

Abstract
Background:
Plant sterols are the major source of micronutrients and have not shown any obvious side effects in human. β-sitosterol is one of the most prevalent phytosterols which have been recorded in ancient medicinal history for its use in the treatment of many chronic diseases, especially cancer. The modulations of mitogen-activated protein kinases’ (MAPKs’) play a crucial role in the development of human renal cell carcinoma.
Objective:
The aim of the current study is to evaluate the antigenotoxic and anticancer role of β-sitosterol against renal carcinogen.
Materials and Methods:
The extent of DNA damage was assessed by the comet assay. The status of p-p38 MAPK, p-c-Jun N-terminal kinase, p-extracellular-signal regulating kinase (ERK), c-fos, c-jun, and endothelial growth factor receptor (EGFR) were analyzed by western blot and polymerase chain reaction techniques. To further confirm the inhibition of ERK-2 by β-sitosterol, molecular docking study was performed.
Results:
Extensive DNA damage in acute study and a significant increase in levels of p-MAPKs’, c-fos, c-jun, and EGFR was observed in N-diethylnitrosamine (200 mg/kg bw) and ferric nitrilotriacetate (9 mg/kg bw) alone treated rats. Rats which are pretreated with 20 mg/kg bw of β-sitosterol reduced the DNA damage and restored the elevated levels of above-mentioned markers (p < 0.05). The binding free energy obtained for β-sitosterol for ERK-2 was found to be-5.578.
Conclusion:
Therefore, it has been concluded that β-sitosterol has a strong potential against genotoxic as well as suppress neoplastic transformation in experimental renal cancer.
SUMMARY
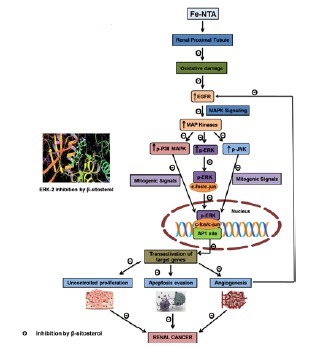
Alterations of EGFR system and MAPKs’ play a major role in the development and progression of RCC. In the present study, the blockade of the Fe-NTA promoted EGFR signaling and sustained ERK activity with β-sitosterol leads to impede tumor promotion and maintenance.
Rats which are pre-treated with 20 mg/kg bw of β-sitosterol significantly reduced the elevated expression of p-p38 MAPK, p-JNK, p-ERK, c-fos and c-jun in carcinogen induced rats, which suggest that β-sitosterol might protect renal tissue from neoplastic transformation. The interaction of β-sitosterol with the ATP binding site of ERK-2 by molecular docking studies also validates the inhibitory effect of β-sitosterol on ERK-2. The results of the present study reveal that β-sitosterol inhibit oncogenic MAPK signaling, to abrogate hyper cell proliferation, angiogenesis, and to induce apoptosis thereby prevent DEN and Fe-NTA induced renal carcinogenesis. Thus,β-sitosterol that modulates signal transduction pathways and their downstream events may serve as a potential cancer chemopreventive and therapeutic agent.
Abbreviation used: AP-1: Activator protein-1,DEPC: Diethyl pyrocarbonate,EDTA: Ethylenediaminetetraacetic acid,EGFR: Endothelial growth factor receptor,ERK: Extracellular-signal regulating kinase,Fe-NTA: Ferric nitrilotriacetate,GAPDH: Glyceraldehyde-3-phosphate dehydrogenase,HBSS: Hank's balanced salt solution,JNK: c-Jun N-terminal kinase,MAPK: Mitogen-activated protein kinase,DEN: N-diethylnitrosamine,RCC: Renal cell carcinoma,SDS-PAGE: Sodium dodecyl sulfate polyacrylamide gel electrophoresis
Keywords: DNA damage, endothelial growth factor receptor (EGFR), mitogen-activated protein kinases', renal carcinogenesis, β-sitosterol
INTRODUCTION
Renal cell carcinoma (RCC) is representing about 3% of all adult cancers. In India, renal cancer death rates are high about 6,000 people will die each year out of the 10,000 new cases which were more frequent in younger people.[1] Men have the doubled risk to this disease than women because of the high consumption of tobacco smoking. Most of the currently available cytostatics are ineffective and often just 5-15% response rates for the treatment of metastasized RCC.[2] Induced DNA damage and alterations of mitogen-activated protein kinases (MAPKs′) seem to play an exclusive role in the development of RCC.[3] The kidney is a prominent site of intense oxidative DNA damage caused by ferric nitrilotriacetate (Fe-NTA) generated radicals.[4]
Research evidence indicates that p38 MAPK activity is critical for normal immune and inflammatory responses.[5] c-Jun N-terminal kinase (JNK) and p38 MAPK is responsible for apoptosis in many cellular systems, but in somewhere it has been implicated in proliferation and differentiation also. Extracellular-signal regulating kinase (ERK) have been involved in several kinds of human cancers that are implicated in the proliferation and prevention of cell death processes and are activated in response to growth factors and phorbol esters, while the JNK is activated in response to different types of stress stimuli.[6,7] The JNK activation is needed for the activation of activator protein-1 (AP-1).[8] AP-1, family of transcriptional activators, consists of dimeric combinations of Jun and Fos proteins that regulate a broad range of biological processes, including the immune response, cell proliferation, apoptosis, and tumorigenesis.[9] c-Jun, a transcription factor, is a major substrate for JNK. Phosphorylation of c-jun results in the enhancement of AP-1 transcriptional activity. The previous report has been proposed that iron can induce early signaling pathways that modulate the activities of oxidative-responsive factor,AP-1[10] which leads to upregulate the expression of cytokine genes, resulted in inflammation and carcinogenesis.[11]
