

Abstract
Although Michael acceptors display a potent and broad spectrum of bioactivity, they have largely been ignored in drug discovery because of their presumed indiscriminate reactivity. As such, a dearth of information exists relevant to the thiol reactivity of natural products and their analogs possessing this moiety. In the midst of recently approved acrylamide-containing drugs, it is clear that a good understanding of the hetero-Michael addition reaction and the relative reactivities of biological thiols with Michael acceptors under physiological conditions is needed for the design and use of these compounds as biological tools and potential therapeutics. This perspective provides information that will contribute to this understanding, such as kinetics of thiol addition reactions, bioactivities, as well as steric and electronic factors that influence the electrophilicity and reversibility of Michael acceptors. This perspective is focused on α,β-unsaturated carbonyls given their preponderance in bioactive natural products.
Introduction
Targeted covalent modification has emerged as a validated approach to drug discovery with the recent FDA approvals of afatanib (2013), ibrutinib (2013), and osimertinib (2015); drugs that were designed to undergo an irreversible hetero-Michael addition reaction with a unique cysteine residue of a specific protein. This strategy has expanded the druggable landscape by enhancing the ligand binding selectivity for proteins in the same family and by increasing the binding affinities for target proteins with shallow binding sites. Moreover, compounds that operate via a targeted covalent inhibition mechanism have been shown to overcome drug resistance (some examples are discussed below in this perspective).
For this perspective, we have drawn inspiration from natural products and other biologically relevant compounds whose activity is presumably dependent upon a hetero-Michael addition reaction with thiols. An understanding and characterization of their thiol reactivity is vital to the continued development of covalent inhibitors as drug candidates and biological probes. Accordingly, this perspective focuses on the thiol reactivity of Michael acceptors possessing an α,β-unsaturated carbonyl group and is organized by functional group roughly in order of increasing reactivity with thiols. Functional groups discussed include α,β-unsaturated amides (acrylamides) and lactams, α,β-unsaturated esters and lactones, acyclic and cyclic α,β-unsaturated ketones (enones), α,β-unsaturated aldehydes (enals), dually activated Michael acceptors, and other unsaturated carbonyls including quinones, acylfulvenes, viridins, α-haloacryloyl compounds, and α,β-unsaturated carboxylic acids. When possible, the kinetic reactivity of these functional groups and how this reactivity correlates to biological activity is included. To the best of our knowledge, kinetic data for reactivity with thiols has not been comprehensively reviewed for biologically active and molecularly complex compounds with α,β-unsaturated carbonyl groups, although a recent toxicological review amasses thiol reactivity data for simple compounds.1 Finally, spectroscopic and computational methods for predicting and analyzing the reactivity of α,β-unsaturated carbonyls with thiols is presented.
This perspective is meant to serve as a complement to reviews focused on biological targets and mechanisms of action,2 design and pharmacological features,3 and strategies to assess off target reactivity.4 Another recent review in toxicology highlights the major reaction mechanisms for protein adduct formation (Sn2, Sn1, acylation, Schiff base formation, hetero-Michael addition, and SnAr), chemical reactivity assays for evaluating electrophilic compounds, and computational parameters for predicting chemical reactivity; this review compiled a listing of chemical reactivity data containing 3089 entries for simple (non-drug like) molecules.1
1.1 Interpretation of the Data Compiled in this Perspective
The following sections discuss the thiol reactivity of α,β-unsaturated carbonyls through a variety of parameters that are provided when available. Reaction conditions utilized for thiol addition reactions are provided along with kinetic data (rate constants, half-lives), which are commonly measured by UV, NMR, HPLC, and fluorescence. The hetero-Michael addition reaction between a thiol and an α,β-unsaturated carbonyl is a second order reaction, with the rate constant having units of M−1s−1 (or equivalent). Thus, comparisons of second order rate constants are the most accurate method for comparing the relative reactivity of two electrophiles. Pseudo-first order reaction rates were commonly measured using an excess of thiol for the studies discussed in this perspective, and these rate constants have units of s−1 (or equivalent). The second order rate constant can be calculated from the pseudo-first order rate constant with the expression: k2 =kobs/[thiol]. Reaction half-lives (t1/2) presented herein represents the time it takes for half of the electrophile to react with excess thiol under pseudo-first order conditions; t1/2 is related to the pseudo-first order rate constant by the expression: t1/2 = ln2/kobs. The values of rate constants are significantly affected by experimental conditions such as solvent, temperature, concentrations of reagents, and the pKa of the thiol reactant. Thus, the reactivities of α,β-unsaturated carbonyls can only be directly compared when the rates of the hetero-Michael addition reaction were measured under identical conditions.
The potency and selectivity of covalent drugs is affected by both their noncovalent affinity for their target (initial binding specificity, Ki) and the second order rate constant for covalent bond formation (k2 or kinact) described by equation 1; for example kinact may be optimized by placing an electrophilic moiety in close proximity to a nucleophile in the binding site. The specificity constant is equal to kinact/Ki and provides a more reliable measure than IC50 values for irreversible inhibitors; kinact/Ki can be measured using a Wilson-Kitz analysis.5 Since irreversible inhibitors display time-dependent target inhibition and IC50 values do not explicitly account for time dependency, the utilization of IC50 measurements for the characterization of irreversible target inhibition is not recommended. Nonetheless, IC50 measurements for the characterization of irreversible inhibitors are commonly reported in biochemical assays with an associated time (i.e. IC50 for target inhibition after 30 min treatment with an inhibitor) and, more appropriately, in cellular assays where the activity of the irreversible inhibitor is dependent on multiple factors (i.e. cellular uptake, compound stability, and target inhibition).
 |
(1) |
Although this perspective focuses on the kinetic, thiol reactivity of α,β-unsaturated carbonyls under biologically-relevant conditions, such as buffered, aqueous solutions, cells, or purified enzymes, limited information of this type is available for many classes of Michael acceptors. In these cases, solution-phase, synthetic chemistry for thiol addition to α,β-unsaturated carbonyls is presented, including the reaction conditions, isolation, and yield of thiol adducts. Additionally, thiol adduct reversibility, measured by the stability of adducts, crossover experiments, and equilibrium or dissociation constants, is presented. While we are aware that this solution phase reactivity is not representative of the reactivity of compounds in enzyme active sites, these conditions provide insight into the ease or difficulty with which an electrophile can undergo the hetero-Michael addition reaction, even though this information cannot be directly extrapolated to reactivity in biological systems.
An emphasis is placed on steric and electronic contributions to the electrophilicity of α,β-unsaturated carbonyls. This perspective highlights the biological activity of Michael acceptors through their cytotoxicity, inhibition of target proteins (kinact/Ki, time-dependent IC50 values), and comparisons to the activity of structurally related compounds lacking a Michael acceptor; covalent protein modification sites and how those sites were determined (crystal structures, LC-MS of tryptic peptides, mutation studies) are also presented. Comparisons of the rates of thiol addition to the biological activities or toxicities of α,β-unsaturated carbonyls and computational methods for predicting electrophilicity are provided. α,β-Unsaturated carbonyls are arranged roughly in order of increasing electrophilicity, although this oversimplification does not take into account the large influence of substituents on the electrophilicity of Michael acceptors. We begin this perspective by presenting examples demonstrating that targeted covalent inhibition provides an excellent approach for gaining specificity to a target protein.
1.2 Success Stories in Targeted Covalent Inhibition and Drug Design
The discovery and utility of the reversible covalent drug telaprevir (1) inspired the development of irreversible hepatitis C viral (HCV) protease inhibitors 3 and 4 by installing an acrylamide motif onto the structurally related scaffold 2 (Figure 1).6 Compound 1 targets a catalytic serine that is common across many viral and human proteases (Ser139 in HCV protease) by forming a hemiacetal with the ketoamide functionality.7 Inhibitors 3 and 4 were designed to target Cys159, a unique amino acid to HCV protease identified using structural bioinformatics.6 In a fixed time point enzymatic assay, compound 5, lacking the acrylamide group, showed weak inhibition of HCV protease (IC50 = 1147 nM), while the compounds with a Michael acceptor were potent inhibitors (IC50 = 4 and 2 nM for 3 and 4, respectively).6 A targeted covalent inhibition mechanism for compounds 3 and 4 was supported by mutation of Cys159 to serine, which resulted in a reduction in potency of 3 and 4, whereas 5 maintained a similarly weak potency compared to the non-mutated protein. Co-crystallization of inhibitor 3 with HCV protease confirmed a covalent C–S bond between Cys159 and the acrylamide group.6
Figure 1.

HCV protease inhibitors.
Gefitinib (6) is a noncovalent, small molecule inhibitor that targets mutated forms of epidermal growth factor receptor (EGFR) in cancer cells and represents the first generation of EGFR inhibitors. After FDA approval in 2003, it was discovered that EGFRs acquire resistance through specific mutations within the binding pocket; nearly half of the patients treated with erlotinib or 6 had a T790M single point mutation responsible for resistance to therapy.8 To combat this acquired resistance, structural modifications were made to 6 that included replacement of the 3-morpholino propyl group with a 4-dimethylaminobutenamide to produce afatinib (7), a compound designed to undergo a hetero-Michael addition with Cys797 in the active site of EGFR (Figure 2). The second generation EGFR inhibitor 7 was active at the nanomolar level against lung cancer cells containing this mutation.8-9 X-ray crystallography and in situ labeling followed by LC-MS/MS analysis confirmed a covalent bond between 7 and Cys797.10 However, it has been speculated that the T790M mutation lowers the affinity of the initial binding event of 7 before covalent linkage to Cys797 based on X-ray co-crystal analyses, which may have led to toxicity and lack of efficacy in the clinic.11 This led to the development of third-generation EGFR inhibitors osimertinib (8) and rociletinib (9) that selectively targeted EGFR containing the T790M mutation.12 Acrylamide 8 (also known as AZD9291, Tagrisso) is an irreversible EGFR inhibitor developed by AstraZeneca, which was approved in November 2015 for treatment of non-small cell lung cancer in patients with the T790M mutation of EGFR; 8 has an IC50 value of 12 nM for inhibition of EGFR with the L858R/T790M mutation and an IC50 value of 480 nM for wild-type EGFR.13 Acrylamide 9 (also known as CO-1686, AVL-301) is another mutant selective covalent inhibitor of L858R/T790M EGFR, currently in clinical trials, with a Ki of 21 nM against this mutation and a Ki of 303 nM against wild type EGFR (Figure 2).14 Another mutation, C797S, which replaces the cysteine that forms a covalent bond to irreversible EGFR inhibitors with a less nucleophilic serine, has been discovered as a common mechanism of resistance to these covalent inhibitors.15
Figure 2.

Reversible and irreversible EGFR inhibitors.
Irreversible inhibitors of related tyrosine kinases, fibroblast growth factor receptors (FGFR1-4) and Bruton's tyrosine kinase (Btk), were designed using a similar targeted covalent inhibition strategy. The FGFR inhibitor PD173074 (10) led to the design of FIIN-1 (11) possessing an acrylamide group, which reacts to form a hetero-Michael adduct with Cys486 in the ATP binding site of FGFR1 (Figure 3).16 The importance of the acrylamide group for effective inhibition was demonstrated by reduction of the acrylamide double bond, resulting in a 24-fold decrease in activity for blocking proliferation and survival of FGFR1-transformed Ba/F3 cells (EC50 = 14 nM vs 340 nM) and a 100-fold decrease in activity against FGFR3-transformed Ba/F3 cells (EC50 = 10 nM vs 1040 nM).16
Figure 3.

FGFR and Btk inhibitors.
The Btk noncovalent inhibitor PCI-29732 (12) was used in the design of targeted covalent inhibitor 13 (ibrutinib, PCI-32765), a compound showing a 500-fold increase in selectivity for Btk over related kinases Lck/Yes-related novel tyrosine kinase (Lyn), and spleen tyrosine kinase (Syk) in Ramos cells; 12 showed only a 4-fold selectivity for Btk in the same assay (Figure 3).17 This increase in selectivity results from a covalent bond between the acrylamide group and Cys481 in the ATP-binding site of Btk.18 Acrylamide 13 shows >100-fold selectivity for kinases containing a cysteine or serine at position 481, inhibiting kinases Btk, B lymphocyte kinase (Blk), and bone marrow kinase on chromosome X (Bmx) with subnanomolar potencies.18 There is no evidence of serine undergoing covalent bond formation to 13; Lou et al. propose that amino acids larger than cysteine or serine at the position equivalent to Cys481 of Btk clash with the pyrazolidine ring, leading to increased selectivity over kinases lacking a cysteine or serine at this position. Covalent inhibitor 13 was approved by the FDA in 2013 for the treatment of mantle cell lymphoma and in 2014 for the treatment of chronic lymphocytic leukemia; it is predicted to reach peak annual sales of $5 billion a year.19 As for the EGFR inhibitors 7-9, a cysteine to serine mutation (C481S) has been shown to confer resistance to therapy with 13.20 Spebrutinib (14, AVL-292, CC-292) is another irreversible Btk inhibitor under investigation for the treatment of chronic lymphocytic leukemia, as well as rheumatoid arthritis and multiple myeloma (Figure 3).21
Covalent kinase inhibitors for Bmx (a non-receptor tyrosine kinase also known as ETK), cyclin dependent kinase (Cdk) 7, and c-jun NH2-terminal kinase (JNK) have also been developed.22 Targeted covalent inhibitor acrylamides 15-17 represent examples of pan JNK inhibitors (Figure 4).23 JNK-IN-11 (15), the most potent inhibitor tested with IC50 values of 0.5-1.3 nM for inhibition of JNK1/2/3, also showed significant inhibition against several other kinases; however, JNK-IN-8 (16) showed excellent selectivity for JNK1/2/3 over other kinases with only a small decrease in activity (IC50 of 1-19 nM for inhibition of JNK1/2/3). Incorporation of an acrylamide motif was necessary for in vivo potency and selectivity for JNK1/2/3 in a panel of 200 kinases using a fixed time point enzymatic assay; without this group, the activity of each inhibitor was reduced 100-fold.23 Mutation of Cys116 of JNK2 to serine also resulted in a 10-fold loss in activity for 15 and a 100-fold loss in activity for 16. When JNK-IN-2 (17) was incubated with recombinant JNK1, the protein displayed a mass increase of 493 daltons, consistent with addition of a single molecule of 17; upon protease digestion, the peptide containing Cys116 was shown to be modified by inhibitor 17 as detected by LC-MS. A co-crystal structure of 17 with JNK-3 showed that Cys154 of JNK3 was covalently bound to the β position of the acrylamide group.
Figure 4.

Pan JNK 1/2/3 inhibitors.
Celgene Avilomics used the pan-phosphoinositide 3-kinase (PI3K) inhibitor GDC-0941 (18) as a lead in the design of the covalent inhibitor of PI3Kα, CNX-1351 (19, Figure 5).24 Analysis of the four class I isoforms of PI3Ks, α, β, γ, and δ, showed that PI3Kα contained a cysteine (Cys862) in the ATP binding site not present in the other isoforms.24 Cys862 was targeted for covalent inhibition by replacing the methylsulfonyl group on the nitrogen of the piperazine ring of 18 with a 6-methylhept-5-ene-1,4-dione to give 19.24 This sole substitution resulted in a 20-400 fold increase in selectivity of compound 19 for inhibition PI3Kα activity in fixed time point enzymatic assays with IC50 values of 7, 166, 240, and 3020 nM for the α, β, γ, and δ isoforms, respectively.24 PI3Kα inhibitor 19 was also tested against 60 other major classes of kinases and none were inhibited more than 40% at a 1 μM dose.24 When inhibitor 19 was incubated with PI3K enzymes, MS analysis confirmed that PI3Kα, but not the other isoforms, formed a covalent adduct with 19; co-crystallography of 19 with PI3Kα showed that Cys862 was covalently linked to the β position of the enone.24 Demonstrative of its low thiol reactivity, PI3Kα inhibitor 19 did not form any detectable adducts with 500-fold excess glutathione (GSH) or with any proteins in albumin-depleted human plasma at 37 °C over 1-3 h by MS analysis.24
Figure 5.

PI3Kα and VEGFR-2 inhibitors.
Wyeth Research prepared a series of quinone-containing inhibitors based upon a computational binding model of ZD-4190 (20) and the vascular endothelial growth factor receptor-2 (VEGFR-2) that showed Cys1045 in the proximity of the 4-anilino substituent (Figure 5).25 Many of these analogs showed good potency against VEGFR-2 in enzymatic assays, but their activity was significantly reduced in the presence of ATP, GSH, plasma, or in cells.25 However, quinone 21 maintained its inhibition of VEGFR-2 phosphorylation with IC50 values of 3.7-11 nM in the presence of 10-1000 μM ATP, 100 μM GSH, or 5% plasma, and an IC50 of 190 nM in KDR15 cells (Figure 5).25
The Src family of tyrosine kinases contains nine members, all of which contain a catalytic lysine (Lys295 of c-Src) in the ATP binding site, but only three members contain a proximal, noncatalytic cysteine residue (Cys277 of c-Src).26 Taunton and coworkers designed inhibitors 23 and 24 based on the structure and reactivity of ATP mimic p-fluorosulfonylbenzoyl 5'-adenosine (FSBA, 22), a compound that covalently binds to all nine Src kinases via a mechanism involving the addition of Lys295 to the fluorosulfonyl group and fluoride elimination (Figure 6).26-27 This work showed that substituting a vinylsulfone for the fluorosulfonyl group led to a covalent bond between 23 and three of the Src kinases, only those containing the conserved cysteine (Cys277). Additionally, vinylsulfone 23 was 40-fold less potent than fluorosulfonyl benzoate 24 against off-target kinases lacking this G-loop cysteine.26
Figure 6.

