

ABSTRACT
Satisfactory therapeutic strategies for septic shock are still missing. Previously we found elevated levels of Wnt5A in patients with severe sepsis and septic shock. Wnt5A is released by activated macrophages but knowledge of its effects in the vascular system remains scant. Here we investigate the response of human coronary artery endothelial cells (HCAEC) to Wnt5A. We used a genome-wide differential expression approach to define novel targets regulated by Wnt5A. Gene ontology analysis of expression profiles revealed clusters of genes involved in actin cytoskeleton remodeling as the predominant targets of Wnt5A. Wnt5A targeted Rho-associated protein serine/threonine kinase (ROCK), leading to phosphorylation of LIM kinase-2 (LIMK2) and inactivation of the actin depolymerization factor cofilin-1 (CFL1). Functional experiments recording cytoskeletal rearrangements in living cells showed that Wnt5A enhanced stress fiber formation as a consequence of reduced actin depolymerization. The antagonist Wnt inhibitory factor 1 (WIF1) that specifically interferes with the WIF domain of Ryk receptors prevented actin polymerization. Wnt5A disrupted β-catenin and VE-cadherin adherens junctions forming inter-endothelial gaps. Functional experiments targeting the endothelial monolayer integrity and live recording of trans-endothelial resistance revealed enhanced permeability of Wnt5A-treated HCAEC. Ryk silencing completely prevented Wnt5A-induced endothelial hyperpermeability. Wnt5A decreased wound healing capacity of HCAEC monolayers; this was restored by the ROCK inhibitor Y-27632. Here we show that Wnt5A acts on the vascular endothelium causing enhanced permeability through Ryk interaction and downstream ROCK/LIMK2/CFL1 signaling. Wnt5A/Ryk signaling might provide novel therapeutic strategies to prevent capillary leakage in systemic inflammation and septic shock.
KEYWORDS: endothelium, sepsis, systemic inflammation, vascular permeability, Wnt5A
Sepsis is a systemic inflammatory response of the organism to an infection. The most serious manifestations of the disease are severe sepsis and septic shock with organ dysfunction and profound hypotension. The pathophysiology is highly complex and includes deregulation of the immune response, severe coagulopathy and endothelial dysfunction.1 A small number of randomized clinical trials showed improved survival in septic patients; however three of these have been disproven in larger studies. The latest example has been trials with activated protein C, which lead to withdrawal of the drug from the market.2 Despite optimized therapies with replacement of organ function using artificial systems, mortality remains high with a rate of 40–70%.1 The clinical manifestations of sepsis have a strong vascular component, predominantly seen as enhanced vascular leakage due to endothelial barrier dysfunction.3,4
During systemic inflammation, activated immune cells communicate with the vascular endothelium by release of and response to an array of inflammatory mediators.5 Inflammatory mediators such as tumor necrosis factor and interleukin (IL)-1 were identified as responsible for disturbed endothelial function with increased leukocyte adhesion and transmigration, loss of control of systemic vascular tone, increased pro-coagulant activity and pronounced vascular leakage. Therapeutic trials targeting these inflammatory mediators have not been efficacious in treating human sepsis.6-8
Recently, Wnt5A was identified as a novel chemokine highly induced and released following Toll-like receptor dependent inflammatory activation of human macrophages and was found in substantial quantities in sera of patients with severe sepsis.9,10 Wnt5A is one of the members of the Wnt family of secreted lipid modified signaling proteins. Wnt5A is capable of sustaining the inflammatory phenotype of macrophages through an autocrine mechanism involving Frizzled (Fzd)-5/CaMKII signaling. This pathway is a target for anti-inflammatory interventions in human macrophages.9,11
In systemic inflammation and sepsis, inflammatory mediators secreted by activated immune cells act paracrinically on endothelial cells.1,12,13 Therefore, Wnt5A secreted by activated macrophages might act in a similar manner and contribute to endothelial dysfunction. A recent report indicates inflammatory activation of vascular endothelial cells may occur following Wnt5A exposure14 but the exact mechanism by which Wnt5A contributes to the inflammatory response of vascular endothelial cells still remain unclear. In the present study, we applied a genome-wide transcriptome analysis to define the targets and pathways of paracrine Wnt5A signaling in our established system of primary vascular endothelial cells, human coronary artery endothelial cells (HCAEC).5 IL-1β, a leading inflammatory cytokine in the vascular system,5,15 was used in parallel as a comparison to Wnt5A and a control for integrity and proper biological function of our HCAEC system. Our study revealed that Wnt5A principally regulated genes involved in cytoskeleton rearrangement. This contrasted with IL-1β, which predominantly affected pathways and genes associated with an immune response. We found that Wnt5A critically affects endothelial barrier function and cell migration. We identified the receptor Ryk and downstream Rho-associated protein serine/threonine kinase (ROCK)/LIM kinase-2 (LIMK2)/cofilin-1 (CFL1) signaling as the pathway responsible for Wnt5A-induced cytoskeletal changes in HCAEC. Targeting the Wnt5A/Ryk interaction by either knockdown of Ryk expression or using the Ryk specific soluble antagonist Wnt inhibitory factor 1 (WIF1) abolished cytoskeletal changes and endothelial hyperpermeability. Wnt5A/Ryk signaling might provide novel strategies for therapeutic intervention in systemic inflammation and sepsis.
Results
Differential gene expression profiling reveals that Wnt5A predominantly affects genes involved in cytoskeleton rearrangements
To define the genes regulated by Wnt5A treatment in HCAEC, we performed competitive 2-color differential gene expression profiling. The transcriptome profiles of HCAEC treated with Wnt5A for 8 h were compared with that of untreated cells (cultured in parallel) using human whole genome oligomicroarrays. Transcriptome profiling of IL-1β treated HCAEC was performed using the same approach. The well-defined cytokine IL-1β was used to compare its inflammatory properties with the suspected but not elucidated effects of Wnt5A in the HCAEC system. GeneSpring analysis of the Wnt5A transcriptome in HCAEC revealed 9854 genes, of which 3317 genes were upregulated and 6537 genes were downregulated. GeneSpring analyses comparing Wnt5A and IL-1β transcriptomes showed that 3188 genes were commonly regulated by both treatments. The presence of common targets for Wnt5A and IL-1β points to an inflammatory action of Wnt5A in HCAEC.
The list of 2 hundred genes highly regulated by Wnt5A and IL-1β is provided in Tables 1 and 2 of the Online Supplement. Complete data sets for Wnt5A and IL-1β are available in the NCBI GEO data repository with accession number GSE62281.
Table 1.
Genes regulated by both Wnt5A and IL-1β in “Cytoskeleton remodeling_TGF, WNT and cytoskeletal remodeling” and “Cell adhesion_Chemokines and adhesion” pathways.
| Regulation |
||||
|---|---|---|---|---|
| Gene symbol | Accession number | Sequence description | Wnt5A | IL-1β |
| LIMK2*,†,*,† | NM_001031801 | Homo sapiens LIM domain kinase 2 | Down | Up |
| LRP5* | NM_001291902 | Homo sapiens low density lipoprotein receptor-related protein 5 | Down | Down |
| MAPK12* | NM_002969 | Homo sapiens mitogen-activated protein kinase 12 | Down | Down |
| MAPK13* | NM_002754 | Homo sapiens mitogen-activated protein kinase 13 | Down | Down |
| MMP1† | NM_002421 | Homo sapiens matrix metallopeptidase 1 | Up | Up |
| PLAU*,†,*,† | NM_001145031 | Homo sapiens plasminogen activator, urokinase | Down | Up |
| TAB1* | NM_006116 | Homo sapiens TGF-β activated kinase 1 | Down | Down |
Genes regulated in “Cytoskeleton remodeling_TGF, WNT and cytoskeletal remodeling” pathway.
Genes regulated in “Cell adhesion_Chemokines and adhesion” pathway.
Table 2.
