

Abstract
To date, reperfusion with tissue plasminogen activator (tPA) remains the gold standard treatment for ischemic stroke. However, when tPA is given beyond 4.5 hours of stroke onset, deleterious effects of the drug ensue, especially, hemorrhagic transformation (HT), which causes the most significant morbidity and mortality in stroke patients. An important clinical problem at hand is to develop strategies that will enhance the therapeutic time window for tPA therapy and reduce the adverse effects (especially HT) of delayed tPA treatment. We reviewed the pharmacological agents which reduced the risk of HT associated with delayed (beyond 4.5 hours post-stroke) tPA treatment in preclinical studies, which we classified into those that putatively preserve the blood-brain barrier (e.g., minocycline, cilostazol, fasudil, candesartan, and bryostatin) and/or enhance vascularization and protect the cerebrovasculature (e.g., coumarin derivate IMM-H004 and granulocyte colony-stimulating factor). Recently, other new therapeutic modalities (e.g., oxygen transporters) have been reported which improved delayed tPA-associated outcomes by acting through other mechanisms. While the above-mentioned interventions unequivocally reduced delayed tPA-induced HT in stroke models, the long-term efficacy of these drugs are not yet established. Further optimization is required to expedite their future clinical application. The findings from this review indicate the need to explore the most ideal adjunctive interventions that will not only reduce delayed tPA–induced HT, but also preserve neurovascular functions. While waiting for the next breakthrough drug in acute stroke treatment, it is equally important to allocate considerable effort to find approaches to address the limitations of the only FDA-approved stroke therapy.
Keywords: Tissue plasminogen activator, Hemorrhage, Vasculature, Blood-brain barrier
Introduction
Thrombolysis via treatment with tissue plasminogen activator (tPA) has been shown to produce neurological and functional improvement in stroke patients who were given the drug within 3 hours of stroke onset [1]. Currently, a new time window for tPA treatment in stroke has been proposed in that tPA can still be administered at 4.5 hours after stroke onset in certain eligible patients [2]. Despite the expanded therapeutic time window, many patients still do not qualify for tPA therapy since they present for evaluation beyond 3-4.5 hours after stroke onset [3]. Moreover, initiating tPA treatment beyond 4.5 hours (i.e., delayed tPA treatment) has been associated with deleterious side effects, notably, hemorrhagic transformation (HT) which could lead to high mortality in stroke patients [4].
The limitations of tPA for stroke management indicate the need for alternative and more ideal stroke therapies. A variety of drugs ranging from those that enhance neurogenesis [5] and other thrombolytic agents [6,7] have been tested with poor clinical outcomes. As reperfusion with tPA remains, to date, the gold standard treatment for ischemic stroke, an important clinical problem at hand is to develop strategies that will expand the therapeutic time window for tPA therapy and reduce the adverse effects (especially HT) of tPA treatment. Indeed, equally important as discovering new drugs for acute ischemic stroke is identifying interventions that will address the above limitations of tPA therapy. In this mini review, we discuss some of the strategies that were examined to enhance the overall efficacy of tPA, with emphasis on pharmacological agents that were used to attenuate HT due to delayed tPA therapy.
We conducted a systematic PubMed search of all English-language materials related to the topic using the following search terms: “stroke”, “delayed-tPA”, “hemorrhage”, and “hemorrhagic transformation”. This process yielded 57 papers covering both preclinical and clinical reports, with 16 of them highly relevant to the topic. However, as we defined “delayed tPA” as beyond 4.5 hours of tPA treatment after stroke onset, and focused only on preclinical studies, the number of English language articles that were reviewed for this paper was 8 (Figure 1). These pharmacological agents, their effects and proposed mechanism of action are summarized in Figure 2 and Table 1.
Figure 1.
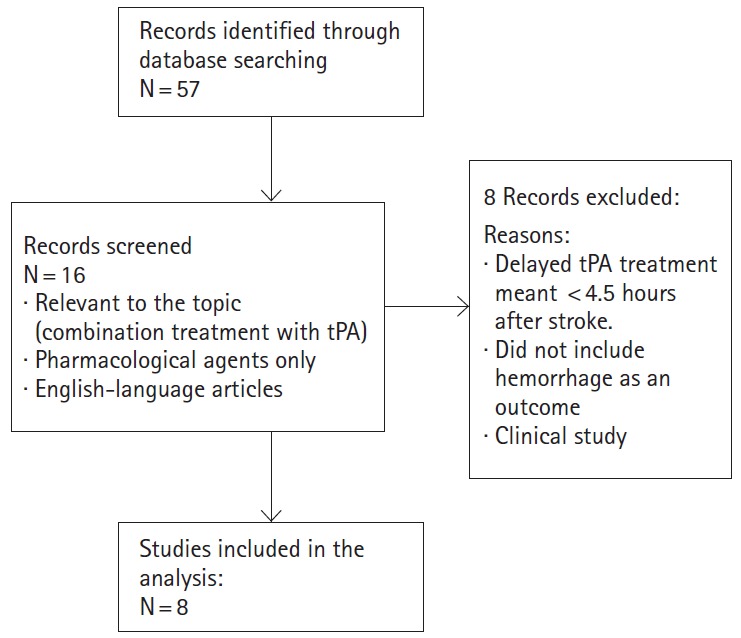
Flowchart for the selection of studies.
Figure 2.

Molecular targets of pharmacological agents tested to attenuate hemorrhagic transformation (HT) after delayed tPA treatment. (A) The HT that ensues after delayed tPA treatment has been ascribed to increased reperfusion and on tPA’s effect on metalloproteinase (MMP) activity and other signaling pathways including the lipoprotein receptor protein (LRP) signaling. In particular, tPA’s signaling actions in the neurovascular unit increases risk of blood-brain barrier (BBB) leakage, neurovascular cell death and HT. Minocycline, cilostazol, GM6001, fasudil, candesartan, bryostatin and IMM-H004 reduces the HT by preserving the BBB through their actions on various MMPs and tight junction (TJ) proteins. (B) Aside from restoring BBB integrity, enhancement of neovascularization or blood vessel formation may also counteract delayed tPA-induced HT. G-CSF and IMM-H004 may reduce the HT by enhancing neurovascularization. G-CSF’s therapeutic effects has been attributed to mobilization of EPCs which reconstitute the BBB. EPC, endothelial progenitor cell; G-CSF, granulocyte-colony stimulating factor; HMGB1, high-mobility-group-box-1; ROS, reactive oxygen species.
Table 1.