Inhibitors of Src kinases.
A similar structure-based design was implemented using crystallographic data that showed human centrosomal kinase NIMA-related kinase (Nek2) bound to reversible inhibitor SU11652 (25, Figure 7). For this study, oxindoles containing electrophilic groups poised to form a covalent bond with Cys22 of Nek2 were prepared.28 The two acrylamide-containing derivatives, 26 and 27 were not effective, but the more reactive propiolamide 28 and chloromethylketone 29 functioned as high nanomolar inhibitors of Nek2 and were used in further studies (Figure 7).28 Although the chloromethylketone 29 was more active than propiolamide 28 for inhibition of Nek2 in enzymatic assays (IC50 = 130 nM vs 770 nM), 29 was less active in cells (50% of cells exiting mitosis vs 4% for 28 after treatment with 5 μM of inhibitor for 45 min); this is likely due to the increased reactivity of 29 with thiols (t1/2 = 3 min for 29 vs. 60 min for 28 for the reaction with β-mercaptoethanol).28 Cys22 was confirmed as the target for propiolamide 28 and chloromethylketone 29 by MS analysis of tryptic peptides of the Nek2 kinase domain and also loss of covalent modification (and activity) when Cys22 was mutated to a valine.28 It is important to note that propiolamide 28 was the first selective Nek2 inhibitor with cellular activity, and was subsequently used as a tool to study spindle assembly and chromosome congression in mitosis without inhibiting other kinases critical to mitosis, such as Cdk1, Plk1, AurB, and Mps1.28
Figure 7.

Nek2 inhibitors.
Finally, by combining the covalent inhibition strategies discussed above, the dual EGFR/VEGFR-2 inhibitor 30 was designed to include two electrophilic groups, an acrylamide and a quinone (Figure 8). Quinone 30 inhibits the phosphorylation of recombinant EGFR and VEGFR-2 with an IC50 of 4.1 and 113 nM, respectively, in the presence of 1 μM ATP.29
Figure 8.

Dual EGFR/VEGFR-2 inhibitor.
The examples presented above demonstrate that targeted covalent inhibition provides an excellent approach for gaining specificity to a target protein. Targeted covalent inhibitor strategies often take advantage of an acrylamide group as the Michael acceptor because it is weakly electrophilic and requires close proximity to the cysteine residue for covalent bond formation to occur, minimizing side reactions with other cellular thiols such as GSH and solvent-exposed cysteines on proteins.3, 30 However, other Michael acceptors have been used successfully to increase inhibitor selectivity and potency. More information on irreversible kinase inhibitors can be found in other excellent reviews.2b, 4b, 30a, 31
Dimethyl fumarate (31, Tecfidera), originally used for treating psoriasis and recently approved for treating multiple sclerosis, is a Michael acceptor that readily forms adducts with GSH. Although its mode of action is still under investigation, 31 is known to rapidly hydrolyze under physiological conditions to mono-methyl fumarate (32), which is considered to be the active metabolite and reacts more slowly with GSH (t1/2 = 4 h for 32 and 9 min for 31, Scheme 1).32
Scheme 1.

Dimethyl Fumarate Hydrolysis
An alternative strategy to incorporating weakly electrophilic groups at key locations in an inhibitor is the inclusion of strong electrophiles that form rapidly reversible covalent bonds in a non-biological environment. Examples include, entacapone (33) and ribosomal s6 kinase (RSK) inhibitor 34, both possessing an α-cyanoacrylamide motif, and bardoxolone methyl (35, CDDO-Me) with an α-cyanoenone (Figure 9).33 Cyanoacrylamide 33 is a selective and reversible inhibitor of catechol-O-methyltransferase (COMT) that has been approved for Parkinson's disease, while bardoxolone methyl (35) inhibits nitric oxide production and the NF-κB pathway.33d The additional electron-withdrawing cyano group increases the rate of thiol addition, but in turn decreases the equilibrium constant, possibly by decreasing the pKa of the α-proton on the Michael adduct.33b Compound 33 reacts rapidly and reversibly with β-mercaptoethanol, but conjugation to COMT has not been confirmed.33a RSK inhibitor 34 also reacts reversibly with β-mercaptoethanol and has been confirmed to form a covalent bond to Cys436 of RSK2 through co-crystallography; additionally, mutation of Cys436 to valine resulted in a 1000-fold decrease in activity. Cyanoacrylamide 34 also displayed inhibition of RSK2 in cells even after washout, with RSK2 activity not recovering for 48 h, and the duration of RSK2 occupancy was indistinguishable from a known irreversible inhibitor containing a fluoromethyl ketone in place of the cyanoacrylamide of 34. However, protein unfolding led to rapid dissociation of the RSK2-34 adduct.33a The cyanoenone of 35 reacted as a Michael acceptor both reversibly and chemoselectively with small molecule thiols such as β-mercaptoethanol.33c
Figure 9.

Rapidly reversible nitrile-containing Michael acceptors.
Small-molecules bearing a variety of different Michael acceptors have also been used as chemical biology tools for elucidating biological pathways. Activity based protein profiling (ABPP) is a method that enables target identification for compounds that form covalent protein adducts and has been reviewed extensively.34 ABPP probes have been used to determine the selectivity of nine approved kinase inhibitors and to identify off-targets of the approved drug orlistat and other lipstatin analogs.35 Mechanism-based covalent crosslinkers have also been used to study protein-protein interactions in the nonreducing fungal polyketide synthase (NR-PKS) pathway as well.36
2. α,β-Unsaturated Amides (Acrylamides) and Lactams
2.1. Acrylamides
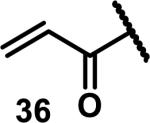
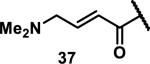
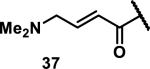
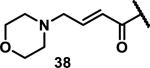
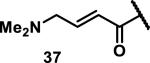
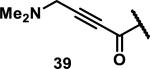
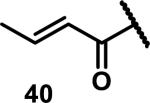
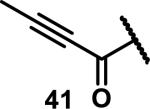
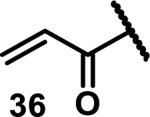
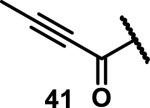
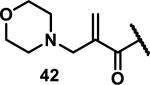
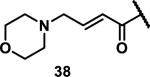
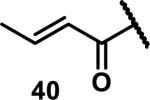
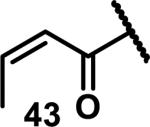
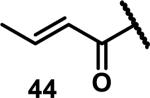
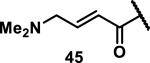
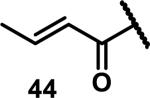
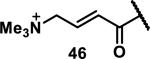
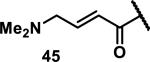
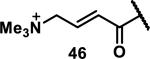
Unfunctionalized acrylamides are weakly electrophilic and relatively unreactive towards thiols; this has led to acrylamides being the most successful electrophile used in targeted covalent inhibitors. However, there are exceptions and these include the placement of an additional electron withdrawing group at the α position of the acrylamide.33a The relative thiol reactivity of acrylamides possessing different substituents were compared in a competition assay with a limiting amount of GSH.37 Two acrylamides (2.5 mM of each) were reacted with GSH (1.25 mM) in a THF-H2O-MeOH mixture at rt for 20-24 h and the amount of adduct formation was determined by integration of HPLC peaks. These conditions were chosen to give low conversion to GSH adducts so that the ratios of GSH adducts formed would reflect kinetic control. A Lewis basic aminomethylene group at the β position 37 afforded more of the GSH adduct relative to an unsubstituted acrylamide 36 (entry 1, Table 1). Competition experiments between acrylamide 37 and propynamide 39 showed the acrylamide to be more reactive towards GSH (entry 3, Table 1). However, methyl substituted propynamide 41 is more reactive than β-methyl acrylamide 40 (entry 4, Table 1).37b Placement of the morpholinomethylene group at the α position of the acrylamide affords 42 that is more reactive than β-morpholinomethylene acrylamide 38 (entry 6, Table 1).37b For unreactive samples, excess base (diisopropylamine or triethylamine) was added to the reaction mixture to promote GSH adduct formation (entries 4,5, and 7, Table 1). Utilizing a different heterocyclic scaffold, β-methyl substituted acrylamide 44 showed no adduct formation with glutathione whereas 45 possessing a N,N-dimethylmethanamine at the β position showed increased reactivity (entry 8, Table 1). The authors initially proposed that under aqueous conditions, the dimethylamino group would exist primarily in its protonated form, which would lead to an increase in the electrophilicity of the Michael acceptor through induction. However, this hypothesis was revised because inhibitor 46 bearing a trimethylmethanium ion did not react with glutathione, therefore it was proposed that an intramolecular base catalysis is operating to increase the reactivity of GSH (entries 9 and 10, Table 1).37a
Table 1.
Kinetic Competition Experiments Between Acrylamides and GSH


| ||||
|---|---|---|---|---|
| Entry | R (A) | R (B) | % Conversion | |
| A | B | |||
| 8 |
|
|
0 | 19 |
| 9 |
|
|
0 | 0 |
| 10 |
|
|
15 | 0 |
Two inhibitors (2.5 mM each) were reacted with a limiting quantity of GSH (1.25 mM), 20-24 h, rt and the percent conversion to the adducts was measured
12 equiv diisopropylethylamine were added
1000 equiv triethylamine were added
Recently, scientists at Amgen examined the GSH reactivity of a series of arylacrylamides.38 In their study, 34 N-arylacrylamides (1 μM) were incubated with 5000 μM GSH at 37 °C in buffer (pH = 7.4) containing 1-1.5% DMSO. The progress of the reactions was monitored by LC-MS with the percent of remaining acrylamide determined by MS using an internal standard.39 The relative rates of GSH addition to N-arylacrylamides are arranged in decreasing order in Scheme 2 and range from 3.38 × 10−5 s−1 (t1/2 = 343 min) for the slowest reacting p-methoxyphenyl acrylamide to 1.92 × 10−3 s−1 (t1/2 = 6 min) for the fastest reacting o-nitrophenyl acrylamide. Substitution of the aryl ring of N-arylacrylamides at the ortho and para positions impacted the reaction rate more than substituents at the meta position.38 These rates were correlated to the chemical shifts of Hβ1 and Cβ, as well as calculated kinetic reaction barriers, which are discussed in Section 9, Spectroscopic and Computational Predictors of Thiol Reactivity for Michael Acceptors, in this perspective.
Scheme 2.

Relative Rates of GSH Addition to N-Arylacrylamides
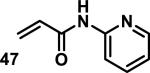
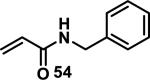
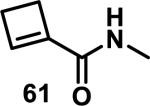
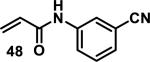
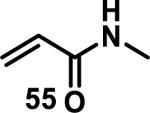
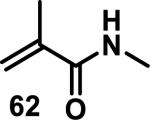
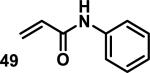
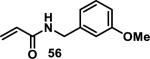
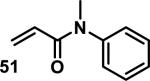
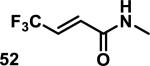
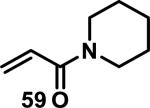
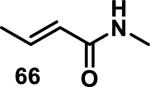
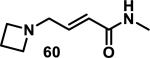
Similarly, scientists at Pfizer studied a more diverse set of acrylamides substituted with N-aryl, N-alkyl, and/or substituents at the α or β position of the double bond (Table 2).40 The acrylamide (1 mM) was incubated with GSH (10 mM) in buffer (pH = 7.4) containing 10% acetonitrile at 37 °C using either LC-MS or 1H NMR to measure the consumption of the acrylamide. The researchers found that the rate of GSH addition using 10% dimethylacetamide in buffer (pH of 7.4) was slower than the rate measured using 10% acetonitrile as the cosolvent for five samples tested.40 The most reactive acrylamides 47-51 had no substitution at the α or β carbon and were substituted with aryl or heteroaryl groups at the nitrogen (entries 1-5, Table 2). Substitution of the β carbon with a trifluoromethyl group 52 increased reactivity relative to the unsubstituted acrylamide 55 (compare entries 6 and 9, Table 2). Alkyl substitution at the α or β carbons greatly reduced reactivity, but some reactivity was regained with certain β-aminomethyl substituents (compare entries 12 and 14 to entries 15-21, Table 2).40 The half-lives of three nonreactive acrylamides 61-63 were measured at a higher temperature (60 °C), but only the half-life of cyclobutene containing acrylamide 61 decreased significantly, demonstrating the low reactivity of these compounds towards thiols (entries 15-17, Table 2).
Table 2.
Half-Lives for the Reaction of Various Acrylamides with GSH

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Reactive t1/2 < 8 h | Mildly Reactive t1/2 = 15 - 41 h | Non-Reactive t1/2 > 60 h | ||||||
| Entry | Acrylamide | t1/2 (h)a | Entry | Acrylamide | t1/2 (h)a | Entry | Acrylamide | t1/2 (h)a |
| 1 |
|
0.13 | 8 |
|
15 | 15 |
|
> 60/20b |
| 2 |
|
0.44 | 9 |
|
17 | 16 |
|
> 60/58b |
| 3 |
|
0.88 | 10 |
|
25 | 17 |
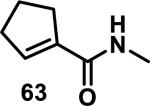
|
> 60/>60b |
| 4 |
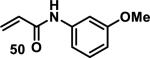
|
1.6 | 11 |
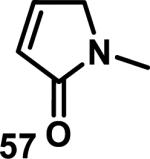
|
27 | 18 |
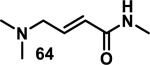
|
> 60 |
| 5 |
|
3.5 | 12 |
|
28 | 19 |
|
> 60 |
| 6 |
|
4.0 | 13 |
|
33 | 20 |
|
> 60 |
| 7 |
|
8.0 | 14 |
|
41 | 21 |
|
> 60 |
Half-lives (t1/2) of acrylamides (1 mM) when reacted with GSH (10 mM) at pH = 7.4 at 37 °C with 10% MeCN
t1/2 measured at 60 °C.40
2.2. α,β-Unsaturated Macrocyclic Lactams
Macrocyclic lactams containing an α,β-unsaturated amide are less common than other types of Michael acceptors in natural and synthetic compounds, and a large portion of these macrocycles contain peptidic backbones. Microcystins are the most prevalent members of the α,β-unsaturated macrocyclic lactam class, and they are heptapeptides containing a dehydroalanine moiety; one member from this class of compounds, 68, is depicted in Figure 10. Microcystins have been reported to form covalent bonds with noncatalytic cysteine residues in serine/threonine protein phosphatases (PP1 and PP2A).41 Moreover, GSH and cysteine have been shown to add to the exocyclic double bond of the unsaturated amide of microcystins; these thiol adducts were tested and showed a 16-fold (GSH adduct) or 7-fold (cysteine adduct) reduction in toxicity compared to free microcystins in mice.41b Mutation studies involving the conversion of Cys273 to alanine resulted in the inability of 68 to form a covalent adduct, but the inhibition of a C273A mutant PP1 by 68 was not significantly affected, suggesting there is considerable noncovalent affinity of 68 for PP1. Covalent targeting by 68 was further confirmed by a co-crystal structure of the compound covalently bound to Cys273 of PP1.41c, 41d
Figure 10.

Example of a microcystin.
Rakicidin A (69) shows cytotoxicity against some cancer cell lines and contains a 4-amido-2,4-pentadienoate moiety that is critical for bioactivity (Scheme 3). A simplified analog of 69 was prepared, 70, and shown to form adduct 71 with methyl thioglycolate through 1,6-addition after 72 h at rt in DMSO-d6.42
Scheme 3.

Rakicidin A and Analog that Forms a 1,6-Addition Product with Methyl Thioglycolate
Thalassospiramides are a family of natural products that inhibit human calpain 1 protease (HCAN1) with nanomolar activity (IC50 values between 3 and 79 nM for inhibition of HCAN1 activity in enzymatic assays).43 Thalassospiramides all contain a conserved 12-membered macrocycle with an α,β-unsaturated lactam, which is crucial for biological activity. Hydrolytic cleavage of the macrocycle at the ester position or hydrogenation of the alkenes in thalassospiramide A (72) resulted in compounds with more than a 100-fold reduction in HCAN1 inhibition (Figure 11). Macrocycle 72 was recently shown to form covalent adducts to catalytic Cys115 of HCAN1 by forming a 1:1 HCAN1:72 adduct detected by MS. Additionally, after incubation of the C-terminal domain of HCAN1 with 72 followed by trypsin digestion, the peptide containing Cys115 covalently bound to 72 was detected by LC-MS.43
Figure 11.

