

Abstract
The natural products colchicine and combretastatin A-4 (CA4) have been inspirational for the design and synthesis of structurally related analogues and spin-off compounds as inhibitors of tubulin polymerization. The discovery that a water-soluble phosphate prodrug salt of CA4 (referred to as CA4P) is capable of imparting profound and selective damage to tumor-associated blood vessels paved the way for the development of a new therapeutic approach for cancer treatment utilizing small-molecule inhibitors of tubulin polymerization that also act as vascular disrupting agents (VDAs). Combination of salient structural features associated with colchicine and CA4 led to the design and synthesis of a variety of fused aryl-cycloalkyl and aryl-heterocyclic compounds that function as inhibitors of tubulin polymerization. Prominent among these compounds is a benzosuberene analogue (referred to as KGP18), which demonstrates sub-nM cytotoxicity against human cancer cell lines and functions (when administered as a water-soluble prodrug salt) as a VDA in mouse models. Structure activity relationship considerations led to the evaluation of benzocyclooctyl [6,8 fused] and indene [6,5 fused] ring systems. Four benzocyclooctene and four indene analogues were prepared and evaluated biologically. Three of the benzocyclooctene analogues were active as inhibitors of tubulin polymerization (IC50 < 5 μM), and benzocyclooctene phenol 23 was comparable to KGP18 in terms of potency. The analogous indene-based compound 31 also functioned as an inhibitor of tubulin polymerization (IC50 = 11 μM) with reduced potency. The most potent inhibitor of tubulin polymerization from this group was benzocyclooctene analogue 23, and it was converted to its water-soluble prodrug salt 24 to assess its potential as a VDA. Preliminary in vivo studies, which utilized the MCF7-luc-GFP-mCherry breast tumor in a SCID mouse model, demonstrated that treatment with 24 (120 mg/kg) resulted in significant vascular shutdown, as evidenced by bioluminescence imaging at 4 h post administration, and that the effect continued at both 24 and 48 h. Contemporaneous studies with CA4P, a clinically relevant VDA, were carried out as a positive control.
Keywords: benzocyclooctene analogues, indene analogues, vascular disrupting agents (VDAs), inhibitors of tubulin polymerization, small-molecule synthesis, bioluminescence imaging (BLI)
Graphical Abstract
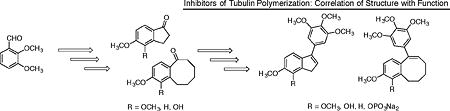
Synthesis of benzocyclooctene and indene analogues inspired by colchicine and combretastatin A-4.
1. Introduction
The colchicine site located on the β-subunit of the α,β-tubulin heterodimer continues to serve as a rich and productive target for a wide-range of structurally diverse small-molecule anticancer agents. Through a direct binding interaction at the colchicine site, these compounds inhibit the polymerization of the tubulin heterodimer into microtubules. Disruption of the inherent dynamic relationship between microtubules and tubulin heterodimers leads to two mechanistically distinct, yet complementary, cancer treatment strategies. One approach centers on the well known cytotoxic effect that results when disruption of the tubulin-microtubule system, caused by treatment with small-molecule inhibitors of tubulin polymerization or depolymerization, impacts the ability of cancer cells to divide. A wide-range of small-molecule therapeutic agents has been discovered and developed that function as antiproliferative agents through this mechanism (Fig. 1).
Figure 1.

Representative small-molecule anti-proliferative agents including colchicine,1 paclitaxel,2,3 CA4,4 vinblastine,5,6 OXi6196,7,8 KGP18,9–11 KGP156,10,12,13 OXi8006,14 and BNC105.15,16
This approach, while productive as an anticancer strategy, has limitations and challenges since both neoplastic and healthy cells are impacted. Targeting strategies such as the use of antibody-drug conjugates (ADCs) bearing active payloads have been successful with two FDA approved ADCs currently available.17–19 In another example, the targeting of tumor hypoxia with bioreductively activatable prodrug conjugates (BAPCs) has been explored20,21 with recent clinical trials involving PR-10422,23 and TH-30224–26 serving as representative examples (Fig. 2).
Figure 2.

Structures of PR-104 and TH-302.
PR-104 is hydrolyzed to its alcohol parent compound by available phosphatases and, under hypoxic conditions, is activated by NADPH-cytochrome P450 reductases to the reactive nitrogen mustards that crosslink to DNA and ultimately cause tumor cytotoxicity.22 TH-302 utilizes a similar hypoxia reduction strategy in order to release its 2-nitroimidazole trigger leaving the bromo-isophosphoramide mustard to cross link with DNA.26 The targeting of tumor hypoxia has also been examined with BAPCs that incorporate the tubulin-active natural product combretastatin A-4 (CA4).27 These strategies, along with others, continue to represent areas of ongoing research effort.