Endothelial growth factor receptor (EGFR) belongs to the ErbB family of tyrosine kinase receptors, which includes ErbB-/EGFR/HER-1 (EGFR itself),HER-2/neu (ErbB-2),HER-3 (ErbB-3), and HER-4 (ErbB-4). Over expression of EGFR in RCC is largely recognized, and its signaling is mitogenic for both malignant and normal renal tubular cells. ErbB signaling activates downstream consequences like Ras/Raf/MAPK and AKT pathways, as well as act as signal transducer and transcription signaling, which mediate cellular growth and proliferation.[12] Nowadays, anti-cancer agents are developed against specific molecular targets because of its crucial importance to elucidate optimal target candidates. EGFR expression is considered as an important drug target as well as having potential prognostic relevance. The US Food and Drug Administration currently approve a variety of anti-EGFR drugs.[13] Research studies have shown that the over expression of epidermal growth factor receptor (EGFR) plays a major role in the initiation and progression of RCC and has been associated with a high tumor grade.[14]
Moreover,Protein-Ligand interaction plays a significant role in structural based drug designing. The analysis of the docking result allowed us to know the efficiency of the natural bioactive compound to control the cancer. Research evidences have shown that the involvement of β-sitosterol in various cell signaling pathways, including cell cycle, apoptosis, proliferation, survival, invasion, angiogenesis, metastasis and inflammation.[15,16,17] The previous study has shown that 20 mg/kg bw of β-sitosterol protected renal cells against nephrotoxicants.[18] There was no research evidence available about the involvement of β-sitosterol in MAPKs′ behavior in experimental renal carcinogenesis. Induce renal carcinogenesis (RCC) by N-diethylnitrosamine (DEN) and Fe-NTA exposition, which sustains their participation in renal cells malignization, offers an opportunity to elucidate the molecular mechanisms at different stages of the RCC.[3]
Based on the above information, it would be of great interest to evaluate the antigenotoxic effect and the potential of β-sitosterol against renal carcinogenesis by explored MAPKs′,AP-1 transcription factors,EGFR expression, and docking analysis.
MATERIALS AND METHODS
Experimental drugs and chemicals
β-sitosterol,DEN, nitrilotriacetic disodium salt,Trizol reagent, and primers for EGFR, c-fos, c-jun and Red Taqman Amplification Kit were purchased from Sigma-Aldrich Chemical Pvt. Ltd. (St. Louis,MO,USA). Antibodies for β-actin, p-p38 MAPK, p-JNK, and p-ERK were purchased from Santa Cruz Biotechnology,USA. All other chemicals used were of analytical grade and purchased from Hi-media Laboratories Pvt,Ltd.,Mumbai,India.
Fe-NTA preparation
Fe-NTA was prepared by the mixing of 0.16 mM ferric nitrate solution and 0.64 mM disodium salt of NTA and the pH was adjusted to 7.4 with a sodium bicarbonate solution.[19]
Animals
Healthy adult male albino Wistar rats (8–10 weeks old; weighing 120-180 g) were purchased from the National Institute of Nutrition,Hyderabad and were maintained in the Central Animal House,Department of Experimental Medicine,Rajah Muthaiah Medical College and Hospital,Annamalai University. The animals were housed in polypropylene cages and were provided with a standard pellet diet and water ad libitum. The animals were maintained under controlled conditions of temperature (23 + 2ºC) and humidity (65-70%) with a 12 h light/dark cycle. The animal treatment and protocol employed was approved by the Institutional Animal Ethics Committee (Registration number 160/1999/CPCSEA; Proposal No. 1041: dated 06.08.2013),Annamalai University. The animals were kept in compliance with the “Guide for the care and use of laboratory animals” and committee for the purpose of control and supervision on experimental animals.
Experimental design
Genotoxicity Study
A total number of 16 animals were divided into four groups and each group contained four animals. Group I animals served as control [0.1% carboxymethyl cellulose (CMC)]; Group II animals were treated with DEN (200 mg/kg bw i.p. on 15th day) and Fe-NTA (9 mg/kg bw i.p. on 30th and 32nd day); Group III animals were treated with β-sitosterol (20 mg/kg bw in 0.1% CMC, p.o. for 32 days) 2 weeks prior to the exposure to the nephrotoxicant; Group IV animals were received β-sitosterol alone and did not receive DEN and Fe-NTA. The experiment was terminated after the 24 h of last dosage of Fe-NTA and all the animals were sacrificed by cervical dislocation for the assessment of DNA damage.
Renal cancer study
Wistar rats were randomized into four groups (control and experimental) of six rats in each. The Group I rats which served as the vehicle control (0.1% CMC). The Groups II and III rats were treated with N-diethylnitrosamine (200 mg / kg bw single i.p., injection) and ferric nitrilotriacetate [9 mg Fe / kg bw i.p.,(2 weeks after DEN exposure) twice a week for 16 weeks]. Group II rats received no other treatment. Group III rats were orally administered with β-sitosterol (20 mg / kg bw in 0.1% CMC p.o., thrice a week for 24 weeks) starting 2 weeks before the exposure to the carcinogens. Group IV rats were orally administered with β-sitosterol alone throughout the experimental period. At the end of 24 weeks, all the animals were sacrificed by cervical dislocation.
