

Abstract
The efficacy of peroxisome proliferator-activated receptor α-agonists (e.g., fibrates) against nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH) in humans is not known. Pemafibrate is a novel selective peroxisome proliferator-activated receptor α modulator that can maximize the beneficial effects and minimize the adverse effects of fibrates used currently. In a phase-2 study, pemafibrate was shown to improve liver dysfunction in patients with dyslipidaemia. In the present study, we first investigated the effect of pemafibrate on rodent models of NASH. Pemafibrate efficacy was assessed in a diet-induced rodent model of NASH compared with fenofibrate. Pemafibrate and fenofibrate improved obesity, dyslipidaemia, liver dysfunction, and the pathological condition of NASH. Pemafibrate improved insulin resistance and increased energy expenditure significantly. To investigate the effects of pemafibrate, we analysed the gene expressions and protein levels involved in lipid metabolism. We also analysed uncoupling protein 3 (UCP3) expression. Pemafibrate stimulated lipid turnover and upregulated UCP3 expression in the liver. Levels of acyl-CoA oxidase 1 and UCP3 protein were increased by pemafibrate significantly. Pemafibrate can improve the pathogenesis of NASH by modulation of lipid turnover and energy metabolism in the liver. Pemafibrate is a promising therapeutic agent for NAFLD/NASH.
The incidence of nonalcoholic fatty liver disease (NAFLD) is increasing worldwide. NAFLD is an important cause of chronic liver injury1. NAFLD ranges from nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma2. First-line treatment for NAFLD is lifestyle modification to achieve weight reduction, particularly through diet and exercise3. However, weight reduction is very difficult to achieve and maintain and therapeutic agents approved for NAFLD treatment are lacking.
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear receptor superfamily of ligand-activated transcription factors4. PPARs contain three isotypes encoded by PPARα (NR1C1), PPARβ/δ (NR1C2), and PPARγ (NR1C3) genes. Each gene exhibits isoform-specific distribution patterns and functions in tissues5. PPARs have important roles in regulation of metabolic homeostasis, inflammation, as well as the growth and differentiation of cells6,7.
PPARα expression is enriched in hepatocytes. PPARα has key roles in regulation of fatty-acid transport as well as peroxisomal and mitochondrial β-oxidation in the liver8,9. PPARα knockout mice increase the susceptibility to liver steatosis, inflammation, and hepatocellular carcinoma10,11,12,13. Therefore, it has been suggested that PPARα has a protective role against NAFLD pathogenesis. PPARα-agonists have been shown to improve the pathologic condition of NAFLD in various preclinical models11,13,14,15,16,17,18,19. However, fibrates such as PPARα-agonists are weak and their efficacy is limited (at least in part) by dose-dependent side effects such as elevation of levels of transaminases, homocysteine and creatinine20,21. Fibrates increase the risk of myopathy, and have been associated with rhabdomyolysis. In addition, the efficacy of fibrates on NASH in humans is not known22,23,24,25,26.
The next generation of PPARα-agonists is called “selective PPARα modulators” (SPPARMα). They maximize the beneficial effects and minimize the adverse effects of fibrates27. Pemafibrate is the first of the SPPARMα to be developed, and has been shown to be safe and efficacious against dyslipidaemia in a phase-2 study28. Pemafibrate has not been associated with rhabdomyolysis in Caucasian or Japanese subjects.
Here, we investigated the effect of pemafibrate on rodent models of NASH (methionine choline-deficient (MCD)-fed db/db mice and amylin liver NASH model (AMLN)) in comparison with fenofibrate. AMLN is a diet-induced model of NASH and elicits obesity, insulin resistance, and the three stages of NAFLD (steatosis, steatohepatitis with fibrosis, and cirrhosis) without reliance on genetic mutations, use of toxins, or nutrient deficiency29.
Results
Effect of pemafibrate on physiologic and biochemical characteristics
We began our studies in MCD-fed db/db mice. Supplementary Table 1 shows the data of MCD-fed db/db mice with or without 4 weeks treatment of pemafibrate. db/db (MCD) mice had increased levels of total cholesterol and aspartate aminotransferase (AST) as well as decreased liver weight and levels of triglycerides. Pemafibrate decreased levels of triglycerides and AST in PEMA-L (db/db) and PEMA-H (db/db) mice. Alanine aminotransferase (ALT) levels tended to decrease upon pemafibrate administration, but not in a significant manner (p = 0.09). Pemafibrate increased liver weight in PEMA-H (db/db) mice. This increase might have been the results of PPARα activation. db/db (MCD) mice increased steatosis, hepatocyte ballooning and the NAFLD Activity Score (NAS). However, they exhibited weak lobular inflammation and showed very little fibrosis.
We continued our investigations using an AMLN because we could not recognize a phenotype of NASH in MCD-fed db/db/mice. Tables 1 and 2 show the characteristics of mice and liver pathology score, respectively. After 20 weeks of feeding of an AMLN diet, CTRL mice exhibited obesity, dyslipidaemia, insulin resistance and liver injury (Table 1). In addition, NASH was clearly evident in CTRL mice, as indicated by steatosis, lobular inflammation, hepatocyte ballooning, and fibrosis. NAS was 7.2 ± 0.2 and fibrosis stage was 2.3 ± 0.2 in CTRL mice (Table 2, Figs 1, 2 and 3).