A second mechanistic approach to cancer therapy involving inhibition of the tubulin-microtubule protein system centers on selective disruption of existing vasculature feeding tumors, leading to limitation of oxygen and nutrient delivery to the cells and impedance of the ability of these cells to clear cellular waste products. This ultimately leads to tumor necrosis. This approach to targeting existing tumor-feeding vasculature utilizes anticancer agents referred to as vascular disrupting agents (VDAs) and is unique and mechanistically distinct from the fairly well-established and more commonly described therapeutic approach employing angiogenesis inhibiting agents (AIAs) that impede the formation of new vessels.28–30 Rapid neovascularization occurs when tumors grow larger than about 1 mm3, as they can no longer acquire sufficient nutrients from the surrounding vasculature.31 Such tumors require their own vascular network to supply oxygen and nutrients and remove waste products.28,29,32 Because of their rapid growth and development, these vessels feature irregular branching and diameter, poor wall structure, abnormal bulges, and blind ends.31,33–36 This immature vasculature provides an attractive target for cancer therapy. Therapeutic agents that target tumor vasculature are referred to as vascular targeting agents, which can be divided into AIAs and VDAs.28,33,30 AIAs inhibit the growth of new vasculature to the tumor, while VDAs damage already sprouted vessels.35–40 VDAs are sub-divided into biological agents and small-molecule therapeutics (Fig. 3),40 and treatment with either of these entities leads to tumor necrosis.
Figure 3.

Several clinically relevant VDAs including CA4P,41–45 CA1P,43,46 BNC105,15,16,43,45,47 AVE8062,43,48–50 and ZD6126.42,43,51
Colchicine, a natural product originally isolated from the autumn crocus, Colchicum autumnale, and the namesake for the colchicine site on tubulin,52,53 is a potent inhibitor of tubulin polymerization, but colchicine has a narrow therapeutic window limiting its development as an anticancer agent.35,54,55 Another natural product binding to the colchicine site on tubulin is CA4 (Fig. 1).4 Originally isolated from the African bushwillow tree, Combretum caffrum, and synthesized by Pettit and co-workers, CA4 binds tightly to the colchicine site on tubulin and functions as a VDA.55–58 Combretastatin A-4P [(CA4P), Fig. 3] was synthesized as a water-soluble prodrug salt for therapeutic use,41,55,56,58 and both CA4 and CA4P are now being evaluated in clinical trials.42,45,59–62
Drawing upon the key structural motifs of colchicine and CA4, the Pinney Research Group (Baylor University) has a long-standing interest in the discovery and development of new small-molecule anticancer agents that function as highly cytotoxic agents and/or as potent VDAs. Representative examples within this library include combretastatin,63–66 benzo[b]thiophene,67 dihydronaphthalene,9 benzosuberene,9–12 and indole-based analogues.14,68 The Pinney Research Group had previously shown that expansion of the fused alkyl ring of the dihydronaphthalene anticancer agent known as OXi6196 (Fig. 1) by one carbon to the fused seven-membered ring benzosuberene anticancer agent known as KGP18 (Fig. 1) was an effective design paradigm, since KGP18 was highly cytotoxic against human cancer cell lines and strongly inhibits tubulin polymerization.
To further elucidate the structure activity relationship (SAR) considerations associated with functionalized fused aryl-carbocyclic ring systems that mimic colchicine and CA4 and their interaction with the tubulin-microtubule system, benzocyclooctene (fused 6,8 ring system) analogues and corresponding indene (fused 6,5 ring system) analogues were prepared by chemical synthesis and subjected to preliminary biological evaluation.
2. Results and Discussion
2.1 Synthesis
Four benzocyclooctene analogues and four indene analogues were synthesized to further investigate SAR considerations associated with fused aliphatic ring structures and corresponding cytotoxicity and inhibition of tubulin polymerization. These analogues were prepared utilizing a synthetic strategy reminiscent of that employed for a variety of benzosuberene analogues developed in the Pinney Research Group.10,11 The synthesis of each benzocyclooctene analogue was initiated with a Wittig olefination followed by catalytic hydrogenation to afford carboxylic acids 3 and 4 (Scheme 1). A Friedel-Crafts intramolecular annulation was accomplished using Eaton’s reagent (7.7 weight percent P2O5 in CH3SO3H),69 and the reaction was diluted and cooled in order to facilitate the construction of benzocyclooctanones 5 and 6 (Scheme 1). Without dilution and cooling, the desired product was not formed and only starting material remained.
Scheme 1.

Synthesis of benzocyclooctanone analogues 5 and 6.
A similar catalytic reduction was utilized with starting material trans-2,3-dimethoxycinnamic acid to afford carboxylic acids 9 and 10, which were cyclized with Eaton’s reagent to their corresponding indanone analogues 11 and 12 (Scheme 2).
Scheme 2.

Synthesis of indanone analogues 11 and 12.
Benzocyclooctanone 5 and indanone 11 bearing the ortho dimethoxy motif were each subjected to a selective demethylation10,11,70 to afford phenolic analogues 13 and 15, which were subsequently protected with TBS to yield 14 and 16 (Scheme 3).
Scheme 3.

Synthesis of TBS-protected benzocyclooctanone 14 and indanone 16.
