

Abstract
Cardiac myosin-binding protein C (cMyBP-C) is an integral part of the sarcomeric machinery in cardiac muscle that enables normal function. cMyBP-C regulates normal cardiac contraction by functioning as a brake through interactions with the sarcomere’s thick, thin, and titin filaments. cMyBP-C’s precise effects as it binds to the different filament systems remain obscure, particularly as it impacts on the myosin heavy chain’s head domain, contained within the subfragment 2 (S2) region. This portion of the myosin heavy chain also contains the ATPase activity critical for myosin’s function. Mutations in myosin’s head, as well as in cMyBP-C, are a frequent cause of familial hypertrophic cardiomyopathy (FHC). We generated transgenic lines in which endogenous cMyBP-C was replaced by protein lacking the residues necessary for binding to S2 (cMyBP-CS2−). We found, surprisingly, that cMyBP-C lacking the S2 binding site is incorporated normally into the sarcomere, although systolic function is compromised. We show for the first time the acute and chronic in vivo consequences of ablating a filament-specific interaction of cMyBP-C. This work probes the functional consequences, in the whole animal, of modifying a critical structure-function relationship, the protein’s ability to bind to a region of the critical enzyme responsible for muscle contraction, the subfragment 2 domain of the myosin heavy chain. We show that the binding is not critical for the protein’s correct insertion into the sarcomere’s architecture, but is essential for long-term, normal function in the physiological context of the heart.
Keywords: Myosin, Cardiac, Myosin-binding protein C, Heart, Sarcomere, Myofilament
Introduction
Cardiac myosin-binding protein C, which can interact with the thick, thin, and titin filament systems of the sarcomere, can modulate the speed and force of cardiac contraction on a beat-to-beat basis, regulating the probability of cross-bridge interaction with actin by binding with either actin, myosin, or both [18]. Although it is not essential for sarcomere contractility, functionally, it appears to operate as an internal viscous load through its interactions with the thick and thin filaments [22]. Cardiac myosin-binding protein C’s (cMyBP-C’s) role in normal cardiac function has attracted extensive attention, as mutations in MYBPC3, which encodes the cardiac-specific isoform, are responsible for a substantial number of the total mutations found in FHC [9]. Despite extensive studies using a combination of co-sedimentation binding assays [12], small angle solution X-ray scattering [33], electron microscopy [11], and NMR spectroscopy [14] that implicate cMyBP-C’s regulation of cardiac muscle contraction by actin and/or myosin-S2 binding, the physiological consequences of the precise interactions of cMyBP-C with the sarcomere’s filament systems in vivo remain obscure.
In light of this protein’s long history of inquiry, spanning almost 40 years, it is surprising that the field lacks clarity with respect to the nucleotide composition for the site or sites that are responsible for thick and thin filament interactions and/or binding. While biochemically based assays pointed to the importance of the C0 domain (Fig. 1a) [12, 20] for filament binding, other data indicated that the Pro-Ala-rich linker between the C0 and C1 domains was critical [30], while other investigators defined a combination of sites within the C0–C2 domains or even MyBP-C’s C-terminal half (C5–C10) as being the specific actin binding site [23, 33, 29, 11, 15, 19, 24]. Considering all the different lines of data, it was therefore surprising when van Dijk and colleagues recently reported that a mouse made with a deletion in the Pro-Ala and C1 domains of cMyBP-C exhibited normal hemodynamics [31].
Fig. 1.
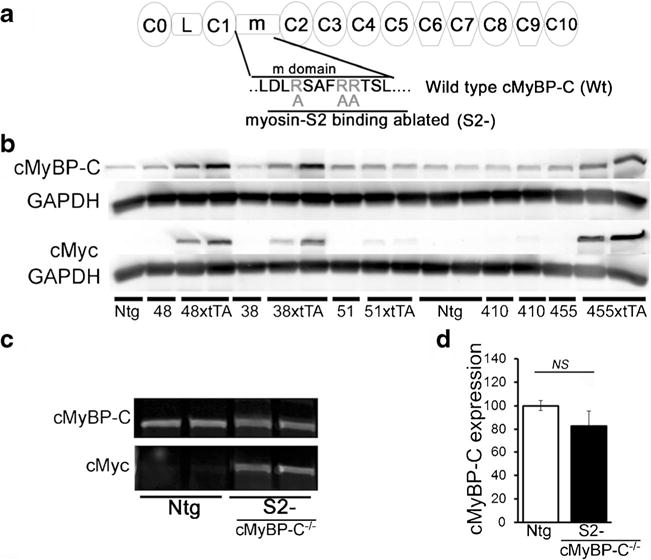
Expression of inducible, mutated cMyBP-C protein. a Schematic diagram showing domain organization and sequences of wild-type cMyBP-C (Wt) and cMyBP-CS2− (m domain; R266, R270, R271 mutated to alanine). b Expression of different lines of the S2- transgene in the S2-xtTA mouse hearts. Representative Western blots using anti-cMyBP-C and anti-c-myc antibodies. α-Sarcomeric actin was used as a loading control. c Western blot analyses of cMyBP-C levels in the heart. To obtain complete replacement of endogenous cMyBP-C with the Tg mutant proteins, the individual cMyBP-CS2−-xtTA double Tg lines were bred into the cMyBP-C−/− background. Transgenic expression of the cMyBP-CS2− protein approximated normal levels of the protein in the Ntg hearts. d Quantitation of transgene expression via Western blotting confirmed equivalent expression of cMyBP-CS2− protein relative to normal cMyBP-C protein in the Ntg hearts
Previously, we used a combination of genetic and biochemical experiments employing individual as well as combinations of domains in cMyBP-C to define critical regions and nucleotides in cMyBP-C that bind to the different filament systems of the sarcomere. Using yeast genetics, we identified cMyBP-C’s actin binding sites (C1 and m domain) and S2-MyHC binding site (m domain) [2]. We were also able to define critical residues in cMyBP-C’s m domain as being both necessary and sufficient for interactions with S2-MyHC. We subsequently confirmed that the mutation of three arginine residues (R266, R270, R271) within the m domain abolished S2 binding with no effect on actin binding [2].