Strategies to attenuate delayed tPA-induced hemorrhagic transformation (HT)
| Strategy (dose, method and timing of treatment) | tPA dose, method & timing of treatment | Species & stroke Model | Parameter/ molecular Target | Outcome | Timing of evaluation | Ref. |
|---|---|---|---|---|---|---|
| Minocycline (antibiotic; 3 mg/kg, intravenous [i.v.], 4 hours after stroke) | 10 mg/kg., i.v., 6 hours post-stroke | Male SHR; embolic | HT | Decreased | 24 hours post stroke | [16] |
| Infarct volume | Decreased | |||||
| MMP-9 (plasma) | Decreased | |||||
| Cilostazol (PDEIII- inhibitor; 10 mg/kg, i.p., before tPA) | 0 mg/kg., i.v., 6 hours post-stroke, before reperfusion | Male ddY (22-26 g) 4 weeks old; intraluminal filament/reperfusion | HT | Decreased | 18 hours post-reperfusion | [14] |
| Infarct volume | Decreased | |||||
| MMP-9 | Decreased | |||||
| claudin 5 | Enhanced | |||||
| locomotor behavior | Improved | 7 days post stroke | ||||
| GM6001 (MMP inhibitor; 100 mg/kg, i.p., alongside tPA) | 10 mg/kg., i.v., 6 hours post-stroke, after reperfusion | Male ddY mic (22-30 g) 4 weeks old; intraluminal filament/reperfusion | HT | Decreased | 48 hours post-stroke/reperfusion | [23] |
| Infarct volume | Not examined | |||||
| MMP-9 | Decreased | |||||
| claudin (in vitro, in vivo) | Not changed | |||||
| occludin (in vitro, in vivo) | Enhanced | |||||
| ZO-1 (in vitro, in vivo) | Enhanced | |||||
| Fasudil (ROCK inhibitor; 3 mg/kg, i.p., before tPA) | 10 mg/kg., i.v., 6 hours post-stroke, after reperfusion | Male SD rats (250-330 g); intraluminal filament/reperfusion | HT | Decreased | 18 hours post-reperfusion | [13] |
| Infarct volume | Not changed | |||||
| MMP-9 (in vitro) | Decreased | |||||
| locomotor behavior | Improved | 7 days post stroke | ||||
| Candesartan (AT1R blocker; 1 mg/kg, i.v., 3 hours after stroke) | 10 mg/kg., i.v., 6 hours post-stroke | Male Wistar rats (330-350 g); embolic | HT | Decreased | 24 hours post stroke | [27] |
| Infarct volume | Not changed | |||||
| MMP-9 | Not changed | |||||
| MMP-2 | Not changed | |||||
| MMP-3 | Decreased | |||||
| NF-κB | Decreased | |||||
| TNF-α | Decreased | |||||
| p-eNOS | Decreased | |||||
| Bryostatin (PKC modulator; 2.5 mg/kg., i.v., alongside tPA) | 5 mg/kg, i.v., 6 hours post-stroke | Female SD rats, 18-20 months old; embolic | HT | Decreased | 24 hours post stroke | [31] |
| Infarct volume | Not changed | |||||
| MMP-9 | Decreased | |||||
| MMP-2 | Not changed | |||||
| PKCɛ | Increased | |||||
| PKCα | Not changed | |||||
| PKCδ | Not changed | |||||
| IMM-H004 (Coumarin derivative; 6 mg/kg, i.v., alongsidetPA) | 10 mg/kg, i.v., post-stroke | Male SD rats (300-320 g); embolic | HT | Decreased | 18 hours post-stroke | [36] |
| Infarct volume | Decreased | 24 hours post-stroke | ||||
| Neurological functions | Improved | 1,2,3 days post-stroke | ||||
| Male SD rats (260-280 g); intraluminal filament/reperfusion | HT | Decreased | 24 hours post-stroke | |||
| Infarct volume | Decreased | |||||
| Neurological functions | Improved | 1-7 days post-stroke | ||||
| pro-MMP-9 | Decreased | 24 hours post-stroke/reperfusion | ||||
| Akt (in vitro) | Decreased | |||||
| Ang-1 | Increased | |||||
| CD31CD31+Ki67 | Increased | 7 days post-stroke/reperfusion | ||||
| MMP-2 | Increased | |||||
| occludi | Not co-localized in astrocytes | |||||
| Tie2 | Decreased | |||||
| Increased | ||||||
| G-CSF (300 µg/kg, i.v., alongside tpa) | 10 mg/kg., i.v., post stroke, before reperfusion | Male SD rats, (200-250 g) 9-10 weeks old; intraluminal filament/reperfusion | HT | Decreased | 24 hours post-drug treatment | [38] |
| Infarct volume | Not changed | |||||
| Neurological functions | Improved | |||||
| Ang-1 | Not changed | |||||
| Ang-2 | Increased | |||||
| CD34 | Increased | |||||
| eNOS | Increased | |||||
| VEGFR2 | Increased | |||||
| vWF | Increased |
tPA, tissue plasminogen activator; SHR, spontaneously hypertensive rat; HT, hemorrhagic transformation; PDEIII; phosphodiesterase III; MMP, matrix metallopeptidase; ZO, zonula occludens; ROCK, Rho-associated protein kinase; SD, Sprague Dawley; AT1R, angiotensin II type 1 receptor, NF-κB; nuclear factor-κB; TNF-α, tumor necrosis factor; eNOS, endothelial nitric oxide synthase; PKC, protein kinase C; Akt or protein kinase B; Ang, angiotensin; CD, cluster of differentiation; Tie, tyrosine kinase with Ig and EGF; G-CSF, granulocyte-colony stimulating factor; VEGF2, vascular endothelial growth factor; vWF, Von Willebrand factor.
Mechanisms of delayed-tPA induced hemorrhagic transformation in ischemic stroke
The mechanisms of delayed tPA-induced HT have been associated with blood-brain barrier (BBB) breakdown, damage to microvessels and also non-thrombolytic actions of tPA [7-10]. Hence, delayed tPA-induced HT may be countered by strategies that interfere with the above-mentioned events, including pharmacological agents that target the molecules that contribute to BBB breakdown and promote vascularization (Figure 2). Here, we discuss some of the strategies examined in preclinical studies to attenuate HT due to delayed (>4.5 hours after stroke onset) tPA therapy.