Table 1. Characteristics of model mice.
| Parameter | BD | CTRL | PEMA-L | PEMA-H | FENO |
|---|---|---|---|---|---|
| n | 5 | 10 | 10 | 10 | 7 |
| Food intake (g/day) | — | 3.28 ± 0.21 | 3.54 ± 0.25 | 3.43 ± 0.14 | 3.02 ± 0.22 |
| Body weight (g) | 29.4 ± 0.5† | 41.3 ± 0.3 | 39.6 ± 0.4* | 36.2 ± 0.3* | 38.7 ± 0.4* |
| Liver weight (g) | 1.29 ± 0.05† | 3.80 ± 0.12 | 4.00 ± 0.07 | 3.88 ± 0.09 | 3.86 ± 0.09 |
| Epididymal adipose tissue (g) | 0.32 ± 0.04† | 1.15 ± 0.04 | 0.92 ± 0.03* | 0.66 ± 0.03* | 0.84 ± 0.03* |
| Subcutaneous adipose tissue (g) | 0.19 ± 0.03† | 0.65 ± 0.03 | 0.53 ± 0.04* | 0.37 ± 0.02* | 0.58 ± 0.02 |
| Total cholesterol (mg/dL) | 96.8 ± 4.4† | 283.5 ± 10.2 | 263.8 ± 6.6 | 199.5 ± 1.8* | 205.0 ± 5.1* |
| Triglycerides (mg/dL) | 61.0 ± 7.5† | 25.1 ± 1.5 | 12.6 ± 1.1* | 9.1 ± 1.0* | 8.6 ± 0.6* |
| Free fatty acids (uEQ/L) | 393.6 ± 42.1† | 673.2 ± 58.9 | 494.7 ± 50.2* | 332.1 ± 32.4* | 274.9 ± 19.5* |
| Fasting plasma glucose (mg/dL) | 95.2 ± 11.4† | 236.0 ± 9.8 | 242.6 ± 11.1 | 194.3 ± 10.9* | 223.7 ± 15.9 |
| Insulin (ng/mL) | 0.024 ± 0.007† | 0.096 ± 0.015 | 0.077 ± 0.002 | 0.048 ± 0.003* | 0.075 ± 0.008 |
| HOMA-IR | 0.13 ± 0.04† | 1.44 ± 0.21 | 1.13 ± 0.05 | 0.61 ± 0.07* | 0.97 ± 0.16 |
| AST (IU/L) | 49.0 ± 6.2† | 197.6 ± 12.1 | 210.8 ± 16.0 | 162.6 ± 14.3 | 227.1 ± 22.7 |
| ALT (IU/L) | 23.2 ± 1.9† | 285.7 ± 21.0 | 250.9 ± 16.2 | 174.3 ± 9.7* | 198.7 ± 30.7* |
| Liver ATP content (μmol/g)§ | 6.59 ± 1.02† | 4.61 ± 0.34 | 6.09 ± 0.28* | 6.47 ± 0.24* | 5.88 ± 0.40* |
| VO2 (mL/min/kg)§ | 53.3 ± 1.7† | 46.6 ± 0.3 | 48.1 ± 2.7 | 52.6 ± 2.2* | 48.4 ± 1.6 |
| VCO2 (mL/min/kg)§ | 45.0 ± 1.2† | 36.7 ± 0.8 | 37.3 ± 1.5 | 43.5 ± 3.0* | 38.6 ± 0.8 |
| RQ§ | 0.85 ± 0.04 | 0.79 ± 0.01 | 0.78 ± 0.01 | 0.83 ± 0.02 | 0.80 ± 0.03 |
HOMA-IR, the homeostasis model assessment of insulin resistance; AST, aspartate aminotransferase; ALT, alanine aminotransferase; ATP, Adenosine triphosphate; VO2, oxygen uptake; VCO2, production of carbon dioxide; RQ, respiratory quotient. Data are the mean ± SE (§n = 5). Significance was determined using Student’s t-test (†p < 0.05 versus CTRL mice) or Dunnett’s test (*p < 0.05 versus CTRL mice).
Table 2. Liver pathology score.
| Parameter | BD | CTRL | PEMA-L | PEMA-H | FENO |
|---|---|---|---|---|---|
| n | 5 | 10 | 10 | 10 | 7 |
| NAS | 0.2 ± 0.2† | 7.2 ± 0.2 | 6.2 ± 0.2 | 3.9 ± 0.5* | 5.7 ± 0.4* |
| Steatosis | 0† | 3 | 3 | 1.3 ± 0.2* | 2.4 ± 0.2* |
| Lobular inflammation | 0.2 ± 0.2† | 2.1 ± 0.2 | 1.2 ± 0.2 | 1.3 ± 0.4 | 1.4 ± 0.3 |
| Hepatocyte ballooning | 0† | 2 | 2 | 1.3 ± 0.2* | 1.9 ± 0.1 |
| Fibrosis | 0.4 ± 0.2† | 2.3 ± 0.2 | 1.6 ± 0.2 | 1.4 ± 0.2* | 1.3 ± 0.3* |
Nonalcoholic fatty liver disease activity score (NAS) and fibrosis stage were scored according to the method described by Kleiner et al.63, as outlined in (Supplementary Table 2). Significance was determined using Student’s t-test (†p < 0.05 versus CTRL mice) or Dunnett’s test (*p < 0.05 versus CTRL mice).
Figure 1. Effect of pemafibrate and fenofibrate on hepatic steatosis.

(A) Liver sections from BD, CTRL, PEMA-L, PEMA-H, and FENO mice. Haematoxylin and eosin staining, (B) oil red O staining. Magnification, 100×. Scale bars: 200 μm. (C) Areas of oil red O staining in the liver of BD, CTRL, PEMA-L, PEMA-H, and FENO mice (n = 5). (D) Triglycerides content were measured in the livers of BD, CTRL, PEMA-L, PEMA-H, and FENO mice (n = 5). Results are the mean ± SE. Significance was determined using Student’s t-test (†p < 0.05 versus CTRL mice) or Dunnett’s test (*p < 0.05 versus CTRL mice).
Figure 2. Effect of pemafibrate and fenofibrate on hepatic inflammation.

