

Abstract
Propionic acidemia is an inborn error of metabolism caused by deficiency of the mitochondrial enzyme propionyl-CoA carboxylase. Sensorineural deafness and severe hearing loss have been described as long-term complications of this disease, however, the mechanism has not yet been elucidated. We have recently shown by patch clamping experiments and Western blots that acute and chronic effects of accumulating metabolites such as propionic acid, propionylcarnitine and methylcitrate on the KvLQT1/KCNE1 channel complex cause long QT syndrome in patients with propionic acidemia by inhibition of K+ flow via this channel. The same KvLQT1/KCNE1 channel complex is expressed in the inner ear and essential for luminal potassium secretion into the endolymphatic space. A disruption of this K+ flow results in sensorineural hearing loss or deafness. It can be assumed that acute and chronic effects of accumulating metabolites on the KvLQT1/KCNE1 channel protein may similarly cause the hearing impairment of patients with propionic acidemia.
Keywords: Propionic acidemia, Propionic aciduria, Hearing loss, Long QT syndrome, Sensorineural deafness, KvLQT1, KCNE1, Voltage-gated potassium channel, Inner ear
Propionic acidemia (PA) is an inborn error of metabolism caused by deficiency of the mitochondrial enzyme propionyl-CoA carboxylase. This enzyme converts propionyl-CoA to D-methylmalonyl-CoA and is involved in the metabolism of branched-chain amino acids, odd-numbered fatty acids, cholesterol side chains, thymine, and uracil [1]. Patients with PA are prone to acute life-threatening metabolic decompensations with severe metabolic acidosis and hyperammonemia, and most affected children already present with symptoms during the neonatal period. With progression of the disease, long-term complications involving the neurological, cardiological, hematological, immunological and gastrointestinal system are common [2]. One of the main cardiological problems is the development of an acquired long QT syndrome (LQTS), which can be found in up to 70% of patients beyond childhood and puts patients at risk for potentially lethal ventricular arrhythmia [2, 3]. Among the neurological complications, sensorineural hearing impairment has been reported in several patients with PA [2, 4, 5]. In a study with 55 PA patients from Germany, Switzerland and Austria, hearing loss was apparent in 13% of the patients [2]. Similarly, Brosch et al. described 4 patients with PA and sensorineural deafness or severe hearing loss [5]. Mutation analyses of the PCCA and PCCB genes, encoding the two subunits of the propionyl-CoA carboxylase, have been performed in the families of these patients to elucidate whether mutations primarily responsible for PA could also be the underlying cause for a genetic form of deafness. Brosch et al. concluded that no connection can be assumed between the mutations found in the patients and the severe sensorineural hearing loss. So far, the mechanism of hearing impairment in PA has not yet been elucidated. However, our studies on the effects of metabolites, that typically accumulate in body fluids of patients with PA, on the KvLQT1/KCNE1 channel give new insights on the possible mechanism of hearing loss associated with this disorder.
The same combination of LQTS and sensorineural deafness as seen in PA patients is also found in Jervell and Lange-Nielsen syndrome. This genetic disorder with an autosomal recessive inheritance is characterized by congenital LQTS with syncopal attacks due to ventricular arrhythmias and congenital profound bilateral sensorineural hearing loss [6]. It is caused by mutations in either the KCNQ1 or KCNE1 genes, encoding the alpha and beta subunits (KvLQT1 and KCNE1/minK) of a potassium channel carrying the so-called delayed rectifier potassium current IKs [7]. About 90% of cases of Jervell and Lange-Nielsen syndrome are caused by mutations in the KCNQ1 gene [8–10]. The KvLQT1/KCNE1 voltage-gated potassium channel is expressed not only in cardiomyocytes but also in cells of the stria vascularis of the inner ear [11, 12]. In the heart, this channel is essential for cardiac repolarization, and mutations in either the KCNQ1 or KCNE1 gene lead to a reduction of the IKs current and therefore prolongation of the action potential [13, 14]. In the inner ear, this channel enables luminal secretion of K+ into the endolymphatic space [15, 16]. A disruption of this K+ flow results in sensorineural hearing loss or deafness. We therefore hypothesize that sensorineural hearing loss and long QT syndrome seen in propionic acidemia share a common pathogenic mechanism.