DNA damage in kidney tissues (COMET assay)
The DNA damage in the kidney tissues was assessed by the method of Tice et al.[20] and the cells were prepared by the method of Kamp et al.[4] The kidney tissues were excised and placed in 1 mL of ice-cold hank's balanced salt solution (HBSS) (pH 7.4, containing 20 mM ethylenediaminetetraacetic acid (EDTA) and 10% dimethyl sulfoxide (DMSO). Tissue was minced into small pieces, incubated with collagenase (Type IV,5 U/mL) for 30 min and trypsin for 30 min. Cells were prepared by mechanical disintegration using a microliter pipet and washed with HBSS containing fetal calf serum. Aliquots of cell suspensions were centrifuged (400 g), cells resuspended in 200 µL of 0.5% low melting point agarose, spread onto a frosted glass microscope slide, precoated with a layer of 1% normal melting point agarose, and then coverslipped and kept at 48ºC for solidification. After removing the cover slip, slides were immersed in the chilled lysing solution (containing 2.5 M Nacl,100 mM Na2+ EDTA,100 mM Tris-HCL, pH 10 and 1% DMSO,1% Triton × 100 and 1% sodium sarcosinate) for 1 h at 4ºC and followed by alkaline buffer (1N NaOH,0.5 M EDTA and DMSO; pH > 13) for 20 min. The electrophoresis was carried out for 20 min at 25v and 300 mA. The slides were stained with 50 µL of ethidium bromide (20 µg/mL) and analyzed under 20× objectives on fluorescence microscope and images were viewed under high performance nikon camera (Nikon Coolpix 4500).
DNA damage, as reflected by % DNA in tail (tail intensity), tail length, tail moment (product of tail DNA/ total DNA by the centre of gravity), and olive tail moment (the product of the distance between the barycentre of the head and tail and the proportion of DNA in the tail) of the stored images, was investigated using CASP software (CASP1.2.3.beta1, http://casplab.com)
Western blotting
Homogenates containing equal amounts of protein were resolved by 8-12% Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and processed for western blotting and electrotransferred onto polyvinylidene fluoride membranes. The membranes were then incubated overnight at 4°C with antibodies specific to p-p38 MAPK, p-JNK, and p-ERK (Santa Cruz Biotechnology,USA). The membranes were washed with TBST [blocking buffer (containing 5% skimmed milk powder in 0.5 M Tris-buffered saline, pH 7.5 containing 0.1% Tween-20)] and incubated with respective secondary antibody for 2 h at room temperature. Protein band detection was performed by enhanced chemiluminescence assay. The band density of each phosphorylated MAPK was normalized to that of total MAPK. Quantitative comparisons of protein expression between various groups were performed using Image J software (from the US National Institutes of Health)
RT-PCR
RNA was extracted from renal tissue using Trizol reagent. The concentration and purity of RNA preparation were checked by using a Nano spectrophotometer (Implen,Germany) measuring the absorbed at 260 and 280 nm. Total RNA (2.0 µg) was reverse transcribed to cDNA in a reaction mixture containing 1 µL of oligo (dT) primer (0.2 µg/mL),1 µL of RNase inhibitor (10 U/mL),1 µL of 0.1 M DTT,4 µL of 5× reaction buffer,2.0 µL of 30 mM dNTP mix (7.5 mM each),0.5 µL of M-MuLV reverse transcriptase (50 U/µL) and made up to 20 µL with diethyl pyrocarbonate (DEPC) water and kept at 37°c for 1h and then heated at 95°c for 2 min. The primer sequences for c-fos, c-jun,EGFR, and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were listed in Table 1. Reactions were run in a total volume of 50 µl, including 25 µL polymerase chain reaction (PCR) reactions Mix (RED Taq-Ready Mix),2 µl of each primer at 10 µM concentrations,5 µL of the previously reverse transcribed cDNA template and 16 µL of DEPC water. The PCR conditions were as follows: 95°c for 5 min,40 cycles of 30 sec at 95°c,30 sec at 52 to 60°c (based on the target), and 60 sec at 72°c. All PCR products were run on 1.5% agarose stained with ethidium bromide and visualized by ultraviolet (UV) transilluminator. Semiquantitative determination of PCR products was performed using the gel documentation system (UV Tech,Genie). Relative expression of each studied gene (R) was calculated using the following formula,R = densitometrical units of each studied gene / densitometrical units of GAPDH.
Table 1.
Primer sequences are used in RT-PCR

Molecular docking
Grid-Based Ligand Docking With Energetics (Glide) was used for Docking studies, which is a part of Schrodinger (version9.3.518) suites, with a system configuration of Intel (R) Xeon (R) Quadcore CPU E3-1225 V2 @3.20 GHz with Linux Operating system. For ligand and protein preparation,Maestro v9.0 Graphical User Interface workspace was used. The ligand structure utilized for the docking calculation was retrieved as Mol file from the ChemSpider Database that contains information on small molecules.
The LigPrep module of Schrodinger suites was used for developing various tautomers of the ligand molecule. The preotein structure for ERK2 (PDB ID: 1TVO) was retrieved from Protein Data Bank. The PDB structure was edited by removing water molecules and by adding polar hydrogen atoms for satisfying their apt valancies. The edited protein structure was then prepared using Protein Preparation Wizard in the Schrodinger suites, which utilizes the Optimized Potential for Liquid Simulations-All Atoms (OPLS-AA) force fields for energy minimization. Following protein preparation,Grid preparation was carried out for assigning the appropriate area of interest for protein ligand docking. The Ligand docking was carried out using Extraprecision (XP) mode. The docking results were visualised using Maestro 10.3 visualiser and Discovery studio 4.1 visualiser (Dassault Systèmes BIOVIA,Discovery Studio Modeling Environment,Release 4.1,San Diego: Dassault Systèmes,2013).
Statistical analysis
Statistical analysis was performed using one-way analysis of variance, followed by Duncan's multiple range test using SPSS version 17.0 for Windows (Tokyo, Japan). Values are represented as mean ± standard deviation and p < 0.05 were considered statistically significant.