(A) Liver sections from BD, CTRL, PEMA-L, PEMA-H, and FENO mice. F4/80 staining. Magnification, 200x. Scale bars: 100 μm. (B) The number of macrophages was counted in the livers of BD, CTRL, PEMA-L, PEMA-H, and FENO mice (n = 5). (C) Expression of tumour necrosis factor α (TNFα) mRNA in BD, CTRL, PEMA-L, PEMA-H, and FENO mice (n = 5–10). Results are the mean ± SE. Significance was determined using Student’s t-test (†p < 0.05 versus CTRL mice) or Dunnett’s test (*p < 0.05 versus CTRL mice).
Figure 3. Effect of pemafibrate and fenofibrate on hepatic fibrosis.

(A) Liver sections from BD, CTRL, PEMA-L, PEMA-H, and FENO mice. Sirius red staining. Magnification, 100x. Scale bars: 200 μm. (B) Areas of Sirius red staining in the livers of BD, CTRL, PEMA-L, PEMA-H, and FENO mice were measured (n = 5). (C) Expression of collagen 1α1 mRNA in BD, CTRL, PEMA-L, PEMA-H, and FENO mice (n = 5–10). Results are the mean ± SE. Significance was determined using Student’s t-test (†p < 0.05 versus CTRL mice) or Dunnett’s test (*p < 0.05 versus CTRL mice).
Food intake was not different among CTRL, PEMA-L, PEMA-H, and FENO mice. Adenosine triphosphate (ATP) content in the liver (p < 0.05) as well as oxygen uptake (VO2) (p < 0.01) and production of carbon dioxide (VCO2) (p < 0.001) were reduced significantly in CTRL mice. These results showed a reduction of energy expenditure in CTRL mice. There were no differences in liver weight among CTRL, PEMA-L, PEMA-H, or FENO mice. Pemafibrate treatment reduced body weight, as well as the weight of epididymal and subcutaneous adipose tissue. Fenofibrate treatment reduced body weight and the weight of epididymal adipose tissue. PEMA-H and FENO mice showed lower levels of total cholesterol, triglycerides, and free fatty acids. Hyperglycaemia (p < 0.05), hyperinsulinemia (p < 0.05), and insulin resistance (p < 0.01) were improved significantly in PEMA-H mice, but these parameters were unchanged in FENO mice. Treatment with pemafibrate or fenofibrate reduced ALT levels and increased ATP content in the liver. Pemafibrate treatment augmented VO2 (p < 0.05) and VCO2 (p < 0.05) significantly in PEMA-H mice. However, fenofibrate treatment tended to increase VO2 (p = 0.66) and VCO2 (p < 0.67), but not significantly. These results suggested that pemafibrate treatment increased energy expenditure markedly.
Pemafibrate improved the pathogenesis in a rodent model of NASH
Treatment with pemafibrate (p < 0.0001) or fenofibrate (p < 0.05) reduced NAS significantly, but it was lower in PEMA-H mice than in FENO mice. Steatosis grade was significantly lower in PEMA-H (p < 0.001) and FENO (p < 0.01) mice. The area of staining by oil red O was smaller in PEMA-L, PEMA-H, and FENO mice (Fig. 1C). Triglyceride content in the liver decreased significantly in PEMA-H mice (p < 0.05, Fig. 1D). Grade of lobular inflammation tended to decrease in PEMA-L (p = 0.06), PEMA-H (p = 0.11), and FENO (p = 0.28) mice, but these differences were not significant. Fenofibrate treatment did not reduce the grade of hepatocyte ballooning, but pemafibrate treatment reduced it significantly in PEMA-H mice (p < 0.001). The number of infiltrating macrophages and tumour necrosis factor α (TNFα) messenger RNA (mRNA) expression decreased in PEMA-L, PEMA-H, and FENO mice (Fig. 2B,C).
The fibrosis stage decreased significantly in PEMA-H (p < 0.05) and FENO (p < 0.05) mice. The area of Sirius-red staining was smaller and mRNA expression of collagen 1α1 reduced in PEMA-L, PEMA-H, and FENO mice (Fig. 3B,C). These results suggested that pemafibrate and fenofibrate had therapeutic effects on the pathogenesis of NASH.
Pemafibrate stimulated lipid turnover and upregulated expression of uncoupling protein 3 (UCP 3) in the liver
We analysed the mRNA and protein expressions of genes involved in fatty-acid transport, lipogenesis, fatty-acid oxidation, and export of very-low-density lipoprotein. We also analysed UCP3 expression in the liver. mRNA expressions of fatty acid transport protein 4 (FATP4) (p < 0.001), PPARα (p < 0.001), acyl-CoA oxidase (ACOX) (p < 0.001), carnitine palmitoyltransferase 1 A (CPT1A) (p < 0.001), and microsomal triglyceride transfer protein (MTTP) (p < 0.001) were significantly lower in CTRL mice (Fig. 4A). mRNA expression of sterol regulatory element-binding protein 1c (SREBP1c), acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), and stearoyl-CoA desaturase 1 (SCD1) did not differ between BD and CTRL mice. Levels of SREBP1 protein (p < 0.05), ACOX1 protein (p < 0.001) and UCP3 protein (p < 0.001) decreased significantly in CTRL mice (Fig. 4B).
Figure 4. Pemafibrate modulated lipid turnover and upregulated expression of UCP3 in the liver.

(A) mRNA expressions involved in lipid metabolism (fatty acid transport protein 4 (FATP4), sterol regulatory element-binding protein 1c (SREBP1c), acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), stearoyl-CoA desaturase 1 (SCD1), peroxisome proliferator activated receptor α (PPARα), acyl-CoA oxidase (ACOX), carnitine palmitoyltransferase 1 A (CPT1A), and microsomal triglyceride transfer protein (MTTP)) and expression of uncoupling protein 3 (UCP3) in BD, CTRL, PEMA-L, PEMA-H, and FENO mice (n = 5–10). (B) SREBP1, ACOX1, and UCP3 protein levels in BD, CTRL, PEMA-L, PEMA-H, and FENO mice (n = 5). Results are the mean ± SE. Significance was determined using Student’s t-test (†p < 0.05 versus CTRL mice) or Dunnett’s test (*p < 0.05 versus CTRL mice).