However, in light of van Dijk’s recent manuscript [31], we wished to extend our studies to the whole animal, in order to determine the consequences of site-specific ablation on cMyBP-C’s functions. In this manuscript, we have focused on cMyBP-C’s S2 binding site and created an inducible transgenic (Tg) mouse in which endogenous cMyBP-C was replaced by cMyBP-C that was unable to bind to myosin’s amino terminal domain.
Methods
DNA constructs and Tg mice
The complementary DNA (cDNA) for mouse cMyBP-C was subjected to site-directed mutagenesis to generate cMyBP-C’s myosin-S2 binding ablated construct (cMyBP-CS2−). Three arginine residues within cMyBP-C’s m domain, R266, R270, and R271, were mutated to alanines to generate the myosin-S2 binding ablated construct [2]. We made cardiomyocyte-specific, inducible S2 binding ablated Tg mice by inserting the mutated cMyBP-C cDNA into the modified α-myosin heavy chain (MyHC) promoter cassette to generate responder Tg mice [28]. Tg mice were identified by PCR analysis of genomic DNA isolated from ear clips. Different lines of the responder actin and S2 binding ablated Tg mice were crossed with tetracycline-controlled transcriptional activator mice (tTA) to generate the double Tg cMyBP-CS2− mice. An N-terminal c-myc tag encoding the human c-myc peptide (EQKLISEEDL) was inserted after the initiation methionine codon to differentiate Tg from endogenous protein. Earlier studies confirmed that the introduction of the c-myc epitope is benign [26, 27, 36]. These constructs were used to generate multiple Tg founders with cardiomyocyte-specific expression; those Tg animals were subsequently bred into a cMyBP-C null background (cMyBP-C−/−) [16]. All animals used were in the FVB/N background and were equally mixed gender. All experiments were carried out in cMyBP-C homozygous nulls containing one copy (heterozygous) of the transgenic cMyBP-CS2 allele. Animals were handled in accordance with the principles and procedures of the Guide for the Care and Use of Laboratory Animals. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. The Institutional Animal Care and Use Committee at Cincinnati Children’s Hospital approved all experimental procedures.
Echocardiography
Cardiac ultrasound was performed on isoflurane-anesthetized, age-matched (3 months) mice with a VisualSonics (Toronto, Ontario, Canada) Vevo 2100 Imaging System using a 40-MHz transducer [3]. Sonographers were blinded as to genotypes being analyzed. Two-dimensional directed M-mode echocardiographic images along the parasternal short axis were recorded to determine left ventricular (LV) size and systolic function. M-mode measurements included the LV internal dimensions in systole and diastole (LVIDs and LVIDd, respectively) as well as the diastolic thickness LV posterior wall (LVPWd) and the diastolic interventricular septum thickness (IVSd). Percent fraction shortening was calculated as [(LVIDd − LVIDs)/LVIDd] × 100. The apical four-chamber view was used to assess the mitral valve using both pulsed wave and tissue Doppler. In pulsed wave Doppler mode, we measured the peak velocities of early (E) and late diastolic flow (A), and calculated the E/A ratio, and the contribution of atrial contraction to diastolic filling [A/(E + A) × 100].
In vivo hemodynamics
Invasive hemodynamic studies were performed in the intact animals at 4.5 months as described [13]. The right femoral artery and vein were cannulated with polyethylene tubing (PE-90) for measuring systemic arterial pressure. To assess myocardial performance, closed-chest animals were studied using a high-fidelity pressure catheter (Transonic Scisense, London, Ontario, Canada) under baseline conditions and in response to graded doses of the β-adrenergic agonist dobutamine. At the end of the experiment, residual cardiac reserve was evaluated by complete β-blockade through supramaximal doses of propranolol. Online heart rate, telemetry, LV developed pressure, and ±dP/dt were archived on a Macintosh computer using Maclab software [13].
Histological analysis
The hearts were collected at approximately 3 months of age, fixed in 10 % buffered formalin, and embedded in paraffin. Serial 5-μm heart sections from each group were stained with hematoxylin and eosin or Masson’s trichrome [3].
Immunofluorescence microscopy
Paraffin-embedded heart sections were used for immunofluorescence analyses as described [32]. The following primary antibodies were used: anti-c-myc monoclonal antibody (1:1000 dilution, Cell Signaling Tech), anti-cMyBP-C rabbit polyclonal antibody raised against the C0–C1 domains (1:400 dilution) [6], and mouse anti-Troponin I (1:1000, Millipore, Billerica, MA). Alexa 488- or Alexa 568-conjugated secondary antibody (Molecular Probes, ThermoFisher, Waltham, MA) directed against mouse or rabbit IgG was used as secondary antibodies, and DAPI (Invitrogen, Carlsbad, CA) was used to identify nuclei.
Transmission electron microscopy
For electron microscopic ultrastructural analysis, 3-month-old mice were anesthetized with isofluorane and the hearts were perfused with 1 % paraformaldehyde and 2 % glutaraldehyde in cardioplegic buffer, then with 1 % paraformaldehyde and 2 % glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2), postfixed in 1 % OsO4, and processed for thin sectioning [3]. Sections were counterstained with uranium and lead salts and examined with a Hitachi 7600 transmission electron microscope. Images were acquired with an AMT digital camera. Multiple sections were cut from two to four mice of mixed gender and >50 fields were observed by a blinded observer.
RNA isolation and quantitative real-time PCR analysis
Total RNAwas isolated from the hearts at 3 months post-birth with TRI reagent (Molecular Research Center, Cincinnati, OH) according to the manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was performed with a CFX-96 instrument (Bio-Rad) using Taqman probes (Applied Biosystems, Foster Drive, CA) for periostin transcripts (Postn, Applied Biosystems), smooth muscle actin (Acta2, Applied Biosystems), and cardiac fetal genes (Nppa and Nppb, Applied Biosystems). All data were normalized to gapdh transcript levels (Applied Biosystems) content and are expressed as fold increase compared to the control group [3].