Genes regulated only by Wnt5A in “Cytoskeleton remodeling_TGF, WNT and cytoskeletal remodeling” and “Cell adhesion_Chemokines and adhesion” pathways.
| Gene symbol | Accession number | Sequence description | Regulation |
|---|---|---|---|
| AKT2*,†,*,† | NM_001626 | Homo sapiens v-akt murine thymoma viral oncogene homolog 2 | Down |
| ARPC1B*,†,*,† | NM_005720 | Homo sapiens actin related protein 2/3 complex, subunit 1B | Down |
| CDKN1A*,† | NM_078467 | Homo sapiens cyclin-dependent kinase inhibitor 1A | Down |
| CFL1*,†,*,† | NM_005507 | Homo sapiens cofilin-1 | Down |
| CRK*,†,*,† | NM_016823 | Homo sapiens v-crk sarcoma virus CT10 oncogene homolog (avian) | Down |
| FLNA† | NM_001110556 | Homo sapiens filamin A | Down |
| MAPK3*,†,*,† | NM_002746 | Homo sapiens mitogen-activated protein kinase 3 | Down |
| MAPK11* | NM_002751 | Homo sapiens mitogen-activated protein kinase 11 | Down |
| MAPK14* | NM_139013 | Homo sapiens mitogen-activated protein kinase 14 | Down |
| TSC2* | NM_000548 | Homo sapiens tuberous sclerosis 2 | Down |
Genes regulated in “Cytoskeleton remodeling_TGF, WNT and cytoskeletal remodeling” pathway.
Genes regulated in “Cell adhesion_Chemokines and adhesion” pathway.
To identify the biological processes regulated by Wnt5A or IL-1β in HCAEC, Metacore gene ontology (GO) analyses were conducted. Using this tool, genes regulated at least 2-fold in their expression were clustered on the basis of their function to generate statistically significant cellular pathways.
Gene ontology cluster analysis of genes regulated by Wnt5A treatment of HCAEC were mainly associated with cytoskeleton remodeling pathways. The “Cytoskeleton remodeling_TGF, WNT and cytoskeletal remodeling” appeared as the most significant pathway for Wnt5A (Fig. 1A), but appeared as only 25th out of 50 pathways significant for IL-1β (not shown). Genes regulated by both Wnt5A and IL-1β (Table 1), such as MAPK13 and LIMK2, were mainly concerned with actin polymerization and cytoskeleton remodeling. Among the genes specifically regulated by Wnt5A (Table 2), ARPC1B and CFL1 are involved in actin polymerization, CRK is involved in cell motility and several other genes are involved in angiogenesis. The “Cell adhesion_Chemokines and adhesion” pathway was the second most statistically significant pathway for Wnt5A (Fig. 1A), but it appeared as only 45th out of 50 pathways significant for IL-1β (not shown). In this pathway, Wnt5A-regulated genes were mainly associated with the cytoskeleton targets described above. Additionally, FLNA, a gene involved in branching of actin filaments, was downregulated by Wnt5A (Table 2).
Figure 1.

Whole genome expression survey of the inflammatory response of HCAEC to Wnt5A and IL-1β. GO pathway maps of the 10 most significant processes regulated by (A) Wnt5A (250 ng/mL) and (B) IL-1β (20 U/mL). Genes are clustered according to biological processes. Pathways represented as histograms are ranked by the –log value (P value). Length of histogram corresponds to the number of genes associated with that specific pathway.
In contrast to Wnt5A, gene ontology cluster analysis of genes regulated by IL-1β were mainly associated with immune response pathways. The “Immune response_HMGB1/RAGE signaling pathway” and “Immune response_HSP60 and HSP70/TLR signaling pathway” were the first and second most significant pathways for IL-1β in HCAEC (Fig. 1B) which involved the upregulation of NFκB (Table 3). IL-1β significantly upregulated the expression of inflammatory mediators (IL-6, IL-8, CCL2; Online Supplement Table 2) and cell adhesion molecules (ICAM; Table 3) whereas the expression of these genes was not regulated by Wnt5A. This correlates with our qRT-PCR and flow cytometry analyses and thereby confirms that Wnt5A does not regulate the expression of cell adhesion molecules or proinflammatory cytokines in HCAEC (Online Supplement Figs. 1 and 2).
Table 3.
Genes regulated by IL-1β in “Immune response_HMGB1/RAGE signaling pathway” and “Immune response_HSP60 and HSP70/TLR signaling pathway.”
| Gene symbol | Accession number | Sequence description | Regulation |
|---|---|---|---|
| ATF2† | NM_001880 | Homo sapiens activating transcription factor 2 | Up |
| CCL4* | NM_002984 | Homo sapiens chemokine (C-C motif) ligand 4 | Up |
| ICAM1*,† | NM_000201 | Homo sapiens intercellular adhesion molecule 1 | Up |
| IL-1α* | NM_000575 | Homo sapiens interleukin 1, α | Up |
| MAP2K6*,†,*,† | NM_002758 | Homo sapiens mitogen-activated protein kinase kinase 6 | Down |
| NFκB1*,†,*,† | NM_003998 | Homo sapiens nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 | Up |
| NFκB2*,†,*,† | NM_001077493 | Homo sapiens nuclear factor of kappa light polypeptide gene enhancer in B-cells 2 | Up |
| NFκBIA*,†,*,† | NM_020529 | Homo sapiens nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, α | Up |
| NFκBIE*,†,*,† | NM_004556 | Homo sapiens nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, epsilon | Up |
| SERPINE1* | NM_000602 | Homo sapiens serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1 | Up |
| TLR2*,†,*,† | NM_003264 | Homo sapiens toll-like receptor 2 | Up |
Genes regulated in “Immune response_HMGB1/RAGE signaling pathway”.
Genes regulated in “Immune response_HSP60 and HSP70/TLR signaling pathway”.
Figure 2.

Expression of Fzd5 and Ryk receptors in HCAEC. Expression of mRNA levels of (A) Fzd5 and (B) Ryk in HCAEC treated with Wnt5A and IL-1β (IL-1b) for 8 h as outlined in Methods. Data were obtained from 3 independent qRT-PCR experiments with duplicate samples and expressed as the mean ± SEM. **P < 0.01, IL-1β versus untreated. (C) Quantitative ratio of constitutive levels of Fzd5 and Ryk mRNAs in HCAEC. ***P < 0.001, Ryk vs. Fzd5 by Student's t-test. (D) Protein expression of Fzd5 and Ryk receptors in HCAEC either untreated (left panel) or treated with IL-1β (IL-1) for 24 h. Images show immunofluorescence staining using specific antibodies for Fzd5 and Ryk (red), F-actin (phalloidin, green) and nuclei (DAPI, blue). Zeiss Axioskope, original magnification 200x.
Wnt5A receptors in HCAEC
We previously described Wnt5A/Fzd5 signaling as an essential pathway maintaining the production of inflammatory mediators IL-1β, IL-6 and IL-8 in human macrophages.9 Since HCAEC did not respond to Wnt5A stimulation with the production of these “Classical” inflammatory molecules or adhesion molecules, and Ryk and Ror2 are other receptors known to be involved in Wnt signaling pathways,16 we checked which of these 3 receptors (Fzd5, Ryk or Ror2) is responsible for Wnt5A signaling in HCAEC. Using qRT-PCR, we measured Fzd5, Ryk and Ror2 mRNAs expressed constitutively and upon Wnt5A or IL-1β treatment. Wnt5A treatment did not affect the mRNA levels of Fzd5 (Fig. 2A) or Ryk (Fig. 2B). In HCAEC treated with IL-1β, Ryk mRNA was increased approximately 2-fold while no change was observed in the Fzd5 mRNA level (Fig. 2A). We then measured Fzd5 and Ryk mRNA transcripts constitutively expressed in HCAEC. As shown in Figure 2C, Ryk mRNA expression is greater than 6 times the expression level of Fzd5 mRNA, the latter being very low. Ror2 mRNA expression was undetectable (data not shown).
Immunofluorescence staining was performed to corroborate this finding and to verify if IL-1β upregulates Ryk receptor protein in HCAEC. HCAEC constitutively express substantial amounts of Ryk protein and, consistent with regulation at the mRNA level, HCAEC treated with IL-1β expressed significantly more Ryk protein compared with the non-treated cells (Fig. 2D, upper panel). Constitutive Fzd5 protein expression was scant and remained unchanged with IL-1β treatment (Fig. 2D, lower panel). CamKII, which is a downstream component of Wnt5A signaling through Fzd receptors9,17 was not expressed in HCAEC (data not shown).