Hepatic fatty-acid transport, fatty-acid oxidation, and export of very-low-density lipoprotein were ameliorated and hepatic lipogenesis were facilitated by treatment with pemafibrate or fenofibrate, as indicated by the increase in mRNA expressions of FATP4, SREBP1c, ACC, FAS, SCD1, ACOX, CPT1A, and MTTP (Fig. 4A). Expression of SREBP1 protein increased in PEMA-H mice (Fig. 4B). Expression of ACOX1 protein increased in PEMA-H mice, but not in FENO mice (Fig. 4B). These results suggested that pemafibrate improved lipid turnover and promoted fatty-acid oxidation notably. Pemafibrate treatment provoked a significant increase in expression the mRNA (p < 0.001) and protein (p < 0.001) of UCP3 in PEMA-H mice (Fig. 4A,B). Fenofibrate treatment tended to increase such expression, but the difference was not significant (p = 0.06 and p = 0.11, respectively).
Fibroblast growth factor 21 (FGF21) is produced mainly by the liver and has insulin-sensitizing activity30. K-877 treatment tended to increase serum levels and mRNA expression of FGF21, but not significantly (p = 0.26 and p = 0.91, respectively, Supplementary Fig. 1).
Discussion
Fibrates (PPARα-agonists) are used to treat dyslipidaemia. It has been suggested that PPARα has a protective role against the pathogenesis of NAFLD. However, the efficacy of fibrates on NAFLD/NASH treatment in humans has not been demonstrated because of study limitations and adverse effects. Pemafibrate is a novel SPPARMα designed to have highly selective and tissue-specific activity without the unwanted side effects of fibrates used currently, and has been developed for dyslipidaemia treatment. Pemafibrate is a more potent PPARα-agonist than fenofibrate (effective concentration inducing 50% response = 1 vs. 14000–22400 nM for fenofibrate)27. In a phase-2 study, pemafibrate reduced plasma concentrations of liver enzymes (ALT and γ-glutamyl transferase) in patients with dyslipidaemia28. Therefore, pemafibrate may have therapeutic efficacy against NAFLD/NASH. Here, we first investigated the effect of pemafibrate on rodent models of NASH.
In the present study, MCD-fed db/db mice and AMLN were used. AMLN is a diet-induced model of NASH. AMLN exhibits the three stages of NAFLD (steatosis, steatohepatitis with fibrosis, and cirrhosis) without reliance on genetic mutations, use of toxins, or nutrient deficiency29. The MCD-fed db/db mice model decreased body weight and insulin resistance, and is a useful rodent model for non-obese NASH31,32. However, fibrosis in db/db (MCD) mice was barely seen, so our main investigations were based on the AMLN.
Supplementary Fig. 2 is a schematic diagram illustrating the effects of pemafibrate on NASH in our study. Fat accumulation in the liver results from imbalanced metabolism of lipids33. Recently, it was reported that expression of the PPARα gene in the human liver is correlated negatively with NASH severity. In addition, histologic improvement is associated with an increase in PPARα expression34. We have reported that reduction in lipid outflow, fatty-acid oxidation and export of very-low-density lipoprotein are key factors in NASH pathogenesis35. In our study, expression of PPARα mRNA decreased and lipid turnover was inhibited strongly in CTRL mice. In addition, CTRL mice exhibited the pathologic condition of NASH. Pemafibrate and fenofibrate improved the pathologic condition of NASH, reduced ALT levels, and inhibited expression of pro-inflammatory and pro-fibrotic genes (F4/80, TNFα, collagen 1α1). Treatment with pemafibrate or fenofibrate increased the expression of PPARα and its target genes, ACOX and CPT1A, significantly. This treatment was thought to increase ATP content in the liver by promoting fatty-acid oxidation, and to improve NASH pathogenesis by stimulating lipid turnover. Therefore, pemafibrate could be a therapeutic agent in NAFLD/NASH, as well as fenofibrate.
Obesity and insulin resistance are important risk factors for NAFLD/NASH. Reports have demonstrated that fenofibrate prevents gain in body weight in genetic or diet-induced models of obesity in rodents36,37. In the present study, CTRL mice exhibited obesity and insulin resistance. In addition, energy expenditure was reduced, as indicated by a decrease of O2 consumption and CO2 production in CTRL mice. Pemafibrate and fenofibrate prevented weight gain. Furthermore, pemafibrate increased energy expenditure markedly.
UCPs are members of the mitochondrial anion carrier family. The function of UCPs is to separate oxidative phosphorylation from ATP synthesis by increasing the permeability of the inner membrane of a mitochondrion. UCP3 is expressed primarily in skeletal muscle. Lanni et al. reported that fenofibrate increases mRNA levels of UCP3 in the liver38. The function of the UCP3 gene is incompletely understood. However, it has been suggested that UCP3 has a protective role against obesity and insulin resistance because it contributes to energy metabolism39,40,41,42,43,44,45,46,47,48. It has also been reported that UCP3 polymorphisms are associated with NAFLD49. Camara et al. investigated the roles of UCP3 in the mitochondria of mouse livers50. The presence of UCP3 specifically enhanced an increase of mitochondrial respiratory activity in the presence of palmitate. Camara et al. suggested that UCP3 expression increased oxidative capacity and enhanced enzymatic machinery for lipid catabolism in mitochondria. We observed that VO2 and VCO2 were decreased in CTRL mice compared with BD mice. Pemafibrate provoked a significant increase in expressions of the mRNA and protein of UCP3 in PEMA-H mice. In addition, the respiratory parameters of PEMA-H mice increased to those seen for CTRL mice. This effect might be attributed (at least in part) to improvements in oxidative capacity in mitochondria. These effects participated synergistically to improve energy metabolism and improvement of the pathogenesis of NASH.