Structure of thalassospiramide A.
Pyrrocidine A (73) is a natural product with a 13-membered macrocycle containing a pyrrolinone (α,β-unsaturated-γ-lactam) that shows antimicrobial activity and is cytotoxic to human promyelocytic leukemia HL-60 cells with an IC50 of 0.12 μM (Figure 12).44 Lactam 73 reacts rapidly with N-acetyl-cysteine methyl ester in less than an hour in a 70% MeOH/water solution at 37 °C. The reaction was monitored by HPLC and the adduct confirmed by LC-MS.
Figure 12.
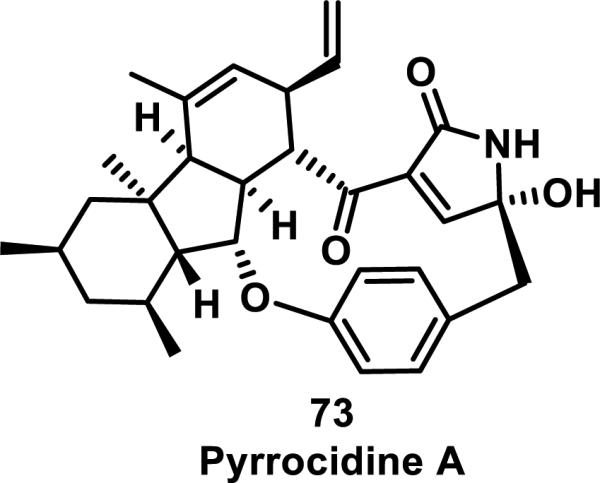
Structure of pyrrocidine A.
Small molecule pyrrolinones 74 and 75, that are not dually activated with two conjugated ketones as in 73, undergo hetero-Michael additions with 2-mercaptoaniline to afford the corresponding thiol adducts 76 and 77 in 80 and 70% yields, respectively; when R was bulky, such as the silyl ether in 75, the diastereomeric ratio afforded in the conjugate addition was greater than 5:1 (Scheme 4).45
Scheme 4.

Thiol Addition to Pyrrolinones
2.3. α-Methylene Lactams
α-Methylene-γ-lactams or 3-methylenepyrrolidin-2-ones are much less common in nature than the related α-methylene-γ-lactones. Naturally occurring examples of α-alkylidene-γ-lactams include pukeleimide (78), possessing an α-methylene group, anatin (79) with an α-benzylidene group, and gelegamine (80) possessing an α-ethylidene-δ-lactam (Figure 13).46 In general, α-methylene lactams are less reactive towards thiols than the corresponding lactones and formation of the thiol adducts usually requires more forcing conditions.47 For example, α-methylene butyrolactam 81 required thioacetic acid solvent and heating to 50 °C to obtain adduct 82 in 58% yield;47e for lactams 83, 84, and oxindole 87, thiols were used in excess, and base additives were needed to effect thiol addition in good yields (Scheme 5).47a, 47b For oxindole 90, thiol addition occurred with an acidic pH between 5 and 5.5 to give adduct 91 in 93% yield (based on glutathione).47c α-Methylene-γ-lactams are a relatively unexplored Michael acceptor that we expect could serve as an isostere for omnipresent α-methylene-γ-lactones that are discussed later in this perspective.
Figure 13.

Natural products containing α-methylene, α-benzylidene, or α-ethylidene lactams.
Scheme 5.

Thiol Adduct Formation with α-Methylene-γ-lactams and Oxindoles
3. α,β-Unsaturated Esters and Lactones
3.1. Acyclic α,β-Unsaturated Esters
Simple α,β-unsaturated, acyclic esters have generally demonstrated less reactivity with GSH than structurally similar acrylamides in toxicological assays. Comparisons of acyclic esters to other electrophiles are discussed later in this perspective.1, 48 Esterase cleavage of acyclic esters is common because of the diversity of esterase enzymes found throughout the body,49 which has led to esters being the most common functionality used in prodrugs.50 The drug dimethyl fumarate (31) rapidly forms adducts with GSH but is also rapidly hydrolyzed by esterases to monomethyl fumarate (32), which reacts significantly slower with GSH, as discussed above (Scheme 1).32a Rupintrivir (92), developed by scientists at Agouron Pharmaceuticals, is an acyclic α,β-unsaturated ester that irreversibly inhibits human rhinovirus 3C protease (HRV-3CP) with a kobs/[I] value of 1.47 × 106 M−1s−1 against serotype 14 HRV-3CP and bears antiviral activity against 48 rhinovirus serotypes with EC90 values of 18-261 nM in H1-HeLa or MRC-5 cells with low toxicity (CC50 > 100 μM, Figure 14).51 Rhinoviruses are the primary cause of the common cold, with at least 100 different serotypes known; their 3C proteases have amino acid sequence identities as low as 50%.51 Although the active site is mostly conserved among HRV-3CPs, the diversity among serotypes and the shallow binding pocket make noncovalent inhibition of a large number of serotypes difficult. Covalent inhibition allows for the targeting of many different HRV-3CPs by forming an adduct with the conserved, catalytic cysteine, even when noncovalent interactions may change among the active sites of diverse HRV-3CPs. Contrary to 32, the acyclic ester of 92 was stable in human and dog plasma (t1/2 > 60 min) despite being rapidly hydrolyzed in rat plasma (t1/2 < 2 min).51 Covalent adduct formation of α,β-unsaturated ester 92 with Cys147 of serotype 2 HRV-3CP was demonstrated by co-crystallography.51 Compound 92 made it through initial Phase II trials with a good safety profile and moderate efficacy, but failed in larger Phase II trials due to low efficacy. Recently, there has been a renewed interest in 92 and similar peptidic compounds with attached Michael acceptors, for the treatment of enterovirus-71, noroviruses, and foot and mouth disease that have 3C proteases resembling HRV-3CP.52
Figure 14.

Inhibitors of human rhinovirus 3C protease (HRV-3CP).
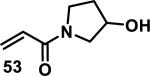
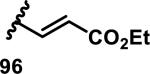
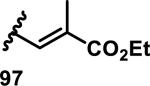
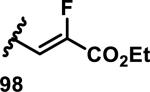
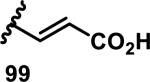
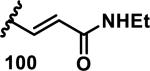
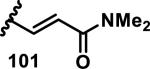
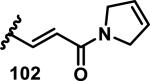
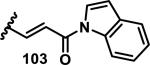
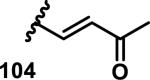
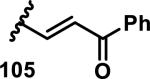
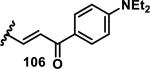
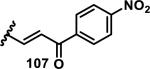
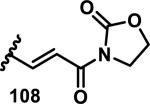
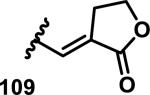
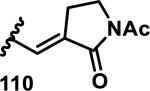
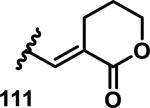
The development of 92 began with discovery that peptidic aldehyde 93 is a reversible 6 nM inhibitor of HRV-3CP (Figure 14).53 Peptidic aldehydes inhibit cysteine proteases by reversible covalent inhibition due to the formation of thiohemiacetals with the catalytic cysteine residue, but these aldehydes frequently have poor pharmacological properties.54 During the optimization of 92, various Michael acceptors were studied. α,β-Unsaturated esters of the lead peptide, such as 94, gave the best combination of HRV-3CP inhibition (kobs/[I] = 2400-39400 M−1s−1), antiviral activity in cells (EC50 = 0.50-3.6 μM), and low cellular toxicity (CC50 > 100 μM; representative examples are shown in entries 1-4, Table 3).53 Notably, the cis methyl propenoate 95 was 10-fold less active than the trans isomer 94 for both HRV-3CP inhibition and antiviral activity (entries 1-2, Table 3). Surprisingly, ester 98 with an α-fluoro substituent on the alkene had no antiviral activity, while an α-methyl ester 97 showed similar antiviral activity (EC50 > 320 μM) to the cis ester 95 (entries 2, 4-5, Table 3). α,β-Unsaturated carboxylic acid 99 was inactive for both HRV-3CP inhibition and antiviral activity (entry 6, Table 3). α,β-Unsaturated amides 100-103 all showed reduced HRV-3CP inhibition (kobs/[I] = 350-8500 M−1s−1) and reduced antiviral activity (EC50 = 16 to >320 μM), with the exception of a pyrrole or indole 103 attached to the carbonyl carbon (representative examples, entries 7-10, Table 3). α,β-Unsaturated acyl indole 103 with a kobs/[I] of 53,000 M−1s−1, along with all α,β-unsaturated ketones 104-107 tested, displayed reduced activity when pretreated with dithiothreitol (DTT) in a separate experiment, suggesting that these Michael acceptors are reactive toward nonenzymatic thiols (representative examples, entries 11-14, Table 3). Generally, Michael acceptors that showed deactivation by DTT had large kobs/[I] values and were commonly cytotoxic (CC50 < 100 μM).53 α-Methylene-γ-lactones and lactams were tested, with γ-lactone 109 having 10-fold reduced antiviral activity compared to the ethyl propenoate 96, while the N-acetyl-γ-lactam 110 had similar antiviral activity and a better HRV-3CP inhibition (kobs/[I] = 155,000 M−1s−1) than ethyl propenoate 96; the activity of the lactone 109 or lactam 110 were unaffected by treatment with DTT (compare entry 3 with entries 16-17, Table 3).53 Interestingly, δ-lactone 111 behaved as a reversible HRV-3CP inhibitor with a Ki of 30 nM, indicating that lactone 111 does not undergo hetero-Michael addition to the catalytic cysteine of HRV-3CP, or that hetero-Michael addition to lactone 111 is reversible (entry 18, Table 3). Further SAR studies used the ethyl propenoate Michael acceptor and optimized the rest of the peptidic structure to reach 92, which showed good selectivity for HRV-3CP compared to a panel of human proteases.51
Table 3.
Comparison of Different Michael Acceptors for Inhibition of HRV-3CP

| |||||
|---|---|---|---|---|---|
| Entry | R = | kobs/[I] (M−1s−1)a | DTT deactivationb | EC50 (μM)c | CC50 (μM)d |
| 1 |
|
20000 | No | 1.3 | >320 |
| 2 |
|
2400 | No | 4.2 | >320 |
| 3 |
|
25000 | No | 0.54 | >320 |
| 4 |
|
7300 | No | 3 | >320 |
| 5 |
|
60 | ND | >320 | >320 |
| 6 |
|
30 | ND | >100 | >100 |
| 7 |
|
350 | No | >320 | >320 |
| 8 |
|
1300 | ND | 56 | >100 |
| 9 |
|
8500 | No | >44 | 44 |
| 10 |
|
53000 | Yes | 1.8 | 29 |
| 11 |
|
54000 | Yes | 2.0 | 60 |
| 12 |
|
500000 | Yes | 4 | 16 |
| 13 |
|
4500 | Yes | >25 | 25 |
| 14 |
|
310000 | Yes | >32 | 32 |
| 15 |
|
600000 | Yes | 1.6 | >100 |
| 16 |
|
10900 | No | 5.2 | >320 |
| 17 |
|
155500 | No | 0.71 | >100 |
| 18 |
|
Ki = 30 nMe | ND | 16 | >100 |
Enzyme inhibition reported as kobs/[I] (M−1s−1)
DTT deactivation indicates partial or complete loss of HRV-3CP inhibition if a compound was pretreated with dithiothreitol for 3 min (ND = not determined)
Antiviral activity reported as EC50 (μM) in H1-HeLa cells
Cellular toxicity of compound to H1-HeLa cells reported as CC50 (μM)
Reversible inhibition of HRV-3CP
In an effort to discover nonpeptidic cysteine protease inhibitors, Kathman et al. utilized a fragment based screening method, where different drug-like fragments were attached to an electrophilic fragment and tested for their ability to inhibit papain, a model cysteine protease.55 The rates of N-acetyl-cysteine-methyl ester addition to N-arylacrylamides 112-114, N-aryl vinyl sulfonamides 115-117, amidomethyl methacrylates 118-120, and vinyl sulfones 121-123 were measured under pseudo-first order conditions (78 mM thiol, 10 mM electrophile) in 2:1 PBS to DMSO-d6 to find an electrophile that would be relatively unaffected by electronically diverse substituents (Figure 15). This allows for optimization of the binding affinity (Ki) while the rate of covalent bond formation (kinact) remains constant; the use of covalent tethering fragments was recently reviewed by Kathman and Statsyuk.56 N-arylacrylamides 112-114 showed a 2044-fold difference in the rate of thiol addition depending on the aromatic substituent. Vinyl sulfonamides 115-117 showed an 8-fold difference, but the acrylates 118-120 and vinylsulfones 121-123 were the least affected by various substituents displaying only a 1.6- or 1.4-fold difference between the most reactive and the least reactive acrylate (compare 120 to 119) or sulfone (compare 123 to 122, Figure 15). Amidomethyl methacrylates were used as the electrophilic fragment in further studies due to their lower reactivity compared to the vinyl sulfones.
Figure 15.

Michael acceptors and pseudo-first order reaction rates with N-acetyl-cysteine methyl ester.
Compound fragments were screened by attaching 100 structurally diverse fragments to an aminomethyl methacrylate. Ten compounds at a time (100 μM each) were incubated with papain (10 μM) for 1 h, and whole protein ESI-MS was used to identify compounds that covalently bound to papain, with hits defined as any compound that labeled papain by more than 50%.55 Three reaction mixtures (10 compounds each) contained a single compound that displayed strong covalent labeling of papain, while no significant covalent labeling of papain was detected in the other 7 reaction mixtures. Compounds 124 and 125 were confirmed to irreversibly inhibit papain with kinact/Ki values of 1.2 and 0.5 M−1s−1, respectively, similar to known peptidic papain inhibitors 126 and 127 with kinact/Ki values of 6.6 and 0.4 M−1s−1, respectively (Figure 16). A similar fragment based approach utilizing a small 10-member acrylamide library was utilized to screen for inhibitors of thymidylate synthase.57
Figure 16.

Irreversible papain inhibitors.
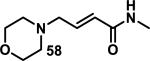
3.2. δ-Valerolactones, Chromones, Coumarins, and Butenolides
Unsaturated δ-valerolactones are a class of Michael acceptors found in bioactive natural products such as leptomycin B (LMB, 128), anguinomycin D (129), and other members of this family of antibiotics (Figure 17).58 LMB (128) inhibits CRM1-mediated (chromosome region maintenance 1; also referred to as exportin1) protein transport across the nuclear membrane completely at 1 nM, 129 completely inhibits at 10 nM, and structurally truncated synthetic derivative 130 completely inhibits at 25 nM, demonstrating the importance of the unsaturated δ-valerolactone (Figure 17).59 Lactone 128 inhibits the binding of CRM1 through covalent modification of Cys529.60 The target residue Cys529 was confirmed through mutation studies, whereas mutation C529S conferred resistance to 128. Labeling studies with 128 using an 18-residue synthetic peptide (residues 513-530), which mimics the N-terminal domain of CRM1, in Tris-HCl buffer (pH = 7.5, 18 h) showed a peptide-probe adduct via LC-MS, whereas no reaction with GSH (6.5 mM) or cysteine (16 mM) occurred under similar conditions (1.8 mM 128).60 The authors attribute this selectivity to the hydrophobicity of 128 and hydrophilicity of cysteine and GSH. Biotinylated LMB also shows high specificity for CRM1; after biotinylated LMB was incubated with HeLa cells, a Western blot of the lysed cells detected CRM1 as the only covalently bound protein.60-61 A crystal structure of 128 covalently bound to CRM1 revealed that the lactone of 128 was hydrolyzed to the carboxylic acid. It is expected that hydrolysis stabilizes the covalent adduct by decreasing the reversibility of adduct formation.62 It was postulated that the reverse Michael reaction, governed by the acidity of the hydrogen α to the carbonyl, is slower for the carboxylate than the lactone.
Figure 17.

Inhibitors of CRM1-mediated nucleocytoplasmic transport and their IC50 values.
Structure-activity relationship (SAR) studies on anguinomycins (e.g 129) and kozusamycins (e.g. 131) have found that the unsaturated δ-lactone is critical for potent cytotoxicity (Figure 18).59, 63 Ando et al. showed that changes made to the lactone of 131 could modulate the cytotoxicity of this compound. Analog 132 with a gem-dimethyl substituent at the γ-position of the lactone showed cytotoxicity towards HPAC cells with an IC50 of 0.04 nM, which is similar to the cytotoxicity of 131 of 0.08 nM. Analog 133 with no methyl groups at the γ-position, but a new methyl substituent at the β-position showed about a 10-fold reduction in cytotoxicity from 132. Analog 134 with a methyl group at the β and γ-positions of the lactone showed a large reduction in activity with an IC50 of 180 nM, while analog 135, with a methyl group at the β-position and a gem-dimethyl at the γ-position of the lactone, showed the lowest cytotoxicity with an IC50 of 16,000 nM (Figure 18). It was hypothesized that the decreased cytotoxicity is a result of decreased reactivity of compounds 132-135 as Michael acceptors (both steric and electronic contributions); however, the chemical reactivities of these analogs were not examined.63
Figure 18.