We have recently shown that the acquired LQTS observed in patients with PA is due to acute and chronic effects of accumulating metabolites on the KvLQT1/KCNE1 channel and its function. Namely, propionic acid, propionylcarnitine and methylcitrate, the key metabolites accumulating in PA, have acute blocking effects on this potassium channel and thereby reduce IKs current and prolong action potential duration (Fig. 1) [17]. Additionally, the expression of KvLQT1 is altered in the presence of these metabolites: Chronic exposure (24 h) to propionylcarnitine and methylcitrate decreases KvLQT1 expression - in line with the acute IKs-blocking effects [17]. We hypothesize that the hearing impairment of patients with PA may be caused by the same mechanisms that lead to acquired LQTS (Fig. 2). In the inner ear the KvLQT1/KCNE1 channel is critical for the production and homeostasis of the endolymph, the potassium-rich fluid surrounding the neuroepithelium of the inner ear [6]: Potassium ions are absorbed from the perilymph by basal cells, further transported through gap junctions to intrastrial fluids, and then pumped into intermediate cells through the Kir4.1 potassium channel, which creates a high intracellular potassium concentration [6]. The consequential concentration gradient between intrastrial fluid and intermediate cells generates the endocochlear potential [18]. From the intrastrial fluid, potassium ions are transported across the basolateral membrane of marginal strial cells by the Na+/K+-ATPase and Na+/K+/Cl− cotransporter [19–22]. Finally, K+ crosses the apical membrane through the KCNQ1/KCNE1 potassium channel and is secreted into the endolymph [11, 19]. The endocochlear potential is essential for audition [20] and disturbances of the endocochlear potential can lead to the immediate loss of hearing, which may be irreversible [23].
Fig. 1.
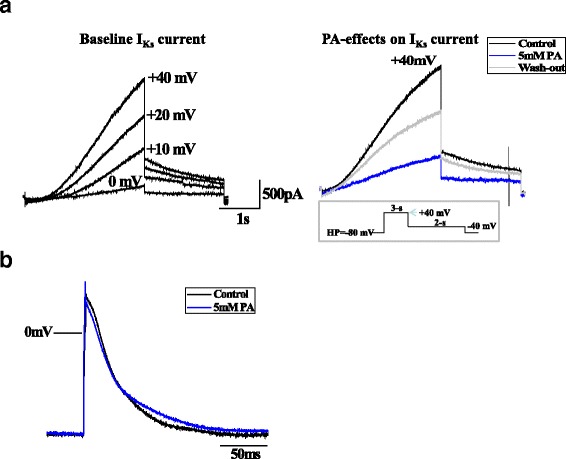
a. Propionic acid effects on IKs current in KCNQ1/KCNE1-CHO cells. Representative whole cell current traces at baseline, after the application of PA (5 mM, blue), and after brief wash-out (grey), demonstrating an acute reduction of IKs by 50%. Voltage clamp protocol is shown in the inset. b. Propionic acid effects on action potential in human iPSC-CMs. Superimposed representative AP traces at baseline and after PA (5 mM, blue), indicating an AP-prolongation
Fig. 2.
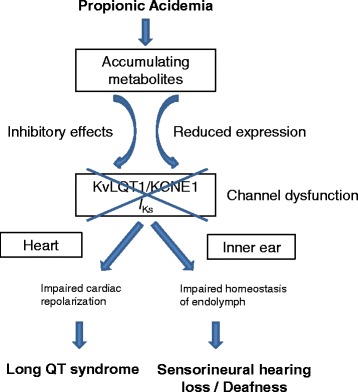
Proposed pathophysiologic mechanism of long QT syndrome and sensoneurinal hearing loss in propionic acidemia. Accumulating metabolites such as propionic acid, methylcitrate and propionylcarnitine cause dysfunction of the KvLQT1/KCNE1 channel by acute blocking effects as well as reduced expression of the channel protein
In addition to altered KCNQ1/KCNE1 channel function, histopathological changes in the inner ear - causatively linked to the altered KCNQ1/KCNE1 function - have been described in patients with Jervell and Lange-Nielsen syndrome: As early as in the 1960ies, temporal bone histology has been investigated and profound histopathologic abnormalities including the collapse of Reissner’s membrane and membranes surrounding the saccule, utricle and ampullae were found, resulting in the obliteration of the scala media and endolymphatic compartments of the vestibular end organs [6, 24, 25]. KCNQ1 knockout mice similarly exhibit histopathologic findings that are comparable to those reported in patients with Jervell and Lange-Nielsen syndrome with marked atrophy of the stria vascularis, contraction of the endolymphatic compartments, collapse and adhesion of surrounding membranes, complete degeneration of the organ of Corti and an associated degeneration of the spiral ganglion [26]. These histopathological changes underline the importance of the KvLQT1/KCNE1 channel for the structural integrity of the inner ear.
