

Abstract
Arthrogryposis, renal dysfunction and cholestasis syndrome (ARC) is a multisystem disorder associated with abnormalities in polarized liver and kidney cells. Mutations in VPS33B account for most cases of ARC. We identified mutations in VIPAR (also called C14ORF133) in individuals with ARC without VPS33B defects. We show that VIPAR forms a functional complex withVPS33B that interacts with RAB11 A. Knockdown of vipar in zebrafish resulted in biliary excretion and E-cadherin defects similar to those in individuals with ARC. Vipar- and Vps33b-deficient mouse inner medullary collecting duct (mIMDC-3) cells expressed membrane proteins abnormally and had structural and functional tight junction defects. Abnormal Ceacam5 expression was due to mis-sorting toward lysosomal degradation, but reduced E-cadherin levels were associated with transcriptional downregulation. The VPS33B-VIPAR complex thus has diverse functions in the pathways regulating apical-basolateral polarity in the liver and kidney.
Generation of distinct apical and basolateral plasma membrane domains is essential for various epithelial functions, including lumen formation1–3. Cohesive epithelial layers act as physical ion-selective barriers. This barrier function is highly dependent on the integrity of apical junctional complexes (AJCs), comprising tight junctions and adherens junctions, that link adjacent epithelial cells4,5. Generation of AJCs is one of the steps in a complex mechanism of cell polarization that is dependent on multiple regulatory pathways6. Better insight into these pathways is essential for understanding the development and physiology of the many organs that rely on intact cell polarity for their function. Investigation of the molecular pathology of human diseases can shed new light on the intra-cellular pathways in mammalian cells, and many common disorders are associated with defects in apical-basolateral polarity7–10.
ARC (MIM208085) is a severe autosomal recessive multisystem disorder that affects the development and function of several organs, particularly the liver and kidneys. Germline mutations in VPS33B are found in approximately 75% of individuals with ARC11. VPS33B encodes VPS33B, the homolog of yeast class C vacuolar protein sorting (vps) protein Vps33p, from the Sec1-Munc18 family of proteins regulating SNARE-mediated membrane fusion. Class C vps proteins participate in at least two multiprotein complexes, class C core vacuole/endosome tethering (CORVET) and homotypic fusion and vacuole protein sorting (HOPS), which interact with yeast orthologs of Rab5 and Rab7 GTPases and are crucial for several steps in vesicular trafficking pathways12–15. Recent studies suggested that mammalian homologs of the yeast constituents of HOPS can be localized to early and late endosomes and lysosomes and regulate a number of intracellular processes16–18. Whereas the Vps33a homolog of yeast Vps33p forms part of the mammalian HOPS complex, the pathway involving Vps33b remains unknown, although interaction with other class C vps protein homologs has been proposed16,19,20. To elucidate the molecular basis of ARC and gain insights into the role of VPS33B in epithelial function, we investigated individuals with ARC and characterized cellular and zebrafish models of the disease.
We identified mutations in C14orf133 (here named VIPAR), which encodes the VPS33B-interacting protein VIPAR (VPS33B-interacting protein involved in polarity and apical protein restriction) in individuals with an ARC phenotype. Our data suggest that VPS33B and VIPAR function as part of a complex that interacts with an active form of RAB11A involved in apical membrane protein sorting. Rab11a also regulates adherens junction expression of E-cadherin in Drosophila and polarized-cell models21,22. We found abnormal expression of E-cadherin and the apical membrane protein CEACAM5 (carcinoembryonic antigen; CEA, CD66e) in liver samples from individuals with ARC and in mouse inner medullary collecting duct (mIMCD-3) cells with stable knockdown of Vps33b and Vipar. Although mis-sorting of Ceacam5 into the lysosomal degradation pathway underlies reduced levels of endogenous Ceacam5, low E-cadherin levels were associated with E-cadherin transcriptional downregulation.
We suggest that the VPS33B-VIPAR complex is involved in stabilization of apical membrane protein content, possibly via the RAB11A-dependent apical recycling pathway, and in transcriptional regulation of E-cadherin, either directly or indirectly. Consequences of disordered apical protein restriction in ARC include mis-sorting of some apical proteins to basolateral membrane and into late endosomes and lysosomes, resulting in cholestasis and in urinary wasting of sugars and amino acids. Reduced expression of E-cadherin underlies disordered formation of the AJCs essential for generation and maintenance of lumenal structures such as bile ducts and renal tubules.
RESULTS
Mutations in VIPAR cause the ARC phenotype
To gain insight into VPS33B function and to identify new genes involved in ARC pathogenesis, we performed a yeast two-hybrid screen for VPS33B-interacting proteins. Human fetal brain and adult kidney cDNA libraries yielded 18 candidates prioritized by bioinformatic analyses of homology and putative function. A protein encoded by C14ORF133, here named VIPAR, received highest priority. A peptide-sequence BLAST search revealed similarity to VPS16 and a golgin A5 domain occupying most of the protein (amino acid residues 12–493). ClustalW alignment showed 15% identity between VPS16 and VIPAR. Individual VPS16 N- and C-terminal-domain alignments found proportionate identities of 5% and 16%, respectively (Supplementary Fig. 1). Bioinformatic analysis of the VIPAR sequence using Pfam and SMART databases found only the C-terminal VPS16 domain. Two splice variants (one bypassing exon 16) were identified, resulting in proteins 493 and 480 amino acid residues long. The larger transcript (predicted unglycosylated weight 57 kDa) was the reference sequence for DNA and protein.
Next, we used transfections of epitope-tagged constructs to confirm yeast two-hybrid data (Fig. 1a). Coimmunoprecipitation identified interaction between overexpressed VPS33B and VIPAR (and between endogenous VPS33B and overexpressed VIPAR; Fig. 1b) similar to findings recently reported elsewhere20; no significant coimmunoprecipitation was found between VPS33B and the HOPS protein VPS16 or between VIPAR and the HOPS protein VPS33A (Fig. 1a,b). When individually overexpressed in cells from the HEK293 line, VIPAR and VPS33B showed generalized cytoplasmic distribution (Fig. 1c and Supplementary Movie 1). However, overexpression of both proteins together led to their colocalization in clusters consistent with formation of VPS33B-VIPAR complexes at cytoplasmic organelles (Fig. 1d). No such colocalization was observed when VIPAR was overexpressed with VPS33A (Fig. 1d). These results collectively demonstrated the specificity of the VPS33B-VIPAR interaction.
Figure 1.
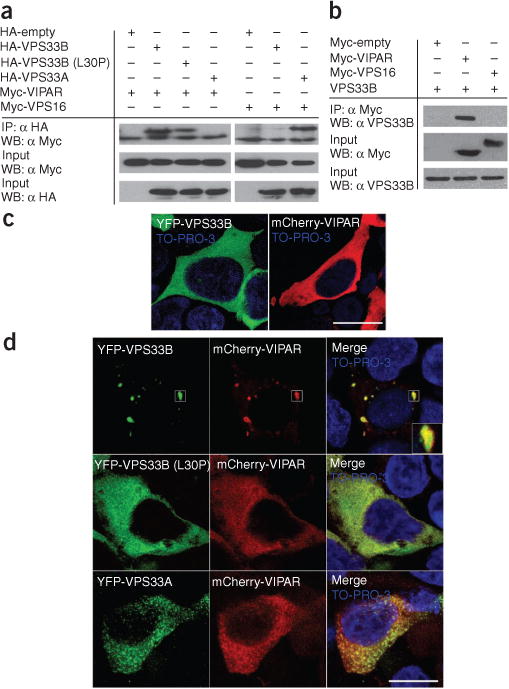
VPS33B interacts with VIPAR. (a) HEK293 cells were co-transfected with hemagglutinin (HA)-tagged VPS33B, VPS33B (L30P) mutant or VPS33A and with Myc-tagged VIPAR or VPS16 constructs. Coimmunoprecipitation experiments showed that HA-VPS33B and HA-VPS33B (L30P) interact with Myc-VIPAR and that HA-VPS33A interacts with Myc-VPS16. No interaction was seen between HA-VPS33B and Myc-VPS16 or between HA-VPS33A and Myc-VIPAR. Quantification revealed that 28.6% ± 1.5% (s.e.m.) (VPS33B), 23.8% ± 0.8% (VPS33B-L30P) and 28.2% ± 1.1% (VPS33A) of the Myc-VIPAR or Myc-VPS16 input were recovered (n = 3, ± 1 s.e.m.). (b) HEK293 cells were transfected with either Myc-VIPAR or Myc-VPS16, and a coimmunoprecipitation experiment was carried out to assess for interaction with endogenous VPS33B. Again, VPS33B interacted with Myc-VIPAR but not with Myc-VPS16. (c,d) Confocal fluorescence photomicrographs of HEK293 cells transfected with YFP-VPS33B and mCherry-VIPAR constructs individually (c) or with both mCherry-VIPAR and YFP-VPS33B, YFP-VPS33B-L30P or YFP-VPS33A (d). Nuclei are stained with TO-PRO-3. Scale bars, 15 μm. Individual overexpression of VPS33B or VIPAR demonstrated generalized cytoplasmic distribution. When both are overexpressed, mCherry-VIPAR and YFP-VPS33B form cytoplasmic clusters. No clusters were seen when mCherry-VIPAR was overexpressed with YFP-VPS33B-L30P or HA-VPS33A.
To investigate whether the VIPAR locus was directly involved in ARC pathogenesis, we analyzed DNA from consanguineous families affected by ARC without detectable mutations in VPS33B or disease linkage to the VPS33B locus. A genome-wide linkage screen using 10K and 250K Affymetrix SNP microarrays in four unrelated probands of consanguineous parentage identified homozygosity at the VIPAR locus in three probands from Turkish and Israeli Arab backgrounds (Table 1). Prompted by these findings, we sequenced the VIPAR coding exons and flanking intronic sequence in all available unrelated VPS33B mutation-negative cases (n = 7) with the classical ARC phenotype and detected a total of eight mutations (six nonsense, one frameshift and one missense, changing the start ATG codon to AGG; Table 1). We verified mutations by segregation with disease status within families and by screening of at least 200 chromosomes from ethnically matched controls for each mutation. All mutations were predicted to eliminate normal protein production because of early message termination and nonsense-mediated mRNA decay or because of translation failure (as with the first-codon mutation). The severity of the phenotypes associated with specific VIPAR mutations correlates well with data regarding VPS33B in that only one (T89C; L30P) of 31 VPS33B-proven pathogenic mutations detected so far in ARC is a missense mutation. No tested tissues from classical ARC cases with VPS33B mutations had any detectable VPS33B protein on protein blotting11. Mutations in VIPAR accounted for all cases of classical ARC in whom mutations in and linkage to VPS33B were excluded. We detected no differences in clinical symptoms, signs or disease course between subjects in VPS33B- and VIPAR-mutated ARC subsets.