Kozusamycin A with analogs and cytotoxicity towards HPAC cells.
Glycanolactones such as 136 are reactive towards thiols at room temperature in aqueous media, forming adducts in 30 min without an added base and are stable to chromatographic purification.64 The use of carbohydrate derived thiol 137 resulted in diastereoselective formation of product 138 in 72% yield, while simple thiols such as thiophenol (not shown) gave mixtures of diastereomers in 78% yield (Scheme 6).64 α-Aculosides, a class of compounds that are structurally related to the glycanolactones, are found in antibiotics of the vineomycin and urdamycin family. These cyclic enones readily react with thiols, as demonstrated by α-aculosides 139 and 140 that form adducts 141 or 142 with N-Boc-cysteine methyl ester in 80 or 67% yield, respectively, and in less than 1 min at 37 °C (Scheme 6).65
Scheme 6.

Thiol Addition to Unsaturated Sugars
Generally, thiol adduct formation with α,β-unsaturated δ-valerolactones is performed in the presence of a weak base. For example, lactone 143 and mevinolin analog 144 both form adducts with thiol acetic acid stereoselectively; lactone 143 forms adducts 145-148 with various other thiols in 0.5 to 2 h and 65-97% yields in the presence of catalytic triethylamine (Scheme 7).66 Goniothalamin derivative 150, and lactone 151 undergo a highly diastereoselective thiol addition, where the thiol adds anti to the trifluoromethyl or chloromethylene groups giving adducts 152 or 153 in 86 or 92% yields, respectively.67 Lactone 154 containing a bulky tert-butyl dimethylsilyl group adjacent to the Michael acceptor required heating to 70 °C to effect thiol addition, and thiols added anti to the silyl group to afford thiol adducts 155-157; bulkier thiols such as tert-butyl thiol and electron poor p-nitrothiophenol failed to react with 154, demonstrating that the electronic and steric nature of the thiol also impacts the Michael addition reaction.68
Scheme 7.

Thiol Addition to α,β-Unsaturated-δ-valerolactones
Thiol addition reactions to 2- and 4-pyrones are scarcely reported, but there are examples of the addition to structurally related chromones 160 and 161.69 Initial formation of an imine between the aldehyde of the chromone and an amine source, followed by hetero-Michael addition of a second amine, thiol, or oxygen nucleophile gives products such as benzothiazapinone 162, via intramolecular thiol addition, or compounds such as 163-167, via intermolecular thiol addition in good yields of 53-89% (Scheme 8).69
Scheme 8.

Thiol Addition to Dually Activated Chromones
There are few reports of coumarins reacting with thiols. Coumarin 168 reacts with p-toluenethiol yielding adduct 169, and styryl coumarin 170 gives the 1,6-addition products 171 and/or 172 in good yields (only elemental analyses characterization data was provided, Scheme 9).70 Coumarins have been employed as fluorophores in fluorescent probes for thiol detection. However, a thiol reactive group is incorporated onto these probes, which typically includes a maleimide, unsaturated malonate, benzoquinone, or enone;71 the coumarin group has not been reported to react with the thiols in these assays.
Scheme 9.

Thiol Addition Reactions to Coumarins
Butenolides are found in the cardenolide family of natural products, with two examples, digitoxigenin (173) and ouabagenin (174) shown in Figure 19.72 Cardenolides display cardiac toxicity, which is lost or reduced upon reduction of the lactone ring.
Figure 19.

Examples of cardenolide natural products containing a butenolide.
Kupchan et al. studied thiol reactivity and the effects of methyl substitution at the α, β, and γ positions of butenolides using propanethiol, benzyl thiol, cysteine, and N-acetyl-cysteine methyl ester.73 Butenolide 175 containing no methyl substituents and butenolide 176 containing a methyl substituent at the γ position formed adducts 177-178 with propanethiol (Scheme 10). However, butenolides 180 and 181, containing a methyl substituent at the α or β position, respectively, only formed adducts with cysteine and the yields for formation of adducts 182 or 183 were low (38 or 12%, respectively).73,74 This trend is further supported by the reluctance of the cardenolides 173 and 174 to undergo thiol addition under a variety of forcing conditions.72
Scheme 10.

Effect of Alkyl Substitution on Thiol Addition to Butenolides
Thiol adducts have been utilized to protect double bonds of α,β-unsaturated carbonyls for synthesis applications. For example, in Trost's synthesis of iso-cladospolide B 186, thiophenol reacted selectively with the double bond of the butenolide group of 184 and the corresponding sulfide was oxidized to the sulfone to give 185 (Scheme 11). The butenolide was then re-installed following base-mediated elimination of the sulfone to yield 186.75
Scheme 11.

Thiol Adducts as Double Bond Protecting Groups
Butenolides such as 187, containing a second methylene conjugated at the γ position, form 1,6-addition products with benzyl thiol and propanethiol (not shown); butenolide 187 reacted with thiophenol in 10 min at 0 °C giving 1,6-addition product 188 and bis addition product 189 in 48 and 16% yield, respectively (Scheme 12).76 Additionally, ring opened products were observed in small amounts with thiol nucleophiles.76a Adenine-containing butenolide 190 formed thiol adduct 191 at the γ position in 73% yield when treated with N-acetyl-L-cysteine (NAC, Scheme 12).77 Hakimelahi explained this anomalous result by a competing oxonium ion formation, protonation, and 1,4-addition sequence to give 191.77 The second order rate constant for NAC addition to butenolide 190 was 84.9 M−1s−1, and it was shown to be a time-dependent inhibitor of S-adenosyl-L-homocysteine hydrolase with a kinact/Ki of 152 M−1s−1.
Scheme 12.

Thiol Addition to γ-Methylene or γ-Alkylidene Butenolides
3.3. Exocyclic α-Methylene-γ-lactones
Sesquiterpene lactones (SLs) are a major class of natural products with a wide range of biological activities including antiproliferative, fungicidal, antibiotic, and antiinflammatory effects. The main pharmacophore contained in many of these molecules is the α-methylene-γ-lactone, which is found in ~3% of all natural products and contributes to much of the observed biological activities of SLs.78 The exocyclic methylene lactone can undergo a hetero-Michael addition with thiols found in cells (e.g. solvent accessible cysteine side chains on proteins, endogenous thiols such as GSH) resulting in covalent adduct formation. Reduction of the exocyclic methylene routinely eliminates biological activity, with some exceptions, even at high micromolar concentrations.79 Several reviews have covered the biological importance and therapeutic potential of α-methylene-γ-lactone-containing compounds.80 Therefore, the thiol reactivity of these Michael acceptors with cysteines and GSH will be the focus here.
A kinetic study by Kupchan et al. compared the reactivity of three α-methylene-γ-lactone-containing natural products, elephantopin (192), eupatundin, (193), and vernolepin (194), with various endogenous nucleophiles in aqueous buffer (pH = 7.4, Figure 20).81 It was determined that each compound underwent a slow addition with lysine, where 75% of the starting material was recovered after 6 days at ambient temperature. Moreover, these compounds were all found to be completely unreactive towards guanine. However, 192-194 reacted completely with cysteine within minutes. Compounds 193 and 194 were found to preferentially react with cysteine at the exocyclic methylene when one equivalent of cysteine was used. The rates of conjugate addition with one equivalent of cysteine to the α-methylene-δ-lactone and α-methylene-γ-lactone of vernolepin were the same according to 1H NMR studies (~200 M−1s−1). Second order reaction kinetics are presented in Table 4 and show that all compounds react at similar rates to that of iodoacetamide, but 10-80 times slower when compared to N-ethylmaleimide. Addition of cysteine to the methacrylate of 195 (confirmed with 1H NMR) occurs 12 times slower (1.7 M−1s−1) when compared to Michael addition to the α-methylene-γ-lactone of the parent compound, 192 (20 M−1s−1).
Figure 20.

Structures of α-methylene-γ-lactone-containing natural products.
Table 4.
Rate Constants for Reaction of α-Methylene-γ-lactones with Cysteine
| Compound | Rate Constant (M−1s−1)a |
|---|---|
| 195 | 1.7 |
| Elephantopin (192) | 43 |
| Iodoacetamide | 25 |
| Eupatundin (193) | ~42b |
| Vernolepin (194) | ~200b |
| N-ethylmaleimide | 3750 |
Reported second order rate constants with cysteine (2 equivalents) in aqueous buffer (pH = 7.4)
The kinetics for the addition of cysteine to 193 and 194 were nonlinear, so the rate constants were calculated from initial rates.
Helenalin (196) is a natural product containing two Michael acceptors, a cyclopentenone and exocyclic methylene butyrolactone. Tenulin (197) is also a natural product but only contains one Michael acceptor, a cyclopentenone (Figure 21). Thiol adducts of 196 and 197 were first reported by Hall et al., who showed that the bioactivity of a tenulin-cysteine adduct was 6-fold lower than the parent compound for inhibition of Ehrlich ascites tumor growth in mice.82 The GSH and cysteine adducts of 196 were further characterized and the rates of adduct formation measured. These studies showed that GSH addition preferentially occurred at the cyclopentenone, while cysteine addition occurred at the α-methylene-γ-lactone.83 Reaction with excess GSH or cysteine led to addition to both double bonds, forming bis-adducts. The second order rate constant for GSH addition to the cyclopentenone (1.3 × 10−3 M−1s−1) was 10 times faster than GSH addition to the α-methylene-γ-lactone (1.3 × 10−4 M−1s−1) as measured by monitoring the disappearance of the olefinic resonances by 1H NMR. Cysteine addition to helenalin (196) showed the opposite chemoselectivity, adding to the α-methylene-γ-lactone in less than 5 min, while requiring approximately 17 hours to completely react with the cyclopentenone olefin to form a bis-adduct. Computational results suggest a stabilization of the cysteine-adduct by coordination of the amine to the carbonyl of the lactone ring.
Figure 21.

Structures of pseudoguaianolides.
Glutathione addition to pseudoguaianolides 198 and 199 containing only the cyclopentenone or α-methylene-γ-lactone, respectively, showed pH dependency, with a faster addition occurring at a more basic pH.84 Under physiological conditions, GSH was found to react very slowly with the exocyclic methylene group of 199. First-order rate constants were not calculated for 199 due to the lack of reactivity, however, the apparent second order rate of addition for 199 was 10-fold less than that of 198.84
Many structure-activity relationship (SAR) studies exist for natural products, as well as synthetic and semi-synthetic analogs, bearing α-methylene-γ-lactones.85 In the case of parthenolide (200), one strategy for improving its water solubility and bioavailability involved hetero-Michael addition of dimethylamine to the α-methylene-γ-lactone, yielding a prodrug, DMAPT (LC-1, 201) bearing a tertiary nitrogen that is eliminated in cells (Figure 22).85d Regeneration of the intact α-methylene-γ-lactone is required for the biological activities of SLs of this class, which typically require covalent adduct formation to biological thiols. This prodrug strategy has been recently reviewed,80a and has also been utilized in optimization studies of costunolide (202).86 In a recent study involving arglabin (203), amine (205-206, 210-212), thiol derivatives (208-209), and other derivatives involving small structural changes (204, 207) to the exocyclic methylene-γ-lactone and periphery of the molecule were prepared; their biological activities were analyzed (Figure 23).79c The Michael adducts were all active at low micromolar IC50 values (2-20 μM) for cytotoxicity to cervical, lung, melanoma, ovarian, colon, thyroid, and breast cancer cell lines, similar to the parent compound, where as the structural changes (204, 207, data not shown) abolished activity altogether (Table 5).
Figure 22.

Natural product α-methylene-γ-lactones and prodrug derivative (fumarate salt of 201 not shown for clarity).
Figure 23.

Structures of arglabin and derivatives.
Table 5.
Cytotoxicity of Arglabin and Derivatives Towards Cancer Cell Lines
| Cell line a | 203 | 205 | 208 | 209 | 210 | 211 | 212 |
|---|---|---|---|---|---|---|---|
| 515A2 (melanoma) | 6.72 | 5.54 | 16.9 | 16.9 | 13.2 | 14.3 | 15.0 |
| A431 (cervical) | 4.48 | 5.19 | 12.8 | 13.6 | 16.9 | 18.9 | 4.40 |
| A549 (lung) | 2.21 | 1.95 | 12.8 | 10.1 | 4.76 | 4.76 | 15.7 |
| HT-29 (colon) | 7.00 | 10.8 | 18.6 | 17.8 | 16.2 | 8.1 | 15.1 |
| MCF-7 (breast) | 2.33 | 1.85 | 17.3 | 16.9 | 13.4 | 16.9 | 15.1 |
IC50 values reported in μM
Biological activity can be attenuated by substituent changes at the exocyclic methylene of α-methylene-γ-lactones. For example, substitution of compound 213 (IC50 value of 15 μM) with an iso-propyl (214), a phenyl (215), or a naphthyl (216) group resulted in IC50 values of 90 μM (214), 63 μM (215), and 46 μM (216) when dosed to L-1210 mouse lymphocytic leukemia cells in a 72 h cytotoxicity assay (Figure 24).87 Steric and electronic effects limit the ability of an endogenous nucleophile to undergo a hetero-Michael addition.
Figure 24.

Reduction in cytotoxicity (IC50) when substituents are on the exocyclic methylene.
The reactivity of the α-methylene-γ-lactone to sulfur nucleophiles has been exploited in mechanistic biochemical studies. Attachment of reporter tags (e.g., biotinylation, ‘clickable’ reporters such as alkynes and azides) to natural products or synthetic derivatives containing α-methylene-γ-lactones has allowed for identification of their covalent protein targets.88 Within live cell culture and in cell lysate it has been determined that, under physiological conditions, probes containing α-methylene-γ-lactones irreversibly target thiols and withstand conditions used for protein pulldown studies (see Figure 26). However, detailed kinetic studies of the reactivities of α-methylene-γ-lactones and their thiol targets (e.g., thiol side chains on proteins; cysteine, GSH) are limited. Hetero-Michael addition of cysteamine to the α-methylene-γ-lactone of parthenolide (200) or costunolide (202) occurs in DMSO-d6 and is irreversible upon dilution with CDCl3 (for more information on this assay and other compounds tested, see section 9, Figure 40).
Figure 26.

Library of α-methylene-γ-lactones containing terminal alkynes used as biological probes for the discovery of novel anti-microbial targets.
Figure 40.

Compounds reactive toward cysteamine.
Crews and co-workers performed target identification studies of parthenolide (PTL, 200), using the analogous natural product, melampomagnolide B (MelB, 217), which contains an allylic alcohol that is useful for chemical modifications such as biotinylation (Figure 25).89 Using tryptic digestion of purified protein and single point mutation analyses, it was determined that 200 covalently modified Cys179 in the activation loop of IKKβ, inhibiting phosphorylation of IκB in the NF-κB pathway. With the biotinylated probe 218, they demonstrated a dose-dependent covalent interaction with IKKβ in HeLa cells.89a
Figure 25.

Melampomagnolide B and biotinylated derivative used for pulldown studies.
In related studies, a labeling experiment with biotinylated PTL probe 218 to live normal bone marrow and primary acute myeloid leukemia cells revealed formation of probe-protein adducts (Figure 25). Enrichment of protein-218 adducts using streptavidin beads followed by Western blotting revealed binding to both IKKβ and p65 in primary AML cells.90 A subsequent report identified additional proteins involved in management of AML oxidative stress as direct targets.88c These data as well as unpublished work from one of our laboratories support that many SLs containing α-methylene-γ-lactones are targeting multiple proteins with a cell proteome bearing accessible cysteines.
Protein pulldown and identification studies have been conducted in bacteria using a variety of α-methylene-, α-alkylidene-, and α-benzylidene-γ-lactones (219-227, Figure 26). This compound-centric approach by Sieber and co-workers showed that these compounds reacted with many proteins within bacterial proteomes,88b which is consistent with related studies in human cells. A select number of proteins were identified with LC-MS/MS and proteomic analysis, including katG, ahpC, and thil; another study found that 220 and 221 labeled TrxA, staphylococcal disulfide reductase, and three transcriptional regulators in bacteria (SarA, SarR, and MgrA).91
4. α,β-Unsaturated Ketones (Enones)
4.1. Styryl Ketones and Chalcones
Styryl ketones are a class of Michael acceptors that include cinnamates, chalcones, curcuminoids, and bis(benzylidenes). Dinkova-Kostova et al. investigated the thiol reactivity of some bis(benzylidene)-acetones and bis(benzylidene)-cyclopentanones and compared the rates of thiol addition to the ability of these compounds to induce NADPH:quinone reductase (NQO1).92 The CD, defined as the concentration of a compound that doubles the activity of NQO1, was used for quantitative comparisons of inducer potency, and it was found that compounds that reacted faster with thiols correlated to higher CD values.92 The rates of thiol addition were measured by UV spectroscopy in 1:1 acetonitrile:Tris-HCl buffer (pH = 7.4). For bis(benzylidene) cyclopentenones 228 and 229, an 11-fold increase in the rate of DTT addition (2.3 compared to 29 M−1s−1) and a 9-fold increase for GSH (9.3 compared to 87 M−1s−1) was observed for 229, containing ortho-hydroxyl groups on the aromatic ring, compared to 228 (Figure 27).92 For acyclic bis(benzylidenes) 231-233, ortho-hydroxyl groups (e.g. 232) increased the rate of DTT addition 6-fold from 0.42 M−1s−1 for 231 to 2.5 M−1s−1 for 232 and the rate of GSH addition 34-fold from 0.15 M−1s−1 for 231 to 5.0 M−1s−1 for 232, while para-hydroxyl groups (233) decreased the rate of thiol addition 30 or 145-fold for DTT (0.014 M−1s−1) and GSH (0.001 M−1s−1), respectively, compared to unsubstituted 231 (Figure 27).92 It was postulated that an ortho-hydroxyl group aids in thiol deprotonation by the mechanism shown in Figure 27.92
Figure 27.