RESULTS
Antigenotoxic effect of β-sitosterol
Figure 1 shows the extent of DNA damage (% DNA in the tail (tail intensity), tail moment, olive tail moment, and tail length in renal cells of control and experimental animals in each group. Extensive DNA damage as reflected by an increase in % DNA in tail, tail moment, olive tail moment, and tail length was noticed in rats treated with DEN and Fe-NTA alone. Oral administration of β-sitosterol pretreatment significantly (p < 0.05) reduced DNA damage when compared with DEN and Fe-NTA treated rats. No obvious difference between control and (Group I) and β-sitosterol alone (Group IV) rats.
Figure 1.
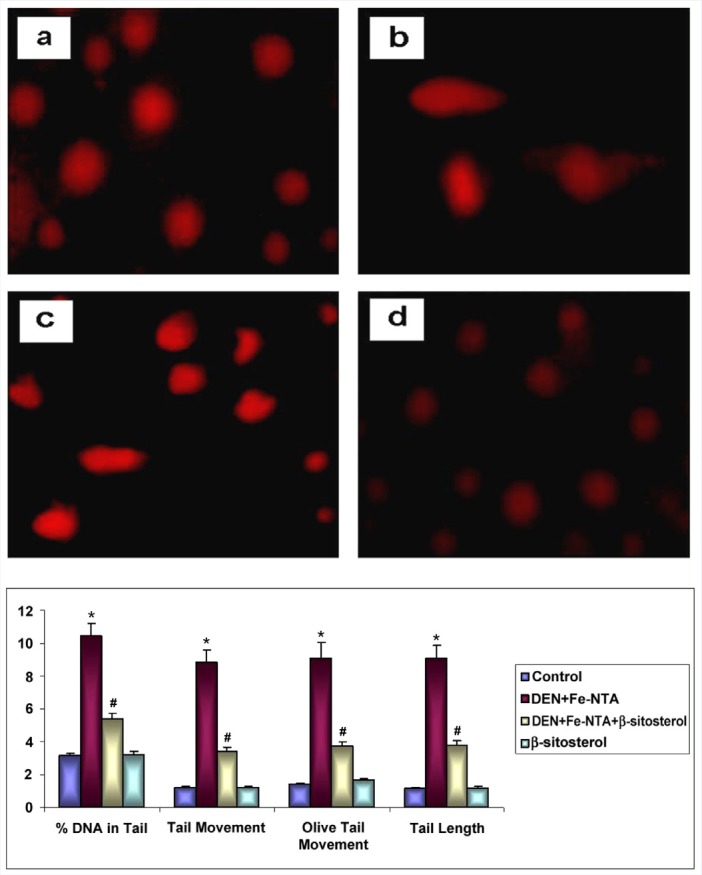
Representative photographs depict the extent of DNA damage in control (a), DEN+Fe-NTA (b), DEN+Fe-NTA+β-sitosterol (c) and β-sitosterol alone (d). (20x magnification). Changes in the levels of DNA damage (% DNA in tail (tail intensity), tail moment, olive tail moment and tail length) in renal cells of control and experimental animals in each group.
Effect of β-sitosterol on MAPKs’ pathway
Western blotting evaluation showed induced expression of p-p38 MAPK, p-ERK, and p-JNK in rats treated with DEN and Fe-NTA alone when compared with control rats [Figure 2]. However,β-sitosterol (20 mg/kg body weight) pretreatment (Group III) significantly reduced the enhanced expression of these phosphorylated proteins in renal tissues when compared with Group II (p < 0.05). There was no significant difference in expression pattern between the control (Group I) and β-sitosterol alone (Group IV) rats.
Figure 2.
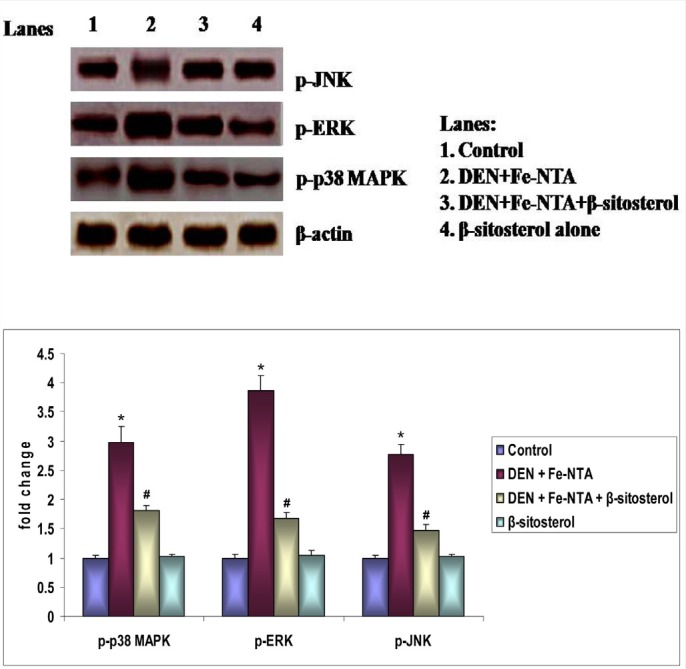
Effect of β-sitosterol on MAPKs′ protein expression in the kidney tissue of control and experimental rats. Bar diagram depicts quantification of six independent experiments (mean ± SD). Phosphorylated MAPKs′ protein expression was normalized to the expression level of total MAPKs′. Values not sharing a common superscript differ significantly at p < 0.05 (DMRT).
Effect of β-sitosterol on proliferative marker
The mRNA expression patterns of EGFR, c-fos, and c-jun in control and experimental rats are depicted in Figure 3. DEN and Fe-NTA induced significant increase in the mRNA expression of EGFR, c-fos, and c-jun in Group II rats when compared with control rats (p < 0.05). β-sitosterol pretreatment regulated EGFR, c-fos, and c-jun expression in Group III. Similar patterns of mRNA expression were observed in control rats (Group I) and rats treated with β-sitosterol alone (Group IV).
Figure 3.