Trimethoxyphenyllithium (prepared from the corresponding aryl bromide) underwent a 1,2-addition reaction to benzocyclooctanone analogues 5, 6 and 14 to afford the corresponding tertiary alcohols 17, 18, and 19. Subsequent dehydration afforded benzocyclooctene analogues 20 and 21 and TBS-protected analogue 22, which underwent deprotection to afford phenolic benzocyclooctene 23 (Scheme 4).
Scheme 4.

Synthesis of target benzocyclooctene analogues 20, 21, and 23.
Phenolic analogue 23 was converted to its corresponding phosphate prodrug salt 24, in order to increase water solubility for in vivo studies (Scheme 5).
Scheme 5.

Conversion of analogue 23 to its corresponding phosphate prodrug salt 24.
Analogous aryl addition reactions were carried out on indanone intermediates 11, 12, and 16 to generate the corresponding tertiary alcohols 25, 26, and 27 (Scheme 6). Subsequent dehydration afforded indene analogues 28 29, and 30. Removal of the TBS group afforded phenolic indene 31.
Scheme 6.

Synthesis of target indene compounds 28, 29, and 31.
The indene phenol 31 was also converted to its corresponding phosphate salt 32 (Scheme 7). While TEA proved to be an acceptable base (due to ease of removal) for use in the synthesis of the eight membered ring phosphate salt 24, in the synthesis of the indene phosphate salt 32 TEA proved to be hard to remove during purification. Altering the base to pyridine solved this problem and allowed the water-soluble phosphate salt 32 to be synthesized in high purity.
Scheme 7.

Synthesis of target indene water-soluble analogue 32.
Due to their increased size, eight membered rings are the first in the homologous series (3, 4, 5, 6, 7 – membered rings) that can accommodate both Z and E double bond configurations.71,72 However, in our hands only the Z configurations were synthesized. X-ray crystal structures were obtained for benzocyclooctene analogues 20 and 23 to confirm their Z double bond configuration (see Supplementary Data).
2.2 Biological Evaluation
The four target benzocyclooctene and four indene analogues (Fig. 4) were evaluated for their cytotoxicity against human cancer cell lines and for their abilities to inhibit tubulin polymerization and colchicine binding to tubulin (Table 1).
Figure 4.

Compilation of synthesized benzocyclooctene analogues 20, 21, 23, and 24 and indene analogues 28, 29, 31, and 32.
Table 1.
Inhibition of tubulin polymerization [expressed as half maximal inhibitory concentration (IC50)] and cytotoxicity [expressed as growth inhibition of 50% (GI50)] of the eight target analogues synthesized.
| Compound | Inhibition of tubulin polymerization IC50(μM) ± SD | % Inhibition of colchicine binding ± SD | GI50 (μM) SRB assaya | ||
|---|---|---|---|---|---|
| NCI-H460 | DU-145 | SK-OV-3 | |||
| CA4 | 1.0b | 98 ± 0.007 | 0.00500c,d | 0.00602c,d | 0.00506d |
| CA4P | >20b | nre | 0.00282c | 0.00336c | 0.00190 |
| KGP18 | 1.4f | nre | 0.0000418g | 0.0000249 g | 0.0000543 g |
| 20 | 4.5 ± 0.5 | 31 ± 0.6 | 0.395 | 0.448 | 0.512 |
| 21 | 3.0 ± 0.1 | 29 ± 5 | 0.431 | 0.570 | 0.400 |
| 23 | 1.2 ± 0.1 | 78 ± 4 | 0.107 | 0.105 | 0.0811 |
| 24 | >20 | ndh | 0.0260 | 0.0410 | 0.0366 |
| 28 | >20 | ndh | 42.2 | 34.6 | 9.46 |
| 29 | >20 | ndh | 4.02 | 4.66 | 6.15 |
| 31 | 11 ± 2 | 17 ± 5 | 0.388 | 0.362 | 0.704 |
| 32 | >20 | ndh | 1.50 | 3.34 | 0.334 |
Among the eight compounds synthesized for this study and analyzed biologically, only the target benzocyclooctene analogues (20, 21, and 23) had activities as inhibitors of tubulin polymerization similar to those of CA4 and KGP18. Benzocyclooctene phenol 23 demonstrated the lowest IC50 value (1.2 μM) among the compounds evaluated in this study. A binding study utilizing radiolabeled colchicine demonstrated that at a concentration of 5 μM, phenol 23 inhibited colchicine binding by 78%, 20% less than the activity obtained with 5 μM CA4 (used as a positive control). Considering the four indene analogues, only phenolic indene 31 demonstrated modest inhibition of tubulin polymerization, with an IC50 value of 11 μM.
Benzocyclooctene phenol 23 and its corresponding phosphate prodrug salt 24 were the most cytotoxic analogues in these series against the three human cancer cell lines examined (for example, GI50 = 0.105 and 0.0410 μM against DU-145 (prostate) for analogues 23 and 24, respectively). While structurally similar to KGP18, differing by the addition of one carbon to the aliphatic ring, the phenolic benzocyclooctene analogue is dramatically less cytotoxic than its benzosuberene counterpart. Compounds 23 and 24 had GI50 values in the submicromolar range, while the values obtained with KGP18 were all subnanomolar.