Table 1.
VIPAR mutations in individuals with ARC
| Subject number | Ethnic origin | Nucleotide alteration | Peptide alteration | Status | Homozygous region sizeaa |
|---|---|---|---|---|---|
| 1b | Turkish | C535T | Q179X | Hom | 0.5 |
| 2 | Croatian | 748_752delACAGA | T250ArgfsX279 | Hom | – |
| 3b | Turkish | C658T | R220X | Het | – |
| C873T | Q291X | Het | |||
| 4 | Turkish | C1273T | Q425X | Hom | – |
| 5 | Italian | C658T | R220X | Hom | – |
| 6b | Israeli Arab | C808T | R270X | Hom | 29 |
| 7b | Turkish | T2G | M1R | Hom | 4 |
Nucleotides are numbered from the A of the ATG initiation codon. None of these mutations was identified in >200 chromosomes from healthy ethnically matched controls. The size of the homozygous region identified in the VIPAR locus is given for subjects who underwent genome-wide scans. Del, deletion; fs, frameshift; X, stop; hom, homozygous; het, heterozygous.
Size in Mb.
Used for genome-wide scan.
Liver phenotype in ARC and zebrafish vipar knockdown larvae
Nonobstructive jaundice is characteristic of ARC liver disease. As shown on radionuclide imaging of ARC-derived samples, labeled compounds normally secreted into bile do not enter the duodenum despite biliary tract patency, reflecting absent or severely reduced intrahepatic bile flow; in addition, peripheral biliary radicles are hypoplastic. Histopathologic features comprise intrahepatocyte pigment accumulation, including lipofuscin (lysosomal degradation products) and hepatocyte giant-cell transformation23,24. Because some apical membrane proteins in ARC liver samples are abnormally localized, VPS33B has been proposed to be involved in trafficking of apical membrane transporters25,26. We used liver biopsy specimens to investigate expression of several membrane proteins that traverse different apical sorting pathways. First, we confirmed that CEACAM5, mislocalized in individuals with VPS33B mutations, was also mislocalized in individuals with VIPAR deficiency (Supplementary Fig. 2). Members of the CEACAM are glycosylphosphoinositol (GPI)-anchored proteins that undergo both N- and O-glycosylation; both modifications contribute targeting signals for apical sorting27. The bile salt export pump (BSEP) is an apical membrane ATP-binding cassette transporter protein with multiple transmembrane domains. It is sorted to the apical membrane of hepatocytes from the trans-Golgi network (TGN) via RAB11A-positive apical recycling endosomes28. Mis-sorting of some BSEP to the basolateral hepatocyte membrane was seen in the livers of individuals with ARC (Supplementary Fig. 2a). We saw no change in localization (Supplementary Fig. 2a) of another multiple-transmembrane domain transporter, MRP2 (multiple drug resistance-associated protein 2), that is known to be sorted directly to the apical membrane and bypass apical recycling endosomes29,30.
Basolateral proteins were shown immunohistochemically to be normally localized in ARC liver samples. We investigated expression of the adherens junction protein E-cadherin, which is crucial for AJC formation31,32. E-cadherin, which is dependent on complexed calcium for normal function, is a cell-adhesion molecule with a functional extracellular domain that forms a part of the adherens junction adhesive belt. Immunohistochemically detectable E-cadherin was decreased in the adherens junctions of ARC-affected livers as compared to controls. We also saw cytoplasmic staining for E-cadherin; however, we did not detect mislocalization at apical membranes (Fig. 2a).
Figure 2.

Intrahepatic defects in individuals with ARC and in Vipar-deficient zebrafish larvae. (a) Immunohistochemical analysis of E-cadherin localization in an individual with ARC26 with VPS33B mutations and in control liver tissue (×400 and ×1,000 magnification; hematoxylin counterstain). Decreased amounts of E-cadherin can be seen at the apical junction complexes in the ARC sample compared with the control. Decreased E-cadherin expression was particularly obvious in hepatocytes cuffing portal tracts (arrow). (b) Lateral and ventral views of vipar in situ hybridization in 5 d post fertilization (d.p.f.) larvae using DIG-labeled antisense and sense (control) vipar probes. High expression in liver (L, outlined) and small intestine (arrow) is seen. (c) Brightfield and green fluorescence images of PED-6–treated non-injected (embryos) larvae and vipar ATG morpholino–injected 5-d.p.f. (embryos) larvae. Liver (white arrow), swimbladder (white arrowhead) and gallbladder samples (red arrowhead) are indicated. (d) A bar graph showing that the amount of PED6 detected in the gallbladder is significantly lower in ATG and exon 3 morpholino–injected larvae than in non-injected control larvae or exon 3–mismatch control larvae. Injecting vipar mRNA, but not an unrelated mRNA (RFP), after morpholino treatment rescued the phenotype (n = 90 for each treatment group (3 independent injections with 30 larvae in each clutch); error bars, ± 1 s.d., *P < 0.001 by z-test). (e) Immunostaining of 5-d.p.f. larvae livers for E-cadherin showing markedly reduced E-cadherin staining in morpholino-injected larvae compared with controls. Scale bars, 200 μm (b,c) or 10 μm (e).
Morpholino knockdown of the zebrafish vps33b ortholog resulted in abnormal biliary tract development, with reduced numbers of terminal branches of intrahepatic bile ducts and reduced excretion of bile into the gallbladder as gauged by labeling with the fluorescent compound PED6. These changes were consistent with the hepatobiliary defects identified in cases of ARC33. We tested whether the VIPAR zebrafish ortholog is also involved in biliary development and function. Tissue expression of zebrafish vipar was comparable to that of vps33b and was substantial in the liver and proximal intestine (Fig. 2b). We evaluated biliary excretion using PED6 in zebrafish larvae subjected to morpholino knockdown of the VIPAR ortholog si:ch211–20b12.1 (vipar)34. Following vipar knockdown, we saw substantial dose-dependent reductions in the volume of PED6 within the gallbladder as compared to controls (Fig. 2c). We obtained this result using two independent morpholinos (targeting the ATG and exon 3 splice site), and the phenotype was rescued with wild-type vipar mRNA (Fig. 2d). In addition, keratin immunostaining patterns 5 d after fertilization in vipar morpholino-injected larvae were abnormal (as was the case in vps33b morphants33). The images we obtained were consistent with abnormal development of terminal biliary ductules, as was seen in cases of ARC (Supplementary Fig. 3). Because AJC expression of E-cadherin, which is essential for lumenal development in epithelial organs, was decreased in ARC liver samples, we immunohistochemically studied E-cadherin expression in liver from zebrafish with morpholino vipar knockdown. Amounts of E-cadherin in the cell membrane were markedly decreased in knockdown larvae, suggesting that the vipar-vps33b complex is involved in regulation of E-cadherin expression during zebrafish development (Fig. 2e).
Vps33b or Vipar knockdown causes epithelial polarization defects
To further investigate the molecular mechanisms affected by ARC using polarized epithelial cell models, we developed clones of mIMCD-3 cells stably transfected with silencing short hairpin (sh) RNAs for Vps33b or Vipar. We achieved a 92% knockdown (detected by protein blot analysis) for Vps33b (Vps33b-shRNA) and a 90% knockdown (detected by quantitative real-time PCR) for Vipar (Vipar-shRNA, Supplementary Fig. 4). We tested expression of apical membrane proteins by immunofluorescence confocal microscopy and immunoblotting. The available antibody to mouse Ceacam5 was suitable only for protein blotting; accordingly, we used cell-surface biotinylation to analyze membrane expression of endogenous Ceacam5. The 180-kDa Ceacam5 band present in the membrane fraction of control cells was not detectable in either membrane fraction or whole-cell lysate of knockdown cells (Fig. 3a). Specific apical localization in mIMCD-3 cells of stably transfected P75 neurotrophin receptor protein, a single transmembrane domain protein that undergoes O-glycosylation35, was also altered, with distribution throughout cell membranes in both cells treated with Vipar-shRNA and cells treated with Vps33b-shRNA (Fig. 3b). Correct basolateral distribution of Na+-K+ ATPase was maintained in knockdown cells (Fig. 3b).
Figure 3.
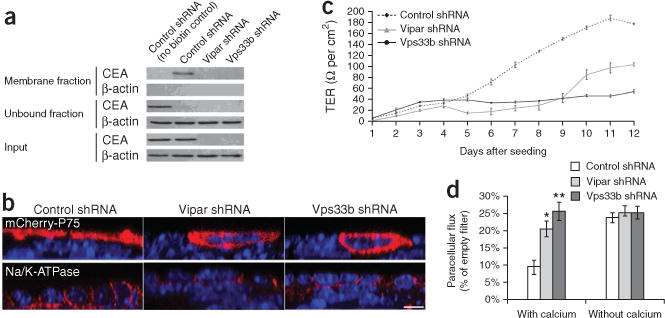
Abnormal membrane polarization of mIMCD-3 cells in Vps33b and Vipar deficiencies. (a) Protein blotting of Ceacam5 (Cea) in biotinylated membrane fraction lysates and whole-cell lysate showing severe reduction of Cea in Vps33b shRNA and Vipar shRNA cells. The unbound fraction contains the unbiotinylated or nonmembrane proteins. (b) Confocal immunofluorescence photomicrographs of cultured cells treated with shRNA (control shRNA, Vipar shRNA and Vps33b shRNA); shown are xz plane images. Loss of apical distribution can be seen for transfected P75. The basolateral protein Na+-K+ ATPase (endogenous) is correctly localized in all three cell types. Nuclei are stained with TO-PRO-3. Scale bar, 10 μm. (c) Average TER readings every 24 h after seeding control and knockdown cells onto Transwell supports (n = 4, error bars, ± 1 s.e.m.). Maximum levels of resistance reached by the knockdown cells are ~50% reduced compared with controls. (d) Paracellular flux of 4 kDa dextran fluorescein isothiocyanate conjugate, with and without calcium in the medium, for all three cell types (n = 4, error bars, ± 1 s.e.m., *P < 0.05, **P < 0.001 by a Student’s t-test). Similar paracellular flux is achieved in control cells during ‘calcium switch’ experiments.