Protein analyses
To identify modifications at the protein level, enriched myofibrillar proteins were isolated from the hearts at 3 months post-birth using F60 buffer (60 mM KCl; 30 mM imidazole; 7.2 mM MgCl2; pH 7.0) with protease/phosphatase inhibitors (Cocktails I and II; Sigma), as described [7]. Tg protein was confirmed by SDS-PAGE (4–15 % criterion Tris-glycine precast gels; Bio-Rad), followed by Western blots using an anti-c-myc monoclonal antibody (1:1000 dilution, Cell Signaling Tech, Danvers, MA) and anti-cMyBP-C rabbit polyclonal antibody raised against the C0–C1 domains (1: 10,000 dilution). Anti-α-sarcomeric actin monoclonal antibody (1:4000 dilution) is used as a loading control. Detection was carried out using fluorescent-conjugated secondary antibodies in combination with an Odyssey CLx Infrared Imaging System (LI-COR BioSciences, Lincoln, NE).
Cell fractionation, SDS-PAGE, and immunoblotting
To prepare whole cell lysates, frozen ventricular tissue derived from the hearts at 3 months post-birth was homogenized in a Bead Beater (Bertin Technologies, Rockvill, MD) in CelLyticM tissue homogenizer buffer (Sigma) freshly supplemented with protease and phosphatase inhibitor cocktails (Roche Applied Sciences) [3]. The heart extracts were centrifuged at 12,000×g for 15 min and the supernatants collected. Protein lysates were separated on SDS-PAGE using precast 4–15 % Criterion gels (Bio-Rad, Hercules, CA) and transferred to PVDF membranes (Bio-Rad). Membranes were blocked for 1 h in 5 % nonfat dried milk and exposed to primary antibodies overnight. The following primary antibodies were used for immunoblotting: anti-c-myc monoclonal antibody (1:1000 dilution, Cell Signaling Tech), anti-cMyBP-C rabbit polyclonal antibody raised against the C0–C1 domains (1:10,000 dilution), and anti-GAPDH (1:7500 dilution). Detection was carried out using fluorescent-conjugated secondary antibodies (LI-COR) in combination with an Odyssey CLx Infrared Imaging System (LI-COR Biosciences).
Fibrosis
Fibrosis areas within sections were measured by using ImageJ software (NIH). Blue-stained areas and nonstained myocyte areas from each section were determined using color-based thresholding. The percentage of total fibrosis area was calculated as the blue-stained areas divided by total surface area from each section.
Statistics
Data are expressed as mean ± SEM. All statistical tests were done with SigmaPlot 9.0 software. Groups of three or more were analyzed with one-way ANOVA, followed by Tukey’s post hoc test. A value of P < 0.05 was considered statistically significant.
Results
Inducible expression of cMyBP-C lacking the S2 binding site
The cDNA for mouse cMyBP-C was subjected to site-directed mutagenesis to generate the cMyBP-C’s myosin-S2 binding ablated construct (cMyBP-CS2−). Three arginine residues within cMyBP-C’s m domain, R266, R270, and R271 (Fig. 1a), were mutated to alanines to generate cMyBP-CS2− [2]. We then made cardiomyocyte-specific, inducible S2 binding ablated Tg mice by inserting the mutated cMyBP-C cDNA into the modified α-myosin heavy chain (MyHC) promoter cassette to generate responder Tg mice [28]. Different lines of the responder cMyBP-CS2− Tg mice were crossed with tetracycline-controlled transcriptional activator mice (tTA) to generate the double Tg cMyBP-CS2− mice with cMyBP-CS2− expression controlled through doxycycline withdrawal [28]. An N-terminal c-myc tag encoding the human c-myc peptide (EQKLISEEDL) was inserted after the initiation methionine codon to differentiate Tg from endogenous protein. Earlier studies confirmed that the introduction of this sequence is benign [26, 27, 36]. These constructs were used to generate multiple Tg founders, and we confirmed that three lines, 38, 48, and 455, could direct robust expression of the transgene when bred into the tTA background (Fig. 1b). After confirming cardiomyocyte-specific expression in all lines, we chose to work with line 48. To ensure the complete replacement of endogenous cMyBP-C with the cMyBP-CS2−, these mice were bred into a cMyBP-C null background (cMyBP-C−/−) [16] and the transgene was expressed from birth, that is, the animals were never treated with doxycyline. The cMyBP-CS2− levels were subsequently measured in normal and Tg cohorts using total protein lysates. Western blot analysis using anti-cMyBP-C and anti-Myc antibodies (the cMyBP-CS2− construct is myc-tagged at its amino terminus) showed normal levels of the mutated proteins compared to endogenous cMyBP-C in non-Tg (Ntg) controls (Fig. 1c, d). Normal cMyBP-C (Wt) was also expressed transgenically and those mice, bred to the nulls, served as control animals (cMyBP-CWt) [35].
cMyBP-CS2− is incorporated normally into the sarcomere
Using immunohistochemical analyses with both anti-myc and polyclonal anti-cMyBP-C antibodies, we first determined whether cMyBP-CS2− protein incorporated normally into the sarcomere. Both the cMyBP-CWt and cMyBP-CS2− proteins gave rise to normal striated patterns (Fig. 2a). As expected, total myofibrillar protein preparations showed essentially normal amounts of transgenically encoded cMyBP-C as compared to NTg samples (Fig. 2b, c). As has been observed in previous experiments when replacement of a sarcomeric protein is driven via transgenic-driven replacement, overall contractile protein expression was unaffected and myofilament protein stoichiometry was maintained in both the cMyBP-CWt and cMyBP-CS2− mice (Fig. 2c). To gain some insight into possible structural alterations as a result of transgenic replacement, we examined sarcomere ultrastructure. As previously determined, the lack of cMyBP-C results in subtle alterations, with diffuse or absent M lines (Fig. 2d). While cMyBP-CWt and cMyBP-CS2− proteins restored normal M line appearance in the null background, the Z lines in cMyBP-CS2− often appeared to be thicker than normal, irregular in shape, or even disorganized. Thus, cMyBP-C lacking a functional myosin S2-binding domain is stably expressed and is able to incorporate normally and maintain grossly normal sarcomere architecture in the sarcomere’s I-A-I region, presumably as a result of the maintenance of its actin, titin, and myosin rod interactions. Abnormal Z lines, however, suggested mechanical stress.