Strategies to extend thrombolytic time window for ischemic stroke treatment: focus on drugs that attenuate delayed tPA-induced HT
The pharmacological agents were classified based on their putative action to preserve the BBB (e.g., minocycline, cilostazol, fasudil, candesartan, and bryostatin) and/or to enhance vascularization and protect the cerebrovasculature (e.g., coumarin derivate IMM-H004 and granulocyte colony-stimulating factor [GCSF]). A third group of drugs represents new therapeutic modalities acting through other mechanisms to improve delayed tPAassociated outcomes.
BBB protectants
One approach suggested to attenuate delayed tPA-associated HT is preserving the integrity of the BBB. Stabilizing the BBB after stroke has also been suggested to substantially improve the overall efficacy of tPA reperfusion therapy [11]. As metalloproteinases (MMPs) participate in the disruption of the BBB [7-10], many studies have targeted various MMPs. As tight junction proteins form the basic structure of the BBB [12-14], effects of therapies known to preserve endothelial tight junction proteins were also examined.
Minocycline
Minocycline is a tetracycline antibiotic clinically used for the treatment of acne vulgaris. Previous studies characterized minocycline as a potent MMP inhibitor [15]. Treatment with minocycline (3 mg/kg, intravenous [i.v.], at 4 hours post-stroke) and tPA (10 mg/kg, i.v., at 6 hours post-stroke) reduced infarction, and ameliorated the brain hemorrhage observed 24 hours after stroke [16]. The combination therapy also decreased plasma MMP-9 levels which correlated with volumes of infarction and hemorrhage [16]. As MMP-9 levels were measured in plasma and not in the brain, further studies were suggested to examine brain MMP-9 levels and correlate them with the extent of infarction and hemorrhage [16]. An exploratory trial was performed to determine safety and efficacy of minocycline when given in combination with tPA [17]. The patients were given loading dose of minocycline within 6 hours time window followed by maintenance dosing for 3 days. No cases of intracerebral hemorrhage was reported in over 60% of patients enrolled in the clinical study. It was also found that tPA-treated subjects in the minocycline trial showed lower plasma MMP-9 levels [18]. Other clinical trials in different populations have been initiated and are awaiting results [19].
Cilostazol
Cilostazol is an FDA-approved medication for the treatment of intermittent claudication [20]. In mice subjected to middle cerebral artery occlusion (MCAO) via the intraluminal filament occlusion model [14], combination therapy with tPA (10 mg/kg, i.v.) plus cilostazol (10 mg/kg, i.v.) given 6 hours post-stroke after reperfusion attenuated HT, reduced brain edema, morbidity and mortality, and neurological deficits at 18 hours and 7 days after the reperfusion. Moreover, treatment with cilostazol reduced delayed tPA-induced upregulation of MMP-9 activity and also prevented the decrease in expression of claudin-5, a molecule involved in the assembly of tight junctions between microvascular endothelial cells [21]. In vitro, cilostazol protected against tPA-induced damage on endothelial cells and pericytes by influencing cyclic AMP (cAMP) activity. Further testing of the neurovascular protective effects of the drug at longer time periods post-stroke has been suggested along with understanding the mechanism by which cilostazol may interact with tPA.
GM6001
GM6001 is a broad-spectrum MMP inhibitor which is capable of binding to the active sites of MMPs and limits the conversion of pro-MMPs to active forms of matrix-degrading MMPs [22]. In a mouse MCAO model, treatment with GM6001 (100 mg/kg, intraperitoneal [i.p.]) and tPA (10 mg/kg, i.v.) at 6 hours poststroke, resulted in reduced tPA-elevated brain hemoglobin indicating its ability to reduce HT [23]. Immunoblots showed that the combination treatment reduced tPA-elevated MMP-9 at 42 hours after the reperfusion, and the degradation of occludin and ZO-1 induced by tPA but not claudin-5 expression [23]. Treatment with GM6001 also significantly improved survival rate and the reduction in locomotor activity at 7 days after ischemia/reperfusion. In vitro, GM6001 prevented tPA-induced damage in endothelial cells and decrease in transendothelial electrical resistance. Other potential mechanisms of the drug need to be explored, such as its influence on tumor necrosis factor-α (TNF-α) converting enzyme (TACE) expression [23], as GM6001 also inhibited TACE, and elevated TNF-α level has been found to correlate with intracerebral hemorrhage in animal models [24].
Fasudil
Fasudil is a Rho kinase inhibitor initially characterized as an intracellular calcium antagonist, and marketed in Japan for the treatment of cerebral vasospasms occurring after subarachnoid hemorrhage [25]. Compared with mice subjected to 6-h MCAO and treated with tPA (10 mg/kg, i.v.) alone, tPA plus fasudil (3 mg/kg, i.p.)-treated subjects showed attenuated HT at 18 hours post-reperfusion. These mice however, did not show reduced infarct volumes when compared with controls and tPA-alone treatment group. Nevertheless, the combination treatment group significantly showed decreased mortality and increased locomotor activity at 7 days after the reperfusion. In vitro, tPA treatment with fasudil prevented injury to human brain microvascular endothelial cells (HBMECs) via reduction of MMP-9 activity [13]. Moreover, lactate dehydrogenase assays also showed that fasudil prevented tPA-induced damage by protecting the endothelial cells. Thus, fasudil counteracted not only acute, but also subacute tPA-induced cerebral injury in the mice with HT and also protected endothelial cells from damage [13]. Further studies have been advised to determine long-term neurovascular protective effects of fasudil, the exact molecular mechanisms in delayed tPA-induced HT and also the optimum dose of the drug when combined with tPA [13].
Candesartan
Candesartan is an angiotensin II type 1 receptor blocker shown to reduce injury due to ischemic stroke [26,27]. In an animal model of embolic MCAO, early treatment with candesartan (1 mg/kg, at 3 hours after stroke onset) and tPA (10 mg/kg, i.v.) at 6 hours after stroke, reduced brain hemorrhage and improved neurological outcomes [27]. The combination therapy, however, increased MMP-9 although it decreased MMP-3 levels. In light of the above findings and the observation that the intracranial bleeding after tPA treatment in stroked mice was attenuated in MMP-3 null, but not MMP-9 null mice compared to wild-type controls [28], the authors of this study suggested that activation of MMP-9 alone is insufficient to cause increased hemorrhage in embolic stroke. Furthermore, candesartan and tPA treatment decreased expression levels of nuclear factor kappa-B, which has previously been shown to mediate MMP-3 expression in endothelial cells after tPA treatment and also to decrease TNF-α expression following activation of nuclear factor kappa-B. Candesartan treatment also enhanced activation of endothelial nitric oxide synthase which is crucial for vascular function and homeostasis, and is considered neuroprotective after stroke [29].