Trans-epithelial resistance (TER) experiments tested whether monolayers of mIMCD3 cells treated with Vipar-shRNA or Vps33b-shRNA generated a barrier to ion movement (Fig. 3c). The maximum achieved TER was substantially lower in both knockdown cell lines than in control cells (50%, Vipar-shRNA treatment; 75%, Vps33b-shRNA treatment), suggesting a defect in tight junctions. As functioning tight junctions are essential to control paracellular flux of macromolecules, we used fluorescently labeled small (4 kDa) and large (70 kDa) dextran assays36 to test whether Vps33b or Vipar deficiency led to abnormal barrier function within mIMCD-3 cell monolayers. Cells treated with either Vipar shRNA or Vps33B shRNA let more than twice as much small dextran through the monolayer as did control cells. No such substantial change was detected for large dextran (data not shown). The formation of tight junctions requires calcium; therefore, to show that changes caused in cell layer permeability and polarity were analogous to physical disruption of tight junctions, we performed TER and dextran experiments in cells grown in a low-calcium medium (‘calcium switch’)37. After control mIMCD-3 cells were cultured in low-calcium medium for 24 h, TER and dextran permeability values approximated those for Vipar-shRNA– and Vps33B-shRNA–treated cells. Both TER and permeability returned to control-cell values after reintroduction of a normal-calcium medium (Fig. 3d).
We used freeze-fracture electron microscopy to assess the three-dimensional configuration of tight junctions in knockdown mIMCD-3 cells (Fig. 4a). Splitting of the layers of frozen cell membranes yielded two half-membrane leaflets whose faces were then metal-replicated and visualized by transmission electron microscopy. The leaflet adjacent to the extracellular space as observed from inside the cell is conventionally the exoplasmic face or ‘E-face’; the leaflet adjacent to the cytoplasm as observed from outside the cell is the protoplasmic face or ‘P-face’38. Compared with controls, tight junctions in both Vipar-shRNA– and Vps33b-shRNA–treated cells showed reduced complexity, disturbed P-face association and an increase of particle-free pores, corresponding to overall tight junction disruption. These findings were consistent with results of TER and dextran-flux studies of tight junction function.
Figure 4.
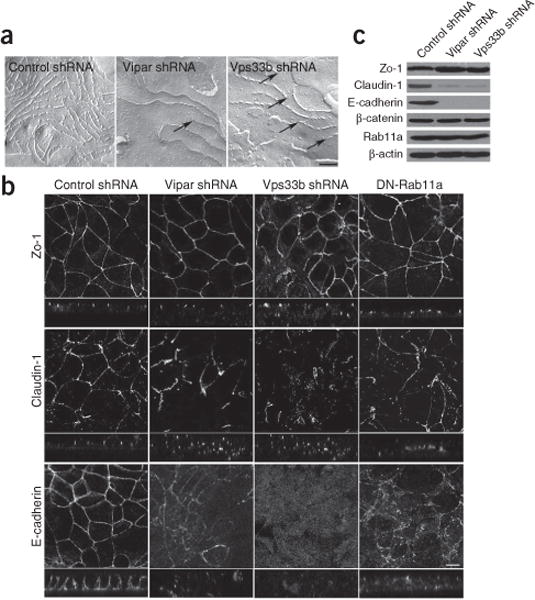
Both Vipar and Vps33b are required for apical junction complex formation. (a) Freeze-fracture images of control, Vipar knockdown and Vps33b knockdown cells showing tight junction strands (scale bar, 0.2 μm). In Vipar shRNA–treated cells and in Vps33b shRNA–treated cells, tight junctions revealed a strong decrease of tight junction strand complexity and/or a decrease of the P-face association, leading to interrupted and blindly ending strands (arrows), when compared with the control cells. Interruptions but no blind ends were seen in the control cells. This can also be seen in the E-face, where continuous networks of grooves with only rare blind ends can be seen (data not shown). (b) Confocal immunofluorescence photomicrographs in the xy and xz planes of zo-1, claudin-1 and E-cadherin in cells treated with control shRNA, Vps33b shRNA or Vipar shRNA and in cells with stable transfection of GDP-locked Rab11a mutant DN-Rab11a (scale bar, 10 μm). The knockdown cells do not grow in a monolayer with constant height, but instead grow partly on top of neighboring cells. Amounts of E-cadherin and, to a lesser extent, claudin-1 are reduced at adherens junctions and tight junctions. (c) Protein blotting shows results compatible with those from immunostaining, with decreased E-cadherin and claudin-1 levels in the knockdown cells. However, levels of another adherens junction protein, β-catenin, were the same as those in controls.
Some AJC proteins were abnormally expressed. Immunohistologic patterns of claudin-1 staining were disordered in knockdown mIMCD-3 cells, with reduced expression at tight junctions and some cytoplasmic marking (Fig. 4b). The same degree of abnormality was not detected on immunostaining for zo-1, which is normally confined to the cytoplasmic surface of tight junctions, responsible for linking the AJC proteins to the actin cytoskeleton (Fig. 4b). Claudin-1, one of a large family of tight junction–forming proteins, has four transmembrane domains and normally colocalizes with zo-1. As its extracellular portion forms an integral part of tight junction strands, Claudin-1 is a key regulator of paracellular permeability. Deficiency of claudin-1 results in a severe autosomal recessive disorder, neonatal ichthyosis–sclerosing cholangitis syndrome, that shares with ARC the features of cholestasis and ichthyosis39,40.
E-cadherin adherens junction expression was markedly decreased in Vipar-shRNA– and Vps33b-shRNA–treated mIMCD-3 cells (Fig. 4b). Immunoblotting for AJC proteins revealed substantial quantitative decreases in E-cadherin and in claudin-1 compared with control cells (Fig. 4c). However, decreases were not seen for zo-1 or for the adherens junction protein β-catenin. In addition, abnormalities appeared in Vps33b-shRNA- and Vipar-shRNA-treated cells cultured on Transwell supports. The knockdown cells formed multilayered structures instead of well-defined monolayers and became flattened rather than columnar (Fig. 5). Wild-type mIMCD-3 cells formed cavities after 5 d of culture on Transwell supports and organized into tubules after 72 h of growth in collagen gels41. Ability to form lumenal structures was lost in cells treated with Vipar-shRNA or Vps33b-shRNA; neither cavity formation nor tubular organization occurred (Fig. 5a–c). As loss of epithelial polarization and of functional AJCs is associated with epithelial-mesenchymal transition, we expected loss of cell-cell interaction and of contact inhibition in cells with reduced expression of E-cadherin and AJC defects42. Indeed, proliferation was significantly increased in cells treated with Vipar-shRNA or Vps33b-shRNA (Fig. 5d).
Figure 5.
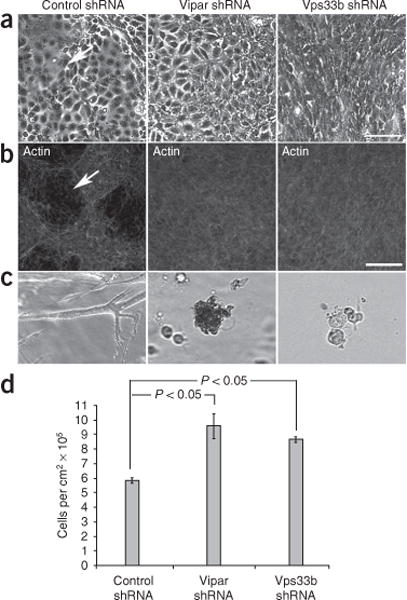
Abnormalities in cell morphology and growth in Vipar and Vps33b deficiencies. (a) Phase contrast images of cells treated with contro shRNA, Vps33b shRNA or Vipar shRNA growing on tissue culture dishes and showing disordered growth in knockdown cells (particularly obvious in Vps33b shRNA-treated cells). Cavitations are seen only in the control cells (arrow). (b) Control shRNA, Vps33b shRNA and Vipar shRNA cells grown on Transwell supports. (β-actin is stained with phalloidin-TRITC conjugate (scale bar, 100 μm). Cavitations (arrow) are present only in control cells. (c) mIMCD-3 cells growing in collagen gels (×10). Cells treated with control shRNA form highly branched tubules after 3 d in culture, whereas cells treated with Vipar shRNA or Vps33b shRNA cells form no tubules. (d) Numbers of cells harvested (mean) from Transwell supports using trypsin after 8 d in culture (n = 3, error bar, ± 1 s.e.m., P < 0.05 by t-test). Knockdown cell numbers are significantly greater than control cell numbers, compatible with loss of contact inhibition. Actual cell proliferation is likely underestimated for knockdown cells, in which ongoing spontaneous detachment led to loss into medium before harvesting.
The Vps33b-Vipar complex interacts with Rab11a
Fluorescence confocal microscopy identified intracellular sites of Vps33b-Vipar complexes. These did not substanitally colocalize (Supplementary Figs. 5 and 6) with either early or late endosomal and lysosomal markers, the Golgi apparatus or the transferrin receptor (a marker of basolateral recycling endosomes). Vps33b-Vipar complexes colocalized with Rab11a in HEK293 and mIMCD-3 cells (Fig. 6a,b) as well as, albeit to a lesser extent, with Rab4a (Supplementary Fig. 5).
Figure 6.