Fig. 2.
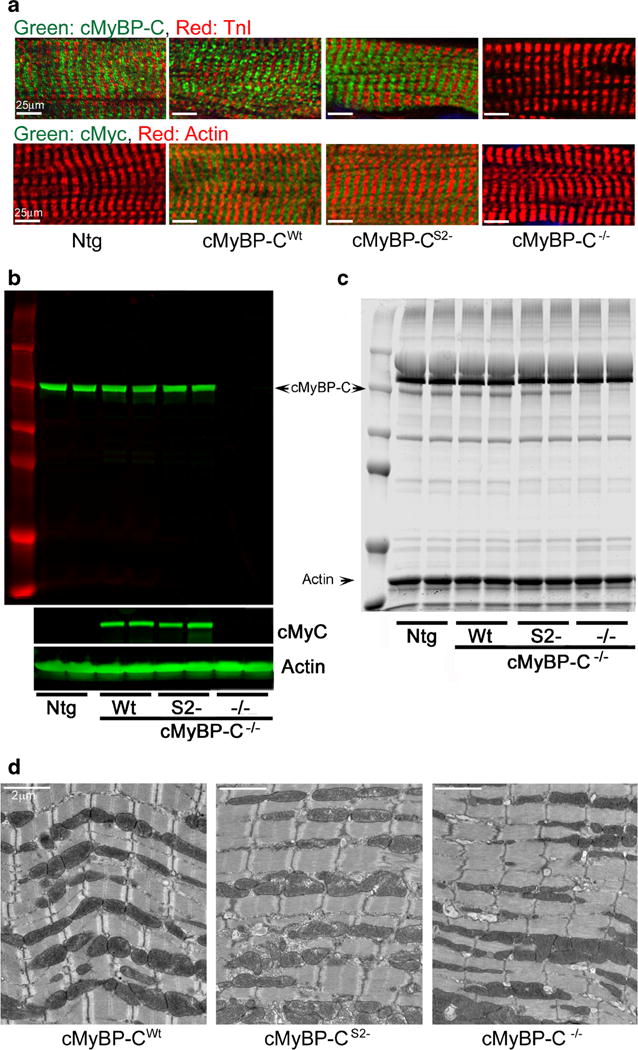
Expression and sarcomere incorporation of cMyBP-CS2−. The S2 − and Wt constructs were each bred into the cMyBP-C−/− backgrounds. a Expression and sarcomere incorporation of the c-myc-tagged, Tg cMyBP-C in the cMyBP-CWt, cMyBP-CS2−, and cMyBP-C−/− mice defined by immunofluorescent staining with anti-cMyBP-C (green) and cardiac TnI antibody (red). All Tg samples were derived from 12-week-old hearts. b Representative Western blots derived from myofilament protein preparations. α-Sarcomeric actin was used as a loading control. c SDS-PAGE analyses of myofibrillar proteins from Ntg, cMyBP-CWt, cMyBP-CS2−, and cMyBP-C−/− hearts. d Transmission electron micrographs of the sarcomeres
cMyBP-CS2− incorporation and cardiac hypertrophy
Cardiac MyBP-C nulls display a progressive heart failure with dilated cardiomyopathy, myocyte hypertrophy, disarray, fibrosis, and calcification [8, 16], but the expression of the cMyBP-CWt protein effectively rescues these mice [35]. In contrast to the cMyBP-CWt mice and despite wild-type levels of cMyBP-CS2− expression, protein lacking theS2 binding site was unable to rescue these aspects of the null phenotype (Fig. 3). The cMyBP-CS2− hearts showed significant septal hypertrophy (Fig. 3a–c). The heart/body weight ratios and left ventricular (LV) mass index were also significantly elevated in the cMyBP-CS2− animals to values similar to cMyBP-C−/− littermates at 3 months. cMyBP-CS2− mice also showed cardiomyocyte disarray and activation of extensive interstitial fibrosis (Fig. 3d), which was quantitated (Fig. 3e) as described in “Methods.” The overt pathology was confirmed at the transcriptional level with upregulation of the genetic markers of cardiac stress such as atrial natriuretic factor (Nppa) and B-type natriuretic peptide (Nppb) in the cMyBP-CS2− mice (Fig. 4a, b) and upregulation of the fibrotic markers periostin and acta2 (Fig. 4c, d).
Fig. 3.
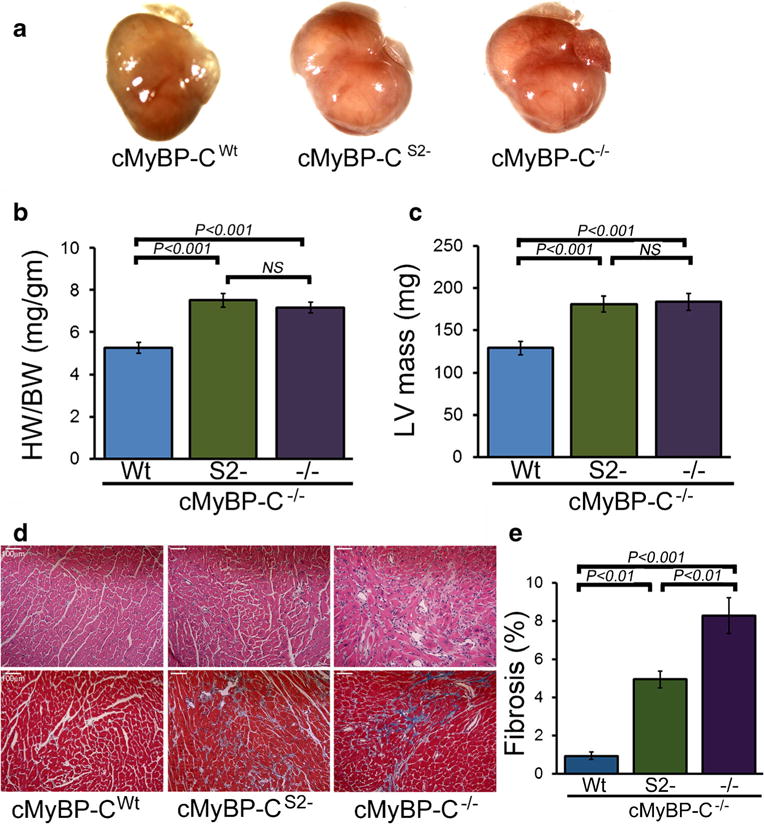
Characterization of the cMyBP-CS2− hearts at 3 months. The S2− and Wt constructs were each bred into the cMyBP-C−/− backgrounds. a Hearts from cMyBP-CWt, cMyBP-CS2−, and cMyBP-C−/− mice. b Heart weight/body weight ratios. c Changes in LV mass index determined by echocardiography (n ≥ 6 mice per group). d H&E (top) and Masson’s trichrome (bottom) staining of cardiac sections. e Blue-stained areas were color-thresholded using NIH ImageJ software and quantitated as described in “Methods.” All analyses were carried out with 3-month-old mice. Scale bars 100 μm
Fig. 4.