2.3 Dynamic bioluminescence imaging
Bioluminescence imaging (BLI) is a highly valuable modality with diverse applications in a wide range of biological models and systems.74–76 BLI is a particularly useful tool for assessing and quantifying in vivo blood flow reduction following treatment with a VDA.54,76–79 In the current study MCF7 human breast cancer cells, which had been previously transfected stably to express the enzyme luciferase, as well as two fluorescent proteins, as designated by MCF7-luc-GFP-mCherry, were implanted in SCID mice, as described previously.78 Since the luciferase substrate luciferin can be delivered to the tumor via the blood stream, damage to the vessels by a VDA can be quantified by measurement of reduced bioluminescence upon injection of luciferin.54 In the study shown in Fig. 5, three mice bearing the MCF7-luc-GFP-mCherry breast tumor were injected ip with analogue 24 (120 mg/kg), and BLI assessment was performed at baseline (0 h, pre-administration) and at 4, 24, and 48 h after administration of 24. In a contemporaneous study, one mouse was treated with CA4P (120 mg/kg) as a positive control, and an additional mouse was treated with saline alone. A limited dose escalation study with analogue 24 in non-tumor bearing SCID mice (60–150 mg/kg, unpublished data) suggested that a dose of 120 mg/kg was tolerated and likely below the maximum tolerated dose (MTD), which has not yet been determined. The optimal CA4P dose in SCID mice for evaluation of VDA efficacy has been previously established at 120 mg/kg.76 Single dose treatment with both analogue 24 and CA4P resulted in a significant reduction in bioluminescence (approximately 80% decrease as compared to baseline or saline control; p<0.05) at 4 h after compound injection. While bioluminescence intensity in the mouse treated with compound 24 recovered somewhat at 24 and 48 h, it remained significantly below baseline values (p<0.005). In this study, the BLI data for both analogue 24 and CA4P at 4 h were statistically different from the saline control, but not from each other or from the data obtained at other time points. Following the 48 h time point BLI mice were sacrificed and tumors excised and prepared for histology. Routine staining using H&E indicated much more extensive necrosis throughout the tumors following administration of 24 or CA4P each at 120 mg/kg, as compared with saline control (Fig. 6). Thus, analogue 24 is a good candidate for further assessment and may be a therapeutically useful VDA.
Figure 5.
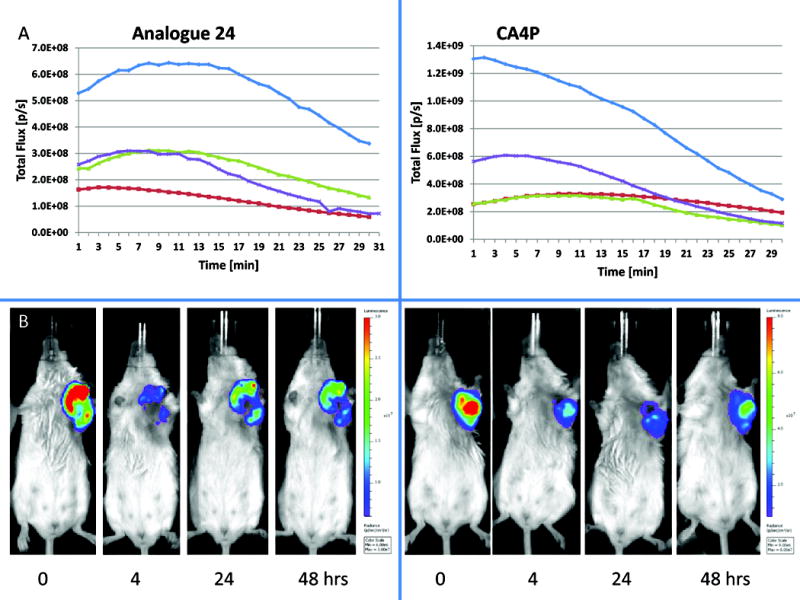
Dynamic bioluminescence with respect to vascular disruption. A) Graphs show evolution of light emission from individual MCF7-luc-GFP-mCherry breast tumors following administration of luciferin substrate subcutaneously in the fore-back region of each mouse at time 0. Respective curves are baseline (blue), 4 h (red), 24 h (green) and 48 h (purple) post administration of VDA. Left hand panel shows a representative mouse with 120 mg/kg analogue 24 and right hand panel shows CA4P (120 mg/kg). B) Photographs show selected images at the 10 min. time point. Left hand group for analogue 24 and right hand group for CA4P for the same mice used to generate the curves for part A. Respective images are each scaled to same values.
Figure 6.

Histological assessment of tumor necrosis. H&E stained tumor cross sections showing necrotic (pink) and viable (purple) regions at 48 h post treatment for A: Analogue 24 (120 mg/kg) B: CA4P (120 mg/kg) C: Saline.