Pemafibrate improved glucose metabolism significantly. FGF21 can sensitize insulin and is produced mainly by the liver30. In a phase-2 study, pemafibrate increased serum levels of FGF21 significantly28. In the present study, pemafibrate tended to increase serum levels and mRNA expression of FGF21. An association of insulin resistance between muscle and adipose tissue cannot be excluded, but weight reduction, increase in energy expenditure, and alternation of FGF21 expression by pemafibrate may contribute to improve glucose metabolism.
Dyslipidaemia is a frequent feature of NAFLD. It has been reported that dyslipidaemia is present in 20–80% of NAFLD patients51. NAFLD is also associated with cardiovascular disease52,53. Fibrates improve dyslipidaemia, reduce levels of triglycerides in plasma, increase levels of high-density lipoprotein-cholesterol in plasma, and lower the risk of major cardiovascular events54. However, as mentioned above, fibrates are known to increase levels of transaminases, creatinine and homocysteine. In a phase-2 study in patients with dyslipidaemia, pemafibrate improved lipid parameters without increasing levels of creatinine or homocysteine. Moreover, pemafibrate strongly reduced levels of liver enzymes, whereas fenofibrate did not28. Based on the results of that human phase-2 study and our animal study, pemafibrate appears to be a promising therapeutic agent for NAFLD, as well as dyslipidaemia and cardiovascular disease associated with NAFLD.
Our study had two main limitations. First, we used a single dose of fenofibrate (50 mg/kg/day) to compare the therapeutic effect of pemafibrate on NASH, and this dose was based on a report by Fatani and colleagues55. Second, there was a discrepancy between improvement of liver steatosis and upregulation of expression of lipogenic transcription factors by treatment with pemafibrate or fenofibrate. Upregulated hepatic lipogenesis is thought to contribute to liver steatosis56,57. However, the genes involved in lipogenesis did not upregulate/downregulate in expression in CTRL mice or in a report by Shindo et al. using fatty liver Shionogi mice58. SREBP1c nor its downstream genes (with the exception of SCD1)59 have been identified as direct PPARα target genes in mice60, but SREBP1c, ACC, FAS, and SCD1 were positively regulated by pemafibrate and fenofibrate in the present study as well in other reports15,59,61,62. Hence, alterations in expression of these lipogenic genes may result in feedback by increase in lipid outflow.
We demonstrated that pemafibrate improved the pathogenesis of NASH by stimulation of lipid turnover and upregulation of UCP3 expression in the liver. Pemafibrate is expected to have more beneficial effects on NAFLD/NASH treatment because of its efficacy and safety. Hereafter, large prospective studies investigating the effect of pemafibrate on human NAFLD/NASH are needed.
Materials and Methods
Drugs and diets
Pemafibrate and fenofibrate were obtained from Kowa Co. Ltd. (Tokyo, Japan). A basal diet (BD) was prepared containing 22% protein, 6% fat, and 47% carbohydrate. MCD (F2MCD; Oriental Yeast Co., Ltd., Tokyo, Japan) were purchased. A diet rich in fat (40% kcal; Primex partially hydrogenated vegetable oil shortening), fructose (22% by weight), and cholesterol (2% by weight) (catalogue number D09100301; Research Diets, New Brunswick, NJ, USA) was purchased. This diet (the AMLN diet) has been shown to induce all pathologic stages of NAFLD for >20 weeks in C57BL/6J mice29.
Animal experiments
The study protocol was in accordance with the Guidelines for the care and use of laboratory animals set by Yokohama City University Medical School (Yokohama, Japan) and was approved by the Committee on the Ethics of Animal Experiments of the same institution.
Surgical procedures were carried out after the induction of anaesthesia using sodium pentobarbital. All efforts were made to minimize animal suffering. Mice were fasted for 12 h and fasting blood glucose measured (Glutest Neo Sensor; Sanwa Kagaku Kenkyusho, Aichi, Japan). The experimental protocol is outlined in Supplementary Fig. 3. Nine-week-old db/db mice (BKS.Cg−+Leprdb/+Leprdb/Jcl, female) were obtained from CLEA Japan (Tokyo, Japan). After a 2-week acclimatization period, mice were divided into four groups: BD (db/db) mice (fed BD and treated with 0.5% aqueous methylcellulose solution (MC); MCD (db/db) mice (fed MCD and treated with 0.5% MC); PEMA-L (db/db) mice (fed MCD and treated with 0.03 mg/kg pemafibrate); PEMA-H (db/db) mice (fed MCD and treated with 0.1 mg/kg pemafibrate). The drug-free solvent or the dosing solution was administered to animals (5 mL/kg body weight, p.o.) once daily (in the morning) for 4 consecutive weeks (Supplementary Fig. 3A). Six-week-old male C57BL/6J mice were obtained from CLEA Japan. After a 2-week acclimatization period, mice groups were fed according to different regimens. BD mice were fed a BD for 20 weeks. CTRL mice were fed D09100301 for 20 weeks. PEMA-L and PEMA-H mice were fed D09100301 for 12 weeks followed by D09100301 with 0.4 mg and 1.3 mg pemafibrate/kg of the diet for 8 weeks, which corresponded to 0.03 mg/kg/day and 0.1 mg/kg/day, respectively. FENO mice were fed D09100301 for 12 weeks followed by D09100301 with 666.7 mg fenofibrate/kg of the diet for 8 weeks, which corresponded to 50 mg/kg/day (Supplementary Fig. 3B). Pemafibrate and fenofibrate were incorporated into the AMLN diet. Animals were housed under conventional conditions with controlled temperature, humidity, and light (12-h light–dark cycle) and provided with food and water.