Wnt5A phosphorylates LIMK2 and CFL1 proteins through ROCK activation
Wnt5A mainly regulated the functional cluster of genes involved in endothelial actin cytoskeleton remodeling including the ROCK downstream targets LIMK2 and CFL1. LIMK2 is a serine/threonine/tyrosine kinase which, when phosphorylated by ROCK activation, phosphorylates the actin depolymerization factor CFL1.18-20 As Wnt/Planar Cell Polarity signaling involves the activation of ROCK17 and Wnt5A regulated LIMK2 and CFL1, we next checked if LIMK2 and CFL1 proteins are phosphorylated upon treatment with Wnt5A. HCAEC were treated with Wnt5A and the phosphorylated forms of both endogenous LIMK2 and CFL1 were detected by immunofluorescence staining using specific antibodies to phosphorylated LIMK2 and CFL1. Amounts of fluorescent phosphorylated LIMK2 and CFL1 in cells were further quantified using Fiji software. Compared with non-treated cells, a substantial increase in phosphorylated LIMK2 and CFL1 was observed in HCAEC treated with Wnt5A for 1 h and 4 h, respectively (Fig. 3A–C).
Figure 3.
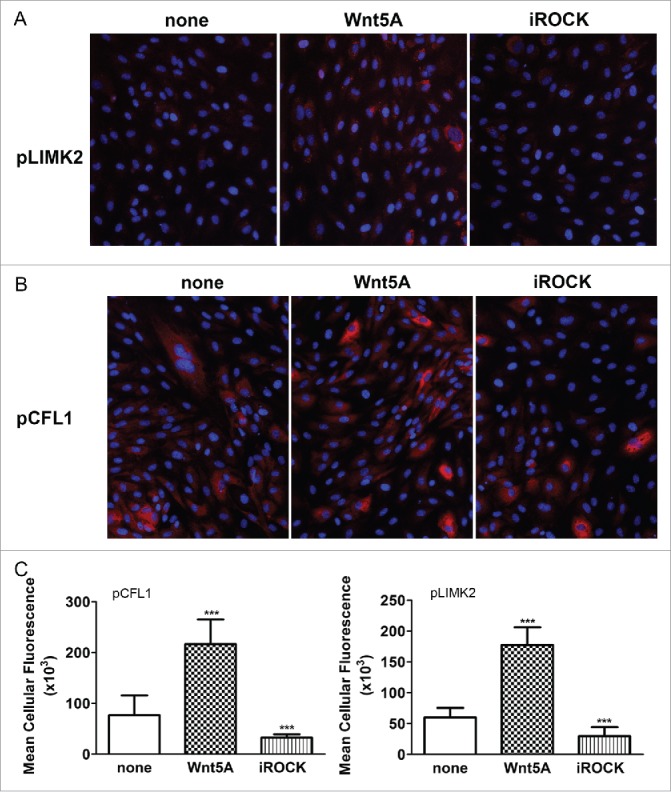
Effects of ROCK inhibition on Wnt5A-induced phosphorylation of LIMK2 and CFL1. Immunofluorescence staining of (A) phosphorylated LIMK2 and (B) phosphorylated CFL1 (both in red) and nuclei (blue) in HCAEC treated with either Wnt5A alone or in combination with the ROCK inhibitor Y-27632 (iROCK) for 1 h and 4 h respectively. Zeiss Axioskope, original magnification 200x. (C) Mean cellular fluorescent intensities of phosphorylated LIMK2 and CFL1 quantified by ImageJ based Fiji software. pLIMK2, phosphorylated LIMK2; pCF1, phosphorylated CFL1. ***P < 0.001, Wnt5A versus none and Wnt5A combined with iROCK vs. Wnt5A alone.
As LIMK2 and CFL1 are phosphorylated by the activation of ROCK,18,20 we next checked if inhibiting ROCK prevents their phosphorylation upon treatment with Wnt5A. Wnt5A-induced phosphorylation of both LIMK2 and CFL1 was notably suppressed when HCAEC were treated with a combination of Wnt5A and the ROCK-inhibitor Y-27632 (iROCK, Fig. 3A–C).
Functional experiments in living cells reveal actin stress fiber formation upon Wnt5A treatment
Since ROCK-mediated LIMK2 and CFL1 phosphorylation causes increased actin fiber bundling and stress fiber formation,19-21 and Wnt5A induced phosphorylation of LIMK2 and CFL1, we next checked if Wnt5A causes actin stress fiber formation. Changes in actin arrangement and F-actin stress fiber formation were recorded in actin-RFP transduced cells. In non-treated HCAEC, an accumulation of non-fibrous g-actin in the perinuclear region was obvious with few thin actin filaments present. In contrast to this, live actin-RFP showed significantly increased stress fiber formation in Wnt5A-treated HCAEC. Similar changes were observed in cells treated with IL-1β (Fig. 4A).
Figure 4.
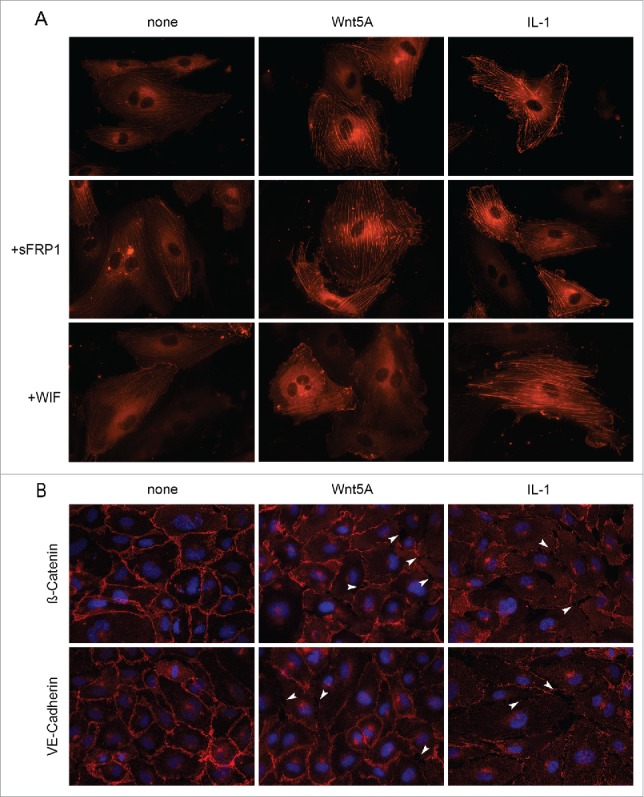
Actin stress fiber formation and adherens junction disruption in HCAEC in response to Wnt5A and IL-1β. (A) Actin stress fibers stained by live actin-RFP in HCAEC either untreated (left panel) or treated with Wnt5A (middle panel) or IL-1β (IL-1, right panel), in the absence or presence of sFRP1 or WIF. Stained cells were captured at 8 h after treatment using a Zeiss Axio Observer.Z1, original magnification 400x. (B) Immunofluorescence staining for β-catenin and VE-cadherin (red) in HCAEC treated with either Wnt5A or IL-1β (IL-1) for 8 h. Nuclei are stained blue. Arrowheads point to inter-endothelial gaps. Zeiss Axioskope, original magnification 400x.
We then checked if Wnt5A-induced stress fiber formation can be prevented by the Wnt antagonists secreted Frizzled-related peptide (sFRP) 1 and WIF1. sFRP1, a member of sFRP family, contains a cysteine-rich domain homologous to the Wnt binding extracellular domain of Fzd and Ror receptors. WIF-Wnt binding domains are homologous to the Wnt binding extracellular domain of Ryk.16 Treatment of HCAEC with Wnt5A in the presence of sFRP1 did not prevent Wnt5A-induced F-actin stress fiber formation, whereas stress fiber formation was notably decreased when HCAEC were treated with a combination of Wnt5A and WIF1 (Fig. 4A). This suggests the involvement of Ryk as an upstream receptor for ROCK in Wnt5A-triggered cytoskeletal rearrangements in HCAEC. Stress fiber formation induced by IL-1β remained unaffected by the presence of sFRP1 and WIF1 (Fig. 4A).
Wnt5A disrupts adherens junction proteins
As increased stress fiber formation disrupts the assembly of β-catenin and VE-cadherin adhesion proteins in adherens junctions or inter-endothelial junctions,22 we next checked if Wnt5A disrupts these adhesion proteins in HCAEC. HCAEC monolayers were treated with Wnt5A for 8 h and the organization of β-catenin and VE-cadherin were examined by immunofluorescence staining. IL-1β treatment served as a positive control in these experiments. In non-treated cells, β-catenin and VE-cadherin were regularly distributed along the cellular periphery and formed tight contacts at cell borders (Fig. 4B). Upon treatment with Wnt5A, β-catenin and VE-cadherin were lost specifically at cell borders resulting in small holes and separations (gaps) between cells. Similar effects were observed in HCAEC treated with IL-1β (arrowheads, Fig. 4B).