3. Conclusions
These studies, which focused on indene and benzocyclooctene ring systems, expanded the known SAR associated with the effect that fused aliphatic ring size plays in regard to cytotoxicity and inhibition of tubulin polymerization. From the group of eight analogues prepared by chemical synthesis, three benzocyclooctene analogues were strong inhibitors of tubulin polymerization and one indene analogue was a modest inhibitor. The most promising compound (benzocyclooctene analogue 23) was prepared and evaluated as its corresponding water-soluble phosphate salt 24. It demonstrated significant in vivo reduction of blood flow (as evidenced by BLI) in an MCF7-luc-GFP-mCherry tumor model in SCID mice, and the results indicated that 24 should undergo further evaluation as a VDA for potential cancer treatment.
Supplementary Material
Acknowledgments
The authors are grateful to the National Cancer Institute of the National Institutes of Health (Grant No. 5R01CA140674 to K.G.P, M.L.T, and R.P.M), the Cancer Prevention and Research Institute of Texas (CPRIT, Grant No. RP140399 to K.G.P., M.L.T., and R.P.M.), and Mateon Therapeutics, Inc. (grant to K.G.P. and M.L.T.) for their financial support of this project. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. The authors would also thank Dr. Craig Moehnke and Dr. Michelle Nemec (Director) for the use of the shared Molecular Biosciences Center at Baylor University, Dr. Alejandro Ramirez (Mass Spectrometry Core Facility, Baylor University) and Dr. Kevin Klausmeyer and Dr. Marissa Penney (X-ray analysis). The authors are grateful to Mr. Jack Littlejohn and Mr. Jake Lofman (Baylor University) for their contributions to the synthesis of certain analogues. John Shelton (JAR Molecular Pathology core, UTSW) prepared the H&E stained sections. Imaging was facilitated with the assistance of resources of the Harold C. Simmons Cancer Center supported through a National Institutes of Health National Cancer Institute Cancer Center Support Grant [Grant 1P30 CA142543], specifically, the Southwestern Small Animal Imaging Resource, and Live Cell Imaging Resource. The IVIS Spectrum was purchased with support of 1S10RR024757.
Footnotes
A significant portion of the work presented in this manuscript was funded through Mateon Therapeutics Inc. (formerly OXiGENE Inc.), and this relationship is properly indicated in the acknowledgement section. One Mateon employee and shareholder Dr. David Chaplin, is included as a co-author on this manuscript for his valuable contributions to the overall project. In addition, one of the authors (KGP) is a paid consultant and shareholder with Mateon. We are pleased with this productive and useful long-term scientific collaboration and funding relationship with Mateon, and it is important to note that there is absolutely no actual conflict of interest associated with the science presented in this manuscript.
Supplementary data including experimental procedures (synthesis, SRB assay, colchicine binding assay, inhibition of tubulin polymerization, in vivo BLI, and histology), 1H NMR, 13C NMR, 31P NMR, HPLC, HRMS for target compounds and intermediates (1H NMR, 13C NMR, only) and X-ray crystallography for compounds 20 and 23 associated with this article can be found in the online Supplementary data file. All animal procedures were approved by the University of Texas
Southwestern Medical Center Institutional Animal Care and Use Committee.
References
- 1.Ludford RJ. J Natl Cancer Inst. 1945;6:89–101. [Google Scholar]
- 2.Schiff PB, Horwitz SB. Proc Natl Acad Sci. 1980;77:1561–1565. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schiff PB, Horwitz SB. Biochemistry (Mosc) 1981;20:3247–3252. doi: 10.1021/bi00514a041. [DOI] [PubMed] [Google Scholar]
- 4.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendal D. Experientia. 1989;45:209–211. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 5.Gorman M, Neuss N, Svoboda GH, Barnes AJ, Cone NJ. J Am Pharm Assoc. 1959;48:256–257. doi: 10.1002/jps.3030480419. [DOI] [PubMed] [Google Scholar]