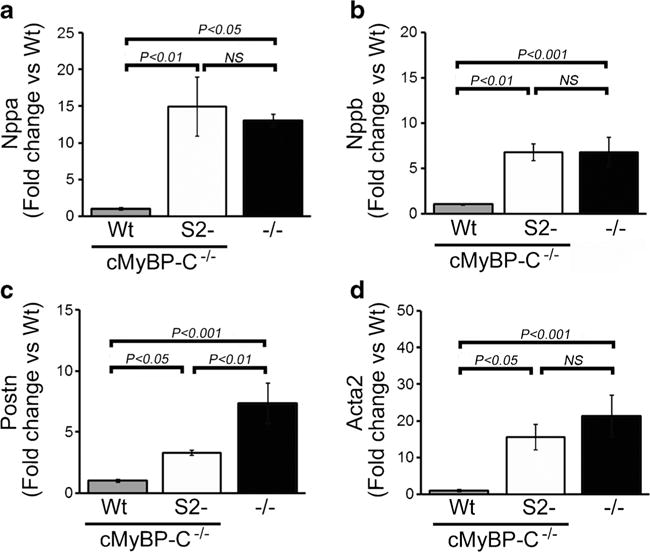
Expression of markers denoting cardiac stress. a, b Nppa and Nppb transcript levels. Values are expressed as fold change versus cMyBP-CWt control (n = 6 per group). c, d mRNA expression of Postn and Acta2. Values are expressed as fold change versus cMyBP-CWt (n = 6 per group). All samples were derived from 3-month-old hearts (n = 4). P values were determined by Tukey’s post hoc test
Cardiac hemodynamics in the cMyBP-CS2− mice
Cardiac function was evaluated both noninvasively and by catheterization in the intact animals at 3–4.5 months. Noninvasive M-mode echocardiographic measurements showed that the cMyBP-CS2− mice had abnormal cardiac dimensions and function at baseline conditions (Fig. 5). The cMyBP-CS2− mice displayed altered LV end-diastolic and end-systolic dimensions and fractional shortening similar to the values observed for the cMyBP-C−/− nulls (Fig. 5a), even though normal amounts of cMyBP-C protein were present, while cMyBP-CWt expression rescued the null phenotype as expected. Intraventricular sepatal thickness at diastole was also increased in the cMyBP-CS2− hearts but appeared normal in the nulls (Fig. 5e). However, cardiac functional deficits in cMyBP-CS2− animals were reflected in terms of survival as the mice aged, and there were no statistically significant differences in survival probabilities between cMyBP-C nulls and cMyBP-CS2− mice (Fig. 5f).
Fig. 5.
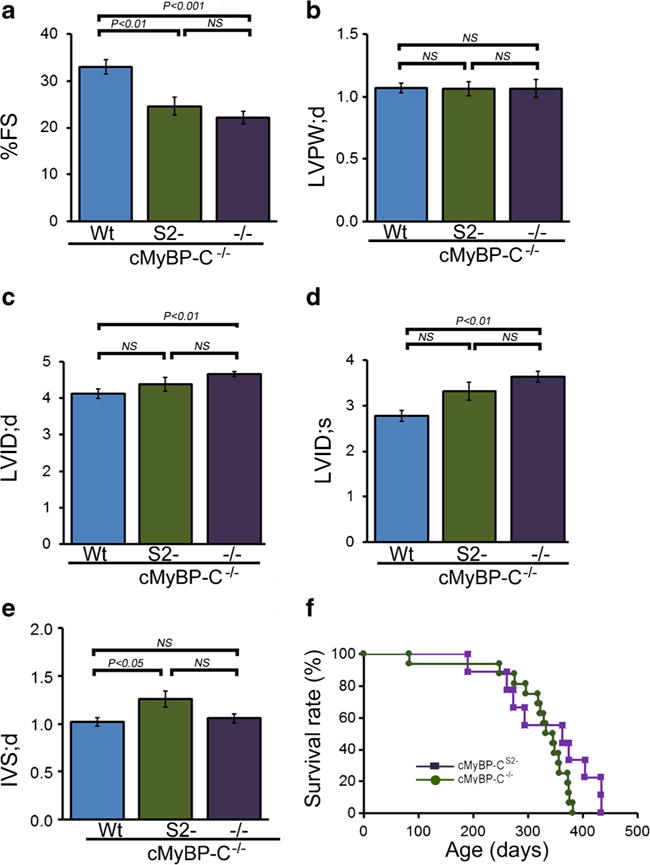
M-mode echocardiography indices of LV end-diastolic diameter and function. a Percent fractional shortening (%FS). b LV diastolic posterior wall thickness. c LV diastolic volume (LV vol;d). d LV systolic volume (LV vol;s). e Diastolic interventricular septum thickness (IVS;d). (n ≧ 10 mice per group, mean ± S.E.M). P value versus cMyBP-CWt mice by Tukey’s post hoc test. All measurements were carried out with 3-month-old mice. f Kaplan-Meier analysis of survival probabilities for the null (n = 16) versus the cMyBP-CS2− (n = 9) animals. There were no statistically significant differences between the two curves
As cMyBP-C is thought to play a major role in diastole [4, 5], we also assessed LV diastolic function using Doppler echocardiographic measurements of mitral inflow velocity during early diastolic filling (E wave) and during atrial contraction (A wave) (Fig. 6). While there were no significant differences in early transmitral flow (i.e., E wave velocity) among the three groups (Fig. 6a), the A wave velocity was decreased in cMyBP-C−/− mice (Fig. 6b) resulting in an increase in the MV E/A ratio, a finding consistent with early diastolic dysfunction (Fig. 6c). The calculated relative contribution of atrial contraction to LV diastolic filling was decreased in cMyBP-C−/− mice, indicating restrictive filling (Fig. 6d). Early diastolic mitral valve septal annulus velocity (MV IVS E′) measured by tissue Doppler was significantly decreased in cMyBP-C−/− mice (Fig. 6e), with an increase in the E/E′ ratio (Fig. 6f), supporting the presence of LV diastolic dysfunction in cMyBP-C−/− mice. For all these parameters, expression of cMyBP-CS2− was sufficient to restore function to a similar degree as expression of cMyBP-CWt. This was confirmed by using invasive catheterization and determining the dobutamine-induced reserve of the hearts (Fig. 6g, h). The complete in vivo hemodynmaic data are included in Online Resource 1. The maximum rates of LV relaxation (dp/dtmin) and relaxation time constant (tau) in cMyBP-CWt, cMyBP-CS2−, and cMyBP-C−/− mice were measured using closed-chest cardiac catheterization at baseline and with increasing adrenergic stimulation via dobutamine infusion. While the cMyBP-C−/− mice had evidence of impaired lusiotropy at baseline and with adrenergic stimulation, cMyBP-CS2− animals did not differ significantly from the controls in these relaxation parameters. Taken together, hemodynamic assessments indicate that the S2 binding site is not necessary for maintaining these parameters and that diastolic dysfunction in cMyBP-C−/− mice is largely rescued by cMyBP-CS2− expression.