Functional experiments targeting the monolayer integrity by live recording of trans-endothelial resistance reveal enhanced permeability through Wnt5A/Ryk signaling
Since Wnt5A disrupted inter-endothelial adherens junctions and induced the formation of intercellular gaps, we proposed that Wnt5A could impair the barrier functions and increase the permeability of an endothelial monolayer. To prove this, HCAEC monolayers cultured in 8W10E+ electrode arrays were treated with Wnt5A and changes in trans-endothelial electrical resistance of the endothelial monolayer were recorded in real time by Electric Cell-substrate Impedance Sensing (ECIS). Upon treatment with Wnt5A, there was a significant decrease in the resistance of HCAEC monolayers to alternating current. The Wnt5A-induced changes in electrical resistivity became significantly evident after 3 h after the addition of Wnt5A and lasted for more than 10 h. Likewise, treatment with IL-1β significantly decreased the resistance of HCAEC monolayers (Fig. 5A). Our data indicate that inflammatory activation of HCAEC with Wnt5A or IL-1β affects endothelial barrier function and increases monolayer permeability.
Figure 5.
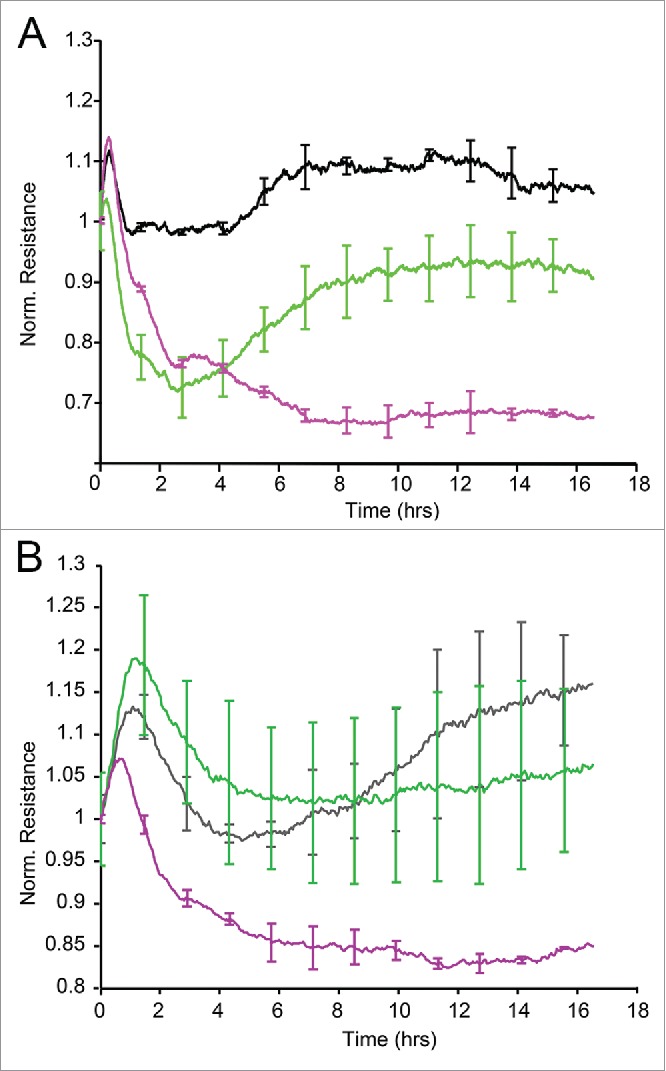
ECIS barrier function assays in HCAEC. Uniform confluent monolayers of HCAEC cultured in stabilized and collagen coated 8W10E+ ECIS culture chambers (see Methods and Figure 5 of Online Supplement) were treated with Wnt5A and IL-1β. Resistance of HCAEC monolayers was continuously recorded in Ohms every 5 min at multiple frequencies ranging from 62.6 Hz to 64 kHz, normalized to its value at time zero, and plotted with respect to time. Resistance measured from duplicate wells were grouped and averaged to plot as a single curve with error bars representing the SD. Data shown are the resistance measurements conducted at 4000 Hz from 4 independent experiments. (A) Un transfected HCAEC. Black, Untreated; Green, Wnt5A; Purple, IL-1β. (B) Ryk siRNA transfected HCAEC. Black, Untreated; Green, Wnt5A; Purple, IL-1β.
Since HCAEC express constitutively high levels of Ryk, and the Ryk specific Wnt antagonist WIF1 diminished Wnt5A-induced endothelial stress fiber formation, we checked if silencing Ryk expression would prevent Wnt5A induced hyperpermeability of HCAEC monolayers. To silence Ryk expression, HCAEC were transfected with Ryk-specific siRNA. Additionally, HCAEC were transfected with highly validated negative control siRNA to detect off target effects caused by siRNA or transfection reagents. To prove the efficiency of Ryk knockdown, Ryk mRNA expression was compared by qRT-PCR in cells transfected with Ryk-siRNA and in cells transfected with negative control siRNA. Ryk mRNA expression was approximately 70% lower in cells transfected with Ryk-siRNA than in cells transfected with negative control siRNA (Online Supplement Fig. 3A). Immunofluorescence staining further confirmed that Ryk-siRNA transfected HCAEC expressed significantly lower amounts of Ryk receptor compared with cells transfected with negative control siRNA (Online Supplement Fig. 3B, 3C). These results prove that efficient and specific silencing of Ryk was achieved in HCAEC.
Next, Ryk-silenced HCAEC were seeded onto 8W10E+ electrode arrays, treated with Wnt5A or IL-1β and changes in trans-endothelial electrical resistance were measured in real time by ECIS. Treatment with Wnt5A did not alter the resistance of Ryk-silenced HCAEC. In contrast, IL-1β significantly decreased the resistance of Ryk-silenced HCAEC (Fig. 5B). This result confirms that Wnt5A specifically signals through Ryk in HCAEC.
Wnt5A-impaired motility of endothelial cells is restored by ROCK inhibition
Since Wnt5A induced stress fiber formation and impaired the barrier function of endothelial monolayers, we hypothesized that Wnt5A could also affect the motility of endothelial cells. An ECIS assisted wounding assay was used to investigate whether Wnt5A influences the motility of HCAEC. HCAEC monolayers on single electrode (8W1E) arrays were treated with Wnt5A and subjected to electric current that killed cells in a defined area of the electrode and created a wound of standard size. The migration of viable cells to the wounded area was recorded in real time by ECIS. Treatment with Wnt5A significantly reduced the number of cells migrating to the wounded area (indicated by a significant decrease in impedance) resulting in delayed wound closure. A similar effect was observed in HCAEC treated with IL-1β whereby cell migration and subsequent wound closure were significantly affected (Fig. 6A).
Figure 6.