Due to the critical role of the KvLQT1/KCNE1 channel, it is well conceivable that blocking effects of circulating metabolites of propionic acid on the IKs current as well as effects on the expression of the channel subunits may have severe consequences and lead to hearing impairment or even deafness. These mechanisms are likely to be the cause of hearing impairment in patients with PA.
Acknowledgements
Not applicable.
Funding
The main research project published by Bodi et al. [17] was funded by the Müller-Fahnenberg-Stiftung, Freiburg.
Availability of data and materials
The datasets supporting the conclusions of this article are included in the cited article “Mechanisms of acquired long QT syndrome in patients with propionic acidemia” published by the same working group [17].
Authors’ contributions
SCG participated in planning and conduction of the study as well as in data interpretation. She drafted the manuscript. IB organized and performed the laboratory investigations of the main research project mentioned above. KEO participated in the study design and coordinated the project. She was involved in the analysis and interpretation of data, supervised the laboratory investigations, and participated in writing this manuscript. All authors have critically read and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- LQTS
Long QT syndrome
- PA
Propionic acidemia
Contributor Information
S. C. Grünert, Phone: +49-761-270-43680, Email: sarah.gruenert@uniklinik-freiburg.de
I. Bodi, Email: Ilona.bodi@uniklinik-freiburg.de
K. E. Odening, Email: Ilona.bodi@uniklinik-freiburg.de
References
- 1.Fenton W, Gravel W, Rosenblatt D. Disorders of propionate and methylmalonate metabolism. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The metabolic and molecular bases of inherited diseases. New York: McGraw-Hill; 2001. pp. 2165–93. [Google Scholar]
- 2.Grünert SC, Mullerleile S, De Silva L, Barth M, Walter M, Walter K, Meissner T, Lindner M, Ensenauer R, Santer R, et al. Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet J Rare Dis. 2013;8:6. doi: 10.1186/1750-1172-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumgartner D, Scholl-Burgi S, Sass JO, Sperl W, Schweigmann U, Stein JI, Karall D. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J Pediatr. 2007;150(2):192–7. doi: 10.1016/j.jpeds.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 4.Williams ZR, Hurley PE, Altiparmak UE, Feldon SE, Arnold GL, Eggenberger E, Mejico LJ. Late onset optic neuropathy in methylmalonic and propionic acidemia. Am J Ophthalmol. 2009;147(5):929–33. doi: 10.1016/j.ajo.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 5.Brosch S, Rauffeisen A, Baur M, Michels L, Trefz FK, Pfister M. [Propionic acidemia and sensorineural hearing loss: is there a connection at the molecular genetics level?] HNO. 2008;56(1):37–42. doi: 10.1007/s00106-007-1560-6. [DOI] [PubMed] [Google Scholar]
- 6.Yanmei F, Yaqin W, Haibo S, Huiqun Z, Zhengnong C, Dongzhen Y, Shankai Y. Cochlear implantation in patients with Jervell and Lange-Nielsen syndrome, and a review of literature. Int J Pediatr Otorhinolaryngol. 2008;72(11):1723–9. doi: 10.1016/j.ijporl.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Crotti L, Celano G, Dagradi F, Schwartz PJ. Congenital long QT syndrome. Orphanet J Rare Dis. 2008;3:18. doi: 10.1186/1750-1172-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyson J, Tranebjaerg L, McEntagart M, Larsen LA, Christiansen M, Whiteford ML, Bathen J, Aslaksen B, Sorland SJ, Lund O, et al. Mutational spectrum in the cardioauditory syndrome of Jervell and Lange-Nielsen. Hum Genet. 2000;107(5):499–503. doi: 10.1007/s004390000402. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, Shkolnikova M, Berul CI, Bitner-Glindzicz M, Toivonen L, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113(6):783–90. doi: 10.1161/CIRCULATIONAHA.105.592899. [DOI] [PubMed] [Google Scholar]