Styryl dienones and proposed mechanism for ortho-hydroxy substituent effect.
Styryl ketones and their corresponding Mannich bases containing different substituents, such as 234-241, were reacted with ethanethiol in 1:1 acetonitrile:phosphate buffer at various pHs (e.g. 7.4, 7.0, or 6.4, Figure 28).93 The second order rate constants were obtained by UV absorbance of diluted aliquots of the reaction mixture at regular time intervals. Decreasing the pH from 7.4 to 7.0 resulted in approximately a 60% reduction in the rate of thiol addition for the styryl ketones 234-237; a 90% reduction in rate was observed when going from a pH of 7.4 to 6.4 for 234-337. The rate of thiol addition to Mannich bases 238-241 decreased approximately 50% when going from a pH of 7.4 to 7.0 and 80% when going from a pH of 7.4 to 6.4.93 Mannich bases 238-241 reacted about 240 times faster than the corresponding ketones 234-237, which was attributed to the protonated amine that could stabilize the intermediate enolate.93 A doubling of the rate was observed going from a hexyl to a methyl substituent for the ketones, which Dimmock et al. attribute to a disruption of solvation because of the larger alkyl group, which can stabilize the developing enolate.93
Figure 28.

Styryl ketones and similar Mannich bases.
1-p-Chlorophenyl-4,4-dimethyl-5-diethylamino-1-penten-3-one hydrobromide (CDDP, 242) is a styryl ketone/Mannich base similar to those above that was reported to react reversibly with β-mercaptoethanol, cysteine, and GSH, but irreversibly with protein thiols.94 The reversibility, or irreversibility, was demonstrated by reacting 242 with GSH, or denatured bovine serum albumin (BSA), in Tris-HCl buffer (pH = 8.8) containing 1 mM ethylenediaminetetracetic acid (EDTA) until complete adduct formation was observed by UV and then iodoacetamide was added which would react with any liberated thiols if the reaction were reversbile.94 For the BSA solution, no recovery of the enone absorbance of 242 was observed, while for the GSH solution, nearly complete recovery of absorbance was observed after 10 min. Mutus et al. provide two possible explanations for this observation, the first being that the reversibility is catalyzed by base and removal of a hydrogen could be more difficult in the protein environment, but as they discuss, this may be unlikely under the denaturing conditions used. The alternative explanation is that the antiperiplanar conformation required to eliminate the thiol is difficult to achieve within the constraints of a large protein, while it is facile with small molecule thiol adducts. No change in UV absorbance occurred when enone 242 was incubated with other reactive amino acids (Arg, Asp, Glu, Lys, Met, Ser, and Tyr), suggesting that 242 is selective for cysteine in biological systems.94
Chalcones are a class of styryl ketones flanked by two aryl rings, with the generic structure of 243, and they are natural products common to a variety of plants. Antiinflammatory, antimalarial, antibacterial, and anticancer activities are among the broad biological effects reported for chalcones.95 Amslinger et al. has examined the thiol reactivity of chalcones and the effects of modifying substituents on the aromatic rings,96 as well as the effects of substituents at the α-position.95 The second order rate constants for the addition of cysteamine, a model protein surface thiol due to its pKa of 8.3, were calculated from the rates observed under pseudo-first order conditions (40 μM chalcone, 480 to 20000 μM cysteamine) by monitoring the decrease in UV absorbance of the enone peak between 350 and 375 nm.96 These reactions were performed in a solution of Tris-HCl buffer (pH = 7.4) and ethylene glycol (1:4), which contained 2 mM EDTA to prevent thiol oxidation, and a 12-500 fold excess of cysteamine.96 A chalcone bearing a 2'-hydroxy group (244) reacted the fastest with cysteamine (k2 = 5.1 M−1s−1) and a chalcone with no aromatic substituents (243, R = H) reacted the second fastest (k2 = 3.0 M−1s−1).96 All other chalcones with hydroxy and alkoxy substituents at various positions on the aromatic rings reacted with a k2 between 0.1 and 1 M−1s−1, and it was observed that a 2'-hydroxy group generally increased the rate of thiol addition while 2'-alkoxy groups reacted more slowly.96 When 2'-hydroxy groups were present, cyclization of the pendant amine from cysteamine into the ketone led to formation of the 1,4-tetrahydrothiazepine 245,96 which has also been reported to occur with unsaturated aldehydes as discussed later (Scheme 13).97 Amslinger et al. postulated that the 2'-hydroxy group engages in an intramolecular hydrogen bond to the carbonyl to activate it towards thiol addition as shown in chalcone 244.
Scheme 13.

Generic Chalcone and Addition of Cysteamine to 2' Hydroxy Chalcone
The rate constants for a series of α-substituted 2',3,4,4'-tetramethoxy chalcones (246-259) were also determined by Amslinger et al. using the method described above, and it was found that varying the substituent at the α position of the enone led to rates of thiol addition spanning 6 orders of magnitude (Table 6).95 As expected, electron withdrawing groups increased the rate of cysteamine addition, while electron donating groups decreased the reactivity.95 Reactivity studies with the α-halogen compounds yielded unanticipated results where the rate of cysteamine addition was α-Br (249) > α-Cl (250) > α-I (252) > α-H (254) > α-F (255), showing that the α-F substitution was deactivating (entries 4-5, 7, 9-10, Table 6).95 The α-CO2H (259) was the slowest reacting compound, whose deactivating nature was attributed to formation of the carboxylate at a pH of 7.4, which would decrease the electrophilicity of the alkene.95 The fastest reacting compounds had the strongest electron withdrawing capacity with α-CN (246) > α-NO2 (247) > α-CF3 (248) and k2 values of 5750, 749, and 17.1 M−1s−1, respectively (entries 1-3, Table 6).95 The rates of β-mercaptoethanol and GSH addition were measured for several compounds and the same trends were observed, although the rates were decreased more than 10-fold compared to cysteamine addition.95 Other α,β-unsaturated carbonyls were tested in this assay including cinnamaldehyde and curcumin (260), which had k2 = 0.64 and 0.066 M−1s−1 respectively, while cinnamic acid and its derivatives, caffeic acid and its phenylethylester, as well as coumarins such as 3-hydroxycoumarin, kaempferol, and quercetin all showed no reaction or exceedingly slow rates of covalent adduct formation (< 0.001 M−1s−1) with 500-fold cysteamine after 63 h.96
Table 6.
Effects of α-Substitution on the Rate of Cysteamine Addition to Chalcones

| ||
|---|---|---|
| Entry | X = | k2 (M−1s−1) |
| 1 | CN (246) | 5700 |
| 2 | NO2 (247) | 750 |
| 3 | CF3 (248) | 17 |
| 4 | Br (249) | 2.9 |
| 5 | Cl (250) | 1.6 |
| 6 | p-NO2-Ph (251) | 0.29 |
| 7 | I (252) | 0.28 |
| 8 | CO2Et (253) | 0.28 |
| 9 | H (254) | 0.19 |
| 10 | F (255) | 0.017 |
| 11 | p-OMe-Ph (256) | 0.0086 |
| 12 | Me (257) | 0.0075 |
| 13 | Ph (258) | 0.0067 |
| 14 | CO2H (259) | 0.0037 |
The influence of these electrophiles (246-259) on the cellular activities of heme oxygenase (HO-1) and inducible nitric oxide synthase (iNOS) was measured in murine macrophage cells (RAW264.7) treated with 246-259 (Table 6). Some correlation between the rates of thiol addition and bioactivity were observed apart from the two most reactive chalcones α-CN 246 and α-NO2 247 which had no significant toxicity, HO-1 induction, or iNOS inhibition, possibly due to inactivation by GSH-conjugation.95 For iNOS inhibition, α-CF3 (248) > α-Br (249) > α-Cl (250) > α-I (252), which correlated to their rates of thiol addition; however, α-p-NO2-C6H4 chalcone 251 did not inhibit iNOS. These results suggest that intermediate electrophilicity may be the most beneficial for biological effects, although other α-CN enones have been shown to act as reversible covalent modifiers and are discussed in the dually activated Michael acceptor section of this perspective.33a, 33c
Curcumin (260), a natural product derived from the spice turmeric, has dienone like character due to its preference for the enol form of the β-diketone and has been shown to form bis-GSH adducts by LC-MS;98 two diastereomeric mono-GSH adducts were also characterized by MS and 1H NMR (Figure 29).99 A number of biological effects have been reported for enone 260, such as inhibition of NF-κB signaling and cell proliferation, induction of apoptosis, and prevention of β-amyloid fibril formation.99 Some biological effects have been shown to be dose dependent, where low doses of curcumin show antioxidant and glutathione-S-transferase (GST) induction, but higher doses are pro-oxidant and suppress GST activity.99 Usta et al. demonstrated that five related GST enzymes could catalyze the conjugation of GSH to curcumin with various rates, which could be a reason for the differential effects of curcumin in cell types with varying GST levels.99
Figure 29.

Structure of curcumin.
Sun et. al. showed that dienones 261 and 264 form covalent adducts with GSH at both double bonds (Scheme 14).100 Only the bis-GSH adducts were detected by LC-MS when 2.1 equiv of GSH were reacted with either 261 or 264, but mixtures of mono and bis adducts were detected when 1 equiv of GSH was used.100 It was observed that 261 formed adducts instantly, while 264 took several hours for complete adduct formation.98-99 The reversibility of the GSH adducts was demonstrated by reacting the bis-GSH adduct 262 with 261, which resulted in GSH transfer, yielding bis-GSH adduct 263 and dienone 264. The cytotoxicity of the GSH adducts was the same as the parent compounds in MDA-MB-435 human breast cancer cells (IC50[218] = 1.5 μM, IC50[215] = 1.0 μM), which also suggests reversible GSH-adduct formation.100
Scheme 14.

Reversibility of Glutathione Adducts of Curcumin Analogs
4.2. Macrocyclic Enones - Resorcylic Acid Lactones
Resorcylic acid lactones (RALs), such as hypothemycin (265), are a family of benzannulated macrolides produced by fungi with a 12- or 14-member macrolactone and many contain a cis-enone. RALs exhibit a variety of biological properties including antifungal, antibiotic, antitumor, and antiviral activities by inhibiting heat shock protein 90 (HSP90), mitogen activated protein kinases (MAPKs), herpes simplex virus 1 (HSV1), and the NF-κB pathway.101 The cis-enone functionality of many RALs leads to irreversible, covalent inhibition of kinases in the MAPK signaling pathway by hetero-Michael addition of a conserved cysteine located in the ATP binding pocket.102 RALs have been used as chemical biology probes, where an alkyne tagged hypothemycin 266 was used to identify the kinase Trypanosoma brucei CDC2-like kinase (TbCLK1) as a target for treating Human African trypanosomiasis (Figure 30).103 Synthetic strategies to access RALs have been reviewed,101a, 101b and SAR studies have observed that nearly all modifications resulted in lower activity compared to the natural product LL-Z1640-2 (267).102 Addition of a methyl group to the α or β position of the enone (268 and 269) completely abolished activity for the inhibition of tumor necrosis factor phospholipase A2-activating protein (TNFα-PLAP), while an α-fluoro substituent (270) resulted in a small 3-fold decrease in activity relative to the parent natural product 267 (Figure 30).102
Figure 30.

Resorcylic acid lactones with IC50 values for inhibition of TNFα-PLAP.
Fluoroenone 270 and the parent compound lacking a fluorine, 267, were reacted with DTT and it was observed that 270 reacted more slowly than 267 and showed reduced potency against kinases VEGFR-2 and tyrosine-protein kinase KIT (CD117 or proto-oncogene c-Kit).104 Studies with 265 have shown ATP-competitive, time-dependent inhibition of a subset of kinases containing a conserved cysteine residue in the ATP binding pocket.105 Schirmer et al. screened 124 kinases for inhibition by 265 and found that 18 out of 19 kinases containing the conserved cysteine were inhibited while only three of the 105 kinases lacking the conserved cysteine were targeted, demonstrating the utility of covalent targeting to discriminate between structurally related proteins. It was also shown that 265 formed stable adducts with Cys166 of extracellular signal-related kinase 2 (ERK2).105 The kinact/Ki of hypothemycin (265) ranged from 160-120,000 M−1s−1 for various kinases and was much greater than the k2 values for reactions with small molecule thiols such as β-mercaptoethanol and GSH (3.6 and 6.6 M−1s−1, respectively), showing that reactions with kinase targets are faster than reactions with free thiols.105
Incorporating an α,β-unsaturated lactam in place of the enone of RALs led to inhibitors of MAPK interacting kinases (MNK1/2), where the reactivity of the unsaturated lactam could be tuned by substitution of electron-withdrawing acetyl group on the nitrogen.106 The thiol reactivity of RAL 271 (NH) was compared to the N-acyl derivative 272, and it was shown that lactam 271 did not react with cysteamine or with MNK1/2 as evidenced by 1H NMR and MS (Figure 31). However, the corresponding N-acyl analog 272 formed thiol adducts with cysteamine and MNK1/2.106 Incorporation of an unsaturated lactam and a trans-enone in the macrocyclic ring for RAL analog 273 resulted in a loss of potency towards various kinases; selectivity for platelet derived growth factor receptor α (PDGFRα) was also lower compared to the trans-enone with an alkene 274, and both 273 and 274 were less potent than the natural product 267.107 FMS-like tyrosine kinase 3 (FLT3), a receptor tyrosine kinase in the same family as PDGFRα, is also inhibited by RAL natural products such as 265 and 267 at low nanomolar concentrations. Simpler, non-macrocyclic RAL analogs 275-277 were prepared with acrylamide or maleimide Michael acceptors and inhibited FLT3, albeit with lower potency than the natural products.108 These compounds were also shown to form adducts with cysteamine by 1H NMR. The effects of substituents on the ester or aromatic ring were also examined, with the morpholine and isopropyl ester shown in 275-277 generally giving the most potent RAL analogs. Maleimide 277 was about 10-fold lower in activity than natural product RALs, despite being a more reactive electrophile than the cis-enone RALs, while acrylamides 275-276 were about 100-fold less active than natural products, demonstrating the importance of noncovalent recognition.
Figure 31.

Synthetic resorcylic acid lactone analogs.
4.3. Cycloalkenones-Cyclopentenones and Cyclohexenones
Cyclopentenone prostaglandins (CyPGs) are a family of compounds possessing rich biological activities including antiinflammatory, antiproliferative, antiviral, and neuroprotective effects that are therapeutically beneficial.109 However, CyPGs may also promote less desirable effects such as tumor cell proliferation or neurodegeneration.109 Proteomic studies of the electrophilic action of CyPGs, their signaling targets, and their potential in overcoming tumor resistance have been reviewed.109 This section of this perspective focuses mainly on the reported thiol reactivity of CyPGs.109a Several CyPGs are depicted below (278-289) that show the variability in substitution patterns, mainly the degree of unsaturation in the cyclopentanone and side chains, as well as the placement of the alkene and hydroxyl groups (Figure 32).
Figure 32.