Fig. 6.
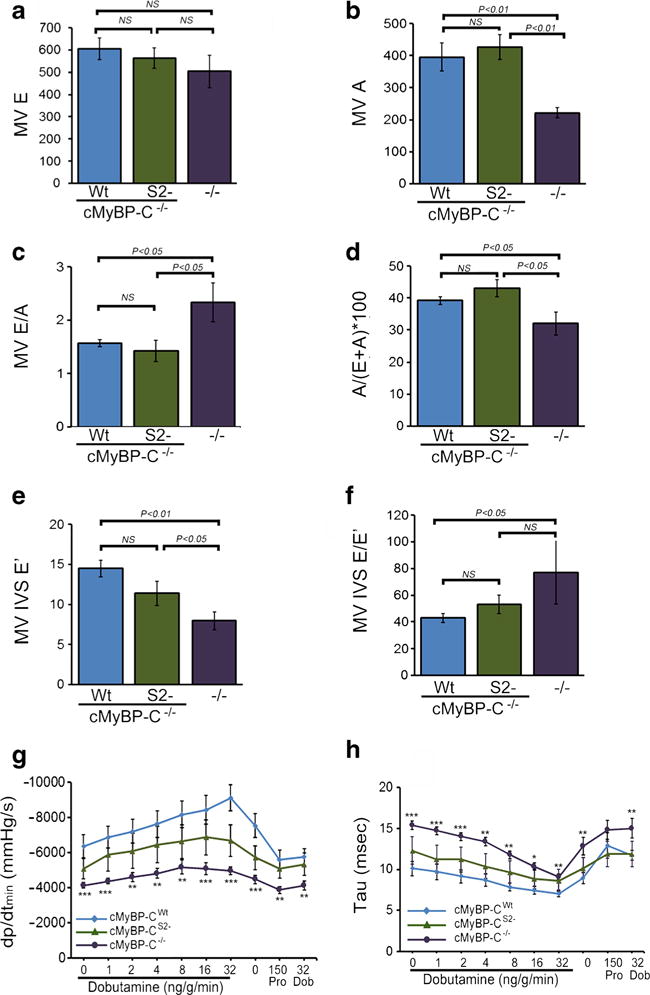
Diastolic function. a, b Doppler echocardiographic measurements of mitral inflow velocity during early diastolic filling (E wave) and mitral inflow velocity during atrial contraction (A wave). c Absolute values for early-to-late diastolic flow velocity ratio (E/A), used as an index of LV diastolic filling. d Contribution (in percent) of atrial contraction to diastolic filling (n ≥ 6 mice per group, values are means ± SEM). e Tissue Doppler measurements of early diastolic mitral annular velocities (E′) at the septal (IVS) wall. f Relationship between early diastolic inflow velocity and early diastolic tissue velocity (E/E′) at the septal (IVS) wall (n ≥ 6 mice per group, values are means ± SEM). g, h Maximum rate of LVrelaxation (dP/dtmin and relaxation time constant (tau) in 4.5-month-old cMyBP-CWt, cMyBP-CS2−, and cMyBP-C−/− mice during closed-chest cardiac catheterization, measured at baseline and with increasing adrenergic stimulation via dobutamine infusion. Propranolol was then delivered to produce complete β-adrenergic blockade (n ≥ 6 mice per group, values are means ± SEM). *P < 0.05 and ***P < 0.001 versus cMyBP-CWt animals
Discussion
The present study highlights the structure-function relationships with respect to the recently identified S2 binding site and its role in maintaining normal cardiac function. These are the first in vivo data that bear on the structural and functional implications of the cMyBP-C interactions with the myosin S2 head. A recent paper by van Dijk et al. described a mouse in which cMyBP-C was replaced by cMyBP-C lacking the Pro-Ala rich and C1 domains, which lie immediately upstream of the m domain [31]. Functional parameters such as rates of force development, calcium sensitivity isovolumic contraction times, as well as fractional shortening and EF were all unaffected. These studies were not consistent with in vitro studies studying the function of these domains [9, 10] and, as such, emphasize the need for directed, precise mutation of the critical structural sites in an in vivo context to truly appreciate the functional and structural consequences of this site. We chose to pursue that strategy, making directed mutations in three residues in the m domain (Fig. 1) that we had identified as playing a necessary role in cMyBP-C’s interactions with myosin S2 [2]: these mutations do not affect the ability of the N terminus to bind actin, and this substitution therefore represents the first attempt to precisely determine the effect of ablating a specific filament interaction while leaving the rest of the molecule intact.