Bryostatin
Bryostatin is a potent protein kinase C (PKC) modulator which reduced ischemic brain injury in aged-female rats when administered at 6 hours following MCAO [30]. In aged (18- to 20-month-old) female rats subjected to MCAO via injection of autologous blood clots, bryostatin (2.5 mg/kg, i.v., given 2 hours post-MCAO) reduced the delayed tPA (5 mg/kg, i.v.)-induced cerebral swelling, hemorrhage as well as mortality at 24 hours post-MCAO [31]. Treatment with bryostatin also reduced BBB disruption and HT, and downregulated MMP-9 expression while upregulating PKCɛ expression. The study’s authors suggested that the bryostatinmediated decrease in MMP-9 may account for outcome improvement post-stroke. Upregulation of PKCɛ by the drug may confer decreased damage to tight junctions within the BBB and attenuate HT [31]. PKCɛ regulation of MMP-9 [32] may also play a role in the beneficial effect of bryostatin to reduce delayed tPA-induced hemorrhage and BBB disruption. As the study was conducted in female animals, experiments on male subjects were suggested [31].
Promoters of vascularization
Aside from restoring BBB integrity, enhancement of neovascularization or blood vessel formation may also prove beneficial in reversing delayed tPA-induced HT. Vascular disruption is the main cause of intracerebral hemorrhage and BBB dysfunction is a secondary consequence [33]. Furthermore, studies showed that angiogenesis emerges in the ischemic region after vascular occlusion and contributes to improvements following infarction, blood flow and neuronal recovery [34].
Coumarin derivative IMM-H004
Coumarin compounds are an important class of organic heterocyclic compounds with multiple biological activities [35]. Combination therapy with tPA (10 mg/kg, i.v., 6 hours post-stroke) and IMM-H004 (6 mg/kg, i.v.) reduced hemorrhage, ischemic infarction and cerebral edema in rats subjected to embolic stroke [36]. The combination therapy also inhibited tPA-mediated HT and exaggeration of infarct volume in rats subjected to stroke using the intraluminal filament method [36]. IMM-H004 also exerted a protective role by decreasing MMP-9/MMP-2, the co-localization of MMP-2 with astrocytes and IgG leakage, and also increased levels of occludin indicating enhancement of BBB integrity. At 7 days post-stroke, IMM-H004 promoted vascularization and improved cerebral perfusion by improving the integrity of vascular endothelial cells. In vitro, IMM-H004 increased levels of ATP and the protein kinase A (PKA)- and PI3K-dependent activation of Akt in HBMECs and PC12 cells, suggesting the involvement of cAMP/PKA and PI3K/Akt signaling [36]. The authors concluded that IMM-H004 may have reduced delayed tPA-induced HT not only by preserving the integrity of the BBB but also by enhancing neurovascularization [36].
G-CSF
G-CSF is a key member of the hematopoietic growth factor family, which regulates the survival, proliferation, and differentiation of hematopoietic stem cells/hematopoietic progenitor cells [37]. We showed that G-CSF (300 µg/kg, i.v.) prevented delayed (6 hours post-MCAO) tPA (10 mg/kg, i.v.)-induced HT as evidenced by reduced hemoglobin content in the brains of rats subjected to the combination therapy compared with rats administered with tPA only [38]. G-CSF plus tPA treatment also increased levels of angiogenesis marker Ang-2 but not Ang-1, vasculogenesis marker vWF, phosphorylated-eNOS, and endothelial progenitor cell markers CD34+ and VEGFR-2 in the ischemic hemispheres of stroked rats compared with tPA treatment alone. Moreover, in rats subjected to the combination therapy, improvement in neurological outcomes at 24 hours post-drug treatment was observed. Thus, G-CSF reduces delayed tPA-induced HT and consequently improves neurological improvement post stroke via angiogenic and vasculogenic activities of G-CSF and/or proliferative or regenerative actions of G-CSF-recruited endothelial progenitor cells [38]. While vascularization may require several days to be completed, drugs that enhance vascularization in stroke, such as those that promote vasculogenesis and angiogenesis, may accelerate the process and allow preservation of a patent vasculature against tPA-induced HT. Since evaluation of G-CSF efficacy was limited to acute time points post-stroke, G-CSF effects on cerebrovascular protection at longer periods of recovery remain unknown. Finding the optimum dosage of G-CSF, and testing the effects of the combination therapy in other experimental stroke models is suggested in order to enhance future clinical application of this approach. We are currently addressing some of the above-mentioned issues in keeping with our goal to expedite the development of G-CSF as a therapeutic agent to attenuate delayed tPA-induced HT. Of note, in a clinical study investigating whether growth factors such as VEGF, Ang-1 and G-CSF can enhance arterial recanalization and improve clinical outcomes in acute ischemic stroke patients treated with tPA, it was found that all three growth factors enhanced recanalization [39]. Interestingly, Ang-1 but not VEGF or G-CSF enhanced HT. Moreover, high serum levels of G-CSF was associated with good functional outcomes even at 90 days post treatment [39]. These findings recapitulate the potential of G-CSF to reduce the HT associated with delayed tPA treatment.
Emerging strategies
As delayed tPA treatment has been associated with accumulation of free radicals [40], the effects of antioxidants have also been investigated. Moreover, oxygen transporters, which held promise in preclinical stroke studies have also been recently investigated for their ability to expand therapeutic window of tPA. While the following interventions have been shown to improve stroke volumes, their impact on HT are still undetermined.
Ascorbic Acid
Considering the decrease in glutathione and ascorbic acid levels coupled with increase in free radical formation after ischemic brain injury, ascorbic acid supplementation has been proposed to improve outcomes after ischemic brain injury [40]. The effects of vitamin C, an ascorbic acid that could neutralize free radicals especially in brain parenchyma and protect endothelial function against ischemic oxidative injury in diabetes, on delayed tPA-induced adverse effects have been evaluated [41]. Rats subjected to permanent MCAO and treated with low dose tPA (1 mg/kg) and vitamin C (500 mg/kg) at 5 hours after stroke showed reduction in infarct volume and edema at 48 hours post-stroke compared with rats given tPA only. ascorbic acid treatment in conjunction with tPA also reduced delayed tPA-induced increase in MMP-9 levels and counteracted delayed tPA-induced BBB disruption. Oxidative stress is an early trigger for the upregulation of MMP-9 which, in turn, facilitates BBB damage after ischemia-reperfusion [41]. The efficacy of vitamin C to attenuate adverse effects and exert neuroprotection indicates its potential as an adjuvant therapeutic approach to expand therapeutic window of tPA treatment.