Selected CyPGs structures.
CyPGs were first reported to react with thiols by Lederle Labs in 1973, when they showed that 15-O-acetylprostaglandin A2 methyl ester (280) reacted with excess thioacetic acid to give the conjugate addition adduct in 90% yield, the stereochemistry of the newly formed thiol ester at C11 was undetermined.110 It was subsequently shown by Merck Laboratories that in red blood cells, containing 2.5 mM exogenous GSH, polar adducts of PGA1 (278) were formed, and these adducts were partially characterized using MS by Pisano et al. in 1976.111 Both of these studies measured the extent of polar adduct formation in red blood cells using radioactive (3H-labeled) prostaglandins. After incubating the prostaglandins with cysteine or GSH in PBS or Tris-HCl buffer (pH = 7.4), adduct formation was stopped by acidification of the solution to pH 3-4, which protonates the remaining thiolates to halt nucleophilic attack, followed by removal of free prostaglandin with an ethyl acetate extraction.111 Polar adducts of PGs with cysteine or glutathione remained in the aqueous layer and the amount of radioactivity extracted into ethyl acetate was measured to determine the amount of free prostaglandin.111 PGA1 (278), PGA2 (279), PGB1 (281), PGE2 (287), and PGF2α (289) were reacted with GSH, but only 278 and 279 formed water soluble adducts.111b PGE2 (287) and PGF2α (289) both lack enones, and therefore do not form covalent adducts to thiols, while the lack of reactivity of 281 is attributed to the less reactive tetra-substituted enone. The extent of polar adduct formation was also measured by reacting a solution of red blood cells with 3H-PGA1 (278).111a After centrifugation, 36% of the original radioactivity was observed in the supernatant and 55% remained in the aqueous layer.111a The extent of adduct formation never reached 100% suggesting that adduct and free prostaglandin exist in equilibrium in solution. It was also found that raising the pH > 9 resulted in quantitative extraction of the free PG from the aqueous solution, demonstrating the reversibility of these adducts under basic conditions.111 These experiments were the first to measure the reactivity of biologically relevant thiols with CyPGs in a cellular environment.
Roberts et al. compared the rates of GSH addition to Δ12-PGJ2 (283) and Δ12-PGD2 (286).112 Δ12-PGJ2 (283) formed covalent adducts with GSH faster than 286, with the cross-conjugated dienone reacting to 70% completion in two min, while the alkylidene cyclopentanone reacted to 38% completion in 60 min.112 The reaction between the CyPGs and GSH was measured by extractable radioactivity as described above, and the corresponding adducts were characterized by 1H NMR and MS.112 The rate of cysteine addition to both 283 and 286 was faster than that of GSH, with 85% adduct formation for 283 and 60% adduct formation for 286 after two min.112 GST was shown to increase the rate of GSH addition, which was most significant for the formation of GSH adducts to 286, but had no effect on the rate of cysteine addition.112 Depletion of GSH from the media of cultured hepatoma and CHO cells resulted in a two-fold enhancement of the antiproliferative activity of 283; Roberts et al. suggested that GSH aids in the removal of CyPGs from cells.113
The thiol reactivity of enone and cross-conjugated dienone CyPGs was compared by Noyori et al. where Δ7-PGA1 methyl ester (290) and PGA1 methyl ester (291) were reacted with various thiols including methanethiol, butanethiol, β-mercaptoethanol, cysteine, and GSH (Scheme 15).114 The rates and equilibrium constants were measured by UV spectroscopy and the thiol adducts were characterized by 1H NMR, 13C NMR, and MS. It was found that 290 reacted 6-8 times faster with thiols than 291.114 However, at equilibrium the dienone 290 generally favored the free PG with Keq ranging from 0.35-1.6 M−1 for different thiols, while the enone 291 favored adduct formation with Keq ranging from 6.1-10.8 M−1 for different thiols (Scheme 15).114
Scheme 15.

Equilibrium Formation of Thiol Adducts of PGA1 and Δ7-PGA1 Methyl Esters
Although CyPG adducts of free, soluble thiols such as cysteine, GSH, or other non-biological thiols, have been shown to be reversible, the adducts of CyPG and polymer-bound, immobilized thiols are stable under physiological conditions and require basic conditions (pH > 9) to dissociate.114 The stability of these immobilized thiol adducts is thought to be representative of the stability of CyPG-protein adducts; moreover, their detection using proteomic methods was recently reviewed by Pérez-Sala et al.109a Survival of CyPG-protein adducts to cell lysis, gel electrophoresis, tryptic digestion, and LC-MS purification and fragmentation further demonstrates the stability of the CyPG-protein adducts.109a
The use of recombinant proteins allows for the determination of covalently modified sites, and in many cases, multiple cysteine residues form adducts with CyPGs.109a However, CyPGs can show site selectivity. For example, Keap1 was modified at Cys273, Cys297, and Cys489 by enone PGA1 (278), but it was modified at Cys273 and Cys288 by dienone 15d-PGJ2 (284).115 Modification of purified proteins by CyPGs can lead to different cysteine modifications than are observed in intact cells or cell lysates.116 Differential cysteine modification of proteins by structurally similar CyPGs was lost with cell free extracts but was partially recovered by addition of GSH to these extracts.117 The formation of reversible GSH-CyPG adducts could explain why addition of GSH to cell extracts leads to a partial recovery of the cysteine selectivity observed in cells. In the case of GSTP1-1, an enzymatic assay identified covalent adduct formation of dienone 284 with Cys47 and Cys101 by MS analysis of tryptic digests, but experiments with intact cells showed only addition at Cys101.117 The kinact/Ki for 284 inhibition of GSTP1-1 was 1.4 × 10−5 M−1s−1. GSTP1-1 undergoes oligomerization in response to stress, which affects enzyme activity, and 284 induces irreversible oligomerization of GSTP1-1. Mutation of Cys47 to serine of GSTP1-1 in Jurkat cells increased 284 induced GSTP1-1 oligomer formation relative to the wild-type as measured by Western blot analysis of GSTP1-1, while mutation of Cys101 to serine abolished oligomerization.118 Pérez-Sala et al. attribute 284 induced oligomerization to the crosslinking of two cysteine residues, as prostaglandins containing a single enone (either the endocyclic or exocyclic) do not induce oligomerization.
Cross-conjugated dienone CyPGs have been shown to function as cross-linkers with nearby cysteine residues through bis-adduct formation. For example, H-Ras protein adducts with dienones 283 and 284 were detected in cells incubated with these prostaglandins by Western blot. When recombinant H-Ras was treated with 284, the major adduct detected by MS after trypsinization was K170-K185 (KLNPPDESGPGCMSCK), which was synthesized and used for further studies. Crosslinking of two cysteine residues in the C-terminal peptide K170-K185 of H-Ras was observed for 284, which formed a 1:1 adduct with peptide K170-K185 containing Cys181 and Cys184 but left no free cysteine residues. A proposed structure for the crosslinked peptide is shown in Figure 33, where a single molecule of a CyPG with two Michael acceptors forms covalent bonds to two cysteine residues.119 When a control peptide K170-K185, was reduced with DTT and alkylated with iodoacetamide, peptide adducts with one or two molecules of acetamide were detected by MS, corresponding to alkylation of one or two cysteine residues. However, after treatment of peptide K170-K185 with dienone 284, the peptide was reduced with DTT, and treated with iodoacetamide, and no alkylation by iodoacetamide was observed. Known cysteine crosslinking agent dibromobimane, or dienone 283, behaved similarly giving the peptide K170-K185 as a 1:1 adduct with no free cysteine residues. In contrast, when peptide K170-K185 was treated with enone 282, a CyPG containing a single Michael acceptor, a 2:1 ratio of 282:peptide K170-K185 was detected by MS. Crosslinking of two monomeric units of GSTP1-1 by a single molecule of a dienone CyPGs (either 283 or 284) was also detected; this oligomerization led to inhibition of this enzyme.118
Figure 33.

Proposed structure of CyPG crosslinking H-Ras C-terminal peptide (K170-K185).
Clavulones are a class of natural products structurally similar to the prostaglandins with the exception of a 4-acetoxy group on the cyclopentenone ring; clavulone (295) is one representative of this class (Scheme 16). Clavulones react irreversibly with nucleophiles by conjugate addition followed by β-elimination of the 4-acetoxy group. Simple clavulone analogs such as 296-298 form mono and bis adducts with β-mercaptoethanol when reacted for 24 h in dichloromethane with triethylamine (Scheme 16).120 Clavulone analog 298, with an acetate group afforded bis adduct 304 in 78% yield, with no monoadduct 301 detected, while the alcohol 297 gave 65% of the bis adduct 303 and 14% of the mono adduct 300.120 Compound 296 with a THP ether gave only 6% of the bis adduct 302 with no mono adduct and 85% of the SM recovered.120
Scheme 16.

Formation of Thiol Adducts with Clavulone Derivatives
Other structurally simplified analogs of clavulones were tested for their reactivity with GSH and N-Boc-cysteine methyl ester.121 Enone 305 formed bis adduct 308 with GSH through a pathway presumed to occur through conjugate addition to give 306, followed by a β-elimination of the tert-butyl dimethylsilyl ether to afford enone 307, which then participates in a second conjugate addition reaction, although these intermediates were not isolated (Scheme 17). Formation of this adduct was reversible as shown in a crossover experiment where GSH-adduct 308 was reacted with N-Boc-cysteine methyl ester to produce the thiol adduct 309 in 16% yield.121 Reaction of the cross-conjugated dienone 310 with N-Boc-cysteine methyl ester produced mono-adduct 312 in 88% yield in 2 h, but, after allowing to react for 14 h, bis-adduct 314 was produced in 49% yield (Scheme 17).121 However, when dienone 311 was reacted under identical conditions, only mono adduct 313 was formed in 75% yield, even when extending the reaction to 72 h, thereby supporting their notion that an irreversible loss of the TBS ether was the driving force for formation of the bis-adduct.121 Mono-adducts 312 and 313 displayed similar biological activities as the parent cyclopentenones for activation of heat shock transcription factor (HSF) and inhibition of NF-κB signaling, while bis-adduct 314 showed no activity.121
Scheme 17.

Reactions and Reversibility of Clavulone Derivatives with Thiols
The regiochemical outcome of nucleophilic attack on cross-conjugated cyclopentenones can depend on many factors. Danishefsky used the fused ring system 315 and found that nucleophilic addition by the carbanion of dimethylmalonate 316, silyl enol ether 317, or allyl silane 318 occurred exclusively at the ring fusion carbon (representative of endocyclic attack) to give products 319 or 320 (stereochemistry not determined), which was attributed to energetically favorable rehybridization in the formation of the enolate (Scheme 18).122 However, when monocyclic α-methylene or α-ethylidene cyclopentenones 321 or 322 were reacted with carbanion 316 or phenyl thiolate nucleophiles, exclusive addition to the exocyclic double bond was observed.123 The phenyl substituent on the cyclopentenone likely deactivates the endocyclic position as dienone CyPGs such as Δ7-PGA1 285 formed adducts predominantly at the endocyclic position; alternatively, the additional steric bulk of the alkyl chains on the CyPGs could deactivate the exocyclic position. Another alternative explanation could be that addition to the phenyl substituted endocyclic enone is reversible whereas addition to the exocyclic methylene is irreversible resulting in the more thermodynamically favored adduct. Unfortunately, further experiments were not conducted to determine the potential of the latter hypothesis.114
Scheme 18.

Nucleophilic Addition to Exocyclic vs Endocyclic Enones of Cyclopentenones
The addition of benzenethiol catalyzed by triethylamine to differentially substituted cyclopentenones was reversible in chloroform, but the reaction did not occur without addition of base. 2-Cyclopentenone (327) reacted fastest and favored the product 330 at equilibrium (Keq = 3,900 M−1); for 2-methyl-2-cyclopentenone (328), a methyl group at the α-position decreased the rate by about three orders of magnitude with a small preference for adduct 331 at equilibrium (Keq = 1.28 M−1, Scheme 19). A methyl group at the β-position, as seen in 3-methyl-2-cyclopentenone (329) decreased the rate of thiol addition even more with a slight preference for the starting cyclopentenone 329 at equilibrium (Keq = 0.88 M−1).124
Scheme 19.

Equilibrium Formation of Thiol Adducts with Cyclopentenones
Di- and triquinanes containing cyclopentenones have shown antiproliferative activity with IC50 values between 0.11-5.7 μM in cancer cell lines (P388, L1210, 3LL, and LY cells), with representative compound 333 shown to form adducts 334-335 with cysteine and propanethiol in good yields (Scheme 20).125
Scheme 20.

Cysteine and Propanethiol Addition to a Triquinane
The kaurane diterpenoids are a class of natural products containing over 600 compounds isolated from plants of the genus Isodon (Lamiaceae), and a significant portion of these molecules contain an α-methylene cyclopentanone.126 The biological effects for these compounds include antitumor, antibiotic, antiinflammatory, and antiviral activities.126a More specifically, compounds in this class have been shown to inhibit NF-κB signaling, including the dual Michael acceptor Eriocalyxin B (336).127 Eriocalxyin B (336) also downregulates S phase kinase-associated protein 2 (Skp2) inducing G2/M cell cycle arrest, inhibits HSP70 1A, and intercalates into DNA.106, 128 The identification and biological activities of these compounds have been reviewed extensively.126 Despite the large interest in these compounds, studies to investigate their thiol reactivities are limited. Kubota et al. first reported that enmein (337) and oridonin (339) formed adducts with cysteine while analogs not possessing the exocyclic enone, dihydrooridonin and dihydroenmein (not shown), did not (Scheme 21).129 Fujita et al. characterized propane thiol adduct 340, which formed in DMF in quantitative yield, and cysteine adduct 341, which formed in 1:1 EtOH:PBS buffer (pH = 7.2) with quantitative yield in only 10 min.130 Fujita et al. also showed that 339 derivatives containing a reduced enone lost antitumor and antibacterial activity, demonstrating the importance of the α-methylene cyclopentanone for their biological activities.130
Scheme 21.

Selected Kaurane Natural Products and the Reaction of Oridonin with Thiols
Oridonin (339) is one of the most studied members of the kaurane diterpenoids and has been shown to form a covalent adduct with Cys267 of HSP70 1A,128b sensitize cells to radiation, and deplete GSH levels in cells leading to an increase in reactive oxygen species (ROS).131 The addition of NAC prevents the reduction of GSH levels and also the apoptotic effects of 339, possibly by addition of NAC to the α-methylene cyclopentanone of 339.131b A similar compound, 13-hydroxy-15-oxo-zoapatlin (OZ, 338), was observed to form a reversible adduct with GSH; additionally, a 338 adduct to Cys25 of Trx was detected by MS analysis of tryptic peptides of Trx, which is one of two reactive cysteines in the active site.132 Similar to 339, these effects were prevented by addition of NAC.
Structurally simplified cryptocaryone analogs, such as 342 that contain a cyclohexenone, reacted with cysteamine in PBS buffer (pH = 8) yielding the thiol adduct 343 within 5 min by 1H NMR and MS analysis (Scheme 22). Compounds such as 342 were found to possess NF-κB inhibitory activity and antiproliferative activity to cancer cells in the micromolar range.133
Scheme 22.

Addition of Cysteamine to a Cryptocaryone Analog
Calicheamicin (344) is a DNA intercalator that undergoes thiol activation and subsequent intramolecular hetero-Michael addition causing electrocyclization of the ene-diyne to form a phenylene diradical 347 that promotes DNA strand cleavage.134 One activation pathway involves cleavage of the trisulfide of 344 by a thiol, such as GSH, to form the sulfide 345 which undergoes intramolecular hetero-Michael addition to afford 346 triggering a Bergman cyclization of the ene-diyne to produce 347 (Scheme 23). The phenylene diradical then abstracts hydrogen atoms to provide 348. The structurally simplified analog 349 has been shown to undergo intermolecular thiol addition of p-chlorothiophenol, which similarly triggers a Bergman cyclization of the ene-diyne.134a
Scheme 23.

Mechanism of Thiol Activation of Calicheamicin and Thiol Addition to a Derivative
4.4. Cyclopentenediones
Cyclopentenediones are a class of natural products isolated from plants, which include lucidone (351) and related compounds 352-354 (Figure 34). These compounds possess an array of bioactivity; for example, 351 displays induction of HO-1/Nrf2 antioxidant genes and inhibition of the NF-κB pathway and MAPKs.135 Cyclopentenediones 352-354 display inhibition of human chymase, leishmanicidal activity, antitumor activity, and inhibition of farnesyl protein transferase.136 Synthetic cyclopentenediones 355-357 were developed as irreversible inhibitors of the zinc endopeptidase of botulinum neurotoxin A; these compounds also exhibited fast reaction kinetics with GSH (Figure 34).137
Figure 34.

Natural product (351-354) and synthetic (355-357) cyclopentenediones with biological activity.
The reaction between cyclopentenedione 358 and cysteamine formed adduct 359 in 89% yield (Scheme 24). Biotin was conjugated to the Michael adduct 359 via the primary amine, and this probe (360) was used to label sulfenic acids in proteins.138 GSH also reacted with 358, and the adduct was used in the preparation of Kef bacterial potassium efflux activators.139 However, a series of N-alkyl substituted maleimides reacted with GSH to give adducts in higher yields (74-97%, not shown) than cyclopentenedione 358, and the maleimides were more potent activators of Kef. Cyclopentenediones possess reactivity patterns similar to that of maleimide; for example, dione 363 reacts to 90% completion with NAC in less than 10 min in aqueous buffer at a pH of 5.5, 6.5, or 7.5 (Scheme 24). Adduct 364 showed stability during one month of monitoring by 1H NMR. Moreover, cyclopentenedione 363 was stable at higher pH values compared to the maleimide which suffered hydrolytic ring-opening under basic conditions.140
Scheme 24.

Synthesis of Sulfenic Acid Probe and Thiol Reactivity of Cyclopentenediones
5. α,β-Unsaturated Aldehydes (Enals)
α,β-Unsaturated aldehydes pervade our environment as air pollutants, water contaminants, flavoring agents, and food components and are produced endogenously via lipid peroxidation and drug metabolism.141 Despite this ubiquity and known health risks, little is known about the molecular mechanisms of toxicity. Acrolein 365 reacts with GSH to give 1,4-adduct 367 with a rate constant of 120 M−1s−1 in pH = 7.4 PBS buffer; crotonaldehyde 366 reacts about 150 times slower (k1 = 0.78 M−1s−1) due to the methyl group at the β-position (Scheme 25).142 Acrolein 365 and crotonaldehyde 366 both form bis-adducts with cysteine through a 1,4-addition to give intermediates 369 or 370 with rates of 220 and 8.9 M−1s−1 respectively, followed by rapid condensation with a second cysteine to form a thiazolidines 371 and 372 (Scheme 25).143 These rates were measured by UV spectroscopy, and it was determined that hetero-Michael addition of cysteine was the rate determining step and cyclization to the thiazolidines 371 and 372 was much faster. 4-Hydroxy pentenal (373) reacts with cysteine to form cyclic hemiacetal 374 at a rate of 25 M−1s−1, but 374 slowly reacts with a second equivalent of cysteine to give thiazolidine 375 with a rate of 1.1 M−1s−1 (Scheme 25).143 Although acrolein and other aldehydes form stable cyclic adducts with cysteine and GSH, acrolein adducts between a cysteine embedded within a protein are unstable in vivo due to competing Schiff base formation via a mechanism that involves a release of the thiol; this may explain why most detected aldehyde-protein adducts are those formed with lysine or histidine residues, despite thiol addition being faster.144 The stability of aldehyde-amine adducts compared to aldehyde-thiol adducts could explain the results of Chan et al. who found that the reactivity of aldehydes towards amines correlated much better to hepatocyte toxicity than the corresponding thiol reactivity.145
Scheme 25.