The VPS33B-VIPAR complex interacts with RAB11A. (a) Confocal fluorescence photomicrographs of HEK293 cells cotransfected with HA-VPS33B (not immunostained), with mCherry-VIPAR and with green fluorescent protein (GFP)-tagged RAB11A (GFP-RAB11A) showing colocalization (inset). Scale bar, 15 μm. (b) Confocal immunofluorescence photomicrographs of mIMCD-3 cells stained for endogenous Rab11a (green) and Vps33b (red). Nuclei are stained with TO-PRO-3. Scale bar, 15 μm. Colocalization of both markers is seen (inset). (c) Coimmunoprecipitation of endogenous Vps33b and Rab11a from mIMCD-3 cells after pulldown with Rab11a antibody or Vps33b antibody. Control (IgG molecules) failed to pull down the relevant protein. (d) HEK293 cells were cotransfected with HA-tagged VPS33B or empty vector, Myc-VIPAR or empty vector, and GFP-RAB11A. Coimmunoprecipitation experiments using HA-VPS33B or Myc-VIPAR as bait show that both VPS33B and VIPAR immunoprecipitate in the same complex as RAB11A. Overexpression of both HA-VPS33B and Myc-VIPAR was necessary for interaction with GFP-RAB11A to occur. Quantification of immunoprecipitation revealed that 6.7% (for HA; IP α HA) and 7.9% (for Myc; IP α Myc) of the RAB11A input was recovered. When GFP-tagged dominant negative (DN) RAB11A was used, no interaction between the VPS33B-VIPAR complex and GDP-locked dominant negative RAB11A could be seen.
When overexpressed together in mIMCD-3 cells, Vipar and Vps33b immunoprecipitated with transfected Rab11a (Fig. 6c); however, this was not the case for either protein when transfected individually. Endogenous Vps33b and Rab11a also interacted (Fig. 6c). No interaction could be shown between Vps33b with GDP-locked Rab11a (dominant negative Rab11a (DN-Rab11a)), which lacks GTPase activity. This finding suggests that the VPS33B-VIPAR complex may act as a rab effector. A similar function has been ascribed to the HOPS complex through interaction of Vps41 with the functionally active form of the Rab7 yeast homolog Ypt7 (ref. 43). Consistent with the function of the Vps33b-Vipar complex in the Rab11a pathway, abnormalities of E-cadherin expression and TER (like those in Vps33b-Vipar deficiency) developed in mIMCD-3 cells transfected with a DN-Rab11a construct (Figs. 3c and 4a).
As all the VIPAR mutations we found were expected to eliminate VIPAR expression entirely, we evaluated interactions between VIPAR and a VPS33B-L30P mutant protein (mimicking the results predicted for the only known pathogenic missense mutation identified in VPS33B). The VPS33B-L30P mutant did not co-accumulate with VIPAR at cytoplasmic organelles; instead, both proteins were present throughout the cytoplasm (Fig. 1c). Although VPS33B-L30P coimmunoprecipitated with VIPAR, no interaction with RAB11A was identified (Fig. 6d). These results suggest, as a whole, that VPS33B-VIPAR complex interaction with RAB11A is functionally important and that loss of this interaction contributes to the pathogenesis of ARC.
We used pulse-chase experiments using transfected constructs of basolaterally targeted vesicular stomatitis virus glycoprotein tagged with yellow fluorescent protein (VSVG-YFP) and of an apically targeted mutant tagged with cyan fluorescent protein (A-VSVG-CFP), as well as wild-type E-cadherin, to investigate whether the Vps33b-Vipar complex was involved in post-Golgi trafficking of newly synthesized membrane proteins in wild-type and knockdown mIMCD-3 cells. We saw normal post-Golgi traffic to the cell membrane for all transfected proteins in both knockdown and wild-type cells44 (Fig. 7a,b, Supplementary Fig. 7 and Supplementary Movie 2). Similarly, membrane expression of P75 was normal as seen by membrane-protein biotinylation assay in wild-type and knockdown pre-polarized mIMCD-3 cells stably expressing mCherry-P75, suggesting normal post-Golgi trafficking of P75. However, when protein synthesis was stopped using cycloheximide, the membrane content of mCherry-P75 rapidly fell in Vps33b- and Vipar-deficient cells, becoming undetectable after 4 h (Fig. 7c). To investigate further the causes for abnormal expression of endogenous proteins, we exposed cells treated with Vps33b-shRNA or Vipar-shRNA to inhibitors of protein degradation. This resulted in recovery of Ceacam5 expression after 24 h treatment with leupeptin, a specific lysosomal degradation inhibitor (Fig. 7d). Knockdown cells immunostained more extensively for lysosome-associated membrane protein-1, which is consistent with an increased number or size of late endosomes and lysosomes and accumulation of degradation products (Fig. 7e). Neither leupeptin nor mg132 (a proteasomal degradation inhibitor) affected cell E-cadherin concentrations (data not shown). Although E-cadherin concentrations fell, knockdown cells showed no reduction in half-life of endogenous E-cadherin compared to wild-type cells (Supplementary Fig. 8). No reduction in Rab11a concentration was detected in knockdown mIMCD-3 cells (Fig. 4c). Using quantitative real-time PCR and luciferase reporter assays of E-cadherin promoter activation, we investigated transcriptional regulation of E-cadherin expression in knockdown and wild-type mIMCD-3 cells. Knockdown cell mRNA levels were substantially reduced (<5% of wild-type controls), with a 60–80% reduction in promoter activation (Fig. 7f).
Figure 7.
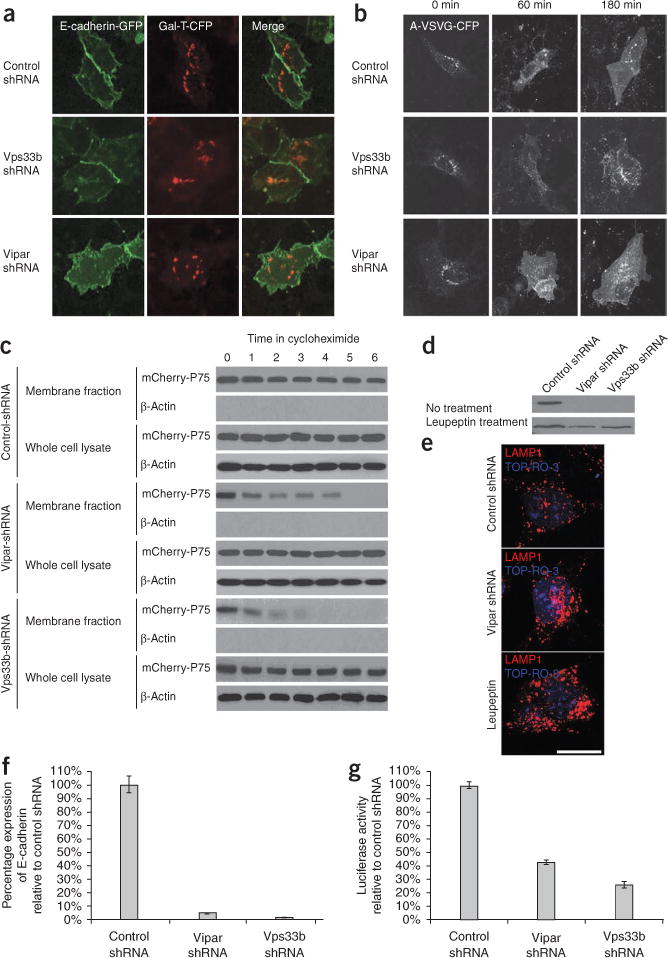
Investigation of the intracellular trafficking defects in Vps33b- and Vipar-deficient mIMCD-3 cells. (a) After transfection with E-cadherin-GFP and with Gal-T (a Golgi-resident protein) linked to CFP (Gal-T-CFP), cells were incubated overnight at 37 °C. Exogenously expressed E-cadherin targeted normally to the plasma membrane in the knockdown and control cells. (b) Cells were infected with apically targeted A-VSVG-CFP and incubated overnight at 40 °C. The cells were next incubated at 20 °C with cycloheximide (10 μg/ml) for 3 h and then, at t = 0, shifted to incubation at 32 °C. At t = 0, A-VSVG-CFP accumulated in the Golgi, but after the temperature switch, A-VSVG-CFP was trafficked normally to the plasma membrane in both knockdown cells and controls. (c) Membrane biotinylation assay showing reduced membrane mCherry-P75 content in the knockdown cell lines compared to the contro cells when incubated with cycloheximide. At t = 0, the membrane content of P75 was similar for all cell lines, suggesting no abnormality in post-Golgi trafficking of P75. No (β-actin was detected in the membrane fraction, and the whole-cell lysates contained mCherry-P75 in all samples. (d) Recovery of the Ceacam5 band after overnight treatment with leupeptin (lysosoma degradation inhibitor) is shown in cells treated with Vps33b shRNA or Vipar shRNA. (e) Lamp-1 immunofluorescent staining of wild-type mIMCD-3 cells, cells treated with Vipar shRNA and cells treated with both leupeptin and Vipar shRNA. Increased Lamp-1 immunostaining is seen in the knockdown cells. Scale bar, 15 μm. (f,g) Quantitative real-time PCR analysis of Cdh1 (E-cadherin) mRNA (f) and Cdh1 promoter activation (g) assessed by luciferase assay in knockdown and wild-type mIMCD-3 cells. Error bars, ± 1 s.e.m. In knockdown cells, mRNA levels were markedly reduced and promoter activity was decreased.
DISCUSSION
We identified mutations of VIPAR in individuals with ARC by combining functional and genetic approaches. VIPAR contains a golgin A5 domain and is weakly homologous to the HOPS complex protein VPS16. Golgins interact with members of the rab family of small GTPases via their coiled-coil motif45, are involved in tethering membranes in intracellular trafficking steps and can link vesicles to the cytoskeleton. Our results suggest that VIPAR, with VPS33B, forms a complex that interacts with RAB11A. RAB11A, primarily known for its role in the apical recycling pathway, is also associated with other steps in vesicle transport, including traffic from early endosomes to the TGN and secretion via recycling endosomes46–48. These diverse functions for RAB11A are thought to depend on RAB11A effectors and to vary with cell type49.