- 10.Tranebjærg L, Samson R, Green G. Jervell and Lange-Nielsen Syndrome. 2014. [Google Scholar]
- 11.Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15(2):186–9. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- 12.Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J. Inner ear defects induced by null mutation of the isk gene. Neuron. 1996;17(6):1251–64. doi: 10.1016/S0896-6273(00)80255-X. [DOI] [PubMed] [Google Scholar]
- 13.Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation of the gene for IsK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of Long-QT syndrome. Circulation. 1998;97(2):142–6. doi: 10.1161/01.CIR.97.2.142. [DOI] [PubMed] [Google Scholar]
- 14.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 15.Bleich M, Warth R. The very small-conductance K+ channel KvLQT1 and epithelial function. Pflugers Arch. 2000;440(2):202–6. doi: 10.1007/s004240000257. [DOI] [PubMed] [Google Scholar]
- 16.Wang W, Flores MC, Sihn CR, Kim HJ, Zhang Y, Doyle KJ, Chiamvimonvat N, Zhang XD, Yamoah EN. Identification of a key residue in Kv7.1 potassium channel essential for sensing external potassium ions. J Gen Physiol. 2015;145(3):201–12. doi: 10.1085/jgp.201411280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bodi I, Grünert SC, Becker N, Stoelzle-Feix S, Spiekerkoetter U, Zehender M, Bugger H, Bode C, Odening KE. Mechanisms of acquired long QT syndrome in patients with propionic academia. Heart rhythm. 2016 doi: 10.1016/j.hrthm.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Salt AN, Melichar I, Thalmann R. Mechanisms of endocochlear potential generation by stria vascularis. Laryngoscope. 1987;97(8 Pt 1):984–91. [PubMed] [Google Scholar]
- 19.Wangemann P, Liu J, Marcus DC. Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro. Hear Res. 1995;84(1–2):19–29. doi: 10.1016/0378-5955(95)00009-S. [DOI] [PubMed] [Google Scholar]
- 20.Hibino H, Nin F, Tsuzuki C, Kurachi Y. How is the highly positive endocochlear potential formed? The specific architecture of the stria vascularis and the roles of the ion-transport apparatus. Pflugers Arch. 2010;459(4):521–33. doi: 10.1007/s00424-009-0754-z. [DOI] [PubMed] [Google Scholar]
- 21.Crouch JJ, Sakaguchi N, Lytle C, Schulte BA. Immunohistochemical localization of the Na-K-Cl co-transporter (NKCC1) in the gerbil inner ear. J Histochem Cytochem. 1997;45(6):773–8. doi: 10.1177/002215549704500601. [DOI] [PubMed] [Google Scholar]
- 22.Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet. 1999;22(2):192–5. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- 23.Wangemann P. Potassium ion secretion and generation of the endocochlear potential in the stria vascularis. HNO. 1997;45(4):205–9. doi: 10.1007/s001060050105. [DOI] [PubMed] [Google Scholar]
- 24.Friedmann I, Fraser GR, Froggatt P. Pathology of the ear in the cardioauditory syndrome of Jervell and Lange-Nielsen (recessive deafness with electrocardiographic abnormalities) J Laryngol Otol. 1966;80(5):451–70. doi: 10.1017/S002221510006552X. [DOI] [PubMed] [Google Scholar]
- 25.Friedmann I, Fraser GR, Froggatt P. Pathology of the ear in the cardio-auditory syndrome of Jervell and Lange-Nielsen. Report of a third case with an appendix on possible linkage with the Rh blood group locus. J Laryngol Otol. 1968;82(10):883–96. doi: 10.1017/S0022215100069620. [DOI] [PubMed] [Google Scholar]
- 26.Rivas A, Francis HW. Inner ear abnormalities in a Kcnq1 (Kvlqt1) knockout mouse: a model of Jervell and Lange-Nielsen syndrome. Otol Neurotol. 2005;26(3):415–24. doi: 10.1097/01.mao.0000169764.00798.84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included in the cited article “Mechanisms of acquired long QT syndrome in patients with propionic acidemia” published by the same working group [17].