- 6.Owellen RJ, Owens AH, Donigian DW. Biochem Biophys Res Commun. 1972;47:685–691. doi: 10.1016/0006-291x(72)90546-3. [DOI] [PubMed] [Google Scholar]
- 7.Pinney KG, Sriram M, George C, Tanpure RP. WO2012068284 (A2) 2012
- 8.Pinney KG, Mocharla VP, Chen Z, Garner CM, Ghatak A, Hadimani M, Kessler J, Dorsey JM. WO2001068654 (A2) 2001
- 9.Sriram M, Hall JJ, Grohmann NC, Strecker TE, Wootton T, Franken A, Trawick ML, Pinney KG. Bioorg Med Chem. 2008;16:8161–8171. doi: 10.1016/j.bmc.2008.07.050. [DOI] [PubMed] [Google Scholar]
- 10.Tanpure RP, George CS, Strecker TE, Devkota L, Tidmore JK, Lin C-M, Herdman CA, MacDonough MT, Sriram M, Chaplin DJ, Trawick ML, Pinney KG. Bioorg Med Chem. 2013;21:8019–8032. doi: 10.1016/j.bmc.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herdman CA, Devkota L, Lin C-M, Niu H, Strecker TE, Lopez R, Liu L, George CS, Tanpure RP, Hamel E, Chaplin DJ, Mason RP, Trawick ML, Pinney KG. Bioorg Med Chem. 2015;23:7497–7520. doi: 10.1016/j.bmc.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanpure RP, George CS, Sriram M, Strecker TE, Tidmore JK, Hamel E, Charlton-Sevcik AK, Chaplin DJ, Trawick ML, Pinney KG. Med Chem Comm. 2012;3:720–724. doi: 10.1039/C2MD00318J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devkota L, Lin C-M, Strecker TE, Wang Y, Tidmore JK, Chen Z, Guddneppanavar R, Jelinek CJ, Lopez R, Liu L, Hamel E, Mason RP, Chaplin DJ, Trawick ML, Pinney KG. Bioorg Med Chem. 2016;24:938–956. doi: 10.1016/j.bmc.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hadimani MB, MacDonough MT, Ghatak A, Strecker TE, Lopez R, Sriram M, Nguyen BL, Hall JJ, Kessler RJ, Shirali AR, Liu L, Garner CM, Pettit GR, Hamel E, Chaplin DJ, Mason RP, Trawick ML, Pinney KG. J Nat Prod. 2013;76:1668–1678. doi: 10.1021/np400374w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn BL, Gill GS, Grobelny DW, Chaplin JH, Paul D, Leske AF, Lavranos TC, Chalmers DK, Charman SA, Kostewicz E, Shackleford DM, Morizzi J, Hamel E, Jung MK, Kremmidiotis G. J Med Chem. 2011;54:6014–6027. doi: 10.1021/jm200454y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kremmidiotis G, Leske AF, Lavranos TC, Beaumont D, Gasic J, Hall A, O’Callaghan M, Matthews CA, Flynn B. Mol Cancer Ther. 2010;9:1562–1573. doi: 10.1158/1535-7163.MCT-09-0815. [DOI] [PubMed] [Google Scholar]
- 17.Lambert JM. Br J Clin Pharmacol. 2013;76:248–262. doi: 10.1111/bcp.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prota AE, Bargsten K, Diaz JF, Marsh M, Cuevas C, Liniger M, Neuhaus C, Andreu JM, Altmann K-H, Steinmetz MO. Proc Natl Acad Sci. 2014;111:13817–13821. doi: 10.1073/pnas.1408124111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma D, Hopf CE, Malewicz AD, Donovan GP, Senter PD, Goeckeler WF, Maddon PJ, Olson WC. Clin Cancer Res. 2006;12:2591–2596. doi: 10.1158/1078-0432.CCR-05-2107. [DOI] [PubMed] [Google Scholar]
- 20.Hay MP, Hicks KO, Wang J. In: Tumor Microenvironment and Cellular Stress. Koumenis C, Hammond E, Giaccia A, editors. Springer; New York: 2014. pp. 111–145. [Google Scholar]
- 21.Wilson WR, Hay MP. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 22.McKeage MJ, Jameson MB, Ramanathan RK, Rajendran J, Gu Y, Wilson WR, Melink TJ, Tchekmedyian NS. BMC Cancer. 2012;12:1–10. doi: 10.1186/1471-2407-12-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abbattista MR, Jamieson SMF, Gu Y, Nickel JE, Pullen SM, Patterson AV, Wilson WR, Guise CP. Cancer Biol Ther. 2015;16:610–622. doi: 10.1080/15384047.2015.1017171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chawla SP, Cranmer LD, Tine BAV, Reed DR, Okuno SH, Butrynski JE, Adkins DR, Hendifar AE, Kroll S, Ganjoo KN. J Clin Oncol. 2014:1–11. doi: 10.1200/JCO.2013.54.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Q, Sun JD, Wang J, Ahluwalia D, Baker AF, Cranmer LD, Ferraro D, Wang Y, Duan J-X, Ammons WS, Curd JG, Matteucci MD, Hart CP. Cancer Chemother Pharmacol. 2012;69:1487–1498. doi: 10.1007/s00280-012-1852-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng F, Evans JW, Bhupathi D, Banica M, Lan L, Lorente G, Duan J-X, Cai X, Mowday AM, Guise CP, Maroz A, Anderson RF, Patterson AV, Stachelek GC, Glazer PM, Matteucci MD, Hart CP. Mol Cancer Ther. 2012;11:740–751. doi: 10.1158/1535-7163.MCT-11-0634. [DOI] [PubMed] [Google Scholar]