Immunostaining of apical membrane proteins in the livers of individuals with ARC showed mislocalization of a bile salt transporter, BSEP, and of an adhesion molecule, CEACAM5, whereas another apical membrane transporter, MRP2, maintained correct localization. The differences in immunolocalization between the canalicular transporters BSEP and MRP2 are noteworthy. Although MRP2 apical trafficking is not completely understood, MRP2 and BSEP canalicular sorting pathways clearly differ. Unlike BSEP, MRP2 contains a PDZ domain (required for membrane retention and/or targeting)50. Apical sorting motifs typically lie in the extracellular domains of proteins or in transmembrane domains. Sorting signals include N- and O-linked oligosaccharides, GPI anchors and transmembrane domains. TGN and recycling endosomes are considered the major sorting-signal recognition sites for both endocytosed and newly synthesized proteins51. Although trafficking mechanisms in simple epithelia such as the Madin-Darby canine kidney (MDCK) renal tubular cell model are well characterized, pathways in hepatocytes are less clear, partly because of practical difficulties with polarized hepatocyte cell models52–55. Differences among trafficking routes in different cell types are likely substantial, precluding extrapolation from one cell model to another56. CEACAM5 is a GPI-anchored adhesion molecule, with seven immunoglobulin-like domains, that undergoes N-glycosylation. How CEACAM5 traverses hepatocytes and renal tubular cells is unknown, but CEACAM5 is bound to galectin3, a molecule thought to be an apical sorting receptor for glycosylated proteins57,58. CEA-family proteins are involved in immune regulation, host-pathogen interaction and cell-cell contact mediation. They are internalized, with recycling to the membrane (as they fulfill these functions) and are candidates for various roles in carcinogenesis59.
In cells treated with Vipar-shRNA or Vps33b-shRNA, we did not detect abnormal post-Golgi trafficking of newly synthesized apically targeted A-VSVG or P75, which is thought to be sorted to the apical membrane via recycling endosomes60. However, after 4 h of culture in the absence of protein synthesis, no membrane-associated P75 could be detected in the Vipar- and Vps33b-deficient cell lines, suggesting a defect in membrane protein stability and/or recycling. Furthermore, in polarized mIMCD3 cells, transfected P75 was mis-sorted to the basolateral membrane and normally sorted to apical membrane domains. These findings may imply correct initial protein delivery to apical membranes with mis-sorting during recycling. As reduced expression of endogenous CEACAM5 in knockdown cells recovered on treatment with an inhibitor of lysosomal degradation, apical membrane proteins that traverse apical recycling endosomes can be inferred to require VPS33B-VIPAR for RAB11A-dependent endocytic recycling. In VPS33B or VIPAR deficiency, these proteins may instead be directed into common recycling endosomes, a step leading to basolateral as well as apical membrane distribution and to mis-sorting to late endosomes and lysosomes, where protein degradation occurs. This effect of VPS33B-VIPAR deficiency in liver and kidney cells is consistent with the increased degradation and mis-sorting of langerin identified in pancreatic Langerhans cells with RAB11A dysfunction61.
AJC expression of an adherens junction protein, E-cadherin, was decreased in livers from individuals with ARC and morpholino-treated zebrafish. The features of knockdown cell lines indicated disturbed apical basolateral polarity, including structural and functional abnormalities of tight junctions, reduced AJC expression of claudin-1 and E-cadherin, loss of contact inhibition and failure of knockdown cells to form tubular structures when grown in gels. Low levels of E-cadherin protein expression were associated with transcriptional downregulation. The generation and maintenance of epithelial tissue polarization is regulated by complex mechanisms involving many molecular interactions. These include interactions of cellular receptors with the extracellular matrix, interaction with neighboring cells via cadherin family proteins and the sensing of various diffusible factors62–64. In combination, these signals stimulate cells to assemble into groups and to orient themselves6. Major intracellular events associated with polarization include asymmetric orientation of polarity complexes with simultaneous reorganization of the cytoskeleton and of membrane trafficking systems6,53,65. Rab11a and recycling endosomes have been implicated in the regulation of E-cadherin expression in polarized cell models and during organogenesis in model organisms, although the links between Rab11a and E-cadherin adherens junction expression are not fully characterized22,66,67. Further work is required to understand the relationships between Rab11a and various molecules (adherens junction and polarity complexes, the cytoskeleton, the extracellular matrix, other vesicular trafficking components, micro-RNA mir-200 and the transcription factors Snail and Zeb) implicated in polarization6. Our finding of transcriptional down-regulation of E-cadherin in Vps33b-Vipar complex deficiency suggests that this complex may also be involved in the Rab11a-dependent regulation of E-cadherin expression and in the development and/or maintenance of lumenal structures. The defects identified here are consistent with the intrahepatic bile duct hypoplasia and the renal defects found in ARC23. That morpholino vipar and vps33b knockdown in zebrafish generated biliary abnormalities similar to those in individuals with ARC also suggests that vipar-vps33b complex function is conserved in vertebrates.
The discovery of pathogenic mutations in VIPAR in individuals with ARC from different ethnic backgrounds provides insight into the pathophysiology of human diseases. The importance of the Rab11a pathway, with its interacting protein myosin Vb, has recently been shown in such diverse cellular functions as brain postsynaptic plasticity and small-intestinal absorption68–70. Abnormal expression of E-cadherin and defective epithelial polarization in mIMCD-3 cells with Vps33b and Vipar knockdown link the human VPS33B-VIPAR complex to common diseases such as cancer, as inactivation of human CDH1 (encoding E-cadherin) is strongly associated with epithelial-mesenchymal transformation, tumorigenesis and increased cancer invasiveness71–73. Our findings advance knowledge of the molecular pathways determining cell polarity and provide new evidence on the role of intracellular trafficking proteins in regulation of epithelial polarization. Further, the fundamental defects in growth and differentiation of epithelial tissues observed in ARC and in knockdown cell lines emphasize the importance of the VPS33B-VIPAR pathway for organ development and function.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
ONLINE METHODS
Antibodies, cDNA and shRNA constructs
Human full-length and truncated VPS33B were cloned into the pCMV-HA vector25. The ‘VPS33B-L30P’construct with the missense mutation T89C (L30P) was created using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s protocol. The nucleotide change was sequence-verified. Both the wild-type and the VPS33B-L30P inserts were subcloned into the pEYFP-C3 vector using EcoRI and KpnI. The full-length integrated molecular analysis of genomes and their expression (IMAGE) cDNA clones encoding human VIPAR (3920459), its zebrafish homolog vipar (7407368) and human VPS16 (6182570) were obtained from Geneservice. Forward and reverse primers were designed to include the native start and stop codons, allowing ligation into various expression vectors. For VIPAR, restriction sites (EcoRI at the 5′ end and KpnI at the 3′ end) were added, allowing ligation into the pCMV-Myc and pEYFP-C3 vectors. For cloning of VPS16, EcoRI and NotI were used for ligation into the pCMV-Myc vector; for vipar, BamHI and EcoRI were used for ligation into the pCS2+ vector. mCherry-VIPAR was made by swapping the mCherry sequence in the pmCherry C1 vector against the YFP sequence in the pEYFP-VIPAR vector using NheI and BsrGI. Human VPS33B was sub-cloned from pEYFP-VPS33B into the pGBT9 vector using EcoRI and BamHI so that the gene was in frame with the DNA-binding domain of GAL4 for the yeast two-hybrid screen. The pCMV-HA-VPS33A plasmid is described in ref. 74. pEGFP-P75 (a gift from E. Rodriguez-Boulan, Cornell University, New York, New York) was sub-cloned into the pmCherryN1 vector using EcoRI and ApaI. pEGFP-CD63 was a gift from J. Jaiswal (The Rockefeller University, New York, New York). Rab4, Rab5 and Rab1 1a constructs were gifts from A. Benmerah and S. Benichou (Institut Cochin, Paris, France). The dominant-negative Rab11a plasmid, pEGFP-DN Rab1 1a, containing the S25N missense mutation was a gift from F. Barr (University of Liverpool Cancer Research Centre, Liverpool, UK). Apical A-VSVG-CFP and basolateral VSVG-YFP adenovirus and E-cadherin-GFP were gifts from I. Mellman (Genentech, South San Francisco, California). All other plasmids were obtained from Clontech. A pSM2c and a pGIPZ plasmid containing shRNA hairpins were used for Vps33b and Vipar, respectively. Non-silencing hairpins were used as controls. Plasmid shRNA constructs (VPS33B; V2MM_91195 and VIPAR; V2LMM_9377) were purchased from Open Biosystems. mIMCD-3 cells were transfected with 4 μg of plasmid with recovery (48 h) before transfected cells were selected using 1.5 (μg/ml puromycin. Individual clones were selected to reduce variability Level of knockdown was assessed by protein blotting (VPS33B) or quantitative real time PCR (VIPAR). Monoclonal antibodies against β-Actin (clone AC-15) and against the HA (clone HA-7) and Myc (clone 9E10) epitope tags were from Sigma-Aldrich. Rabbit polyclonal antibodies against GFP were from Santa Cruz Biotechnology and β-catenin was from Abcam. Rabbit poly-clonal antibodies against zo-1 and claudin-1 and a mouse monoclonal antibody against E-cadherin were from Invitrogen. Rabbit polyclonal antibodies against VPS33B and CEACAM5 were respectively from Proteintech Europe and Dako. The mouse monoclonal antibodies against Na+-K+ ATPase and dsRed were from Millipore and Clontech, respectively. The mouse monoclonal antibody against RAB11A was from BD Transduction Laboratories. The rabbit polyclonal anti-BSEP antibody (a gift from B. Stieger, Department of Medicine, University of Zürich, Zürich, Switzerland) is described75. The rabbit polyclonal antibody against zebrafish E-cadherin was a gift from C.P Heisenberg (Max Planck Institute, Dresden, Germany). The mouse monoclonal antibody against cytokeratin 18 (clone KS18.04) was from Millipore. Goat anti-mouse IgG and anti-rabbit IgG conjugates with Alexa Fluor-488, -546 and -568 were used as secondary antibodies. 1 mg/ml of Texas Red-conjugated transferrin was pulsed into serum-starved HEK293 cells (30 min) and chased for a further 60 min. The cells were then fixed at various times with 4% paraformaldehyde. All fluorescent products, including tetramethyl rhodamine isothiocyanate (TRITC)-conjugated phalloidin and the nuclear stain TOP-RO-3, were from Invitrogen. Cells were mounted in ProLong Gold antifade solution (Invitrogen).
Yeast two-hybrid screen
The Genomics and Proteomics Core Facility Deutsches Krebsforschungszentrum (Heidelberg, Germany) conducted the yeast two-hybrid screen using human fetal brain and adult kidney cDNA libraries as sources of prey.