Biochemical analyses
Serum levels of total cholesterol, triglycerides, free fatty acids, AST, and ALT were measured by a local laboratory (SRL, Tokyo, Japan). Serum levels of insulin were measured using a mouse insulin ELISA kit (Morinaga Institute of Biological Sciences, Kanagawa, Japan). As an alternative method for assessment of insulin resistance, the homeostasis model assessment of insulin resistance (HOMA-IR) was calculated using the following formula:
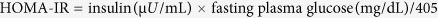 |
ATP contents in liver tissue were measured by a luciferase assay using an ATP assay kit for animal tissues (TOYO B-Net, Tokyo, Japan). Levels of triglycerides in total lipid extracts of the liver were determined by colorimetric assays (Wako Pure Chemical Industries, Osaka, Japan). Serum levels of FGF21 were measured using a mouse and rat FGF21 ELISA kit (Biovendor, Karasek, Czech Republic).
Indirect calorimetry
Energy expenditure, VO2 and VCO2 were measured using a small-animal metabolic measurement system (MK-5000RQ, Muromachi Kikai, Tokyo, Japan). The respiratory quotient was obtained as the ratio of VCO2 to VO2.
Histologic and immunohistochemical analyses
Paraffin-embedded sections were stained with haematoxylin and eosin, or Sirius red. The NAS and fibrosis stage were scored by Y.N. in a blinded manner according to the method of Kleiner et al. (Supplementary Table 1)63. For lipid staining, frozen sections were stained with oil red O and counterstained with haematoxylin. Immunohistochemistry for macrophages was based on F4/80 staining. Immunohistochemistry was carried out on cryostat liver sections (thickness, 7 μm). Sections were incubated with primary antibodies and stained with Alexa Fluor®-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MA, USA). To quantify the area of staining by oil red O and Sirius red, images of five random fields from each section were processed with Photoshop Elements v13 (Adobe Systems, San Jose, CA, USA). Each value was expressed as the percentage of the total area of the section. Numbers of F4/80 positive cells were counted and averaged for five random fields of each section.
RNA isolation and real-time polymerase chain reaction (PCR) analyses
Total RNA was extracted from samples of liver tissue using an RNeasy mini kit (Qiagen, Tokyo, Japan). mRNA of murine TNFα, collagen 1α1, FATP4, SREBP1c, ACC, FAS, SCD1, PPARα, ACOX, CPT1A, MTTP, UCP3, FGF21, and β-actin in liver tissue were determined using a fluorescence-based reverse transcription-PCR and an ABI PRISM 7700 sequence detection system (Life Technologies, Carlsbad, CA, USA).
Analyses of western blotting
Proteins were incubated with primary antibodies and horseradish-conjugated secondary antibody (Cell Signaling Technology). Primary antibodies were SREBP1, ACOX1 and UCP 3 (Abcam, Cambridge, UK).
Statistical analyses
Data are the mean ± standard error (SE). Differences between two groups were assessed using Student’s t-test or Dunnett’s test. p < 0.05 was considered significant. Statistical analyses were carried out using JMP v11.2.0 (SAS Institute, Cary, NC, USA).
Additional Information
How to cite this article: Honda, Y. et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 7, 42477; doi: 10.1038/srep42477 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The skilful technical assistance of Ayaka Miura and Machiko Hiraga is gratefully acknowledged. This study was supported by Kowa Co. Ltd. (Tokyo, Japan).
Footnotes
The authors declare no competing financial interests.
Author Contributions Coordination and design of study: Y.H., T.T., A.N. Undertaking of experiments: Y.H., T.K., Y.O., W.T., K.I. Statistical analyses: Y.H., F.K., M.Y., Y.N., A.N. Data collection, critical review of the manuscript, and approval of the final version of the manuscript: Y.H., S.S., A.N. Writing and approval of the final version of the manuscript: Y.H., T.T., S.S., A.N.
References
- Lazo M. & Clark J. M. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Semin. Liver Dis. 28, 339–350, doi: 10.1055/s-0028-1091978 (2008). [DOI] [PubMed] [Google Scholar]
- Day C. P. & Saksena S. Non-alcoholic steatohepatitis: definitions and pathogenesis. J. Gastroenterol. Hepatol. 17, S377–S384, doi: 10.1046/j.1440-1746.17.s3.31.x (2002). [DOI] [PubMed] [Google Scholar]
- Promrat K. et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 51, 121–129, doi: 10.1002/hep.23276 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aagaard M. M., Siersbæk R. & Mandrup S. Molecular basis for gene-specific transactivation by nuclear receptors. Biochim. Biophys. Acta 1812, 824–835, doi: http://dx.doi.org/10.1016/j.bbadis.2010.12.018 (2011). [DOI] [PubMed] [Google Scholar]
- Kliewer S. A. et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. 91, 7355–7359 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen L. C., Siersbæk M. & Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 23, 631–639, doi: http://dx.doi.org/10.1016/j.semcdb.2012.01.003 (2012). [DOI] [PubMed] [Google Scholar]
- Tailleux A., Wouters K. & Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim. Biophys. Acta 1821, 809–818, doi: http://dx.doi.org/10.1016/j.bbalip.2011.10.016 (2012). [DOI] [PubMed] [Google Scholar]
- Xu J. et al. Peroxisome proliferator-activated receptor alpha (PPARalpha) influences substrate utilization for hepatic glucose production. J. Biol. Chem. 277, 50237–50244, doi: 10.1074/jbc.M201208200 (2002). [DOI] [PubMed] [Google Scholar]