While some cMyBP-C mutations result in stable proteins that compete successfully with normal cMyBP-C, others are incorporated into the sarcomere and function as poison peptides [34, 35]. Alternatively, other mutations lead to unstable proteins that are rapidly degraded, leading to cMyBP-C haploinsufficiency [4, 17]. It is somewhat surprising that the S2 binding region is dispensable for correct sarcomere incorporation of cMyBP-C, but the protein does have an additional myosin binding site at its COOH terminal that interacts with the body of the thick filament and this, together with the titin and actin binding sites, appears to be sufficient for at least overtly normal incorporation (Fig. 2a) and restoration of sarcomeric ultrastructure (Fig. 2d). As noted above, to achieve complete replacement of the normal protein with cMyBP-CS2−, we utilized a previously characterized cMyBP-C functional null by breeding the transgene into that background [16]. However, the cMyBP-CS2− phenotype clearly differs from that of the null, consistent with cMyBP-CS2− being present at wild-type levels and incorporated into the sarcomere (Fig. 1). The null was found to have dilated cardiomyopathy while our mice were characterized by increased septal thickness (Fig. 5), which is often found in hypertrophic cardiomyopathies. It may be that the cMyBP-CS2− animals are exhibiting signs of obstructive hypertrophy, a hypothesis that is consistent with the slight distortion of mitral flow in these animals (Fig. 6).
An important physiological role of cMyBP-C is to increase the dynamic range of the myofilament’s response and “tune” sarcomeric function by allowing complete relaxation under diastolic conditions [4, 5]. We initially hypothesized that the S2 binding site on cMyBP-C was essential for the protein’s incorporation and function. However, essentially normal amounts of cMyBP-CS2− are found associated with the myofilaments (Fig. 1), but the protein’s incorporation resulted in a hypertrophic response, indicating that S2 interaction is essential for the conservation of normal cMyBP-C anatomy over time.
We [25, 36] and others [1, 11, 31, 21] have studied the cardiac-specific m domain extensively, using both in vitro and in vivo approaches, and this region clearly plays both structural and regulatory roles in cMyBP-C’s functions. The phosphorylatable serines present in this domain are a major determinant in its interactions with the other sarcomeric filaments and its overall structure and function [21, 26, 27]. We showed that the region also contains a site crucial to the protein’s ability to interact with myosin’s S2 region and other sequences in this domain clearly play a role in influencing how the protein interacts with actin as well [1].
The S2 binding site, which is distinct from the N-terminal actin binding site [2], is contained within a region of the m domain that bends by virtue of containing a hinge point and this bending is normally caused by phosphorylation, which induces closure of the N terminus [21] and abolishes S2 interaction [27, 26]. Yet, the effects of ablating the S2 binding site clearly differ from those observed when dephosphorylated cMyBP-C was substituted for normal cMyBP-C. Dephosphorylated cMyBP-C was incorporated normally into the sarcomere at wild-type levels but was unable to rescue the hearts when expressed in the cMyBP-C null background [26, 27]. We think it is likely that charge-dependent intramolecular interactions play a critical role in how the N terminus folds, modulating cMyBP-C’s ability to interact with the thick and thin filaments. While reductionist, in vitro experiments can model these molecular interactions, it will be important to validate the conclusions drawn from those data with in vivo approaches as well, in which whole organ function can be assessed longitudinally over an animal’s life span. Despite the conservation of normal hemodynamics and the lack of substantial diastolic dysfunction (Figs. 5 and 6), our data show that the ablation of S2 interaction results in septal hypertrophy and fibrosis. In the absence of S2 interaction, even with actin binding intact [3], cMyBP-C can only partially rescue the null phenotype, emphasizing the importance of S2-cMyBP-C interaction for normal function over an extended period of time.
Supplementary Material
Acknowledgments
Sources of funding This work was supported by the National Institutes of Health grants P01HL69779, P01HL059408, R01HL05924, and R011062927 and a Trans-Atlantic Network of Excellence grant from Le Fondation Leducq (J.R.) as well as National Institutes of Health grant K99 HL122354 and an American Heart Association Postdoctoral Fellowship grant (M.S.B.).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00424-016-1873-y) contains supplementary material, which is available to authorized users.
Disclosures None.