Reactions of Aldehydes with Thiols
6. Toxicological Studies on Simple α,β-Unsaturated Carbonyls
Reactive carbonyl species, which can damage proteins, are generated as intermediates in cellular processes (e.g. sugar auto oxidation, lipid peroxidation, and glycation).146 Compounds containing thiols and amines were tested together for their ability to competitively form adducts with reactive carbonyl species, such as acrolein, in order to develop a method for evaluating reactive carbonyl scavengers with potential therapeutic value.146 In general, thiols were more reactive than amines, and the fastest reacting scavengers contained an α-amino-β-mercaptoethane structure such as cysteine or cysteamine. The reactivity of thiols towards α,β-unsaturated carbonyls decreases in the order of aldehydes > ketones > esters > amides, but small changes to the Michael acceptor structure can have a significant impact on its reactivity. Studies have been conducted correlating thiol reactivity with toxicity in living systems by Schüürmann and co-workers who screened thiol reactive molecules. Initially, SAR studies were correlated to the rate of reaction with GSH in buffer (pH = 7.2) containing 5-20% DMSO.48b RC50 values, defined as the reaction concentration that produced 50% reaction with GSH after 120 min, were assigned to each of these compounds. In general, cyclic α,β-unsaturated ketones reacted faster than acrylates and substituents at the α or β position of the alkene resulted in a significant decrease in the rate of reaction.
The relationship of Michael acceptor reactivity to biological toxicity was further studied in a subsequent paper where the reaction kinetics of GSH addition to α,β-unsaturated carbonyls, including aldehydes, ketones, and esters, in PBS buffer (pH = 7.2) was compared to the ciliate toxicity of these compounds; examples representing the most structurally diverse α,β-unsaturated carbonyls are shown in Table 7.48a, 48b The rate of GSH addition to structurally similar enals and enones was comparable, with enals being slightly faster to react (compare entries 4 to 12, 6 to 14, and 8 to 16, Table 7). α,β-Unsaturated esters were much slower to form adducts with GSH than related enals (compare entry 9 to 27) or enones (compare entries 10 to 22 and 14 to 24, Table 7). In general, conjugation to an alkyne increased the reactivity relative to an alkene (compare entries 1 to 6 for aldehydes, 11 to 12 for ketones, and 19 to 22 for esters, Table 7). Alkyl substituents at the α or β position of the alkene significantly reduced the rate of GSH addition to enones (compare entry 10 to 12), cyclopentenones (compare entry 13 to 17 or 18), and α,β-unsaturated esters (compare entry 22 to 25 or 26, Table 7). Alkyl substituents at the β position of the alkene reduced the rate more than substituents at the α position for enals (compare entry 3 to 4) or cyclopentenones (compare entry 17 to 18), but the opposite was seen for esters (compare entry 25 to 26, Table 7).48a Multiple alkyl substituents at the α and/or β position of the alkene had the largest impact on reaction rates. Alkyl substituents at both the α and β positions of the alkene reduced the rate of GSH addition relative to a single α or β substituent for enals (compare entries 3 and 4 to 9), enones (compare entry 12 to 15), and α,β-unsaturated esters (compare entries 25 or 26 to 27, Table 7). β,β-Dimethyl enal 383 reacted 3.6-fold faster with GSH than α,β-dimethyl enal 384 (compare entry 8 to 9, Table 7). In contrast, α,β-dimethyl enone 390 reacted with GSH 3.7-fold faster than β,β-dimethyl enone 391 (compare entry 15 to 16, Table 7). The alkyl chain length at the β position of the alkene had a small effect on the rate of GSH addition as evidenced by enals 379 and 381 (entries 4 and 6) or enones 387 and 389 (entries 12 and 14, Table 7). Extended conjugation reduced the rate of GSH addition to enals six-fold (compare entry 5 to 7, Table 7). 48a A propargyl or allyl substituent attached to acrylates 395 or 396 increased the rate of GSH addition relative to alkyl ester substituents in 397 or 398, and the propargyl substituent had a more significant impact on the rate (compare entries 20-23, Table 7).
Table 7.
Second Order Rate Constants for GSH Addition to α,β-Unsaturated Carbonyls

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Aldehyde | Ketones | Esters | ||||||
| Entry | Structure | kGSH (M−1s−1) | Entry | Structure | kGSH (M−1s−1) | Entry | Structure | kGSH (M−1s−1) |
| 1 |
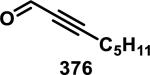
|
487 | 10 |
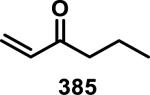
|
1173 | 19 |
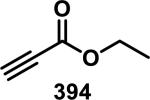
|
105 |
| 2 |
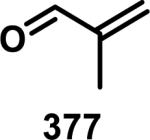
|
203 | 11 |
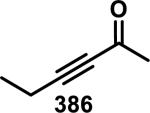
|
80.0 | 20 |
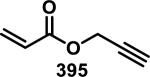
|
51.4 |
| 3 |
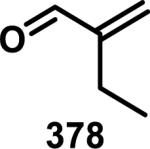
|
59.4 | 12 |
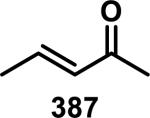
|
26.7 | 21 |
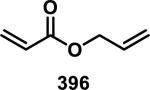
|
19.6 |
| 4 |
|
28.3 | 13 |

|
25.6 | 22 |
|
10.6 |
| 5 |
|
22.8 | 14 |
|
11.4 | 23 |
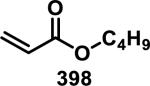
|
8.54 |
| 6 |
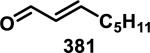
|
18.0 | 15 |
|
0.779 | 24 |
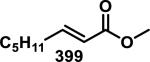
|
0.785 |
| 7 |
|
3.49 | 16 |
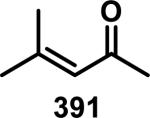
|
0.208 | 25 |
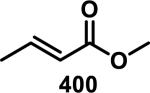
|
0.164 |
| 8 |
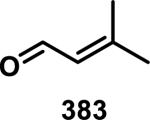
|
1.71 | 17 |

|
0.200 | 26 |
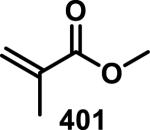
|
0.072 |
| 9 |
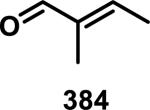
|
0.474 | 18 |

|
0.074 | 27 |
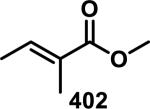
|
0.007 |
The ciliate toxicity of Michael acceptors 376-402 was measured as the 50% growth inhibition over 48 h (EC50) and compared to the rates of GSH addition. Enal ciliate toxicity ranged from an EC50 of 19 μM for 376 to an EC50 of 1380 μM for 384. The most toxic enone 385 (EC50 = 22 μM) and ester 394 (EC50 = 17 μM) had a similar toxicity to enal 376. However, the least toxic enone 393 (EC50 = 20,900 μM) and least toxic ester 401 (EC50 = 16,600 μM) were far less toxic than the least toxic enal 384. A correlation between the rate of GSH addition and the ciliate toxicity was apparent through linear regression analysis (log kGSH vs log EC50). Enals showed a weak correlation between the rate of GSH addition and ciliate toxicity with a correlation coefficient (R2) of 0.44, but enones and α,β-unsaturated esters showed good correlation between rates and ciliate toxicity (R2 = 0.93 and 0.83, respectively). The examples presented in Table 7 were chosen to exemplify the effects of small structural changes on the reactivity of simple Michael acceptors, but a large compilation of reactivity data for electrophilic compounds was compiled in a review by Schwöbel et al.1
7. Dually Activated Michael Acceptors
Dually activated Michael acceptors contain an electron withdrawing substituent at the α-position of an α,β-unsaturated carbonyl. The reactivity of these compounds towards thiols was first reported in 1968, where adduct formation of 403-406 with butanethiol was instantaneous as detected by UV, but the adducts 407-410 could not be isolated due to their reversibility (Scheme 26).147 More recently, unsaturated carbonyls with a nitrile at the α-position have gained attention due to the bioactivity of compounds such as cyanoenone CDDO (411, an inhibitor of nitric oxide production, similar to bardoxolone methyl 35) and cyanoacrylamide 418 (a RSK2 inhibitor),33d discussed previously in the targeted covalent inhibition section.33a
Scheme 26.

Dually Activated Michael Acceptors
The addition of β-mercaptoethanol to singly and dually activated Michael acceptors was compared, and it was observed that the singly activated acceptors 412 and 413 formed stable adducts 414 and 415, respectively, that could be isolated and characterized (Scheme 27). The dually activated acceptor 416 reacted faster with β-mercaptoethanol than the singly activated acceptors, but the adduct 417 could not be isolated due to the rapid reversibility of the reaction.33a Inhibition of RSK2 in MDA-MB-231 cells by dually activated Michael acceptors 418 and 419 (IC50 = 0.005 and 0.007 μM) was more than 200-fold more potent than singly activated pyrrolopyrimidines 420 and 421 (IC50 = 0.75 and 0.25 μM, Scheme 27).33a Singly activated acceptors 420 and 421 slowly formed covalent adducts that were detected by LC-MS, but dually activated acceptors 418 and 419 formed no adducts that were detectable by LC-MS.33a UV measurements revealed disappearance of the cyanoacrylamide absorbance at 304 nm upon incubation of 418 with the C-terminal kinase domain (CTD) of RSK2, but reappearance of this peak was detected within seconds upon protein denaturation with sodium dodecyl suflate (SDS) or guanidine.33a Cyanoacrylamide 418 maintained inhibition of RSK2 after dialysis and prevented the labeling of RSK2 by an irreversible inhibitor containing an α-fluoroketone, suggesting that covalent bond formation to RSK2 is stable under physiological conditions, but the stability of the thiol adduct is disrupted by protein denaturation.33a This is a major difference between cyanoacrylamides and cross-conjugated dienone prostaglandins, which usually maintain their covalent bonds to proteins after denaturation as shown by the detection of peptide-thiol adducts to CyPGs in numerous proteomic studies. A co-crystal structure of 419 and RSK2 showed covalent bond formation between Cys436 and the unsaturated ester of 419; stabilizing noncovalent interactions such as hydrogen bonding of the pyrrolopyrimidine to Thr493 and extension of the p-tolyl group into a hydrophobic pocket were also shown.33a Mutation of Cys436 to valine (C436V) resulted in a 1000-fold decrease in activity for the dually activated acceptors, supporting the importance of covalent bond formation.33a
Scheme 27.

Reversibility of Dually Activated Michael Acceptors and RSK2 Inhibitors
Michael acceptors equipped with α-cyano groups have been shown to undergo rapidly reversible thiol–carbon bond formation with thiol-containing nucleophiles such as cysteine; however, few studies have examined the equilibria effects of other electron-withdrawing groups at the α-position of a Michael acceptor. Recently, Taunton and coworkers reported that the reversibility of the hetero-Michael addition of acrylonitriles can be modulated with heteroaromatic groups. Placement of these electron withdrawing groups at the α-position of the acrylonitrile results in t1/2 values of the adduct ranging from < 1 min for methyl thiazole 423a to > 58 h for the pyrazol-1-yl 427a upon 10-fold dilution with DMSO/PBS buffer (3:1 v/v, Scheme 28).148 These reversible Michael acceptors were used to design kinase inhibitors similar in structure to the α-cyanoacrylamide 418 but with better selectivity for RSK2 over PLK1 and Nek2, despite all three kinases containing a reactive cysteine. This increased selectivity was attributed to steric clashes between the heteroaromatic activating group and the binding pockets of PLK1 and Nek2.148
Scheme 28.

Tunable Reversibility of α-Heteroaromatic-Substituted Acrylonitriles
The equilibrium dissociation constants (KD) for some cyanoacrylamide-β-mercaptoethanol adducts were between 0.2 and 33 mM β-mercaptoethanol for cyanoacrylamides 428-433, including the approved drug entacapone (430) that is used to treat Parkinson's disease therapy; these KD values corresponds to a free energy change between −2 and −5 kcal/mol demonstrating that these β-substituents had a small effect on the reversibility of cyanoacrylamide-thiol adducts.33a The KDs were determined by titration of cyanoacrylamides 428-433 with β-mercaptoethanol and monitoring the disappearance of cyanoacrylamide UV absorbance at 304 nm (Scheme 29). The adducts were then diluted 10-fold with PBS buffer (pH = 7.4) and the reappearance of the absorbance at 304 nm was observed.
Scheme 29.

Effect of β-Substituent on the Reversibility of Thiol Addition to Dually Activated Michael Acceptors
CDDO (411) contains a cyanoenone as part of a pentacyclic steroid skeleton and has gained interest due to the 1000-fold increase in potency over the descyano compound for suppression of inflammatory enzymes iNOS and COX-2.33c The reaction of 411 with GSH or DTT was monitored by the appearance of new UV peaks between 280 and 300 nm, but attempts to isolate the adducts by HPLC were unsuccessful.33c The addition of DTT to 411 was confirmed by 1H NMR monitoring, which showed the disappearance of the cyanoenone alkenyl resonance at 8.7 ppm.33c Variable temperature NMR also demonstrated the reversibility of the reaction, as 411 was regenerated upon heating of the DTT-411 adduct.33c Other cyanoenones, such as tricyclic TBE-31 (434)149 and monocyclic compounds 435-437,150 have shown similar reactivity towards thiols using the same UV and 1H NMR methods (Figure 35). Surprisingly, compounds 439-440 showed no reactivity with DTT and 441-442 showed greatly reduced reactivity requiring 10 equiv of DTT for adduct formation, thereby demonstrating the importance of the surrounding structure on electrophilicity.150
Figure 35.

α-Nitrile cyclohexenone dually activated Michael acceptors.
8. Other α,β-Unsaturated Carbonyls
8.1. Quinones
Quinones have unique redox capabilities that are widely utilized in nature. Mitochondria use ubiquinone as part of the electron transport chain to generate ATP. Several endogenous signaling molecules such as L-dopamine and estrogen contain catechols and are oxidized to quinones as part of their natural elimination processes.151 Vitamin K is a napthoquinone that is involved in blood coagulation. Red wine contains resveratrol and other stilbenes that contain catechols, which can be oxidized autonomously or enzymatically to quinones and serve as antioxidants.152 Despite these advantages, quinones can also cause harm to living systems by undergoing reaction with thiols and eliciting an increase in oxidative stress or alkylation of enzymes and DNA that cause toxicity.153 Quinones can also be a part of the covalent mechanism of action of bioactive molecules.
Para-quinones undergo conjugation with cysteines, but their reactivity can be modulated by the addition of electron donating substituents. For example, irreversible VEGFR-2 inhibitors were designed where R1 was substituted with a methoxy (443) or amino (444-446) group and these compounds did not react appreciably with GSH (entries 1-4, Table 8). However, compounds where R1 was substituted with a chlorine (447), aryl ether (448), or thioether (449) group reacted readily with GSH (entries 5-7, Table 8).25 Despite not reacting appreciably with GSH, para-quinone 443 was shown to covalently modify Cys1045 on VEGFR-2 after incubation for one hour followed by trypsin digestion and LC-MS analysis of the digested protein. It was predicted that GSH and VEGFR-2 are reacting with the six position of the para-quinone.
Table 8.
Effect of Quinone Substitution on their Half-Lives when Reacted with GSH

| |||
|---|---|---|---|
| Entry | R1 | t1/2, 10 μM (h)a | t1/2, 1 μM (h)b |
| 1 | OMe (443) | 80 | 68 |
| 2 | N-piperazinyl-4-CH2Ph (444) | 85 | 73 |
| 3 | N(CH3)Ph (445) | 295 | 341 |
| 4 | N(CH3)2 (446) | 601 | 160 |
| 5 | Cl (447) | < 0.02 | < 0.02 |
| 6 | OPh (448) | < 0.02 | < 0.02 |
| 7 | SCH3 (449) | 3 | 2 |
Half-life (t1/2) at 10 μM and
1 μM of the compound in the presence of 100 μM of GSH in a 1:1 solution of H2O:MeCN (no final pH of the solution was reported).
An assay has recently been developed to assess the kinetics of covalent adduct formation to para-quinones using 4-nitrobenzenethiol as the nucleophile.154 A double-mixing stop-flow spectrophotometer was used to measure the very fast pseudo-first order reaction at pH 7.4 and reaction progress was monitored by the reduction of free thiol at 412 nm. Most benzoquinones reacted relatively quickly where 450 had a rate constant of 1.50 × 103 s−1 compared to 2-methylbenzoquinone (451), 2-tert-butylbenzoquinone (452), and 2,5-dimethylbenzoquinone (453) having pseudo-first order rate constants of 3.2 × 102, 2.3 × 101, and 7.3 × 10−1 s−1, respectively (entries 1-4, Table 9). Clearly, electron-donating and/or sterically hindering alkyl substituents slow hetero-Michael addition by 10 - 10,000-fold. On the other hand, electron-withdrawing substituents such as a chlorine (454) increase the rate of reaction with a rate constant of 2.6 × 105 s−1, and two chloro groups further increase the rate as seen for 2,5-dichlorobenzoquinone (455) with a rate constant of 2.8 × 106 s−1 (entries 5-6, Table 9).
Table 9.
Effect of Quinone Substituents on the Psuedo-first Order Rate Constants for the Reaction with 4-Nitrobenzenethiol

| |||
|---|---|---|---|
| Entry | R1 | R2 | Psuedo-first Order Rate Consant (s−1) |
| 1 | H | H (450) | 1.5 × 103 |
| 2 | H | Me (451) | 3.2 × 102 |
| 3 | H | tBu (452) | 2.3 × 101 |
| 4 | Me | Me (453) | 7.3 × 10−1 |
| 5 | Cl | H (454) | 2.6 × 105 |
| 6 | Cl | Cl (455) | 2.8 × 106 |
8.2. Acylfulvenes
Illudins and their derivatives, acylfulvenes, are cytotoxic alkylating agents that have been shown to react with thiols and DNA. Illudin S (456) and M (457) are highly cytotoxic, limiting their use as therapeutic agents; thus, derivatives have been synthesized, the most prominent being dehydroilludin M (458), acylfulvene (459), and hydroxymethylacylfulvene (HMAF, 460) which made it to phase III clinical trials as an anticancer agent (Figure 36).155 Acylfulvenes have been found to inhibit glutathione reductase (GR), thioredoxin reductase (TrxR), Trx, and alkylate DNA at purine bases.155-156 Compound 459 inhibited GR reversibly while 460 inhibition was irreversible, and 456 was inactive despite being the most reactive towards small-molecule thiols.156a, 156b Tanasova and Sturla recently reviewed the synthesis, bioactivity, and metabolism of acylfulvenes, which included a summary of SAR studies demonstrating the importance of the enone, cyclopropane ring, and hydroxymethyl groups of HMAF for potent activity.155
Figure 36.