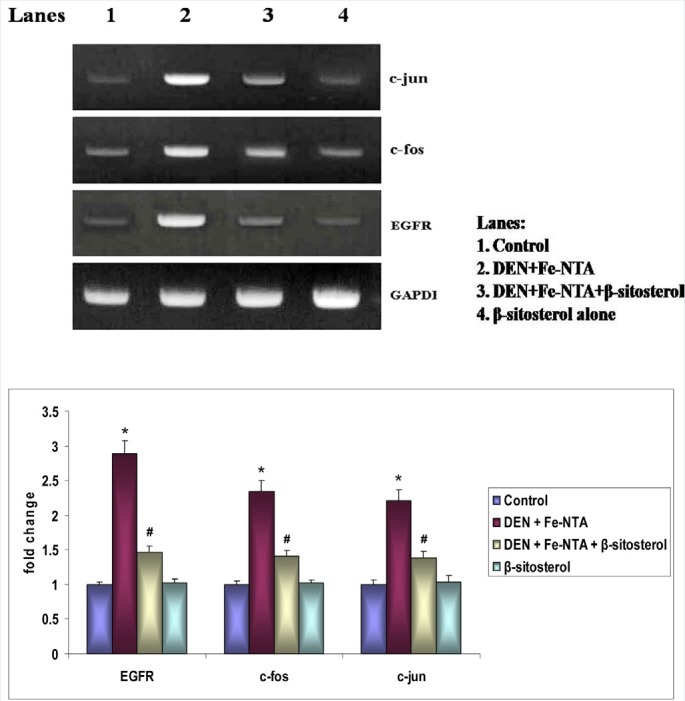
Effect of β-sitosterol on EGFR, c-fos and c-jun mRNA expression in the kidney tissue of control and experimental rats. Bar diagram depicts quantification of six independent experiments (mean ± SD). The mRNA expression of EGFR, c-fos and c-jun was normalized to the expression level of the GAPDH mRNA expression. Values not sharing a common superscript differ significantly at p < 0.05 (DMRT).
Molecular docking
Figure 4 shows the docking analysis of ERK-2 with β-sitosterol. The interaction between ERK2 and β-sitosterol involved one H-bond with MET 108 (Bond length: 1.63), along with one Pi-Alkyl interaction with TYR 36 (Bond length: 4.95), three Alkyl interactions with VAL 39 (Bond length: 4.66,4.73,4.80), two Alkyl interactions with LEU 156 (Bond length: 4.47,4.12) and one Alkyl interaction with ILE 31 (Bond length: 4.25) and CYS 166 (Bond length: 5.36). The binding-free energy obtained for β-sitosterol for the target proteins was found to be -5.578.
Figure 4.
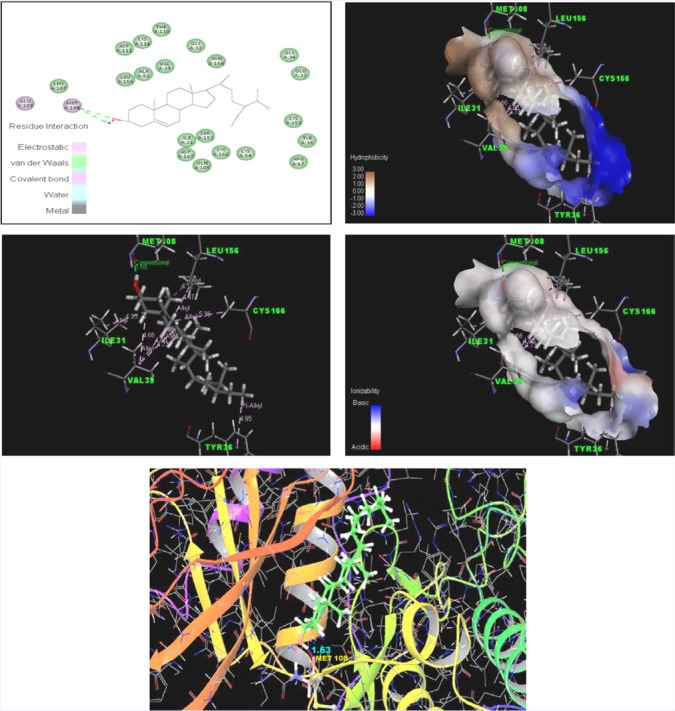
Photographs shows the docking analysis of ERK-2 with β-sitosterol visualised by Maestro 10.3 visualiser and Discovery studio 4.1 visualiser.
DISCUSSION
Induced DNA damage act as an initiating step, in combination with hyperproliferative response, might lead to malignant transformation.[4] Renal cells of rats treated with DEN + Fe-NTA alone have the appearance of a comet, with a head (the nuclear region) and a tail containing DNA fragments or strands. Oral administration of β-sitosterol at a dose of 20 mg⁄kg bw to DEN and Fe-NTA treated rats significantly protected the DNA as evidenced by undamaged and lightly damaged DNA in renal cells. Our results thus indicate that β-sitosterol has a potent antigenotoxic effect against DEN and Fe-NTA-induced DNA damage.
Cancer cells have faulty intracellular circuits that wrongly regulate cell proliferation and homeostasis.[21] The balance and integration of the Jun N-terminal kinase and p38 signals are important for the effective treatment. Alterations of MAPKs′ play a major role in the development and progression of RCC.[3] The JNK predominantly activated by cytokines,UV radiation, growth factor deprivation,DNA-damaging agents, and certain G-protein coupled receptors.[8] JNK activation has been demonstrated in several glomerulonephrities and JNK inhibition suppresses inflammation in rat antiglomerular basement membrane disease.[22,23] An et al.[24] described that the blockade of JNK activation provokes reduced growth and motility of RCC cells in vitro and in vivo. Our results are in good agreement with the previous report that the pretreatment of β-sitosterol reduced the activation of JNK, which suggest that β-sitosterol might protect renal tissue from neoplastic transformation.