- Lefebvre P., Chinetti G., Fruchart J.-C. & Staels B. Sorting out the roles of PPARα in energy metabolism and vascular homeostasis. J. Clin. Invest. 116, 571–580, doi: 10.1172/JCI27989 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmegeed M. A. et al. PPARα expression protects male mice from high fat–induced nonalcoholic fatty liver. J. Nutr. 141, 603–610, doi: 10.3945/jn.110.135210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stienstra R. et al. Peroxisome proliferator-activated receptor α protects against obesity-induced hepatic inflammation. Endocrinology 148, 2753–2763, doi: 10.1210/en.2007-0014 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang N. et al. Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-κB signaling pathway. Oncotarget 5, 8330–8340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalloyer F. et al. Peroxisome proliferator–activated receptor-α gene level differently affects lipid metabolism and inflammation in apolipoprotein E2 knock-in mice. Arterioscler. Thromb. Vasc. Biol. 31, 1573–1579, doi: 10.1161/atvbaha.110.220525 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiri-Sverdlov R. et al. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J. Hepatol. 44, 732–741, doi: http://dx.doi.org/10.1016/j.jhep.2005.10.033 (2006). [DOI] [PubMed] [Google Scholar]
- Larter C. Z. et al. Peroxisome proliferator-activated receptor-α agonist, Wy 14 643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 27, 341–350, doi: 10.1111/j.1440-1746.2011.06939.x (2012). [DOI] [PubMed] [Google Scholar]
- Ip E. et al. Central role of PPARα-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 38, 123–132, doi: 10.1053/jhep.2003.50307 (2003). [DOI] [PubMed] [Google Scholar]
- Ip E., Farrell G., Hall P., Robertson G. & Leclercq I. Administration of the potent PPARα agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 39, 1286–1296, doi: 10.1002/hep.20170 (2004). [DOI] [PubMed] [Google Scholar]
- Baron M. et al. PPARα activation differently affects microparticle content in atherosclerotic lesions and liver of a mouse model of atherosclerosis and NASH. Atherosclerosis 218, 69–76, doi: http://dx.doi.org/10.1016/j.atherosclerosis.2011.03.009 (2011). [DOI] [PubMed] [Google Scholar]
- Kondo K. et al. Fenofibrate, a Peroxisome Proliferator-Activated Receptor α Agonist, Improves Hepatic Microcirculatory Patency and Oxygen Availability in a High-Fat-Diet-Induced Fatty Liver in Mice. Oxygen transport to tissue XXXI (eds Takahashi Eiji & Bruley F. Duane) 77–82 (Springer US, 2010). [DOI] [PubMed] [Google Scholar]
- Keech A. et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 366, 1849–1861, doi: http://dx.doi.org/10.1016/S0140-6736(05)67667-2 (2005). [DOI] [PubMed] [Google Scholar]
- de Lorgeril M., Salen P., Paillard F., Lacan P. & Richard G. Lipid-lowering drugs and homocysteine. Lancet 353, 209–210, doi: http://dx.doi.org/10.1016/S0140-6736(05)77220-2 (1999). [DOI] [PubMed] [Google Scholar]
- Bajaj M. et al. Effects of peroxisome proliferator-activated receptor (PPAR)-α and PPAR-γ agonists on glucose and lipid metabolism in patients with type 2 diabetes mellitus. Diabetologia 50, 1723–1731, doi: 10.1007/s00125-007-0698-9 (2007). [DOI] [PubMed] [Google Scholar]
- Laurin J. et al. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: A pilot study. Hepatology 23, 1464–1467, doi: 10.1002/hep.510230624 (1996). [DOI] [PubMed] [Google Scholar]
- Nakamuta M. et al. Short-term intensive treatment for donors with hepatic steatosis in living-donor liver transplantation. Transplantation 80, 608–612, doi: 10.1097/01.tp.0000166009.77444.f3 (2005). [DOI] [PubMed] [Google Scholar]
- Fernández-Miranda C. et al. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 40, 200–205, doi: 10.1016/j.dld.2007.10.002 (2008). [DOI] [PubMed] [Google Scholar]
- Basaranoglu M., Acbay O. & Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 31, 384, doi: http://dx.doi.org/10.1016/S0168-8278(99)80243-8 (1999). [DOI] [PubMed] [Google Scholar]
- Fruchart J.-C. Selective peroxisome proliferator-activated receptorα modulators (SPPARMα): The next generation of peroxisome proliferator-activated receptor α-agonists. Cardiovasc. Diabetol. 12, 82, doi: 10.1186/1475-2840-12-82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S. et al. Effects of K-877, a novel selective PPARα modulator (SPPARMα), in dyslipidaemic patients: a randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis 249, 36–43, doi: 10.1016/j.atherosclerosis.2016.02.029 (2016). [DOI] [PubMed] [Google Scholar]
- Clapper J. R. et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G483–495, doi: 10.1152/ajpgi.00079.2013 (2013). [DOI] [PubMed] [Google Scholar]
- Inagaki T. et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425, doi: http://dx.doi.org/10.1016/j.cmet.2007.05.003 (2007). [DOI] [PubMed] [Google Scholar]
- Sahai A. et al. Obese and diabetic mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G1035–G1043 (2004). [DOI] [PubMed] [Google Scholar]
- Rinella M. E. et al. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 49, 1068–1076, doi: 10.1194/jlr.M800042-JLR200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning J. D. & Horton J. D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114, 147–152, doi: 10.1172/JCI200422422 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francque S. et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 63, 164–173, doi: http://dx.doi.org/10.1016/j.jhep.2015.02.019 (2015). [DOI] [PubMed] [Google Scholar]
- Fujita K. et al. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 50, 772–780, doi: 10.1002/hep.23094 (2009). [DOI] [PubMed] [Google Scholar]
- Guerre-Millo M. et al. Peroxisome proliferator-activated receptor α activators improve insulin sensitivity and reduce adiposity. J. Biol. Chem. 275, 16638–16642, doi: 10.1074/jbc.M907421199 (2000). [DOI] [PubMed] [Google Scholar]