Dodecafluoropentane emulsion nanodroplets
Dodecafluoropentane emulsion nanodroplets are oxygen transporters shown to protect ischemic brains in stroke models. The efficacy of dodecafluoropentane emulsion (0.3 mL/kg) to extend the time window for tPA treatment (0.9 mg/kg, given 9 hours after stroke) was examined in rabbits subjected to embolic stroke procedures [42]. At 24 hours post-stroke, the combination treatment improved neurological scores and stroke volumes. The authors did not examine the underlying mechanisms of the combined treatment but suggested the involvement of improved oxygen transport without the need to red blood cell flow [42].
Summary and perspectives
The findings of this study indicate that very little work has been done to explore potential strategies to reduce complications (e.g., HT) of delayed tPA treatment. It is also noteworthy, that while all drugs reviewed in this work unequivocally reduced delayed tPA-induced HT, a majority of them did not exert neuroprotection when given alongside tPA. In theory, the most ideal adjunct treatment should not only reduce the risk for hemorrhage but also exert neuroprotective effects.
Some variances in experimental conditions across studies deserve further attention. For instance, the use of different stroke models may provide different information on drug efficacy based on the model’s stability or translational value. While the intraluminal suture technique is widely used, clinically relevant findings can be obtained by testing drug effects in the embolic stroke model. To this end, it is crucial to examine efficacy of the drug across different stroke models, also in line with the STAIR recommendations for effective translational research [43]. In this regard, the same STAIR recommendations [44] for developing potential stroke therapies (e.g., testing efficacy in two or more laboratories and in animal models carrying stroke comorbidities, replication in a second species, and consideration of sex difference) may also be followed in the development of drugs to reduce tPA-induced complications. Notably, the STAIR guidelines have already suggested strategies to maximize thrombolytic efficacy of tPA with the use of intra-arterial and adjunctive neuroprotective therapies [44].
The different doses of tPA used, and timing of administration (before or after reperfusion, post MCAO) may also produce outcomes in experimental investigations that are difficult to compare. Lower tPA doses (1 or 5 mg/kg) may produce lesser adverse effects but as the thrombolytic efficacy of the drug may be altered, the higher tPA dose (10 mg/kg), which is also assumed to be clinically relevant [41], has been employed in many studies. Nevertheless, the 10 mg/kg dose used in animal studies is 10 times higher the dose used in humans (there are fibrin specificity differences between rodents and humans) [45], entailing caution when interpreting results of studies using tPA at such dose. However, some studies showed no difference in fibrinolytic efficacy of tPA at high or low doses [46-48] which begs the question of whether it is more practical to use a lower tPA dose in conjunction with a drug when given beyond the therapeutic window. With regard to the timing of tPA administration in preclinical studies, it is critical to consider a timepoint which closely mimics the clinical scenario of delayed tPA treatment. The 6 hours post-stroke timepoint is an important advance in modeling delayed tPA treatment post-stroke, and the field continues to move forward with current studies which specify delayed tPA treatment as >6 hours after stroke induction [42]. Furthermore, when using the intraluminal filament method for stroke induction, it is ideal that tPA be given before reperfusion to facilitate examination of tPA-associated damage under reperfusion conditions similar to those observed clinically. In the clinical setting, if spontaneous reperfusion is achieved, patients are usually not eligible to receive tPA.
In all studies reviewed, the most common recommendation is to examine the drug’s long-term efficacy, which is a prudent course to take to evaluate further the worth of the drug not only in terms of attenuating tPA-induced complications but also in producing long-term outcome improvements. Accordingly, neurological assessments should also include motor behavior functions examined not only a few days, but even months after drug administration (see STAIR guidelines [43,44]). Moreover, finding the optimal dosage and the right time point of drug administration in relation to tPA is key to enhance possible clinical application of the combined therapy. It is important to consider that the FDA standards require assurance that the drug under investigation does not interfere with the fibrinolytic activity of tPA [43].
While most of the research has focused on BBB preservation to reduce delayed tPA-induced HT, new research is emerging targeting other mechanisms (e.g., vascularization/ vascularization coupled with BBB protection), which will advance the field further in terms of drug development and completely deciphering tPA’s role in the HT after delayed treatment. However, further research is encouraged in order to discover therapeutic approaches that will not only afford neuroprotection, but also attenuate delayed tPA-induced HT by preserving the neurovascular unit, protecting microvessels and enhancing neurovascularization. Considering the multi-pronged effects of stem cells which include the above-mentioned mechanisms [49], testing efficacy of this type of treatment may prove to be a worthwhile research endeavor. Moreover, some stem cell treatments have been shown to produce robust and stable brain and functional improvement not only during acute phases but also at later time-points following stroke onset [50].
Extending the time window for thrombolysis may not only be achieved through pharmacological means and stem cells, but also with other non-drug strategies. For instance, brain imaging has been employed to determine patient subgroups with increased risk for hemorrhage and poor clinical outcomes profile. Identifying at risk patients guided treatment decisions and significantly improved tPA’s therapeutic time window with acceptable safety [51]. Previous trials also showed that endovascular procedures, for instance, intra-arterial thrombectomy, improved stroke outcomes in patients who received intravenous thrombolysis. Thrombectomy plus thrombolysis compared with thrombolysis alone has been shown to improve functional outcomes and reduce mortality in patients with ischemic stroke [52,53].
Aside from HT attenuation, there are other significant clinical implications of expanding tPA’s time window via the combination therapy. This approach will result in improvement of the risk/benefit ratio for thrombolytic therapy and eventually, an increase the number of patients eligible for tPA treatment. An expanded treatment window will also allow more patients more complete stroke recovery [42]. Furthermore, this strategy is relevant in the case of “wake up strokes,” i.e., patients go to sleep normal and awaken with stroke symptoms, which presents a dilemma for acute stroke providers to determine whether patients qualify for tPA therapy [54]. Combining tPA with a pharmacological agent that could expand tPA’s therapeutic time window may be a logical approach in the management of patients with wake up strokes.