ERK signaling plays a pivotal role in mediating renal cell responses to a diverse range of stimuli. Activation of ERK can promote cell proliferation, growth, survival, and blood vessel sprouting and is probably required for the secretion of angiogenic factors from tumor cells.[25] High p-ERK has been implicated in epithelial to mesenchymal transition, which is proposed as an initial event for tumor development.[26] A previous study has shown that altered ERK signaling is involved in renal hypertrophy, glomerular, and tubulointerstitial diseases, including renal cancer. This study also reported that inhibition of ERK pathway leads to the inhibition of cyst-induction in kidney mass and also improve the renal function.[6] Recent research data have been explained that increased p-ERK levels have been reported in both stages of tumor development during Fe-NTA induced RCC development in experimental model which mimics human RCC cases.[3] Our results are in line with previous findings. Repeated administration of Fe-NTA leads to sustained ERK activity, facilitating tumor promotion and maintenance. β-sitosterol decreased RCC proliferation in Fe-NTA induced rats, which were correlated with downregulation of MAPK. Treatment with β-sitosterol to Fe-NTA induced rats in the present study decreased the phosphorylation of all the three MAPKs′ like p38 MAPK,ERK, and JNK. Different studies have indicated that the inhibitory effect of cancer cell growth by β-sitosterol was observed in various cancer cell lines includes oral, prostate, breast, colon, leukemia, stomach, and lung.[14,15,16,27,28,29,30] Taken together, the results from this study, as well as others, suggest that β-sitosterol play a regulatory role in preventing neoplastic transformation in experimental models.
Transcription factor AP-1 complex mediates cellular processes, such as proliferation, differentiation, and stress response by inducing transcription of respective genes. A variety of chemical stresses can activate AP-1 via the MAPK cascades resulted in altered gene expression.[8] c-Fos heterodimerize with c-jun to form AP-1 which binds to specific sites in the regulatory region of target genes. Normally, c-jun expression is low in most cell types, but its expression has been elevated in response to various stimuli includes growth factors, cytokines, and UV irradiation. c-Jun is exclusively activated by JNK and the c-fos activation can be regulated by JNK and ERK signal pathways.[26,31] Research evidence implicates that c-jun involved in the protection of cells from apoptosis.[32] Over expression of c-fos was observed in human laryngeal carcinoma, hepatocellular carcinoma, osteosarcoma, and endometrial carcinoma.[33,34,35,36] The present study showed that DEN and Fe-NTA induces altered expression and activation of the AP-1 subunits c-fos and c-jun, might be via the MAPK pathway especially the JNK and ERK. Various experiments shown that stronger proliferation, malignant transformation, and enhanced aggressiveness are accompanied by changes in AP-1 complex composition.[9,10] Down regulation of tumor-suppressor genes by c-fos regulated genes might be important for tumorigenesis.[9] Our results corroborated with previous data point to a stimulating effect of c-fos on cell invasion in accordance with its oncogenic function. The results of this present study suggest that the DEN and Fe-NTA induced coordinated expression of c-fos and c-jun in RCC may reflect on proliferation and progression of tumor cell. Oral administration of β-sitosterol reduced the elevated levels of c-fos and c-jun induced by renal carcinogen. These results suggest that β-sitosterol exhibit antiproliferative effect against experimental renal carcinogenesis.
Nuclear translocation of EGFR mediates signals to stimulate proliferation, invasion, metastasis in many human cancers, including RCC.[14,37] Previous studies have shown that an enhanced expression of the EGF receptor gene was observed in RCC which involves the transduction of mitogenic signals through the activation of multiple signaling cascades.[38] Promising preclinical evidence also support that the capacity of EGFR inhibition could enhance the antitumor activity.[39] In the present study, the blockade of the Fe-NTA promoted EGFR signaling pathway with β-sitosterol leads to inhibit hyperproliferation.
Furthermore, the interaction of β-sitosterol with the ATP binding site of ERK-2 by molecular docking studies also validates the inhibition of ERK-2 by β-sitosterol. The docking of the ERK protein with β-sitosterol revealed the presence of conventional hydrogen bond (H-bond), Pi-alkyl, and alkyl hydrophobic interactions. Thus,β-sitosterol can inhibit ERK-2 protein involved in cell proliferation by inhibiting ATP-binding site and can act as a competitive inhibitor of ATP.[40] These findings suggested that β-sitosterol block the process of cell proliferation, thereby preventing the tumor invasion and angiogenesis.
CONCLUSION
The present study indicated that the antigenotoxic effect and suppression of MAPKs’ phosphorylation and EGFR signals by β-sitosterol might inhibit the growth of renal cancer in experimental animals. β-sitosterol inhibits the activation and nuclear translocation of these proteins and almost inhibits neoplastic transformation in Fe-NTA-induced experimental renal carcinogenesis
Financial support and sponsorship
This study was funded by Indian Council of Medical Research, India for providing financial support for this research project, in the form of Senior Research Fellowship (ICMR-SRF-IRIS ID: 2013-17190), to Ms. R. Sharmila.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Raja MA. Keep Kidney Cancer at bay. 31 Mar 2015. [Last accessed on 2016 Mar 17]. Available at http://www.newindianexpress.com/cities/chennai/Keep-Kidney-Cancer-at-Bay/2015/03/31/article%20 2738469.ece .
