, Hemers E, McCaig C, Przemeck S, Wang TC, et al. Increased gastric expression of MMP‐7 in hypergastrinemia and significance for epithelial‐mesenchymal signaling. Am J Physiol Gastrointest Liver Physiol 2007; 292: G1133–G1140. [DOI] [PubMed] [Google Scholar]
- 42. Kumar JD, Steele I, Moore AR, Murugesan SV, Rakonczay Z, Venglovcz V, et al. Gastrin stimulates MMP‐1 expression in gastric epithelial cells: putative role in gastric epithelial cell migration. Am J Physiol Gastrointest Liver Physiol 2015; 309: G78–G86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nørsett KG, Steele I, Duval C, Sammut SJ, Murugesan SV, Kenny S, et al. Gastrin stimulates expression of plasminogen activator inhibitor‐1 in gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol 2011; 301: G446–G453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lloyd KA, Moore AR, Parsons BN, O'Hara A, Boyce M, Dockray GJ, et al. Gastrin‐induced miR‐222 promotes gastric tumor development by suppressing p27kip1 . Oncotarget 2016. doi:10.18632/oncotarget.99990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jiang C, Chen X, Alattar M, Wei J, Liu H. MicroRNAs in tumorigenesis, metastasis, diagnosis and prognosis of gastric cancer. Cancer Gene Ther 2015; 22: 291–301. [DOI] [PubMed] [Google Scholar]
- 46. Wander SA, Zhao D, Slingerland JM. P27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res 2011; 17: 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boyce M, David O, Darwin K, Mitchell T, Johnston A, Warrington S. Single oral doses of netazepide (YF476), a gastrin receptor antagonist, cause dose‐dependent, sustained increases in gastric pH compared with placebo and ranitidine in healthy subjects. Aliment Pharmacol Ther 2012; 36: 181–189. [DOI] [PubMed] [Google Scholar]
- 48. Vannella L, Lahner E, Osborn J, Annibale B. Systematic review: gastric cancer incidence in pernicious anaemia. Aliment Pharmacol Ther 2013; 37: 375–382. [DOI] [PubMed] [Google Scholar]
- 49. Rice K, Dickson G, Lane M, Crawford J, Chung SK, Rees MI, et al. Elevated serum gastrin levels in Jervell and Lange‐Nielsen syndrome: a matter of severe KCNQ1 dysfunction. Heart Rhythm 2011; 8: 551–554. [DOI] [PubMed] [Google Scholar]
- 50. Winbo A, Sandström O, Palmqvist R, Rydberg A. Iron‐deficiency anaemia, gastric hyperplasia, and elevated gastrin levels due to potassium channel dysfunction in the Jervell and Lange‐Nielsen syndrome in Sweden. Cardiol Young 2013; 23: 325–334. [DOI] [PubMed] [Google Scholar]
- 51. Winbo A, Stattin E, Diamant U, Persson J, Jensen SM, Rydberg A. Prevalence, mutation spectrum, and cardiac phenotype of the Jervell and Lange‐Nielsen Syndrome in Sweden. Europace 2012; 14: 1799–1806. [DOI] [PubMed] [Google Scholar]
- 52. Winbo A, Stattin E, Nordin C, Diamant U, Persson J, Jensen S, et al. Phenotype, origin and estimated prevalence of a common long QT syndrome mutation: a clinical, genealogical and molecular genetics study including Swedish R518X/KCNQ1 families. BMC Cardiovasc Disord 2014; 14: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Calvete O, Reyes J, Zuñiga S, Paumard‐Hernández B, Fernández V, Bujanda L, et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumour. Hum Mol Genet 2015; 24: 2914–2922. [DOI] [PubMed] [Google Scholar]
- 54. Modlin IM, Lye KD, Kidd M. A 50‐year analysis of 562 gastric carcinoids: small tumor or larger problem? Am J Gastroenterol 2004; 99: 23–32. [DOI] [PubMed] [Google Scholar]
- 55. Hodgson N, Koniaris LG, Livingstone AS, Franceschi D. Gastric carcinoids: a temporal increase with proton pump introduction. Surg Endosc 2005; 19: 1610–1612. [DOI] [PubMed] [Google Scholar]
- 56. Data sheet for octreotide acetate (Sandostatin LAR®). Available at http://www.medsafe.govt.nz/profs/datasheet/s/SandostatinLARinj.pdf (last accessed 28 September 2016).
- 57. Caplin M, Pavel M, Ćwikła J, Phan AT, Raderer M, Sedláčková E, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014; 371: 224–233. [DOI] [PubMed] [Google Scholar]
- 58. Grozinsky‐Glasberg S, Thomas D, Strosberg J, Pape UF, Felder S, Tsolakis A, et al. Metastatic type 1 gastric carcinoid: a real threat or just a myth? World J Gastroenterol 2013; 19: 8687–8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 12‐week studies (n = 16 patients): A. number of tumours; B. size of largest tumour (mm); C. CgA (adjusted); and D. gastrin (pmol l−1)
Table S2 52‐week studies (n = 13 patients): A. number of tumours; B. size of largest tumour (mm); C. CgA (adjusted); and D. gastrin (pmol l−1)
Supporting info item
Supporting info item