Notably, a recent meta-analysis involving 6,756 participants in the 9 clinical trials of intravenous alteplase versus controls showed that the increase in the occurrence of HT was similar irrespective of whether tPA was given early on or beyond the 4.5 h time window [55]. The study also found stroke severity, but not time-point of tPA treatment post-stroke increased the risk of HT. A number of information can be extracted from these results. First, these findings entail more stringent analysis on the benefits and risks of tPA treatment. With regard to improving the benefit/risk ratio, a number of steps could be undertaken, including enhancing safety and efficacy profiles of tPA, developing other thrombolytics with better safety and efficacy, and also finding the optimum dose of tPA for administration to patients. Secondly, these findings may potentially revise the widely accepted concept that early tPA treatment does not lead to HT. Moreover, in light of the role that stroke severity plays in HT, interventions that could mitigate severity of stroke may be given with tPA, thus recapitulating the importance of combination therapy to augment tPA’s safety and therapeutic efficacy. The combination therapy, pertaining to drug or non-drug approaches and tPA, should exert neuroprotection and accelerate the salvage of brain tissue after stroke. A recent clinical study showed that uric acid therapy repressed the early ischemic worsening (EIW) after stroke in patients given tPA [56]. While this study exemplifies beneficial effect of combination therapy to reduce stroke severity, it also emphasizes the importance of including outcome measures in clinical trials that sensitively predicts clinical efficacy of treatments and long-term outcomes [56,57]. Nevertheless, as it has been reported that there were more stroke patients with improved outcomes than those that died from hemorrhage after tPA treatment initiated within 4.5 h post-stroke, early tPA treatment is still vital in stroke management, especially in patients with severe stroke [55].
Conclusions
In comparison with drug development for acute ischemic stroke, research on strategies to expand therapeutic time window and reduce tPA-induced complications is sparse. While waiting for the next breakthrough drug in acute stroke therapy, it is equally important to allocate considerable effort to find approaches to address the limitations of the only FDA-approved stroke therapy. As mentioned in this review, combining tPA with other drugs may attenuate the HT associated with delayed tPA treatment, thus, expanding time window of tPA treatment. Nevertheless, current efforts are also made towards identifying other fibrinolytics or thrombolytic drugs with better reperfusion efficacy than tPA [58,59].
Because of the extended therapeutic time window of 4.5 hours for tPA treatment, only a few preclinical studies were included in this review, although some other drugs have also been shown to reduce the HT after (i.e., at <4.5 hours after stroke) tPA treatment (e.g., annexin A2, fingolimod, progesterone, and uric acid) [9,10]. Of note, erythropoietin, which showed promise as a vascular protective agent has been shown to increase the incidence of HT [60], indicating the need for rigorous preclinical studies to identify potential detrimental interactions of treatments prior to conducting clinical trials [61]. In addition to finding other adjunctive treatments to tPA to reduce complications associated with delayed treatment of the drug, evaluating further the efficacy (both short and long-term) and mechanism of action of the above-mentioned strategies is encouraged. It is also imperative that we completely understand the exact mechanisms of tPA-induced HT (e.g., tPA’s effect on various MMPs) and the primary substrates of tPA in the brain, so we can identify the best targets and speculate drug interactions which could potentiate the benefits of thrombolytic therapy.
Footnotes
This research is supported by the American Heart Association (16POST27520023) and the Loma Linda University School of Pharmacy (LLUSP-360033).
The authors have no financial conflicts of interest.
REFERENCES
- 1.Albers GW. Expanding the window for thrombolytic therapy in acute stroke. The potential role of acute MRI for patient selection. Stroke. 1999;30:2230–2237. doi: 10.1161/01.str.30.10.2230. [DOI] [PubMed] [Google Scholar]
- 2.Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 3.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.NINDS rt-PA Stroke Study Group Intracerebral hemorrhage after intravenous tPA therapy for ischemic stroke. Stroke. 1997;28:2109–2118. doi: 10.1161/01.str.28.11.2109. [DOI] [PubMed] [Google Scholar]
- 5.Greenberg DA. Neurogenesis and stroke. CNS Neurol Disord Drug Targets. 2007;6:321–325. doi: 10.2174/187152707783220901. [DOI] [PubMed] [Google Scholar]
- 6.Adams HP, Jr, Adams RJ, Brott T, del Zoppo GJ, Furlan A, Goldstein LB, et al. Guidelines for the early management of patients with ischemic stroke: a scientific statement from the Stroke Council of the American Stroke Association. Stroke. 2003;34:1056–1083. doi: 10.1161/01.STR.0000064841.47697.22. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Tsuji K, Lee SR, Ning M, Furie KL, Buchan AM, et al. Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke. 2004;35:2726–2730. doi: 10.1161/01.STR.0000143219.16695.af. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Li M, Chen Q, Wang J. Hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke: mechanisms, models, and biomarkers. Mol Neurobiol. 2015;52:1572. doi: 10.1007/s12035-014-8952-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jickling GC, Liu DZ, Stamova B, Ander BP, Zhan X, Lu A, et al. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34:185–199. doi: 10.1038/jcbfm.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lapchak PA. Hemorrhagic transformation following ischemic stroke: significance, causes, and relationship to therapy and treatment. Curr Neurol Neurosci Rep. 2002;2:38–43. doi: 10.1007/s11910-002-0051-0. [DOI] [PubMed] [Google Scholar]