Cell culture and transfection
All tissue culture reagents were from Sigma-Aldrich unless otherwise stated. HEK293 cells were grown in high-glucose (4.5 g/l) DMEM medium supplemented with 10% FBS (PAA Laboratories), 2 mM L-glutamine, MEM nonessential amino acid solution and penicillin-streptomycin. mIMCD-3 cells (American Type Culture Collection CRL2123) were grown in a 1:1 mix of DMEM and Ham’s F-12 medium, supplemented with 10% FBS and penicillin-streptomycin. Where necessary, mIMCD-3 cells were plated onto 0.4-μm-pore Transwell permeable supports (Corning) at 1 × 105 cells/cm2 to allow the cells to polarize fully. When necessary, mIMCD-3 cells were incubated with 250 μM leupeptin overnight. For tubule morphogenesis assays, mIMCD-3 cells were grown in 3D collagen gels as described76. After 72 h of culture, the tubules were visualized using ×10 phase-contrast optics on an Axioplan 2 microscope with images captured on an AxioCam CCD camera (Zeiss). Both HEK293 and mIMCD-3 cells growing on plastic plates were transfected with plasmid DNA using Lipofectamine 2000 according to manufacturer’s protocols (Invitrogen). Stable transfection of mCherry-P75 and DN-Rab11a into mIMCD-3 cells was achieved using 500 μg/ml G418 for selection (PAA Laboratories). For live cell imaging, mIMCD-3 cells were grown in Lab-Tek glass chambered slides (Thermo Fisher Scientific).
Protein extraction
Cells were grown to confluence in either 25-cm2 flasks, Transwell supports or six-well plates before protein extraction. The cells were washed twice with ice-cold PBS and scraped into 250 μl of cell lysis buffer containing 50 mM Tris-HCl (at pH 7.5), 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM EDTA, 1 mM EGTA, 0.27 M sucrose, 1% Triton X-100 and Complete, Mini Protease Inhibitor Cocktail (Roche Diagnostics). Cell lysates were centrifuged (15,000 RPM, centrifuge MIKRO 22R, Hettich Zentrifugen, rotor 1195-L for 15 min at 4 °C); super-natants were removed for immunoblotting.
Coimmunoprecipitation and membrane protein biotinylation
For coimmunoprecipitation, 20 μg of anti-HA or anti-Myc monoclonal antibodies were covalently conjugated to 100 μl of Dynabeads Protein G (Invitrogen) using dimethyl pimelimidate and triethanolamine according to the manufacturer’s instructions. HEK293 cells growing on six-well plastic plates were transfected with a total of 4 μg of appropriate plasmid DNA constructs, with recovery (48 h), before proteins were extracted as described above. Extracted proteins (1 mg) were mixed with 20 μl of antibody-conjugated Dynabeads and incubated on a blood rotor with end-over-end mixing (at 4 °C for 3 h). The complexes were then washed three times using cell lysis buffer supplemented with 150 mM NaCl, after which proteins were eluted by boiling in 2× SDS loading buffer (10 min). The protein samples were loaded directly onto SDS-PAGE gels for analysis by protein blotting. For membrane protein biotinylation, mIMCD-3 cells were seeded onto Transwell supports and grown for 7 d, after which filters were incubated using (30 min at 4 °C) in 500 μl of 0.25 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Thermo Scientific) on either side of the filter. This reaction was quenched by incubating the filter in 1 M NH4Cl for 5 min. Protein was then extracted using the protocol above. The biotin-labeled proteins were separated from the total lysates using streptavidin agarose beads (Sigma-Aldrich) according to the manufacturer’s instructions. The protein samples were loaded directly onto SDS-PAGE gels for analysis by protein blotting. For the P75 recycling assay, mCherry–P75 mIMCD-3 cell lines stably expressing mCherry-P75 seeded onto six-well plates were grown (3 d) and incubated with 20 μg/ml cycloheximide with membrane protein biotinylation (protocol above) hourly for 6 h.
Protein blotting
For immunoblotting, a 20-μg aliquot (extracted protein) or all eluted protein (coimmunoprecipitate) was separated using 12% SDS-PAGE and transferred to transblot polyvinylidene difluoride membranes (Hybond-P; Amersham Biosciences). Proteins were immunodetected by standard protocols77. All details of primary antibodies are given above. Rabbit anti-mouse IgG and goat anti-rabbit IgG HRP conjugates were both purchased from Dako. The bands were detected using Enhanced Chemiluminescence Plus protein blotting (GE Healthcare). Where necessary, after detection of the first antibody, the membranes were stripped using 0.2 M NaOH for 20 min and re-blocked overnight. The second antibody was then immunodetected on the same membrane.
Immunofluorescence confocal microscopy
HEK293 cells were grown on glass coverslips transfected as above with 24-h recovery before fixation (4% paraformaldehyde in PBS) and permeabilization (0.1% Triton X-100 in PBS). Transwell supports containing mIMCD-3 cells were prepared as above, except for immunolabeling for all tight junction and adherens junction proteins, which required ice-cold methanol fixation and 0.1% saponin permeabilization. Microscopic images were captured using an inverted Leica TCS SP2 AOBS confocal microscope with a ×10 dry objective (N/A 0.4) and a ×63 oil immersion objective (N/A 1.4); the pinhole was set to 1 Airy unit. A series of optical sections were collected from xy plane and merged into maximum projection images. Single xz plane images were also obtained where necessary Colocalization was assessed on single optical planes by a scatter plot using Leica software78.
Affected individuals and families
DNA was used from individuals with phenotypically classical ARC in whom mutations in VPS33B could not be found. Informed consent was obtained from all participating families; research ethics committees from all participating institutions approved the study. DNA was extracted from blood samples using standard protocols.
Genome-wide scan and mutation screening
DNA from four individuals with ARC in different consanguineous families was used for a genome-wide scan using 250K (three individuals) and 10K (one individual) SNP chip microarrays (Affymetrix). Primers were designed from the genomic sequence to flank all coding exons of VIPAR, and direct sequencing was carried out using the dideoxy termination method (ABI BigDye v 3.0) on an ABI 3 730 DNA sequencer (Applied Biosystems). Results were analyzed using Chromas v 2.23 software (Technelysium). All mutations were verified bidirectionally None of the mutations found was present in >200 ethnically matched control chromosomes.
Zebrafish experiments
The zebrafish AB strain was used for all experiments. In situ hybridization was carried out as previously described79. Full-length digoxigenin (DIG)-labeled antisense and sense (control) RNA probes were made from 1 μg of linearized pCS2+vipar vector, using Sp6 and T7 RNA polymerases, respectively, from the DIG Labeled RNA Probe Synthesis Kit (Roche) according to manufacturer’s instructions. The optimal hybridization temperature was 65 °C. Alkaline phosphatase–conjugated rabbit anti-DIG was from Roche. Staining was visualized on a binocular SMZ800 microscope with a ×6.3 objective and captured with a DXM200 CCD camera controlled by ACT-1 (version 2.7) software (Nikon). Morpholino oligonucleotides against the transcription initiation codon (ATG) and the splice site for exon 3 of vipar (sequences shown in Supplementary Table 1) were designed and purchased from Gene Tools. Vipar and red fluorescent protein (RFP) mRNA were produced using T7 RNA polymerase from the Ambion MessageAmp mRNA amplification kit per manufacturer’s protocol, with linearized pCS2+-vipar and pCS2+-RFP vectors as templates. The ATG, exon 3 and exon 3 mismatch morpholinos were injected into one-cell embryos in injection solution at concentrations of 10, 100 and 100 μM, respectively. When necessary, vipar and RFP mRNAs were also injected to rescue the phenotype or as a control for the mRNA injection. Liver-function and bile duct assays using the phospholipid reporter PED-6 (Invitrogen) were carried out as described33. Brightfield and PED-6 (green fluorescence) images were visualized using a SMZ1500 binocular microscope with a ×10 objective and were captured using a DSQi1Mc CCD camera controlled by NIS-Elements (version 3.0) software (Nikon), which was also used to measure gallbladder area. For immunostaining of zebrafish larvae, the larvae were oriented in optimal cutting temperature (OCT) embedding medium in shallow moulds, quickly frozen on dry ice and sectioned transversely at 18 μm. The sections were then immunostained using standard protocols as previously described33.
Keratin immuno staining of bile ducts was performed as follows: morpholino-mediated knockdown of zebrafish vipar was performed as described above. At 5 d.p.f., the larvae were fixed in 4% paraformaldehyde (in PBS) on ice for 15 min and then for 75 min at room temperature (approximately 20 °C) with shaking. After fixation, the larvae were transferred stepwise (25%, 50% and 75% methanol in PBS) into 100% methanol and stored overnight at −20 °C. The next day, the larvae were transferred into PBS and had their livers dissected out using fine forceps and needles (BD Microlance 3). The livers were then permeabilized with 0.1% Triton X-100 in PBS for 15 min, blocked for 1 h in 2% BSA and incubated with mouse monoclonal cytokeratin 18 antibody for 1h at room temperature, followed by 1 h incubation with Alexa Fluor 488 secondary antibody conjugate. The whole livers were mounted on coverslips in Citifluor and imaged with an inverted Leica TCS SP2 using a ×40 oil immersion lens (N/A 1.25) and ×1.5 optical zoom. Optical sections (0.4 μm thick) were taken in the xy plane through 50 μm of material and were merged into maximum projection images.
Immunostaining of liver-biopsy specimens from individuals with ARC
Liver-biopsy specimens obtained for clinical purposes were fixed, sectioned, stained and immunostained as described25,26,75 using monoclonal antibodies against MRP2 and polyclonal antibodies against BSEP, Ceacam5 and E-cadherin. Distributions of immunoreactivity were assessed, with all antigens apart from E-cadherin expected to be at the canalicular margins and E-cadherin expected at the hepatocyte and cholangiocyte lateral margins. Variations (such as cytoplasmic or basolateral expression or absence of the antigens) were particularly noted. Control sections of liver were evaluated in parallel. All studies were conducted toward diagnostic ends.