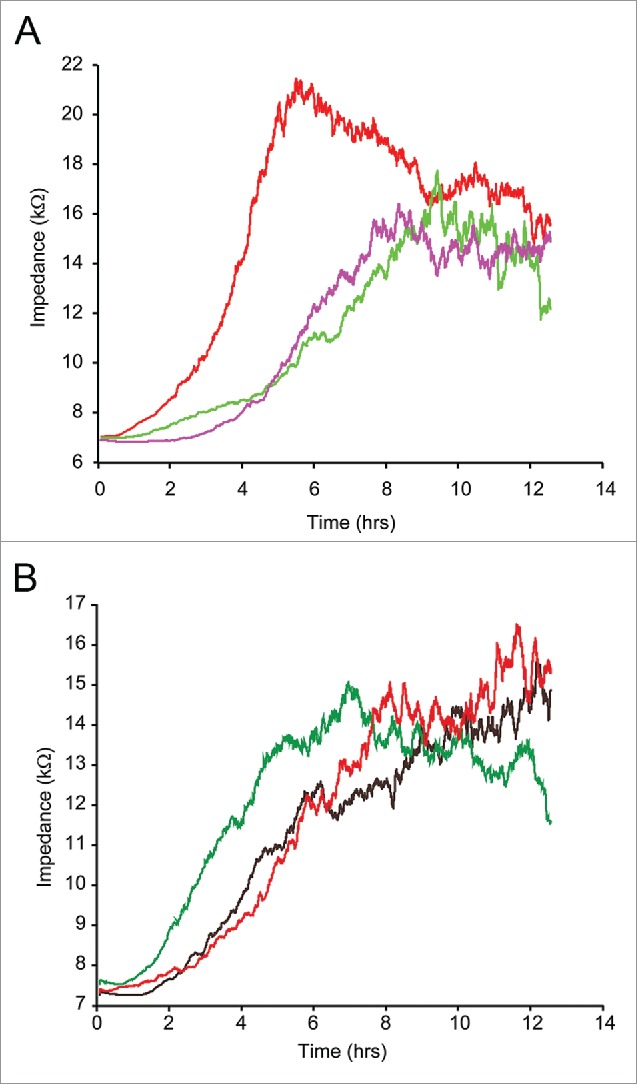
ECIS assisted wound healing assay. Uniform confluent monolayers of HCAEC cultured in stabilized and collagen coated single electrode 8W1E ECIS culture chambers (Methods) were treated with Wnt5A and IL-1β either alone or in combination with the ROCK inhibitor Y-27632 for 3 h. At time 0 h, a defined wound with a diameter of 250 µm was created in each of the 8 wells by applying an elevated electric field at 1400 μA, 60 kHz for 20 sec. Immediately after wounding, impedance of HCAEC was continuously recorded in Ohms every 5 min at multiple frequencies ranging from 62.6 Hz to 64 kHz and plotted with respect to time. Data shown represent the measurements conducted at 4000 Hz from 4 independent experiments. (A) HCAEC without ROCK inhibition. Red, Untreated; Purple, Wnt5A; Green, IL-1β. (B) HCAEC with ROCK inhibition. Black, Control with Y-27632; Red, Wnt5A+Y-27632; Green IL-1β+Y-27632.
We have shown that Wnt5A-induced phosphorylation of LIMK2 and CFL1 could be suppressed by inhibiting ROCK. ROCK is involved in a variety of cellular processes including cell motility, actin cytoskeleton rearrangements and cell adhesion.20 Therefore, we investigated whether inhibiting ROCK prevents Wnt5A-induced impairment of HCAEC motility in an ECIS assisted wounding assay. HCAEC treated with a combination of Wnt5A and the ROCK inhibitor Y-27632 showed significant increase in cell motility and faster wound closure compared with HCAEC treated with Wnt5A in the absence of ROCK inhibitor. Similar effects were observed in HCAEC treated with a combination of IL-1β and Y-27632 (Fig. 6B). This data further indicate that ROCK is involved in Wnt5A/Ryk signaling in HCAEC.
Discussion
Using a genome-wide expression survey, we characterized the targets and determined the biological function of paracrine inflammatory Wnt5A signaling in a tightly controlled system of adult human vascular endothelial cells. Our study reveals that Wnt5A primarily regulates genes involved in actin cytoskeleton rearrangements in endothelial cells. We have identified a novel Wnt5A/Ryk signaling mechanism that targets the ROCK/LIMK2/CFL1 axis to induce cytoskeleton remodeling and barrier dysfunction in vascular endothelial cells. Gene expression profiling of IL-1β performed in parallel showed that IL-1β mainly regulates genes and pathways associated with immune responses, thereby confirming previous findings.5 The response to IL-1β proves the integrity of biological functions in our HCAEC system.
Our previous study revealed that Wnt5A signals through Fzd5 to cause inflammatory activation of human macrophages.9 In this study, we found that HCAEC constitutively express high levels of Ryk, and only trace amounts of Fzd5 (Fig. 2C and D), thereby suggesting that Wnt5A could exert its function without upregulating the level of endogenous Ryk receptors. We further found that Wnt5A-induced stress fiber formation can be prevented by WIF1, thereby pointing to the involvement of Ryk as the receptor for Wnt5A-mediated cytoskeleton remodeling in HCAEC (Fig. 4A). Additionally, the transcriptome analysis of the current study shows that Wnt5A downregulates the expression of LRP5 (Table 1). Since LRP5 acts as a co-receptor for Fzd5 in β-catenin dependent signaling,17,23 the canonical Wnt/β-catenin pathway involving Fzd receptors could be repressed by Wnt5A in HCAEC. This is supported by our finding that β-catenin remains sequestered on the plasma membrane of HCAEC after Wnt5A treatment and is not transported to the nucleus (Fig. 4B) to signal via the β-catenin pathway. The subsequent functional experiments demonstrate that Wnt5A-induced HCAEC barrier impairment can be prevented by silencing Ryk expression, thereby providing direct evidence that Wnt5A signals through Ryk (Fig. 5). Thus, the present study identifies a novel β-catenin independent non-canonical Wnt5A/Ryk signaling mechanism that targets the ROCK/LIMK2/CFL1 axis to impair barrier function in human vascular endothelial cells. Ryk is a member of the family of atypical receptor tyrosine-protein kinases. It comprises an extracellular Wnt binding domain, a PDZ binding motif and an intracellular atypical kinase domain lacking tyrosine kinase activity. Wnt5A/Ryk signaling has been functionally demonstrated in adult rat corticospinal axons.24 Moreover, Derailed, a member of the Ryk family of receptors in Drosophila, has been described as acting as a receptor for Wnt5A in axon guidance.25 Furthermore, abundant expression of the Ryk gene was reported in mouse pulmonary capillary endothelial cells.26 Data obtained in the present study indicate that Wnt5A signaling via ROCK activation induces actin cytoskeletal rearrangements and disrupts VE-cadherin-mediated intercellular adhesion. The involvement of Ryk but not other known Wnt receptors such as Fzd or Ror is further evident from the observation that Wnt5A-induced actin stress fiber formation was prevented by the Ryk specific antagonist WIF1, while sFRP1 was uneffective (Fig. 4A). Previous studies have already established that inactivation of ROCK and RhoA prevents endothelial hyperpermeability.27,28
Our study reveals that Wnt5A decreases the motility of HCAEC and combining Wnt5A with the ROCK inhibitor Y-27632 restores the migratory capacity of HCAEC (Fig. 6). This is in full accordance with the proposed model of Wnt5A/Ryk signaling. The reduction in endothelial cell motility caused by Wnt5A is due to actin cytoskeleton rearrangements and increased stress fiber formation (Fig. 4A) induced by ROCK mediated phosphorylation of LIMK2 and CFL proteins (iROCK, Fig. 3A–C).
Our present study further proves that Wnt5A, unlike IL-1β, does not induce a cytokine storm (IL-6, IL-8, CCL2; Online Supplement Fig. 1) or cell adhesion molecules (ICAM-1, VCAM-1; Online Supplement Fig. 2) in endothelial cells as has been reported previously.14 Data from our genome-wide transcriptome analysis and functional studies indicate that Wnt5A/Ryk signaling in HCAEC is independent of the induction of any other inflammatory mediators. The biological integrity of the cells in our system is proven by the typical inflammatory response to IL-1β (Online Supplement Figs. 1 and 2). An interesting observation during the present study is the induction of Wnt5A by IL-1β on transcriptional and translational level (Online Supplement Fig. 4). That IL-1β triggers endogenous Wnt5A production in HCAEC is in accordance with the concept of immunovascular communication between leukocytes and vascular target cells. Wnt5A and IL-1β, both produced by inflammatory activated monocytes and present in serum in systemic inflammation and sepsis, might act independently and complementarily to each other on vascular endothelial cells. This important observation needs to be further investigated. Wnt5A expression is elevated in sepsis9 and our present study finds Wnt5A as a critical mediator of endothelial hyperpermeablity. This warrants further studies to determine the correlation of Wnt5A levels with vascular leakage in appropriate septic animal models. However, current sepsis animal models fail to mimic the human disease for a number of reasons, and most of the highly successful trials in animal models did not improve patient survival when translated to clinical settings.12,29
In conclusion, the present study shows for the first time that Wnt5A/Ryk signaling impairs the barrier functions and affects the wound healing of a damaged monolayer of human vascular endothelial cells. This suggests that Wnt5A, secreted by activated macrophages during a systemic inflammatory response, could cause capillary leakage and subsequent edema in severe sepsis. Restoring vascular integrity along with reducing edema is an established factor that improves the prognosis of septic shock. Considering the essential role for Rho/ROCK in Wnt5A signaling in adult human vascular endothelial cells, therapeutic approaches targeting the Wnt5A/Ryk/ROCK signaling pathway would probably improve the prognosis of sepsis.