References
- 1.Bezold KL, Shaffer JF, Khosa JK, Hoye ER, Harris SP. A gain-of-function mutation in the M-domain of cardiac myosin-binding protein-C increases binding to actin. J Biol Chem. 2013;288:21496–21505. doi: 10.1074/jbc.M113.474346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhuiyan MS, Gulick J, Osinska H, Gupta M, Robbins J. Determination of the critical residues responsible for cardiac myosin binding protein C’s interactions. J Mol Cell Cardiol. 2012;53:838–847. doi: 10.1016/j.yjmcc.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carrier L, Bonne G, Bahrend E, Yu B, Richard P, Niel F, Hainque B, Cruaud C, Gary F, Labeit S, Bouhour JB, Dubourg O, Desnos M, Hagege AA, Trent RJ, Komajda M, Fiszman M, Schwartz K. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res. 1997;80:427–434. [PubMed] [Google Scholar]
- 5.Carrier L, Knoll R, Vignier N, Keller DI, Bausero P, Prudhon B, Isnard R, Ambroisine ML, Fiszman M, Ross J, Jr, Schwartz K, Chien KR. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res. 2004;63:293–304. doi: 10.1016/j.cardiores.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Decker RS, Decker ML, Kulikovskaya I, Nakamura S, Lee DC, Harris K, Klocke FJ, Winegrad S. Myosin-binding protein C phosphorylation, myofibril structure, and contractile function during low-flow ischemia. Circulation. 2005;111:906–912. doi: 10.1161/01.CIR.0000155609.95618.75. [DOI] [PubMed] [Google Scholar]
- 7.Fewell JG, Hewett TE, Sanbe A, Klevitsky R, Hayes E, Warshaw D, Maughan D, Robbins J. Functional significance of cardiac myosin essential light chain isoform switching in transgenic mice. J Clin Invest. 1998;101:2630–2639. doi: 10.1172/JCI2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 9.Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108:751–764. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herron TJ, Rostkova E, Kunst G, Chaturvedi R, Gautel M, Kentish JC. Activation of myocardial contraction by the N-terminal domains of myosin binding protein-C. Circ Res. 2006;98:1290–1298. doi: 10.1161/01.RES.0000222059.54917.ef. [DOI] [PubMed] [Google Scholar]
- 11.Kensler RW, Shaffer JF, Harris SP. Binding of the N-terminal fragment C0–C2 of cardiac MyBP-C to cardiac F-actin. J Struct Biol. 2011;174:44–51. doi: 10.1016/j.jsb.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulikovskaya I, McClellan G, Flavigny J, Carrier L, Winegrad S. Effect of MyBP-C binding to actin on contractility in heart muscle. J Gen Physiol. 2003;122:761–774. doi: 10.1085/jgp.200308941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorenz JN, Robbins J. Measurement of intraventricular pressure and cardiac performance in the intact closed-chest anesthetized mouse. Am J Phys. 1997;272:H1137–H1146. doi: 10.1152/ajpheart.1997.272.3.H1137. [DOI] [PubMed] [Google Scholar]
- 14.Lu Y, Kwan AH, Trewhella J, Jeffries CM. The C0C1 fragment of human cardiac myosin binding protein C has common binding determinants for both actin and myosin. J Mol Biol. 2011;413:908–913. doi: 10.1016/j.jmb.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 15.Luther PK, Winkler H, Taylor K, Zoghbi ME, Craig R, Padron R, Squire JM, Liu J. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc Natl Acad Sci U S A. 2011;108:11423–11428. doi: 10.1073/pnas.1103216108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–1244. doi: 10.1172/jci7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moolman JA, Reith S, Uhl K, Bailey S, Gautel M, Jeschke B, Fischer C, Ochs J, McKenna WJ, Klues H, Vosberg HP. A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation. 2000;101:1396–1402. doi: 10.1161/01.cir.101.12.1396. [DOI] [PubMed] [Google Scholar]
- 18.Moss RL, Fitzsimons DP, Ralphe JC. Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circ Res. 2015;116:183–192. doi: 10.1161/circresaha.116.300561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mun JY, Gulick J, Robbins J, Woodhead J, Lehman W, Craig R. Electron microscopy and 3D reconstruction of F-actin decorated with cardiac myosin-binding protein C (cMyBP-C) J Mol Biol. 2011;410:214–225. doi: 10.1016/j.jmb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orlova A, Galkin VE, Jeffries CM, Egelman EH, Trewhella J. The N-terminal domains of myosin binding protein C can bind polymorphically to f-actin. J Mol Biol. 2011;412:379–386. doi: 10.1016/j.jmb.2011.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Previs MJ, Mun JY, Michalek AJ, Previs SB, Gulick J, Robbins J, Warshaw DM, Craig R. Phosphorylation and calcium antagonistically tune myosin-binding protein C’s structure and function. Proc Natl Acad Sci U S A. 2016;113:3239–3244. doi: 10.1073/pnas.1522236113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Previs MJ, Prosser BL, Mun JY, Previs SB, Gulick J, Lee K, Robbins J, Craig R, Lederer WJ, Warshaw DM. Myosin-binding protein C corrects an intrinsic inhomogeneity in cardiac excitation-contraction coupling. Sci Adv. 2015;1 doi: 10.1126/sciadv.1400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Razumova MV, Shaffer JF, Tu AY, Flint GV, Regnier M, Harris SP. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: evidence for long-lived cross-bridges. J Biol Chem. 2006;281:35846–35854. doi: 10.1074/jbc.M606949200. [DOI] [PubMed] [Google Scholar]
- 24.Rybakova IN, Greaser ML, Moss RL. Myosin binding protein C interaction with actin: characterization and mapping of the binding site. J Biol Chem. 2011;286:2008–2016. doi: 10.1074/jbc.M110.170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J, Robbins J. A critical function for Ser-282 in cardiac myosin binding protein-C phosphorylation and cardiac function. Circ Res. 2011;109:141–150. doi: 10.1161/CIRCRESAHA.111.242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, Klevitsky R, Seidman CE, Seidman JG, Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci U S A. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–616. doi: 10.1161/01.RES.0000065442.64694.9F. [DOI] [PubMed] [Google Scholar]
- 29.Shaffer JF, Kensler RW, Harris SP. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J Biol Chem. 2009;284:12318–12327. doi: 10.1074/jbc.M808850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Squire JM, Luther PK, Knupp C. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J Mol Biol. 2003;331:713–724. doi: 10.1016/s0022-2836(03)00781-2. [DOI] [PubMed] [Google Scholar]
- 31.van Dijk SJ, Witt CC, Harris SP. Normal cardiac contraction in mice lacking the proline-alanine rich region and C1 domain of cardiac myosin binding protein C. J Mol Cell Cardiol. 2015;88:124–132. doi: 10.1016/j.yjmcc.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Osinska H, Dorn GW, 2nd, Nieman M, Lorenz JN, Gerdes AM, Witt S, Kimball T, Gulick J, Robbins J. Mouse model of desmin-related cardiomyopathy. Circulation. 2001;103:2402–2407. doi: 10.1161/01.cir.103.19.2402. [DOI] [PubMed] [Google Scholar]
- 33.Whitten AE, Jeffries CM, Harris SP, Trewhella J. Cardiac myosin-binding protein C decorates F-actin: implications for cardiac function. Proc Natl Acad Sci U S A. 2008;105:18360–18365. doi: 10.1073/pnas.0808903105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witt CC, Gerull B, Davies MJ, Centner T, Linke WA, Thierfelder L. Hypercontractile properties of cardiac muscle fibers in a knock-in mouse model of cardiac myosin-binding protein-C. J Biol Chem. 2001;276:5353–5359. doi: 10.1074/jbc.M008691200. [DOI] [PubMed] [Google Scholar]
- 35.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J Clin Invest. 1998;102:1292–1300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res. 1999;85:841–847. doi: 10.1161/01.res.85.9.841. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.