Tight junction and adherens junction experiments
mIMCD-3 cells treated with non-silencing shRNA and with shRNAs against Vipar and Vps33b were seeded onto Transwell supports. TER was measured every 24 h thereafter with an EVOM AC square wave current (± 20 mA, 12.5 Hz) resistance meter (World Precision Instruments). TER values for individual cultures were calculated by subtracting the values measured for filters without cells incubated in the same conditions36. These assays were performed as described37, with low-calcium medium (Spinner culture medium, Sigma-Aldrich) and dialyzed FBS (PAA Laboratories). One milligram per milliliter each of 4-kDa fluorescein isothiocyanate (FITC) dextran and 70-kDa rhodamine B dextran (Sigma-Aldrich) were dissolved in either low-calcium or normal medium and added to the apical chambers of Transwell supports. After 3 h, diffusion of each molecule to the basolateral compartment was assayed simultaneously using a Wallac Victor3 fluorometer (PerkinElmer). Wavelengths were 485 nm and 560 nm (excitation) and 535 nm and 620 nm (emission) for FITC and rhodamine B, respectively. Freeze-fracturing was performed after fixation (2 h) of control and transfected mIMCD-3 cells in 2.5% glutaraldehyde buffered with 0.1 M cacodylate (at pH 7.4). The cells were then scraped from the substrate and impregnated with 30% glycerol in cacodylate buffer for 30 min, shock-frozen in nitrogen slush, transferred into a Balzers BAF 400D freeze-fracturing device, fractured (~10−6 mbar and −150 °C) and shadowed with platinum-carbon (2.5 nm, 45 °C) and carbon (25 nm, 90 °C). The replicas were cleaned in 13% sodium hypochlo-rite, washed several times in distilled water, mounted on pioloform-coated copper grids and observed in a Zeiss EM10 transmission electron microscope. Negatives were digitized and images were arranged in Adobe Photoshop 7.0.
E-cadherin transcriptional regulation analysis
Quantitative real-time PCR (primer sequences available on request) was performed to quantify CDH1 mRNA as described11. CDH1 promoter activity was assessed using the human promoter as described80. The human E-cadherin promoter sequence (−301/+21) cloned into pGL3-basic luciferase reporter was a gift from F. Lock (University of Birmingham, Birmingham, UK). Firefly and Renilla luciferase activities were assayed 24 h post-transfection using the dual reporter assay kit Stop & Glo (Promega).
Intracellular trafficking experiments
For VSVG-YFP and A-VSVG-CFP pulse chase experiments, cells grown in four-well Lab-Tek Chambered Coverglasses (Nunc) and infected with VSVG-YFP (basolaterally targeted) adenovirus were incubated overnight at 40 °C. Live cell imaging used a Zeiss 510 inverted confocal microscope with temperature-controlled stage (×20 oil immersion objective lens, fully open pinhole). Images were taken every minute. Experiments started 10 min after stage temperature was shifted to 32 °C. In addition, cells grown as above were infected with A-VSVG-CFP (apically targeted) adeno-virus. After overnight incubation (40 °C), cells were incubated with cycloheximide (10 μg/ml; 3 h at 20 °C) to permit accumulation of A-VSVG-CFP in the Golgi apparatus. After the temperature was shifted to 32 °C (t = 0), the cells were assessed at t = 60 min and t = 3 h (×63 oil immersion objective lens; pinhole, 1 Airy unit). Series of optical sections were collected from the xy plane and merged into maximum projection images.
Newly synthesized E-cadherin trafficking
Cells were grown as above, transfected with E-cadherin-GFP and the Golgi-resident protein Gal-T-CFP, and incubated at 37 °C for 15 h. Images were captured as described above. A series of optical sections were collected from xy plane and merged into maximum projection images.
Statistical analysis
Student’s two-tailed nonpaired t-tests and normal distribution two-tailed z-tests were carried out using pooled standard error and s.d. values to determine the statistical significance of differences between samples. The significance level was set at P < 0.05.
Supplementary Material
Acknowledgments
A.R.C. holds the Children Liver Disease Foundation PhD Studentship and P.G. is a GlaxoSmithKline Clinician Scientist. A.Z. and F.M. are supported by Framework 6 IP EUTRACC, LSGH CT 2006037445. H.C. is supported by grants from the European Molecular Biology Organization (ASTF 121:2007) and European Science Foundation (Exchange Grant 2008). J.Z.R. is supported by the Biotechnology and Biosciences Research Council (BB/H002308/1). Thanks to F. Lock for her help with the E-cadherin luciferase assay, G. Reynolds for help with E-cadherin immunostaining and R. Knittel for her help with freeze fracture experiments. The authors also wish to thank all the families and clinicians who contributed to this research and thank the ARC syndrome association; Children Living with Inherited Metabolic Diseases (CLIMB); Birmingham Children’s Hospital Research Foundation (BCHRF); WellChild; the Wellcome Trust; and the Intramural Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, US National Institutes of Health, for their generous financial support.
Footnotes
Requests for materials: p.gissen@bham.ac.uk.
Accession numbers. RefSeq, NM_001001836.
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS
A.R.C. designed, conducted and interpreted experiments and wrote the manuscript. A.S.-I., A.Z. and Y.W. designed, conducted and interpreted experiments. C.K.B., G.L., F.R. and H.C. performed experiments. F.G., E.U., T.B.Ö., J.D., J.V., M.D.R., H.M. contributed subjects’ DNA. R.P.M., S.G.T., J.Z.R., I.M.A., H.W., A.S.K., D.A.K., F.M. and E.R.M. designed and supervised experiments. P.G. conceived and directed the project, obtained funding and wrote the manuscript. All authors edited the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Rodriguez-Boulan E, Nelson WJ. Morphogenesis of the polarized epithelial cell phenotype. Science. 1989;245:718–725. doi: 10.1126/science.2672330. [DOI] [PubMed] [Google Scholar]
- 2.Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–235. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- 3.Lubarsky B, Krasnow MA. Tube morphogenesis: making and shaping biological tubes. Cell. 2003;112:19–28. doi: 10.1016/s0092-8674(02)01283-7. [DOI] [PubMed] [Google Scholar]
- 4.Anderson JM, Van Itallie CM, Fanning AS. Setting up a selective barrier at the apical junction complex. Curr Opin Cell Biol. 2004;16:140–145. doi: 10.1016/j.ceb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryant DM, Mostov KE. From cells to organs: building polarized tissue. Nat Rev Mol Cell Biol. 2008;9:887–901. doi: 10.1038/nrm2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan H, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Invest. 1999;104:123–133. doi: 10.1172/JCI5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Izaddoost S, Nam SC, Bhat MA, Bellen HJ, Choi KW. Drosophila Crumbs is a positional cue in photoreceptor adherens junctions and rhabdomeres. Nature. 2002;416:178–183. doi: 10.1038/nature720. [DOI] [PubMed] [Google Scholar]
- 9.Ceteci F, et al. Disruption of tumor cell adhesion promotes angiogenic switch and progression to micrometastasis in RAF-driven murine lung cancer. Cancer Cell. 2007;12:145–159. doi: 10.1016/j.ccr.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 10.Wapenaar MC, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut. 2008;57:463–467. doi: 10.1136/gut.2007.133132. [DOI] [PubMed] [Google Scholar]
- 11.Cullinane AR, et al. Molecular investigations to improve diagnostic accuracy in patients with ARC syndrome. Hum Mutat. 2009;30:E330–E337. doi: 10.1002/humu.20900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seals DF, Eitzen G, Margolis N, Wickner WT, Price AA. Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci USA. 2000;97:9402–9407. doi: 10.1073/pnas.97.17.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starai VJ, Hickey CM, Wickner W. HOPS proofreads the trans-SNARE complex for yeast vacuole fusion. Mol Biol Cell. 2008;19:2500–2508. doi: 10.1091/mbc.E08-01-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nickerson DP, Brett CL, Merz AJ. Vps-C complexes: gatekeepers of endolysosomal traffic. Curr Opin Cell Biol. 2009;21:543–551. doi: 10.1016/j.ceb.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peplowska K, Markgraf DF, Ostrowicz CW, Bange G, Ungermann C. The CORVET tethering complex interacts with the yeast Rab5 homolog Vps21 and is involved in endo-lysosomal biogenesis. Dev Cell. 2007;12:739–750. doi: 10.1016/j.devcel.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Kinchen JM, et al. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol. 2008;10:556–566. doi: 10.1038/ncb1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edinger AL, Cinalli RM, Thompson CB. Rab7 prevents growth factor-independent survival by inhibiting cell-autonomous nutrient transporter expression. Dev Cell. 2003;5:571–582. doi: 10.1016/s1534-5807(03)00291-0. [DOI] [PubMed] [Google Scholar]
- 18.Wilkin M, et al. Drosophila HOPS and AP-3 complex genes are required for a Deltex-regulated activation of notch in the endosomal trafficking pathway. Dev Cell. 2008;15:762–772. doi: 10.1016/j.devcel.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Richardson SC, Winistorfer SC, Poupon V, Luzio JP, Piper RC. Mammalian late vacuole protein sorting orthologues participate in early endosomal fusion and interact with the cytoskeleton. Mol Biol Cell. 2004;15:1197–1210. doi: 10.1091/mbc.E03-06-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu GD, et al. SPE-39 family proteins interact with the HOPS complex and function in lysosomal delivery. Mol Biol Cell. 2009;20:1223–1240. doi: 10.1091/mbc.E08-07-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogard N, Lan L, Xu J, Cohen RS. Rab11 maintains connections between germline stem cells and niche cells in the Drosophila ovary. Development. 2007;134:3413–3418. doi: 10.1242/dev.008466. [DOI] [PubMed] [Google Scholar]
- 22.Lock JG, Stow JL. Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol Biol Cell. 2005;16:1744–1755. doi: 10.1091/mbc.E04-10-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gissen P, et al. Clinical and molecular genetic features of ARC syndrome. Hum Genet. 2006;120:396–409. doi: 10.1007/s00439-006-0232-z. [DOI] [PubMed] [Google Scholar]