- 27.Thomson P, Naylor MA, Everett SA, Stratford MRL, Lewis G, Hill S, Patel KB, Wardman P, Davis PD. Mol Cancer Ther. 2006;5:2886–2894. doi: 10.1158/1535-7163.MCT-06-0429. [DOI] [PubMed] [Google Scholar]
- 28.Siemann DW, Bibby MC, Dark GG, Dicker AP, Eskens FA, Horsman MR, Marmé D, LoRusso PM. Clin Cancer Res. 2005;11:416–420. [PubMed] [Google Scholar]
- 29.Horsman MR, Bohn AB, Busk M. Exp Oncol. 2010;32:143–148. [PubMed] [Google Scholar]
- 30.Thorpe PE. Clin Cancer Res. 2004;10:415–427. doi: 10.1158/1078-0432.ccr-0642-03. [DOI] [PubMed] [Google Scholar]
- 31.Denekamp J. Cancer Metastasis Rev. 1990;9:267–282. doi: 10.1007/BF00046365. [DOI] [PubMed] [Google Scholar]
- 32.Siemann DW, Chaplin DJ, Horsman MR. Cancer. 2004;100:2491–2499. doi: 10.1002/cncr.20299. [DOI] [PubMed] [Google Scholar]
- 33.Fens MH, Storm G, Schiffelers RM. Expert Opin Investig Drugs. 2010;19:1321–1338. doi: 10.1517/13543784.2010.524204. [DOI] [PubMed] [Google Scholar]
- 34.Denekamp J. Br J Cancer. 1982;45:136–139. doi: 10.1038/bjc.1982.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tozer GM, Kanthou C, Baguley BC. Nat Rev Cancer. 2005;5:423–435. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]
- 36.Siemann DW. Cancer Treat Rev. 2011;37:63–74. doi: 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vasudev NS, Reynolds AR. Angiogenesis. 2014;17:471–494. doi: 10.1007/s10456-014-9420-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain RK. Cancer Cell. 2014;26:605–622. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Folkman J. Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 40.Pilat MJ, LoRusso PM. J Cell Biochem. 2006;99:1021–1039. doi: 10.1002/jcb.20783. [DOI] [PubMed] [Google Scholar]
- 41.Pettit GR, Rhodes MR. Anticancer Drug Des. 1998;13:183–191. [PubMed] [Google Scholar]
- 42.Kretzschmann VK, Fürst R. Phytochem Rev. 2014;13:191–206. [Google Scholar]
- 43.Ji Y-T, Liu Y-N, Liu Z-P. Curr Med Chem. 2015;22:1348–1360. doi: 10.2174/0929867322666150114163732. [DOI] [PubMed] [Google Scholar]
- 44.Nathan P, Zweifel M, Padhani AR, Koh D-M, Ng M, Collins DJ, Harris A, Carden C, Smythe J, Fisher N, Taylor NJ, Stirling JJ, Lu S-P, Leach MO, Rustin GJS, Judson I. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18:3428–3439. doi: 10.1158/1078-0432.CCR-11-3376. [DOI] [PubMed] [Google Scholar]
- 45.Baguley BC, McKeage MJ. Clin Investig. 2012;2:985–993. [Google Scholar]
- 46.Pettit GR, Lippert JW. Anticancer Drug Des. 2000;15:203–216. [PubMed] [Google Scholar]
- 47.Rischin D, Bibby DC, Chong G, Kremmidiotis G, Leske AF, Matthews CA, Wong S, Rosen M, Desai J. Clin Cancer Res. 2011;17:5152–5160. doi: 10.1158/1078-0432.CCR-11-0937. [DOI] [PubMed] [Google Scholar]
- 48.Hori K, Saito S, Nihei Y, Suzuki M, Sato Y. Jpn J Cancer Res. 1999;90:1026–1038. doi: 10.1111/j.1349-7006.1999.tb00851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hori K, Saito S. Br J Cancer. 2003;89:1334–1344. doi: 10.1038/sj.bjc.6601261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blay J-Y, Pápai Z, Tolcher AW, Italiano A, Cupissol D, López-Pousa A, Chawla SP, Bompas E, Babovic N, Penel N, Isambert N, Staddon AP, Saâda-Bouzid E, Santoro A, Franke FA, Cohen P, Le-Guennec S, Demetri GD. Lancet Oncol. 2015;16:531–540. doi: 10.1016/S1470-2045(15)70102-6. [DOI] [PubMed] [Google Scholar]
- 51.Davis PD, Dougherty GJ, Blakey DC, Galbraith SM, Tozer GM, Holder AL, Naylor MA, Nolan J, Stratford MRL, Chaplin DJ, Hill SA. Cancer Res. 2002;62:7247–7253. [PubMed] [Google Scholar]
- 52.Lee RM, Gewirtz DA. Drug Dev Res. 2008;69:352–358. [Google Scholar]
- 53.Ludford J. J Natl Cancer Inst. 1945;6:89–101. [Google Scholar]
- 54.Mason RP, Zhao D, Liu L, Trawick ML, Pinney KG. Integr Biol. 2011;3:375–387. doi: 10.1039/c0ib00135j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ. Cancer Res. 1997;57:1829–1834. [PubMed] [Google Scholar]