- 11.Won S, Lee JH, Wali B, Stein DG, Sayeed I. Progesterone attenuates hemorrhagic transformation after delayed tPA treatment in an experimental model of stroke in rats: involvement of the VEGF-MMP pathway. J Cereb Blood Flow Metab. 2014;34:72–80. doi: 10.1038/jcbfm.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ballabh P, Braun A, Nedergaard M. The blood–brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Ishiguro M, Kawasaki K, Suzuki Y, Ishizuka F, Mishiro K, Egashira Y, et al. A Rho kinase (ROCK) inhibitor, fasudil, prevents matrix metalloproteinase-9-related hemorrhagic transformation in mice treated with tissue plasminogen activator. Neuroscience. 2012;220:302–312. doi: 10.1016/j.neuroscience.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 14.Ishiguro M, Mishiro K, Fujiwara Y, Chen H, Izuta H, Tsuruma K, et al. Phosphodiesterase-III inhibitor prevents hemorrhagic transformation induced by focal cerebral ischemia in mice treated with tPA. PLoS One. 2010;5:e15178. doi: 10.1371/journal.pone.0015178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Machado LS, Kozak A, Ergul A, Hess D, Borlongan CV, Fagan SC. Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC Neurosci. 2006;7:56. doi: 10.1186/1471-2202-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murata Y, Rosell A, Scannevin RH, Rhodes KJ, Wang X, Lo EH. Extension of the thrombolytic time window with minocycline in experimental stroke. Stroke. 2008;39:3372–3377. doi: 10.1161/STROKEAHA.108.514026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke. 2010;41:2283–2287. doi: 10.1161/STROKEAHA.110.582601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Switzer JA, Hess DC, Ergul A, Waller JL, Machado LS, Portik-Dobos V, et al. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke. 2011;42:2633–2635. doi: 10.1161/STROKEAHA.111.618215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blacker DJ, Prentice D, Alvaro A, Bates TR, Bynevelt M, Kelly A, et al. Reducing haemorrhagic transformation after thrombolysis for stroke: a strategy utilising minocycline. Stroke Res Treat. 2013;2013:362961. doi: 10.1155/2013/362961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumoto M. Cilostazol in secondary prevention of stroke: impact of the cilostazol stroke prevention study. Atheroscler Suppl. 2005;6:33–40. doi: 10.1016/j.atherosclerosissup.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Koto T, Takubo K, Ishida S, Shinoda H, Inoue M, Tsubota K, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol. 2007;170:1389–1397. doi: 10.2353/ajpath.2007.060693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao JL, Nagano T, Nakamura M, Kumagai N, Mishima H, Nishida T. Galardin inhibits collagen degradation by rabbit keratocytes by inhibiting the activation of pro-matrix metalloproteinases. Exp Eye Res. 1999;68:565–572. doi: 10.1006/exer.1998.0637. [DOI] [PubMed] [Google Scholar]
- 23.Mishiro K, Ishiguro M, Suzuki Y, Tsuruma K, Shimazawa M, Hara H. A broad-spectrum matrix metalloproteinase inhibitor prevents hemorrhagic complications induced by tissue plasminogen activator in mice. Neuroscience. 2012;205:39–48. doi: 10.1016/j.neuroscience.2011.12.042. [DOI] [PubMed] [Google Scholar]
- 24.Mayne M, Ni W, Yan HJ, Xue M, Johnston JB, Del Bigio MR, et al. Antisense oligodeoxynucleotide inhibition of tumor necrosis factor-alpha expression is neuroprotective after intracerebral hemorrhage. Stroke. 2001;32:240–248. doi: 10.1161/01.str.32.1.240. [DOI] [PubMed] [Google Scholar]
- 25.Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, et al. Effect of AT877 on cerebral vasospasm after aneurismal subarachnoid hemorrhage. J Neurosurg. 1992;76:571–577. doi: 10.3171/jns.1992.76.4.0571. [DOI] [PubMed] [Google Scholar]
- 26.Fu H, Hosomi N, Pelisch N, Nakano D, Liu G, Ueno M, et al. Therapeutic effects of postischemic treatment with hypotensive doses of an angiotensin II receptor blocker on transient focal cerebral ischemia. J Hypertens. 2011;29:2210–2219. doi: 10.1097/HJH.0b013e32834bbb30. [DOI] [PubMed] [Google Scholar]
- 27.Ishrat T, Pillai B, Ergul A, Hafez S, Fagan SC. Candesartan reduces the hemorrhage associated with delayed tissue plasminogen activator treatment in rat embolic stroke. Neurochem Res. 2013;38:2668–2677. doi: 10.1007/s11064-013-1185-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki Y, Nagai N, Umemura K, Collen D, Lijnen HR. Stromelysin-1 (MMP-3) is critical for intracranial bleeding after t-PA treatment of stroke in mice. J Thromb Haemost. 2007;5:1732–1739. doi: 10.1111/j.1538-7836.2007.02628.x. [DOI] [PubMed] [Google Scholar]
- 29.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 30.Tan Z, Turner RC, Leon RL, Li X, Hongpaisan J, Zheng W, et al. Bryostatin improves survival and reduces ischemic brain injury in aged rats after acute ischemic stroke. Stroke. 2013;44:3490–3497. doi: 10.1161/STROKEAHA.113.002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan Z, Lucke-Wold BP, Logsdon AF, Turner RC, Tan C, Li X, et al. Bryostatin extends tPA time window to 6 h following middle cerebral artery occlusion in aged female rats. Eur J Pharmacol. 2015;764:404–412. doi: 10.1016/j.ejphar.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding Q, Chai H, Mahmood N, Tsao J, Mochly-Rosen D, Zhou W. Matrix metalloproteinases modulated by protein kinase Cepsilon mediate resistin-induced migration of human coronary artery smooth muscle cells. J Vasc Surg. 2011;53:1044–1051. doi: 10.1016/j.jvs.2010.10.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keep RF, Zhou N, Xiang J, Andjelkovic AV, Hua Y, Xi G. Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS. 2014;11:18. doi: 10.1186/2045-8118-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meenakshisundaram T, Jose AF, Steven ML, John HG, Berislav VZ. Activated protein C promotes neovascularization and neuro-genesis in post-ischemic brain via protease activated receptor 1. J Neurosci. 2008;28:12788–12797. doi: 10.1523/JNEUROSCI.3485-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fylaktakidou KC, Hadjipaclou-Litina DJ, Litinas KE, Nicolaides DN. Natural and synthetic coumarin deriv58atives with antiinflammatory/antioxidant activities. Curr Pharm Design. 2004;30:3813–3833. doi: 10.2174/1381612043382710. [DOI] [PubMed] [Google Scholar]
- 36.Zuo W, Chen J, Zhang S, Tang J, Liu H, Zhang D, et al. IMMH004 prevents toxicity induced by delayed treatment of tPA in a rat model of focal cerebral ischemia involving PKA-and PI3K-dependent Akt activation. Eur J Neurosci. 2014;39:2107–2118. doi: 10.1111/ejn.12551. [DOI] [PubMed] [Google Scholar]
- 37.Hartung T. Anti-inflammatory effects of granulocyte colony-stimulating factor. Curr Opin Hematol. 1998;5:221–225. doi: 10.1097/00062752-199805000-00013. [DOI] [PubMed] [Google Scholar]