- 24.Horslen SP, Quarrell OW, Tanner MS. Liver histology in the arthrogryposis multiplex congenita, renal dysfunction, and cholestasis (ARC) syndrome: report of three new cases and review. J Med Genet. 1994;31:62–64. doi: 10.1136/jmg.31.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gissen P, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36:400–404. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 26.Bull LN, et al. VPS33B mutation with ichthyosis, cholestasis, and renal dysfunction but without arthrogryposis: incomplete ARC syndrome phenotype. J Pediatr. 2006;148:269–271. doi: 10.1016/j.jpeds.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Thompson JA, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Lab Anal. 1991;5:344–366. doi: 10.1002/jcla.1860050510. [DOI] [PubMed] [Google Scholar]
- 28.Wakabayashi Y, Lippincott-Schwartz J, Arias IM. Intracellular trafficking of bile salt export pump (ABCB11) in polarized hepatic cells: constitutive cycling between the canalicular membrane and Rab11-positive endosomes. Mol Biol Cell. 2004;15:3485–3496. doi: 10.1091/mbc.E03-10-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nies AT, Keppler D. The apical conjugate effux pump ABCC2 (MRP2) Pfugers Arch. 2007;453:643–659. doi: 10.1007/s00424-006-0109-y. [DOI] [PubMed] [Google Scholar]
- 30.Bandler PE, Westlake CJ, Grant CE, Cole SP, Deeley RG. Identification of regions required for apical membrane localization of human multidrug resistance protein 2. Mol Pharmacol. 2008;74:9–19. doi: 10.1124/mol.108.045674. [DOI] [PubMed] [Google Scholar]
- 31.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 32.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 33.Matthews RP, et al. Zebrafsh vps33b, an ortholog of the gene responsible for human arthrogryposis-renal dysfunction-cholestasis syndrome, regulates biliary development downstream of the onecut transcription factor hnf6. Development. 2005;132:5295–5306. doi: 10.1242/dev.02140. [DOI] [PubMed] [Google Scholar]
- 34.Farber SA, et al. Genetic analysis of digestive physiology using fuorescent phospholipid reporters. Science. 2001;292:1385–1388. doi: 10.1126/science.1060418. [DOI] [PubMed] [Google Scholar]
- 35.Yeaman C, et al. The O-glycosylated stalk domain is required for apical sorting of neurotrophin receptors in polarized MDCK cells. J Cell Biol. 1997;139:929–940. doi: 10.1083/jcb.139.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matter K, Balda MS. Functional analysis of tight junctions. Methods. 2003;30:228–234. doi: 10.1016/s1046-2023(03)00029-x. [DOI] [PubMed] [Google Scholar]
- 37.Aijaz S, Sanchez-Heras E, Balda MS, Matter K. Regulation of tight junction assembly and epithelial morphogenesis by the heat shock protein Apg-2. BMC Cell Biol. 2007;8:49. doi: 10.1186/1471-2121-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liebner S, Kniesel U, Kalbacher H, Wolburg H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000;79:707–717. doi: 10.1078/0171-9335-00101. [DOI] [PubMed] [Google Scholar]
- 39.Colegio OR, Van Itallie C, Rahner C, Anderson JM. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fbril architecture. Am J Physiol Cell Physiol. 2003;284:C1346–C1354. doi: 10.1152/ajpcell.00547.2002. [DOI] [PubMed] [Google Scholar]
- 40.Hadj-Rabia S, et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: a tight junction disease. Gastroenterology. 2004;127:1386–1390. doi: 10.1053/j.gastro.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 41.Dawe HR, et al. The Meckel-Gruber syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16:173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- 42.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 43.Brett CL, et al. Efficient termination of vacuolar Rab GTPase signaling requires coordinated action by a GAP and a protein kinase. J Cell Biol. 2008;182:1141–1151. doi: 10.1083/jcb.200801001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keller P, Toomre D, Diaz E, White J, Simons K. Multicolour imaging of post-Golgi sorting and trafficking in live cells. Nat Cell Biol. 2001;3:140–149. doi: 10.1038/35055042. [DOI] [PubMed] [Google Scholar]
- 45.Short B, Haas A, Barr FA. Golgins and GTPases, giving identity and structure to the Golgi apparatus. Biochim Biophys Acta. 2005;1744:383–395. doi: 10.1016/j.bbamcr.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Wilcke M, et al. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-golgi network. J Cell Biol. 2000;151:1207–1220. doi: 10.1083/jcb.151.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Savina A, Fader CM, Damiani MT, Colombo MI. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic. 2005;6:131–143. doi: 10.1111/j.1600-0854.2004.00257.x. [DOI] [PubMed] [Google Scholar]
- 48.Ducharme NA, et al. Rab11–FIP2 regulates differentiable steps in transcytosis. Am J Physiol Cell Physiol. 2007;293:C1059–C1072. doi: 10.1152/ajpcell.00078.2007. [DOI] [PubMed] [Google Scholar]
- 49.Prekeris R. Rabs, Rips, FIPs, and endocytic membrane traffic. Sciientific World Journal. 2003;3:870–880. doi: 10.1100/tsw.2003.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kikuchi S, et al. Radixin deficiency causes conjugated hyperbilirubinemia with loss of Mrp2 from bile canalicular membranes. Nat Genet. 2002;31:320–325. doi: 10.1038/ng905. [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez-Boulan E, Müsch A. Protein sorting in the Golgi complex: shifting paradigms. Biochim Biophys Acta. 2005;1744:455–464. doi: 10.1016/j.bbamcr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 52.Fölsch H, Mattila PE, Weisz OA. Taking the scenic route: biosynthetic traffic to the plasma membrane in polarized epithelial cells. Traffic. 2009;10:972–981. doi: 10.1111/j.1600-0854.2009.00927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tuma PL, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev. 2003;83:871–932. doi: 10.1152/physrev.00001.2003. [DOI] [PubMed] [Google Scholar]
- 54.Mellman I, Nelson WJ. Coordinated protein sorting, targeting and distribution in polarized cells. Nat Rev Mol Cell Biol. 2008;9:833–845. doi: 10.1038/nrm2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wakabayashi Y, Arias IM. Transporters on demand: intracellular reservoirs and cycling of bile canalicular ABC transporters. J Biol Chem. 2006;281:27669–27673. doi: 10.1074/jbc.R600013200. [DOI] [PubMed] [Google Scholar]
- 56.Braiterman L, et al. Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. Am J Physiol Gastrointest Liver Physiol. 2009;296:G433–G444. doi: 10.1152/ajpgi.90489.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohannesian DW, et al. Carcinoembryonic antigen and other glycoconjugates act as ligands for galectin-3 in human colon carcinoma cells. Cancer Res. 1995;55:2191–2199. [PubMed] [Google Scholar]
- 58.Delacour D, et al. Requirement for galectin-3 in apical protein sorting. Curr Biol. 2006;16:408–414. doi: 10.1016/j.cub.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 59.Shao L, Allez M, Park MS, Mayer L. Immunomodulatory roles of the carcinoembryonic antigen family of glycoproteins. Ann NY Acad Sci. 2006;1072:194–209. doi: 10.1196/annals.1326.037. [DOI] [PubMed] [Google Scholar]
- 60.Nokes RL, Fields IC, Collins RN, Fölsch H. Rab13 regulates membrane trafficking between TGN and recycling endosomes in polarized epithelial cells. J Cell Biol. 2008;182:845–853. doi: 10.1083/jcb.200802176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uzan-Gafsou S, et al. Rab11A controls the biogenesis of Birbeck granules by regulating Langerin recycling and stability. Mol Biol Cell. 2007;18:3169–3179. doi: 10.1091/mbc.E06-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kass L, Erler JT, Dembo M, Weaver VM. Mammary epithelial cell: infuence of extracellular matrix composition and organization during development and tumorigenesis. Int J Biochem Cell Biol. 2007;39:1987–1994. doi: 10.1016/j.biocel.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 64.Gurdon JB, Bourillot PY. Morphogen gradient interpretation. Nature. 2001;413:797–803. doi: 10.1038/35101500. [DOI] [PubMed] [Google Scholar]
- 65.Goldstein B, Macara IG. The PAR proteins: fundamental players in animal cell polarization. Dev Cell. 2007;13:609–622. doi: 10.1016/j.devcel.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langevin J, et al. Drosophila exocyst components Sec5, Sec6, and Sec15 regulate DE-Cadherin trafficking from recycling endosomes to the plasma membrane. Dev Cell. 2005;9:365–376. doi: 10.1016/j.devcel.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 67.Shaye DD, Casanova J, Llimargas M. Modulation of intracellular trafficking regulates cell intercalation in the Drosophila trachea. Nat Cell Biol. 2008;10:964–970. doi: 10.1038/ncb1756. [DOI] [PubMed] [Google Scholar]
- 68.Wang Z, et al. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Müller T, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40:1163–1165. doi: 10.1038/ng.225. [DOI] [PubMed] [Google Scholar]
- 70.Wakabayashi Y, Dutt P, Lippincott-Schwartz J, Arias IM. Rab11a and myosin Vb are required for bile canalicular formation in WIF-B9 cells. Proc Natl Acad Sci USA. 2005;102:15087–15092. doi: 10.1073/pnas.0503702102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- 72.Guilford P, et al. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 73.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 74.Gissen P, et al. Comparative evolutionary analysis of VPS33 homologues: genetic and functional insights. Hum Mol Genet. 2005;14:1261–1270. doi: 10.1093/hmg/ddi137. [DOI] [PubMed] [Google Scholar]
- 75.Strautnieks SS, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134:1203–1214. doi: 10.1053/j.gastro.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 76.Leung-Hagesteijn C, et al. Integrin-linked kinase mediates bone morphogenetic protein 7-dependent renal epithelial cell morphogenesis. Mol Cell Biol. 2005;25:3648–3657. doi: 10.1128/MCB.25.9.3648-3657.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harlow E, Lane D. Using Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York, USA: 1998. [Google Scholar]
- 78.Rappoport JZ, Taha BW, Lemeer S, Benmerah A, Simon SM. The AP-2 complex is excluded from the dynamic population of plasma membrane-associated clathrin. J Biol Chem. 2003;278:47357–47360. doi: 10.1074/jbc.C300390200. [DOI] [PubMed] [Google Scholar]
- 79.Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 80.Turner FE, et al. Slug regulates integrin expression and cell proliferation in human epidermal keratinocytes. J Biol Chem. 2006;281:21321–21331. doi: 10.1074/jbc.M509731200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.