Materials and methods
Cell culture
HCAEC (Clonetics, Lonza) were propagated in EBM-2 medium (Clonetics, Lonza) supplemented with EGM-2MV Single Quots with 5% FBS (Clonetics, Lonza) as described.5 Cells were cultured under standard conditions (37°C, 5% CO2, 80% humidity) in a Class 100 HEPA air filtered system (SteriCult, Fisher Scientific). To avoid masked low-level contamination in cultures, culture medium without antibiotics was used throughout the study. In experimental settings, cells from passages 3 to 6 were used and serum concentration of the medium was reduced to 2% FBS. HCAEC were treated with recombinant human/mouse Wnt5A (250 ng/mL; CHO-derived Gln38-Lys380, purity > 80%, endotoxin level < 1.0 EU/µg of protein; Cat. No. 645-WN, R&D systems), recombinant human IL-1β (20 U/mL; purity > 98%; PeproTech), recombinant human secreted Frizzled-related peptide (sFRP) 1 (10 μg/mL; purity > 95%; Cat. No.1384-SF, R&D systems), recombinant human WIF1 (15 μg/mL; purity > 97%; Cat. No. 1341-WF-050, R&D systems). For all pipetting steps, Biopure ep Dualfilter T.I.P.S. sterile filter tips (Eppendorf) were used.
RNA isolation and quantitative Real Time-PCR (qRT-PCR) analyses
RNA isolation and qRT-PCR were performed as described9 with the following modifications and primers. RNA was reverse transcribed into cDNA using TaqMan® Kit (Applied Biosystems). qRT-PCR was performed using Fast SYBR® Green Master Mix and a 7500 Fast Real-Time PCR System (Applied Biosystems). Relative quantification analyses were performed with HPRT gene as endogenous control using RQ Study SDS software v1.4 (Applied Biosystems). Sequence specific PCR detection primers used for Wnt5A, IL-6, IL-8, HPRT, MCP-1, ICAM-1, Ryk and Fzd5 are as follows: Wnt5A forward, 5′-AGT TGC CTA CCC TAG C-3′; Wnt5A reverse, 5′-GTG CCT TCG TGC CTA T-3′; IL-6 forward, 5′-CCT GAC CCA ACC ACA AA-3′; IL-6 reverse, 5′-AGT GTC CTA ACG CTC ATA C-3′; IL-8 forward, 5′-AGA CAG CAG AGC ACA CAA GC-3′; IL-8 reverse, 5′-ATG GTT CCT TCC GGT GGT-3′; HPRT forward, 5′-CCA GTC AAC AGG GGA CAT AAA-3′; HPRT reverse, 5′-CAC AAT CAA GAC ATT CTT TCC AGT-3′; MCP-1 forward, 5′-CAG CCA GAT GCA ATC AAT GC-3′; MCP-1 reverse, 5′-GTG GTC CT GGA ATC CTG AA-3′; ICAM-1 forward, 5′-TTG GAA GCC TCA TCC G-3′; ICAM-1 reverse, 5′-CAA TGT TGC GAG ACC C-3′; Ryk forward, 5′-TGC CCT CTC CAG AGA CTT GT-3′; Ryk reverse, 5′-CAT CTC GAA GGG GTC AAT GT-3′; Fzd5 forward, 5′-CT CGA CAT GGA ACG C-3′; Fzd5 reverse, 5′-CGT GAT GGA CTT GAC GC-3′. Temperature cycling conditions of the PCR were as follows; an initial denaturation step (10 min, 95°C) followed by 40 cycles of denaturation (15 sec, 95°C), annealing (30 sec, 55°C), and extension (30 sec, 72°C).
Constitutively expressed mRNA levels of Fzd5 and Ryk in HCAEC were calculated by the 1/ΔCt method and values were given as arbitrary units.
Microarray based differential gene expression profiling
Gene expression profiling by competitive 2-color hybridization of cRNA probes onto 4 × 44K Human Whole Genome Oligonucleotide microarrays (Agilent Technologies) was performed as described.30 Briefly, total RNA was isolated using RNeasy Mini Kit (Qiagen). Total RNA was quantified by Nanodrop spectrophotometry and RNA integrity was confirmed using the Agilent 2100 Bioanalyzer System (Agilent Tech.). Only RNA samples with a RNA integrity number (RIN) >9 were used for microarray experiments. From each sample, 500 ng of total RNA was reverse transcribed and labeled with Cy3- and Cy5-CTP using the 2-color Quick Amp Labeling Kit (Agilent Tech.), containing internal control probes and spike in's (labeled Spike A mix with Cy-3 and Spike B mix with Cy-5) for control of reaction performance and background normalization of arrays. cRNA fragmentation and hybridization onto the Human GE 4×44K V2 Microarray chips were performed as per the Quick Amp Labeling protocol (Agilent Techno., version 5.7, 2008). To avoid ozone-induced deterioration of cyanine dyes, array chips were washed with stabilization and drying solution (Agilent Tech.).
Microarray data analyses
Scanning, feature extraction and preprocessing including data normalization of the microarrays were done by Microarray Scanner and Feature Extraction Software 10.7 (Agilent Tech. Inc.) with default settings for Agilent 4 × 44 K 2-color arrays. The preprocessed data was further analyzed using GeneSpring GX 9.0 Software (Agilent Tech. Inc.) with default settings for 2-color arrays using fold change cutoff of 1.5. Gene ontology analysis was performed using Metacore GeneGO software (Thomson Reuters, http://portal.genego.com). Genes regulated ≥ 2-fold in their expression and satisfying a P value < 0.05 were grouped into pathways according to their biological functions and gene ontology classes.
Immunofluorescence staining
HCAEC were cultured in 4-chamber culture slides coated with rat tail collagen type I (diluted 0.01% in sterile pyrogen-free water; BD Biosciences). To stain for the adherens junction proteins β-catenin and VE-cadherin, HCAEC were grown to a confluent monolayer. After treatment with the stimuli indicated, cells were fixed with 4% formalin in PBS (in case of detecting adherens junction proteins) for 20 min or with a mixture of acetone and methanol (1:1) for 5 min (in case of Fzd5, Ryk and Wnt5A detection). Slides were washed 3 times with PBS (pH 7.4) at room temperature (RT). Slides were treated with blocking solution containing 10% goat serum and 1% BSA in PBS for 1 h at RT in case of secondary antibodies derived from goat. Rabbit blocking solution (10% rabbit serum and 1% BSA in PBS) was used in case of secondary antibodies derived from rabbit. Slides were again washed with PBS at RT and incubated with primary antibodies diluted in blocking solution at 4°C overnight. The following antibodies were used for immunofluorescence: goat-anti-Wnt5A (1:50; Cat. No. AF645, R&D Systems), rabbit-anti-Ryk (1:500; Product No. Ab5518, Abcam), rabbit-anti-Fzd5 (1:100; Product No. Ab14475, Abcam), rabbit-anti-β-catenin (1:150; Product No. 9562, Cell Signaling Technology), rabbit-anti-VE-cadherin (1:100; Product No. 2158, Cell Signaling Technology), rabbit-anti-phosphorylated LIMK2 (1:300; pT505; Product No. Ab131343, Abcam), rabbit-anti-phosphorylated CFL1 (1:500; pS3; Product No. Ab100836, Abcam). After three washes with PBS at RT, slides were incubated with Alexa 568 labeled goat anti-rabbit or rabbit anti-goat secondary antibodies (1:2000; Molecular Probes, Invitrogen) depending on the first antibody's species. Incubations with secondary antibodies were combined with Alexa Fluor 488 Phalloidin (1:80; Molecular Probes, Invitrogen) for 1 h at RT in the dark. After three washes with PBS at RT, slides were counterstained with 10 μg/mL diamidino-phenylindole (DAPI; Sigma-Aldrich) and mounted using ProLong Gold AntifadeR (Life Technologies). Images were captured by Axioskope microscope equipped with an Axio-Cam MRm digital camera using AxioVision Rel.4.6 software (Carl Zeiss). The intensity of cellular fluorescent signals was quantified from 5 different positions (areas; 20 cells/area) of the respective fluorescent images covering approx. 90% of total cells in the field using ImageJ based Fiji software (Fiji is Just ImageJ) and corrected for background fluorescence.