- 2.Hu SL, Chang A, Perazella MA, Okusa MD, Jaimes EA, Weiss RH. The Nephrologist's Tumor: Basic Biology and Management of Renal Cell Carcinoma. J Am Soc Nephrol. 2016;27:2227–37. doi: 10.1681/ASN.2015121335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguilar-Alonso FA, Solano JD, Vargas-Olvera CY, Pacheco-Bernal I, Pariente-Perez TO, Ibarra-Rubio ME. MAPKs’ status at early stages of renal carcinogenesis and tumors induced by ferric nitrilotriacetate. Mol Cell Biochem. 2015;404:161–70. doi: 10.1007/s11010-015-2375-5. [DOI] [PubMed] [Google Scholar]
- 4.Kamp HG, Eisenbrand G, Janzowski C, Kiossev J, Latendresse JR, Schlatter J, et al. Ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Mol Nutr Food Res. 2005;49:1160–7. doi: 10.1002/mnfr.200500124. [DOI] [PubMed] [Google Scholar]
- 5.Rashid S, Ali N, Nafees S, Hasan SK, Sultana S. Amelioration of renal carcinogenesis by bee propolis: a chemo preventive approach. Toxicol Int. 2013;20:227–34. doi: 10.4103/0971-6580.121676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang D, Ding Y, Luo WM, Bender S, Qian CN, Kort E, et al. Inhibition of MAPK Kinase Signaling Pathways Suppressed Renal Cell Carcinoma Growth and Angiogenesis In vivo . Cancer Res. 2008;68:81–8. doi: 10.1158/0008-5472.CAN-07-5311. [DOI] [PubMed] [Google Scholar]
- 7.Jia ZC, Wan YL, Tang JQ, Dai Y, Liu YC, Wang X, et al. Tissue factor/activated factor VIIa induces matrix metalloproteinase-7 expression through activation of c-Fos via ERK1/2 and p38 MAPK signaling pathways in human colon cancer cell. Int J Colorectal Dis. 2012;27:437–45. doi: 10.1007/s00384-011-1351-0. [DOI] [PubMed] [Google Scholar]
- 8.Lee MK, Kim IH, Choi YH, Nam TJ. A peptide from porphyra yezoensis stimulates the proliferation of IEC-6 cells by activating the insulin-like growth factor I receptor signaling pathway. Int J Mol Med. 2015;35:533–8. doi: 10.3892/ijmm.2014.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahner S, Baasch C, Schwarz J, Hein S, Lber WL, Nicke FJ, et al. C-Fos expression is a molecular predictor of progression and survival in epithelial ovarian carcinoma. Br J Cancer. 2008;99:1269–75. doi: 10.1038/sj.bjc.6604650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–58. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 11.Goswami B, Tayal D, Mallika V. Ferritin: A multidimensional bio marker. Internet J Lab Med. 2008;3:1–9. [Google Scholar]
- 12.Costa LJ, Drabkin HA. Renal cell carcinoma: new developments in molecular biology and potential for targeted therapies. Oncologist. 2007;12:1404–15. doi: 10.1634/theoncologist.12-12-1404. [DOI] [PubMed] [Google Scholar]
- 13.Minner S, Rump D, Tennstedt P, Simon R, Burandt E, Terracciano L. Epidermal growth factor receptor protein expression and genomic alterations in renal cell carcinoma. Cancer. 2012;118:1268–75. doi: 10.1002/cncr.26436. [DOI] [PubMed] [Google Scholar]
- 14.Weidner U, Peter S, Strohmeyer T, Hussnattar R, Ackermann R, Sies H. Inverse relationship of epidermal growth factor receptor and HER2/neu gene expression in human renal cell carcinoma. Cancer Res. 1990;50:4504–9. [PubMed] [Google Scholar]
- 15.Park C, Moon DO, Rhu CH, Choi BT, Lee WH, Kim GY, et al. β-Sitosterol induces anti-proliferation and apoptosis in human leukemic U937 cells through activation of caspase-3 and induction of Bax/Bcl-2 ratio. Biol Pharm Bull. 2007;30:1317–23. doi: 10.1248/bpb.30.1317. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Chang SK, Qu G, Li T, Cui H. Beta-sitosterol inhibits cell growth and induces apoptosis in SGC-7901 human stomach cancer cells. J Agric Food Chem. 2009;57:5211–8. doi: 10.1021/jf803878n. [DOI] [PubMed] [Google Scholar]
- 17.Vundru SS, Kale RK, Singh RP. β-Sitosterol induces G1 arrest and causes depolarization of mitochondrial membrane potential in breast carcinoma MDA-MB-231 cells. BMC Complement Altern Med. 2013;13:280–8. doi: 10.1186/1472-6882-13-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharmila R, Sindhu G, Arockianathan PM. Nephroprotective effect of β-sitosterol on N-diethylnitrosamine initiated and ferric nitrilotriacetate promoted acute nephrotoxicity in Wistar rats. J Basic Clin Physiol Pharmacol. 2016;27:473–82. doi: 10.1515/jbcpp-2015-0085. [DOI] [PubMed] [Google Scholar]
- 19.Athar M, Iqbal M. Ferric nitrilotriacetate promotes N-diethylnitrosamine-induced renal tumorigenesis in the rat: implications for the involvement of oxidative stress. Carcinogenesis. 1998;19:1133–9. doi: 10.1093/carcin/19.6.1133. [DOI] [PubMed] [Google Scholar]
- 20.Tice RR, Andrews PW, Hirai O, Singh NP. The single cell gel (SCG) assay: an electrophoretic technique for the detection of DNA damage in individual cells. Adv Exp Med Biol. 1991;283:157–64. doi: 10.1007/978-1-4684-5877-0_17. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka T, Kondo S, Iwasa Y, Hiai H, Toyokuni S. Expression of stress-response and cell proliferation genes in renal cell carcinoma induced by oxidative stress. Am J Pathol. 2000;156:2149–57. doi: 10.1016/S0002-9440(10)65085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stambe C, Atkins RC, Hill PA, Nikolic-Paterson DJ. Activation and cellular localization of the p38 and JNK MAPK pathways in rat crescentic glomerulonephritis. Kidney Int. 2003;64:2121–32. doi: 10.1046/j.1523-1755.2003.00324.x. [DOI] [PubMed] [Google Scholar]