- Mancini F. P. et al. Fenofibrate prevents and reduces body weight gain and adiposity in diet-induced obese rats. FEBS Lett. 491, 154–158, doi: 10.1016/S0014-5793(01)02146-9 (2001). [DOI] [PubMed] [Google Scholar]
- Lanni A. et al. De novo expression of uncoupling protein 3 is associated to enhanced mitochondrial thioesterase-1 expression and fatty acid metabolism in liver of fenofibrate-treated rats. FEBS Lett. 525, 7–12, doi: 10.1016/S0014-5793(02)02828-4 (2002). [DOI] [PubMed] [Google Scholar]
- Chieregatti E. & Meldolesi J. Regulated exocytosis: new organelles for non-secretory purposes. Nat. Rev. Mol. Cell Biol. 6, 181–187 (2005). [DOI] [PubMed] [Google Scholar]
- Silvestri E. et al. Fenofibrate activates the biochemical pathways and the de novo expression of genes related to lipid handling and uncoupling protein-3 functions in liver of normal rats. Biochim. Biophys. Acta 1757, 486–495, doi: 10.1016/j.bbabio.2006.02.016 (2006). [DOI] [PubMed] [Google Scholar]
- Boss O. et al. Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 408, 39–42, doi: 10.1016/S0014-5793(97)00384-0 (1997). [DOI] [PubMed] [Google Scholar]
- Fleury C. et al. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 15, 269–272 (1997). [DOI] [PubMed] [Google Scholar]
- Schrauwen P. & Hesselink M. UCP2 and UCP3 in muscle controlling body metabolism. J. Exp. Biol. 205, 2275–2285 (2002). [DOI] [PubMed] [Google Scholar]
- Ježek P. Possible physiological roles of mitochondrial uncoupling proteins—UCPn. Int. J. Biochem. Cell Biol. 34, 1190–1206, doi: http://dx.doi.org/10.1016/S1357-2725(02)00061-4 (2002). [DOI] [PubMed] [Google Scholar]
- Dulloo A. G. & Samec S. Uncoupling proteins: do they have a role in body weight regulation? Physiology 15, 313–318 (2000). [DOI] [PubMed] [Google Scholar]
- Goglia F. & Skulachev V. P. A function for novel uncoupling proteins: antioxidant defense of mitochondrial matrix by translocating fatty acid peroxides from the inner to the outer membrane leaflet. FASEB J. 17, 1585–1591, doi: 10.1096/fj.03-0159hyp (2003). [DOI] [PubMed] [Google Scholar]
- Moore G. B. T., Himms-Hagen J., Harper M.-E. & Clapham J. C. Overexpression of UCP-3 in skeletal muscle of mice results in increased expression of mitochondrial thioesterase mRNA. Biochem. Biophys. Res. Commun. 283, 785–790, doi: http://dx.doi.org/10.1006/bbrc.2001.4848 (2001). [DOI] [PubMed] [Google Scholar]
- Busiello R. A., Savarese S. & Lombardi A. Mitochondrial uncoupling proteins and energy metabolism. Front. Physiol. 6, 36, doi: 10.3389/fphys.2015.00036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.-P. et al. Association between UCP3 gene polymorphisms and nonalcoholic fatty liver disease in Chinese children. World J. Gastroenterol. 19, 5897–5903, doi: 10.3748/wjg.v19.i35.5897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara Y., Mampel T., Armengol J., Villarroya F. & Dejean L. UCP3 expression in liver modulates gene expression and oxidative metabolism in response to fatty acids, and sensitizes mitochondria to permeability transition. Cell. Physiol. Biochem. 24, 243–252 (2009). [DOI] [PubMed] [Google Scholar]
- Souza M. R., Diniz M. D., Medeiros-Filho J. E. & Araújo M. S. Metabolic syndrome and risk factors for non-alcoholic fatty liver disease. Arq. Gastroenterol. 49, 89–96 (2012). [DOI] [PubMed] [Google Scholar]
- Bhatia L. S., Curzen N. P., Calder P. C. & Byrne C. D. Non-alcoholic fatty liver disease: a new and important cardiovascular risk factor? Eur. Heart J. 33, 1190–1200, doi: 10.1093/eurheartj/ehr453 (2012). [DOI] [PubMed] [Google Scholar]
- Ballestri S. et al. Risk of cardiovascular, cardiac and arrhythmic complications in patients with non-alcoholic fatty liver disease. World J. Gastroenterol 20, 1724–1745, doi: 10.3748/wjg.v20.i7.1724 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels B., Maes M. & Zambon A. Fibrates and future PPAR[alpha] agonists in the treatment of cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 5, 542–553 (2008). [DOI] [PubMed] [Google Scholar]
- Fatani S., Itua I., Clark P., Wong C. & Naderali E. K. The effects of diet-induced obesity on hepatocyte insulin signaling pathways and induction of non-alcoholic liver damage. Int. J. Gen. Med. 4, 211–219, doi: 10.2147/IJGM.S17376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 142, 711–725 e716, doi: 10.1053/j.gastro.2012.02.003 (2012). [DOI] [PubMed] [Google Scholar]
- Gan L., Xiang W., Xie B. & Yu L. Molecular mechanisms of fatty liver in obesity. Front. Med. 9, 275–287, doi: 10.1007/s11684-015-0410-2 (2015). [DOI] [PubMed] [Google Scholar]
- Shindo N. et al. Involvement of microsomal triglyceride transfer protein in nonalcoholic steatohepatitis in novel spontaneous mouse model. J. Hepatol. 52, 903–912, doi: http://dx.doi.org/10.1016/j.jhep.2009.12.033 (2010). [DOI] [PubMed] [Google Scholar]
- Miller C. W. & Ntambi J. M. Peroxisome proliferators induce mouse liver stearoyl-CoA desaturase 1 gene expression. Proc. Natl. Acad. Sci. 93, 9443–9448 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak M., Lefebvre P. & Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 62, 720–733, doi: http://dx.doi.org/10.1016/j.jhep.2014.10.039 (2015). [DOI] [PubMed] [Google Scholar]
- Patel D. D., Knight B. L., Wiggins D., Humphreys S. M. & Gibbons G. F. Disturbances in the normal regulation of SREBP-sensitive genes in PPARα-deficient mice. J. Lipid Res. 42, 328–337 (2001). [PubMed] [Google Scholar]
- Oosterveer M. H. et al. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J. Biol. Chem. 284, 34036–34044, doi: 10.1074/jbc.M109.051052 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321, doi: 10.1002/hep.20701 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.