Live cell imaging of actin
HCAEC grown in rat tail collagen type I coated optical 96 microwell culture plates (Thermoscientific) were transduced with CellLight Actin-RFP probe (Cat. No. C10583, Life Technologies) as per manufacturer's instructions. Cells were treated with Wnt5A or IL-1β alone or in combination with either sFRP1 or WIF1 for 8 h. Images of the cultures were captured with an Axio Observer.Z1 inverted microscope equipped with Axio-Cam MRm digital camera using ZEN 2012 software (Carl Zeiss).
Endothelial barrier function assays
The response of endothelial barrier to a particular stimulus can be assessed non-invasively in a fully standardized manner by continuously recording changes in trans-endothelial electric resistance using Electric Cell-substrate Impedance Sensing (ECIS).31,32 Endothelial barrier function was recorded in real-time using the ECIS Z Theta system (Applied Biophysics) with associated software v.1.2.126 PC, as described33 with the following modifications. 8W10E+ electrode chamber arrays were equilibrated with EBM-2 basal medium (Clonetics, Lonza) for 24 h and coated with rat tail collagen type I (diluted 0.01% in sterile pyrogen-free water; BD Biosciences) for 12 h. Then HCAEC in EGM2MV complete medium (Clonetics, Lonza) were added to the wells with a density of 80,000- 90,000 cells/well and grown up to 24 h to form dense monolayers (Supplemental Fig. 5). Treatments were then conducted by replacing the medium with fresh medium without or with stimuli (Wnt5A, IL-1β). Measurements were conducted at multiple frequencies ranging from 62.6 Hz to 64 kHz. 8W10E+ contains 40 electrodes per each of its 8 wells that traced the cells at 40 different locations in each well and averaged the measurements.
ECIS assisted cell migration assay
Motility/wound healing assays were performed in ECIS single electrode arrays as described34 with the following modifications. 8W1E single electrode chambers were equilibrated with EBM-2 basal medium (Clonetics, Lonza) for 24 h and coated with rat tail collagen type I (BD Biosciences) for 12 h. HCAEC in EGM2MV complete medium (Clonetics, Lonza) were added to the wells with a cell density of 80,000-90,000 cells/well. To attain confluent HCAEC monolayers, single electrode array chambers were further incubated for up to 24 h. Stimuli were then added as described above for barrier function assays. 3 h after stimulation, wounding was activated with default conditions for 8W1E arrays (1400 μA, 60000 Hz, for 20 sec). This fully standardized method produces a highly reproducible wound with an exact diameter of 250 µm surrounded by undamaged living cells in 8W1E chambers. After wounding, impedance was continuously recorded to observe the migration of cells and formation of confluent monolayer within the defined wounded area.
Ryk silencing
Specific and functionally validated small interfering RNA (siRNA) against human Ryk (Hs-RYK-9, Cat No. SI02223074) was obtained from Qiagen. To control for “off target” effects of siRNAs, validated AllStars negative control siRNA (Qiagen) with no homology to any mammalian gene was used. For knockdown of Ryk expression, transfection complexes formed of Ryk-siRNA (5 nM) and 6 μL/mL HiPerFect Transfection reagent (Qiagen) in EBM-2 basal medium were added to 80% confluent cultures of HCAEC in 24-well plates in a total volume of 500 μL/well. After 24 h of incubation, transfected cells were split using Trypsin EDTA, seeded onto collagen coated 6 well plates, retransfected for a second round as above and incubated until the cells attained 90% confluency. At this stage, cells were either stimulated with Wnt5A or IL-1β for 8 h and lysed (for qRT-PCR analyses) or trypsinized and transferred into collagen-coated 8W10E+ arrays (for ECIS barrier function measurements). Alternatively, HCAEC after one round of Ryk-siRNA transfection were harvested by trypsinization and seeded into collagen-coated BD Falcon 4 chamber culture slides (BD Biosciences). There, a second round of Ryk-siRNA transfection was performed as in 24-well plates. After growing to a confluent monolayer, cells were fixed for immunostainings. Stimulations were carried out in complete EGM medium with 2% FBS devoid of transfection complexes.
ROCK inhibition
To inhibit ROCK activity, Y-27632 (Cat. No. 688000, Calbiochem; Millipore) was used35 at an optimal concentration for this system of 10 μmol/L. HCAEC were incubated in medium containing Y-27632 (10 μmol/L) alone or in combination with either Wnt5A or IL-1β for 1 h and 4 h.
Flow cytometry
HCAEC stimulated with Wnt5A, IL-1β or combination of these 2 agents for 24 h were trypsinized and resuspended in PBS supplemented with 0.1% BSA (sample buffer) such that cell density did not exceed more than 105 cells/tube. Cells were incubated on ice with directly labeled mouse antibodies against human VCAM-1 (CD106, fluorescein isothiocyanate label, BD pharmingen), ICAM-1 (CD54, phycoerithrin labeled, BD pharmingen), E selectins (CD62E, allophycocyanin label, BD pharmingen) and human integrin b1 chain (CD29, allophycocyanin label, BD pharmingen) at a final concentration of 2 μg/ml. Isotype controls were performed using mouse IgG polyclonal antibodies. After washing twice with FACS buffer, samples were analyzed using FACS CantoII cell sorting system (BD Biosciences, Allschwil, Switzerland). Cells were gated according to their forward and sidescatter pattern, at least 10'000 gated events were acquired for analysis. The fluorescence intensity of corresponding samples labeled with the isotype control antibody was used to set the individual threshold of antigen positivity. Data were analyzed using FlowJo 7.2.5 software.
Statistical analysis
Data were analyzed using GraphPad Prism software version 5.04 (GraphPad Software, San Diego, CA). An unpaired 2-tailed Student's t-test or for comparison of data among groups, 1-way ANOVA followed by the Newman-Keuls test was used and differences were considered statistically significant at P < 0.05.
Supplementary Material
Abbreviations
- CFL1
cofilin-1
- ECIS
Electric Cell-substrate Impedance Sensing
- Fzd
Frizzled
- HCAEC
human coronary artery endothelial cells
- IL
interleukin
- LIMK2
LIM kinase-2
- qRT-PCR
quantitative Real Time-PCR
- ROCK
Rho-associated protein serine/threonine kinases
- sFRP
secreted Frizzled-related peptide
- WIF1
Wnt inhibitory factor 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Cheryl Pech, PhD, for critical reading of the manuscript.
Funding
This study was supported by the Swiss National Science Foundation No. 31-124861 to Gabriele Schoedon.