- 23.Flanc RS, Ma FY, Tesch GH, Han Y, Atkins RC, Bennett BL, et al. A pathogenic role for JNK signaling in experimental anti-GBM glomerulonephritis. Kidney Int. 2007;72:698–708. doi: 10.1038/sj.ki.5002404. [DOI] [PubMed] [Google Scholar]
- 24.An J, Liu H, Magyar CE, Guo Y, Veena MS, Srivatsan ES, et al. Hyperactivated JNK is a therapeutic target in pVHL-deficient renal cell carcinoma. Cancer Res. 2013;73:1374–85. doi: 10.1158/0008-5472.CAN-12-2362. [DOI] [PubMed] [Google Scholar]
- 25.Prager GW, Poettler M, Unseld M, Zielinski CC. Angiogenesis in cancer: anti-VEGF escape mechanisms. Transl Lung Cancer Res. 2012;1:14–25. doi: 10.3978/j.issn.2218-6751.2011.11.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gui T, Sun Y, Shimokado A, Muragaki Y. The roles ofmitogen-activated protein kinase pathways in TGF-β-induced epithelial-mesenchymal transition. J Signal Transduct 2012. 2012:289243. doi: 10.1155/2012/289243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendilaharsu M, Stefani ED, Deneo-Pellegrini H, Carzoglio J, Ronco A. Phytosterols and risk of lung cancer: A case-control study in Uruguay. Lung Cancer. 1998;21:37–45. doi: 10.1016/s0169-5002(98)00044-0. [DOI] [PubMed] [Google Scholar]
- 28.Awad AB, von Holtz RL, Cone JP, Fink CS, Chen YC. β-Sitosterol inhibits the growth of HT-29 human colon cancer cells by activating the sphingomyelin cycle. Anticancer Res. 1998;18:3. [PubMed] [Google Scholar]
- 29.von Holtz RL, Fink CS, Awad AB. β-Sitosterol activates the sphingomyelin cycle and induces apoptosis in LNCaP human prostate cancer cells. Nutr Cancer. 1998;32:8–12. doi: 10.1080/01635589809514709. [DOI] [PubMed] [Google Scholar]
- 30.Cabeza M, Bratoeff E, Heuze I, Ramírez E, Sánchez M, Flores E. Effect of β-sitosterol as Inhibitor of 5α-reductase in Hamster Prostate. Proc West Pharmacol Soc. 2003;46:153–5. [PubMed] [Google Scholar]
- 31.Milde-Langosch K, Röder H, Andritzky B, Aslan B, Hemminger G, Brinkmann A, et al. The role of the AP-1 transcription factors c-Fos, FosB, Fra-1 and Fra-2 in the invasion process of mammary carcinomas. Breast Cancer Res Treat. 2004;86:139–52. doi: 10.1023/B:BREA.0000032982.49024.71. [DOI] [PubMed] [Google Scholar]
- 32.Sztalmachova M, Hlavna M, Gumulec J, Holubova M, Babula P, Balvan J, et al. Effect of zinc(II) ions on the expression of pro- and anti-apoptotic factors in high-grade prostate carcinoma cells. Oncol Rep. 2012;28:806–14. doi: 10.3892/or.2012.1897. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Y, Chen C, Chen SM, Wang YQ, Xu Y, Wang Y, et al. Telomerase reverse transcriptase promotes the proliferation of human laryngeal carcinoma cells through activation of the activator protein 1. Oncol Lett. 2013;6:75–80. doi: 10.3892/ol.2013.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuen MF, Wu PC, Lai VCH, Lau JYN, Lai CL. Expression of c-Myc, c-Fos, and c-Jun in Hepatocellular Carcinoma. Cancer. 2001;91:106–12. doi: 10.1002/1097-0142(20010101)91:1<106::aid-cncr14>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 35.Gamberi G, Benassi MS, Bohling T, Ragazzini P, Molendini L, Sollazzo MR, et al. C-myc and c-fos in human osteosarcoma prognostic value of mRNA and protein expression. Oncol. 1998;55:556–63. doi: 10.1159/000011912. [DOI] [PubMed] [Google Scholar]
- 36.Bamberger AM, Milde-Langosch K, Rossing E, Goeman C, Loning T. Expression pattern of the AP-1 family in endometrial cancer: Correlations with cell cycle regulators. J Cancer Res Clin Oncol. 2001;127:545–50. doi: 10.1007/s004320100255. [DOI] [PubMed] [Google Scholar]
- 37.Thomasson M, Hedman H, Guo D, Ljungberg B, Henriksson R. LRIG 1 and epidermal growth factor receptor in renal cell carcinoma: a quantitative RT-PCR and immunohistochemical analysis. Br J Cancer. 2003;89:1285–9. doi: 10.1038/sj.bjc.6601208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gallego GA, Villaamil VM, Grande E, Caínzos IS, Antón Aparicio LM. Crossing paths in human renal cell carcinoma (hRCC) Int J Mol Sci. 2012;13:12710–33. doi: 10.3390/ijms131012710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyake K, Tani K, Kakiuchi S, Suzuka C, Toyoda Y, Kishi J, et al. Epidermal growth factor receptor-tyrosine kinase inhibitor (gefitinib) augments pneumonitis, but attenuates lung fibrosis in response to radiation injury in rats. J Med Invest. 2012;59:174–85. doi: 10.2152/jmi.59.174. [DOI] [PubMed] [Google Scholar]
- 40.Ohori M, Kinoshita T, Okubo M, Sato K, Yamazaki A, Arakawa H, et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor–ERK2 complex. Biochem Biophys Res Communications. 2005;336:357–63. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]