- 38.dela Peña IC, Yoo A, Tajiri N, Acosta SA, Ji X, Kaneko Y, et al. Granulocyte colony-stimulating factor attenuates delayed tPA-induced hemorrhagic transformation in ischemic stroke rats by enhancing angiogenesis and vasculogenesis. J Cereb Blood Flow Metab. 2015;35:338–346. doi: 10.1038/jcbfm.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sobrino T, Millán M, Castellanos M, Blanco M, Brea D, Dorado L, et al. Association of growth factors with arterial recanalization and clinical outcome in patients with ischemic stroke treated with tPA. J Thromb Haemost. 2010;8:1567–1574. doi: 10.1111/j.1538-7836.2010.03897.x. [DOI] [PubMed] [Google Scholar]
- 40.Ullegaddi R, Powers HJ, Gariballa SE. Antioxidant supplementation with or without B-group vitamins after acute ischemic stroke: a randomized controlled trial. JPEN J Parenter Enteral Nutr. 2006;30:108–114. doi: 10.1177/0148607106030002108. [DOI] [PubMed] [Google Scholar]
- 41.Allahtavakoli M, Amin F, Esmaeeli-Nadimi A, Shamsizadeh A, Kazemi-Arababadi M, Kennedy D. Ascorbic acid reduces the adverse effects of delayed administration of tissue plasminogen activator in a rat stroke model. Basic Clin Pharmacol Toxicol. 2015;117:335–339. doi: 10.1111/bcpt.12413. [DOI] [PubMed] [Google Scholar]
- 42.Culp WC, Brown AT, Lowery JD, Arthur MC, Roberson PK, Skinner RD. Dodecafluoropentane emulsion extends window for tPA therapy in a rabbit stroke model. Mol Neurobiol. 2015;52:979–984. doi: 10.1007/s12035-015-9243-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Albers GW, Goldstein LB, Hess DC, Wechsler LR, Furie KL, Gorelick PB, et al. Stroke Treatment Academic Industry Roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra-arterial and neuroprotective therapies. Stroke. 2011;42:2645–2650. doi: 10.1161/STROKEAHA.111.618850. [DOI] [PubMed] [Google Scholar]
- 44.Lapchak PA, Zhang JH, Noble-Haeusslein LJ. RIGOR guidelines: escalating STAIR and STEPS for effective translational research. Transl Stroke Res. 2013;4:279–285. doi: 10.1007/s12975-012-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korninger C, Collen D. Studies on the specific fibrinolytic effect of human extrinsic (tissue-type) plasminogen activator in human blood and in various animal species in vitro. Thromb Haemost. 1981;46:561–565. [PubMed] [Google Scholar]
- 46.Haelewyn B, Risso JJ, Abraini JH. Human recombinant tissueplasminogen activator (alteplase): why not use the ‘human’ dose for stroke studies in rats? J Cereb Blood Flow Metab. 2010;30:900–903. doi: 10.1038/jcbfm.2010.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang Y, Fan X, Yu Z, Cheng C, Wang XS, Lo EH, et al. Low dose tPA plus annexin A2 combination attenuates tPA delayed treatment-associated hemorrhage and improves recovery in rat embolic focal stroke. Neurosci Lett. 2015;602:73–78. doi: 10.1016/j.neulet.2015.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu H, Fan X, Yu Z, Liu J, Murata Y, Lu J, et al. Annexin A2 combined with low-dose tPA improves thrombolytic therapy in a rat model of focal embolic stroke. J Cereb Blood Flow Metab. 2010;30:1137–1146. doi: 10.1038/jcbfm.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borlongan CV. Bone marrow stem cell mobilization in stroke: a ‘bonehead’ may be good after all! Leukemia. 2011;25:1674–1686. doi: 10.1038/leu.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.dela Peña I, Antoine A, Reyes S, Hernandez D, Acosta S, Pabon M, et al. Stem cell-based neuroprotective strategies in stroke. In: Shetty AK, editor. Neural Stem Cells in Health and Disease. Singapore: World Scientific Publishing Co.; 2015. pp. 371–408. [Google Scholar]
- 51.Bentley P, Ganesalingam J, Carlton Jones AL, Mahady K, Epton S, Rinne P, et al. Prediction of stroke thrombolysis outcome using CT brain machine learning. Neuroimage Clin. 2014;4:635–640. doi: 10.1016/j.nicl.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berkhemer OA, Fransen PSS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2014;372:1009–1018. doi: 10.1056/NEJMoa1411587. [DOI] [PubMed] [Google Scholar]
- 53.Minnerup J, Wersching H, Teuber A, Wellmann J, Eyding J, Weber R, et al. Outcome after thrombectomy and intravenous thrombolysis in patients with acute ischemic stroke: a prospective observational study. Stroke. 2016;47:1584–1592. doi: 10.1161/STROKEAHA.116.012619. [DOI] [PubMed] [Google Scholar]
- 54.Rubin MN, Barrett KM. What to do with wake-up stroke. Neurohospitalist. 2015;5:161–172. doi: 10.1177/1941874415576204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whiteley WN, Emberson J, Lees KR, Blackwell L, Albers G, Bluhmki E, et al. Risk of intracerebral haemorrhage with alteplase after acute ischaemic stroke: a secondary analysis of an individual patient data meta-analysis. Lancet Neurol. 2016;15:925–933. doi: 10.1016/S1474-4422(16)30076-X. [DOI] [PubMed] [Google Scholar]
- 56.Amaro S, Laredo C, Renú A, Llull L, Rudilosso S, Obach V, et al. Uric acid therapy prevents early ischemic stroke progression: a tertiary analysis of the URICO-ICTUS trial (efficacy study of combined treatment with uric acid and r-tPA in acute ischemic stroke) Stroke. 2016;47:2874–2876. doi: 10.1161/STROKEAHA.116.014672. [DOI] [PubMed] [Google Scholar]
- 57.Toni D, Fiorelli M, Gentile M, Bastianello S, Sacchetti ML, Argentino C, et al. Progressing neurological deficit secondary to acute ischemic stroke. A study on predictability, pathogenesis, and prognosis. Arch Neurol. 1995;52:670–675. doi: 10.1001/archneur.1995.00540310040014. [DOI] [PubMed] [Google Scholar]
- 58.Parsons M, Spratt N, Bivard A, Campbell B, Chung K, Miteff F, et al. A randomized trial of tenecteplase versus alteplase for acute ischemic stroke. N Engl J Med. 2012;366:1099–1107. doi: 10.1056/NEJMoa1109842. [DOI] [PubMed] [Google Scholar]
- 59.Henninger N, Fisher M. Extending the time window for endovascular and pharmacological reperfusion. Transl Stroke Res. 2016;7:284–293. doi: 10.1007/s12975-015-0444-4. [DOI] [PubMed] [Google Scholar]
- 60.Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40:e647–e656. doi: 10.1161/STROKEAHA.109.564872. [DOI] [PubMed] [Google Scholar]
- 61.Ishrat T, Soliman S, Guan W, Saler M, Fagan SC. Vascular protection to increase the safety of tissue plasminogen activator for stroke. Curr Pharm Des. 2012;18:3677–3684. doi: 10.2174/138161212802002779. [DOI] [PMC free article] [PubMed] [Google Scholar]