References
- [1].Russell JA. Management of sepsis. N Engl J Med 2006; 355:1699-713; PMID:17050894; http://dx.doi.org/ 10.1056/NEJMra043632 [DOI] [PubMed] [Google Scholar]
- [2].Thachil J, Toh CH, Levi M, Watson HG. The withdrawal of Activated Protein C from the use in patients with severe sepsis and DIC [Amendment to the BCSH guideline on disseminated intravascular coagulation]. Br J Haematol 2012; 157:493-4; PMID:22225506; http://dx.doi.org/ 10.1111/j.1365-2141.2011.09019.x [DOI] [PubMed] [Google Scholar]
- [3].Lee WL, Slutsky AS. Sepsis and Endothelial Permeability. N Engl J Med 2010; 363:689-91; PMID:20818861; http://dx.doi.org/ 10.1056/NEJMcibr1007320 [DOI] [PubMed] [Google Scholar]
- [4].Mehta D, Ravindran K, Kuebler WM. Perspective: Novel regulators of endothelial barrier function. Am J Physiol Lung Cell Mol Physiol 2014; 307:L924-L35; PMID:25381026; http://dx.doi.org/ 10.1152/ajplung.00318.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Franscini N, Bachli EB, Blau N, Leikauf M-S, Schaffner A, Schoedon G. Gene expression profiling of inflamed human endothelial cells and influence of activated protein C. Circulation 2004; 110:2903-9; PMID:15505101; http://dx.doi.org/ 10.1161/01.CIR.0000146344.49689.BB [DOI] [PubMed] [Google Scholar]
- [6].Reinhart K, Karzai W. Anti-tumor necrosis factor therapy in sepsis: update on clinical trials and lessons learned. Crit Care Med 2001; 29:S121-5; PMID:11445746; http://dx.doi.org/ 10.1097/00003246-200107001-00037 [DOI] [PubMed] [Google Scholar]
- [7].Fisher CJ Jr., Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, Iberti TJ, Rackow EC, Shapiro MJ, Greenman RL, et al.. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. J Am Med Assoc 1994; 271:1836-43; PMID:8196140; http://dx.doi.org/ 10.1001/jama.1994.03510470040032 [DOI] [PubMed] [Google Scholar]
- [8].Opal SM, Fisher CJ Jr., Dhainaut JF, Vincent JL, Brase R, Lowry SF, Sadoff JC, Slotman GJ, Levy H, Balk RA, et al.. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med 1997; 25:1115-24; PMID:9233735; http://dx.doi.org/ 10.1097/00003246-199707000-00010 [DOI] [PubMed] [Google Scholar]
- [9].Pereira C, Schaer DJ, Bachli EB, Kurrer MO, Schoedon G. Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the antiinflammatory action of activated protein C and interleukin-10. Arterioscler, Thromb, Vasc Biol 2008; 28:504-10; PMID:18174455; http://dx.doi.org/ 10.1161/ATVBAHA.107.157438 [DOI] [PubMed] [Google Scholar]
- [10].Schulte DM, Kragelund D, Muller N, Hagen I, Elke G, Titz A, Schadler D, Schumacher J, Weiler N, Bewig B, et al.. The wingless-related integration site-5a/secreted frizzled-related protein-5 system is dysregulated in human sepsis. Clin Exp Immunol 2015; 180:90-7; PMID:25382802; http://dx.doi.org/ 10.1111/cei.12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].George SJ. Wnt pathway: a new role in regulation of inflammation. Arterioscler, Thromb, Vasc Biol 2008; 28:400-2; PMID:18296599; http://dx.doi.org/ 10.1161/ATVBAHA.107.160952 [DOI] [PubMed] [Google Scholar]
- [12].Riedemann NC, Guo R-F, Ward PA. Novel strategies for the treatment of sepsis. Nat Med 2003; 9:517-24; PMID:12724763; http://dx.doi.org/ 10.1038/nm0503-517 [DOI] [PubMed] [Google Scholar]
- [13].Xu H, Ye X, Steinberg H, Liu SF. Selective blockade of endothelial NF-κB pathway differentially affects systemic inflammation and multiple organ dysfunction and injury in septic mice. J Pathol 2010; 220:490-8; PMID:20020511 [DOI] [PubMed] [Google Scholar]
- [14].Kim J, Kim J, Kim DW, Ha Y, Ihm MH, Kim H, Song K, Lee I. Wnt5a induces endothelial inflammation via β-catenin–independent signaling. J Immunol 2010; 185:1274-82; PMID:20554957; http://dx.doi.org/ 10.4049/jimmunol.1000181 [DOI] [PubMed] [Google Scholar]
- [15].Linscheid P, Schaffner A, Blau N, Schoedon G. Regulation of 6-pyruvoyltetrahydropterin synthase activity and messenger RNA abundance in human vascular endothelial cells. Circulation 1998; 98:1703-6; PMID:9788822; http://dx.doi.org/ 10.1161/01.CIR.98.17.1703 [DOI] [PubMed] [Google Scholar]
- [16].Green J, Nusse R, van Amerongen R. The role of Ryk and Ror receptor tyrosine kinases in Wnt signal transduction. Cold Spring Harb Perspect Biol 2014; 6:a009175; PMID:24370848; http://dx.doi.org/ 10.1101/cshperspect.a009175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol 2009; 10:468-77; PMID:19536106; http://dx.doi.org/ 10.1038/nrn2674 [DOI] [PubMed] [Google Scholar]
- [18].Rath N, Olson MF. Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep 2012; 13:900-8; PMID:22964758; http://dx.doi.org/ 10.1038/embor.2012.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999; 285:895-8; PMID:10436159; http://dx.doi.org/ 10.1126/science.285.5429.895 [DOI] [PubMed] [Google Scholar]
- [20].Liao JK, Seto M, Noma K. Rho kinase (ROCK) inhibitors. J Cardiovasc Pharmacol 2007; 50:17-24; PMID:17666911; http://dx.doi.org/ 10.1097/FJC.0b013e318070d1bd [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fazal F, Bijli KM, Minhajuddin M, Rein T, Finkelstein JN, Rahman A. Essential role of cofilin-1 in regulating thrombin-induced RelA/p65 nuclear translocation and intercellular adhesion molecule 1 (ICAM-1) expression in endothelial cells. J Biol Chem 2009; 284:21047-56; PMID:19483084; http://dx.doi.org/ 10.1074/jbc.M109.016444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci 2008; 1123:134-45; PMID:18375586; http://dx.doi.org/ 10.1196/annals.1420.016 [DOI] [PubMed] [Google Scholar]
- [23].Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature 2005; 438:873-7; PMID:16341017; http://dx.doi.org/ 10.1038/nature04185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Miyashita T, Koda M, Kitajo K, Yamazaki M, Takahashi K, Kikuchi A, Yamashita T. Wnt-Ryk signaling mediates axon growth inhibition and limits functional recovery after spinal cord injury. J Neurotrauma 2009; 26:955-64; PMID:19473059; http://dx.doi.org/ 10.1089/neu.2008.0776 [DOI] [PubMed] [Google Scholar]
- [25].Yoshikawa S, McKinnon RD, Kokel M, Thomas JB. Wnt-mediated axon guidance via the Drosophila Derailed receptor. Nature 2003; 422:583-8; PMID:12660735; http://dx.doi.org/ 10.1038/nature01522 [DOI] [PubMed] [Google Scholar]
- [26].Favre CJ, Mancuso M, Maas K, McLean JW, Baluk P, McDonald DM. Expression of genes involved in vascular development and angiogenesis in endothelial cells of adult lung. Am J Physiol Heart Circ Physiol 2003; 285:H1917-H38; PMID:12842817; http://dx.doi.org/ 10.1152/ajpheart.00983.2002 [DOI] [PubMed] [Google Scholar]
- [27].Carbajal JM, Gratrix ML, Yu CH, Schaeffer RC Jr. ROCK mediates thrombin's endothelial barrier dysfunction. Am J Physiol Cell Physiol 2000; 279:C195-204; PMID:10898731 [DOI] [PubMed] [Google Scholar]
- [28].Carbajal JM, Schaeffer RC Jr. RhoA inactivation enhances endothelial barrier function. Am J Physiol 1999; 277:C955-64; PMID:10564088 [DOI] [PubMed] [Google Scholar]
- [29].Fink MP. Animal models of sepsis and its complications. Kidney Int 2008; 74:991-3; PMID:18827799; http://dx.doi.org/ 10.1038/ki.2008.442 [DOI] [PubMed] [Google Scholar]
- [30].Schaer CA, Deuel JW, Bittermann AG, Rubio IG, Schoedon G, Spahn DR, Wepf RA, Vallelian F, Schaer DJ. Mechanisms of haptoglobin protection against hemoglobin peroxidation triggered endothelial damage. Cell Death Differ 2013; 20:1569-79; PMID:23995229; http://dx.doi.org/ 10.1038/cdd.2013.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bernas MJ, Cardoso FL, Daley SK, Weinand ME, Campos AR, Ferreira AJG, Hoying JB, Witte MH, Brites D, Persidsky Y, et al.. Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nat Protocols 2010; 5:1265-72; PMID:20595955; http://dx.doi.org/ 10.1038/nprot.2010.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Szulcek R, Bogaard HJ, van Nieuw Amerongen GP. Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. J Vis Exp 2014; (85):e51300; PMID:24747269; http://dx.doi.org/ 10.3791/51300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One 2013; 8:e59989; PMID:23527291; http://dx.doi.org/ 10.1371/journal.pone.0059989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Keese CR, Wegener J, Walker SR, Giaever I. Electrical wound-healing assay for cells in vitro. Proc Natl Acad Sci U S A 2004; 101:1554-9; PMID:14747654; http://dx.doi.org/ 10.1073/pnas.0307588100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S-i, Muguruma K, et al.. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotech 2007; 25:681-6; PMID:17529971; http://dx.doi.org/ 10.1038/nbt1310 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.