- 56.Kirwan IG, Loadman PM, Swaine DJ, Anthoney DA, Pettit GR, Lippert JW, Shnyder SD, Cooper PA, Bibby MC. Clin Cancer Res. 2004;10:1446–1453. doi: 10.1158/1078-0432.ccr-0518-03. [DOI] [PubMed] [Google Scholar]
- 57.Grosios K, Holwell SE, McGown AT, Pettit GR, Bibby MC. Br J Cancer. 1999;81:1318–1327. doi: 10.1038/sj.bjc.6692174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iyer S, Chaplin DJ, Rosenthal DS, Boulares AH, Li L-Y, Smulson ME. Cancer Res. 1998;58:4510–4514. [PubMed] [Google Scholar]
- 59.Cooney MM, Ortiz J, Bukowski RM, Remick SC. Curr Oncol Rep. 2005;7:90–95. doi: 10.1007/s11912-005-0033-x. [DOI] [PubMed] [Google Scholar]
- 60.Dowlati A, Robertson K, Cooney M, Petros WP, Stratford M, Jesberger J, Rafie N, Overmoyer B, Makkar V, Stambler B, Taylor A, Waas J, Lewin JS, McCrae KR, Remick SC. Cancer Res. 2002;62:3408–3416. [PubMed] [Google Scholar]
- 61.Rustin GJS, Galbraith SM, Anderson H, Stratford M, Folkes LK, Sena L, Gumbrell L, Price PM. J Clin Oncol. 2003;21:2815–2822. doi: 10.1200/JCO.2003.05.185. [DOI] [PubMed] [Google Scholar]
- 62.Nathan P, Zweifel M, Padhani AR, Koh D-M, Ng M, Collins DJ, Harris A, Carden C, Smythe J, Fisher N, Taylor NJ, Stirling JJ, Lu S-P, Leach MO, Rustin GJS, Judson I. Clin Cancer Res. 2012;18:3428–3439. doi: 10.1158/1078-0432.CCR-11-3376. [DOI] [PubMed] [Google Scholar]
- 63.Pinney KG, Mejia MP, Villalobos VM, Rosenquist BE, Pettit GR, Verdier-Pinard P, Hamel E. Bioorg Med Chem. 2000;8:2417–2425. doi: 10.1016/s0968-0896(00)00176-0. [DOI] [PubMed] [Google Scholar]
- 64.Siles R, Ackley JF, Hadimani MB, Hall JJ, Mugabe BE, Guddneppanavar R, Monk KA, Chapuis J-C, Pettit GR, Chaplin DJ, Edvardsen K, Trawick ML, Garner CM, Pinney KG. J Nat Prod. 2008;71:313–320. doi: 10.1021/np070377j. [DOI] [PubMed] [Google Scholar]
- 65.Tanpure RP, Harkrider AR, Strecker TE, Hamel E, Trawick ML, Pinney KG. Bioorg Med Chem. 2009;17:6993–7001. doi: 10.1016/j.bmc.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanpure RP, Nguyen BL, Strecker TE, Aguirre S, Sharma S, Chaplin DJ, Siim BG, Hamel E, Lippert JW, Pettit GR, Trawick ML, Pinney KG. J Nat Prod. 2011;74:1568–1574. doi: 10.1021/np200104t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinney KG, Bounds AD, Dingeman KM, Mocharla VP, Pettit GR, Bai R, Hamel E. Bioorg Med Chem Lett. 1999;9:1081–1086. doi: 10.1016/s0960-894x(99)00143-2. [DOI] [PubMed] [Google Scholar]
- 68.MacDonough MT, Strecker TE, Hamel E, Hall JJ, Chaplin DJ, Trawick ML, Pinney KG. Bioorg Med Chem. 2013;21:6831–6843. doi: 10.1016/j.bmc.2013.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eaton PE, Carlson GR, Lee JT. J Org Chem. 1973;38:4071–4073. [Google Scholar]
- 70.Kemperman GJ, Roeters TA, Hilberink PW. Eur J Org Chem. 2003;2003:1681–1686. [Google Scholar]
- 71.Neuenschwander U, Hermans I. J Org Chem. 2011;76:10236–10240. doi: 10.1021/jo202176j. [DOI] [PubMed] [Google Scholar]
- 72.Cossy J, Arseniyadis S, Meyer C. Metathesis in Natural Product Synthesis: Strategies, Substrates and Catalysts. John Wiley & Sons; 2011. [Google Scholar]
- 73.Pettit GR, Grealish MP, Herald DL, Boyd MR, Hamel E, Pettit RK. J Med Chem. 2000;43:2731–2737. doi: 10.1021/jm000045a. [DOI] [PubMed] [Google Scholar]
- 74.Paley MA, Prescher JA. Med Chem Comm. 2014;5:255–267. doi: 10.1039/C3MD00288H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma X, Hui H, Shang W, Jia X, Yang X, Tian J. Curr Drug Targets. 2015;16:542–548. doi: 10.2174/1389450116666150102112747. [DOI] [PubMed] [Google Scholar]
- 76.Zhao D, Richer E, Antich PP, Mason RP. FASEB J. 2008;22:2445–2451. doi: 10.1096/fj.07-103713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alhasan MK, Liu L, Lewis MA, Magnusson J, Mason RP. PLoS ONE. 2012;7:e46106. doi: 10.1371/journal.pone.0046106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu L, Beck H, Wang X, Hsieh H-P, Mason RP, Liu X. PloS One. 2012;7:e43314. doi: 10.1371/journal.pone.0043314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou H, Hallac RR, Lopez R, Denney R, MacDonough MT, Li L, Liu L, Graves EE, Trawick ML, Pinney KG, Mason RP. Am J Nucl Med Mol Imaging. 2015;5:143–153. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.