Illudin natural products and synthetic derivatives.
Compound 460 was shown to be an irreversible inhibitor of GR as enzyme inhibition was time dependent, while 459 showed establishment of an equilibrium.156a Covalent modification of GR by two molecules of 460 was detected by MS, which showed that 460 reacted with both the active site cysteines of GR.156a Acylfulvenes 459 and 460 were both found to inhibit Trx and TrxR.155, 156b Covalent adducts detected by MS suggest that 459 monoalkylates and 460 bisalkylates these enzymes. For selenocysteine containing enzyme TrxR, it was observed that only the selenocysteine (Sec498, pKa = 5.2) of the active site was alkylated at pH 6.5, but both the selenocysteine and cysteine (Cys497, pKa = 8.3) were alkylated at pH 8.5 by 459 or 460 using a biotin-conjugated iodoacetamide (BIAM) assay previously used to demonstrate curcumin covalent modification of TrxR.157 Glutathione peroxidase (Gpx) is another redox-regulating enzyme containing a reactive selenocysteine residue but was not inhibited by 456, 459, or 460, demonstrating the role of noncovalent enzyme-small molecule interactions to promote covalent bond formation.156b
The thiol reactivity of illudins and acylfulvenes has been studied predominantly by McMorris and coworkers who have measured the thiol reactivity of compounds 456-460 with thiols including β-mercaptoethanol, methyl thioglycolate, GSH, and cysteine.158 These thiol additions occur at acidic and neutral pH, with the ideal pH ranging from 4.8-6.1 depending on the substrate and thiol, which is in contrast to most hetero-Michael additions that are normally faster at more basic pH (mechanism for thiol addition shown in Scheme 30).158a, 158c Dick et al. showed that at a more biologically relevant pH of 7.4, HMAF (460, also called irofulven) is unreactive with GSH while illudin M (457) reacts readily.159 Under more acidic conditions, 460 has been shown to react with other thiols such as those mentioned above, as well as 4-hydroxythiophenol, thioglycerol, thiocresol, and benzylmercaptan.158e McMorris et al. found that strongly acidic conditions (dilute H2SO4, pH = 0) favored thiol additions via substitution of the allylic alcohol to give adducts such as 463 in high yields (70-80%, Scheme 31).158e Moderately acidic to neutral conditions (pH = 4-7) favored hetero-Michael addition (0.24 M 460, acetone/water) to give 464-466 or suspected radical addition (0.011 M, THF/water) to give 467.158e The thiol used also affected the ratio of adducts, with 4-hydroxythiophenol favoring radical addition to give 467 or substitution of the allylic alcohol yielding 463, and methyl thioglycolate favoring hetero-Michael addition and/or substitution to give major products 464 or 466.158e McMorris et al. propose that the preference for acidic pH is a balance between concentration of the thiolate anion and cyclopropane ring opening/loss of water to form the stable aromatic product, where at higher pH the rate is dependent on the elimination of the tertiary alcohol (less protonated) and at lower pH the rate is dependent on the concentration of thiolate (less deprotonated).158a
Scheme 30.

Mechanism for Direct Thiol Alkylation of Hydroxymethylacylfulvene
Scheme 31.

Products Obtained from the Reaction of HMAF with Thiols
8.3. α-Haloacrolyl Compounds
The α-haloacrolyl group is found in cytotoxic natural products such as bromovulone (468), discorhabdin C (469), verongiaquinol (470), brostallicin (471), and the punaglandins 2 (472) and 3 (473, Figure 37).160 In contrast to many thiol conjugations that inactivate reactive electrophiles, thiol addition to a α-haloacrolyl group generates a more reactive electrophile that can alkylate DNA. α-Bromocyclopentenone 474 is believed to form episulfonium ion intermediate 475 that can then react with DNA, with guanine adducts such as 476 being the most commonly detected adduct by MS analysis (Scheme 32).161 When 474 was reacted with β-mercaptoethanol, adduct 477 was obtained in 40% yield supporting the formation of an episulfonium ion intermediate 475 followed by elimination/ring opening; cyclopentenone 474 also reacted to 90% completion in two min with GSH to form two isobaric adducts when monitored by LC-MS. A similar type of activation, where thiol addition generates a more reactive electrophile, was described above for illudins (456-458), acylfulvenes 459-460), and calicheamicin (344).
Figure 37.

Examples of Bioactive α-haloacrolyl compounds.
Scheme 32.

Reactions of α-Bromocyclopentenone with Thiols and DNA
α-Halo butenolide 479 reacts readily with one equivalent of 2-propanethiol in the presence of potassium carbonate and tetrabutyl ammonium bromide to give Michael adduct 480 in 13% yield and hetero-Michael addition-bromine elimination adduct 481 as the major product in 40% yield (Scheme 33). Small amounts of 483 and 484 were obtained and are thought to arise from episulfonium ion 482.162 When a 2:1 ratio of furanone 479 to thiol were used, cyclopropane 484 and α-sulfide butenolide 483 were obtained as the major products in 47 and 25% yields, respectively.162
Scheme 33.

Products of Thiol Addition to α-Halo Butenolide with Proposed Intermediate
8.4. Rhodanines
Rhodanines and their analogs are classified as privileged scaffolds in drug discovery due to their various biological effects, including antibacterial, antiviral, antitumor, and antiinflammatory activities.163 Rhodanines have also been classified as pan-assay interference compounds (PAINS).164 The utility of rhodanines in drug discovery is a topic of debate, and although they frequently appear as high-throughput screening (HTS) hits, only a few studies to elaborate on their mechanisms of action and specificities have been reported.165,166 The most common rhodanine scaffold in screening libraries possesses a 5-alkylidene group in conjugation with the carbonyl, leading to the supposition that these compounds covalently label protein targets.166 This moiety is embedded in the drug, epalrestat (485), which functions as a reversible aldose reductase inhibitor used to treat diabetic neuropathy (Figure 38).167
Figure 38.

Examples of biologically active rhodanines.
Amgen scientists obtained a crystal structure of rhodanine 486, an inhibitor of hepatitis C virus RNA polymerase (NS5B), which showed covalent bond formation to Cys366 in the NS5B binding site; however, this inhibitor displayed noncompetitive, reversible inhibition in enzymatic assays (Figure 38).168 Rhodanine containing inhibitors of JNK-stimulating phosphatase (JSP-1), such as 487, discovered by scientists at Ceptyr, display reversible, competitive inhibition and show selectivity toward JSP-1 over other phosphatases.169 JSP-1 inhibitors are hypothesized to undergo reversible, covalent bond formation to a cysteine in the active site, though this has not been demonstrated experimentally. The reversibility of 5-alkylidenerhodanine inhibitors demonstrates the lability of the corresponding thiol adducts. Reversible inhibition of sortase A (SrtA) and a sphingosine 1-phosphate receptor (S1P) by rhodanine 488 and pseudothiohydantoin 489, respectively, have been reported.170 Optovin (490) reversibly inhibits a transient receptor potential cation channel (TRPA1), until photochemical activation renders the formation of an irreversible adduct.171
The promiscuity of 163 rhodanines and analogs was analyzed by testing their activity in four enzymatic assays (bacterial transferase MurA, metalloprotease methionine aminopeptidase, serine proteases thrombin, and dengue virus protease) measuring aggregate formation and testing for GSH adduct formation.166 Mendgen et al. concluded that the rhodanine scaffold, especially the thiocarbonyl group, can lead to nonselective binding at concentrations commonly used for HTS screening through polar interactions and hydrogen bonds.166 However, rhodanine type scaffolds do not typically form aggregates and are not very electrophilic, presumably due to their aromaticity. This lack of thiol reactivity is supported by the study where fourteen 5-alkylidene rhodanines (491-504) with high activity in one or more of the enzymatic assays mentioned above were treated with GSH in Tris-HCl buffer (pH = 7.5) and no adducts were detected by RP-HPLC (Figure 39).166
Figure 39.

Rhodanines and analogs that did not form detectable adducts with GSH.
Other studies have shown rhodanine-thiol adducts to be reversible. Rhodanine UDP-galactopyranose mutase inhibitor 505 rapidly forms thiol adduct 506 with DTT, detected by UV spectroscopy, which is reversible under neutral, buffered conditions (Scheme 34).172 Rhodanine 507 and thiazolidine-2,4-dione 508 react with thioacetic acid and 4-chlorothiophenol when treated with catalytic triethylamine to form Michael adducts 510-513, but heating these adducts promotes the reverse reaction; pseudothiohydantoin 509 did not form any detectable thiol adducts (Scheme 34).173
Scheme 34.

Reversible Additions of Thiols to Rhodanines and Related Scaffolds
8.5. Viridins (wortmannin)
Wortmannin (514) and other viridins containing an activated furan ring have been shown to undergo 1,6-conjugate addition followed by ring opening with amine and thiol nucleophiles.174 Wortmannin is an inhibitor of PI3K that covalently bonds to Lys802 in the ATP binding pocket of PI3K.175 Compound 514 also inhibits the PI3K-related kinases (PIKKs) ATR, ATM, DNA-PK and PLK1.176 A number of amine and thiol adducts of wortmannin, represented by 515-517, were prepared and showed activity similar to 514 for inhibition of PI3K (IC50 values for 514-517 were 0.8, 0.1, 0.5, and 0.3 nM, respectively, Scheme 35).174b The amine adduct 517 went into Phase I clinical trials in combination with cetuximab or docetaxel for treatment of squamous cell cancer of the head and neck, where it showed a small increase in treatment efficacy relative to either drug alone, but showed no increase in efficacy in larger Phase II trials.177 Wortmannin adducts with NAC and proline are reversible, but the adduct formed from N-Ac-lysine is stable, possibly due to hydrogen bonding between the amine and carbonyl of the δ-lactone; this was shown by crossover experiments where a thiol or amine adduct 518 or 519 was mixed with N- Ac-lysine, and HPLC monitoring showed complete conversion to the N-Ac-lysine adduct 520 in 20 h (Scheme 36).178
Scheme 35.

Reaction of Thiol and Amine Nucleophiles with Wortmannin
Scheme 36.

Crossover Experiments Showing the Reversibility of Wortmannin Adducts
8.6. α,β-Unsaturated Carboxylic Acids
α,β-Unsaturated carboxylic acids are generally not very reactive with thiols and require forcing conditions such as refluxing with excess thiol, or conditions that utilize an iodine or fluoride catalyst to affect adduct formation.179 At biologically relevant pH, carboxylic acids exist primarily as carboxylates, which deactivates them for Michael addition. This was observed for α,β-unsaturated ketone 259, where α-substitution with a carboxylic acid was more deactivating than electron donating substituents like a methyl group 257 or a methoxyphenyl group 256 (Table 6).95 Similarly, the reaction of the drug dimethyl fumarate (31) with GSH at a pH of 7.4 is about 30 fold faster than the reaction of monomethyl fumarate 32 with GSH at the same pH (t1/2 of 9 min for 31 vs 4 h for 32, Scheme 1). Dimethyl fumarate 31 is rapidly hydrolyzed by esterases to monomethyl fumarate 32, but it is still not well understood whether 31, 32 or some other metabolite is the biologically active compound in the treatment for psoriasis.32a Monomethyl fumarate 32 forms thiol adducts 521 and 522, identified by 2D NMR analysis, in approximately equal amounts (Scheme 37). The isolation of 521 suggests that it reacts in its protonated form, which is only present in small concentrations at a pH of 7.4, and this could explain the slower rate of GSH adduct formation.32a
Scheme 37.

Reaction of Monomethyl Fumarate with GSH
The reaction of fumaric acid (523), another metabolite of 32, with GSH has been detected by MS when reacted at 37 °C in buffer (pH = 7.4) for 3 h, although the amount of product 524 formed or rate of adduct formation was not reported (Scheme 38).180 A small amount of metabolite 524, making up 2.2% of GSH species, was detected by LC-MS when excess 523 was incubated with UOK262 FH-null cells demonstrating that α,β-unsaturated carboxylic acids can react with thiols in a cellular environment; free GSH made up 90% of GSH species and oxidized GSH disulfide made up the remaining 7.8% of GSH species detected by LC-MS.180
Scheme 38.

Reaction of Fumaric Acid with GSH
Recently, pH dependent probes 525 and 526 that display “turn-on” fluorescence when reacted with thiols were reported.181 Dicarboxylic acid 525 reacts readily with cysteine at pH values less than 7, while diester 526 reacts readily with cysteine at pH values greater than 7 (Scheme 39). These probes emit weak fluorescence between pH values of 2 and 11 prior to thiol addition, due to photoinduced electron transfer to the Michael acceptor, and the fluorescence is enhanced 96-fold upon addition of cysteine. The rate of thiol addition to α,β-unsaturated carboxylic acid 525 showed pH dependence with two inflection points at a pH of 3.5 and 7.5, corresponding to pKa2 and pKa3 (Scheme 39). The maximum rate of addition was observed at a pH of 5 and rapidly fell to zero between a pH of 7 and 8, suggesting that protonation of the quinoline nitrogen as in structures 529-530 deactivates the Michael acceptor, while deprotonation of both carboxylic acids as in 532 shuts down thiol addition.181 The rate of cysteine addition to diester 526 showed a single inflection point at a pH of 7, reaching a maximum at a pH of 9, suggesting that deprotonation of cysteine (pKa = 8.3) was the main factor that influenced the rate.
Scheme 39.

pH Dependence of Thiol Reactive Quinolines
9. Spectroscopic and Computational Measurements and Predictions of Thiol Reactivity for Michael Acceptors
Spectroscopic methods to measure and predict thiol reactivity of α,β-unsaturated carbonyls include 1H NMR, 13C NMR, fluorescence, and UV, with the latter being the most commonly used method to measure reaction kinetics. 1H NMR has been used to monitor the reactions of thiols with Michael acceptors, but the chemical shifts at the α or β position in 1H and 13C NMR can also be used to predict the thiol reactivity for structurally similar Michael acceptors. Additionally, computational predictions of thiol reactivity, utilizing transition state calculations, Fukui functions, local softness, HOMO and LUMO energies, or electrophilicity indices have been calculated and compared to experimental values. Thiol reactivity assays that utilize fluorescence or 1H NMR, and the predictive power of spectroscopic and computational parameters will be discussed below.
9.1 Spectroscopic Methods
Fluorogenic probes, similar to the dicarboxylic acid probes 526-527 discussed above, with a coumarin as the fluorescent moiety instead of a quinoline, were developed for the imaging of GSH in living cells, and second order rate constants for their reaction with GSH (100-fold excess) were measured in a DMSO/pH 7.4 HEPES buffer (1:1).182 α,β-Unsaturated ester 533 did not react with GSH under these conditions while unsaturated ketone 534 was slow to react (Scheme 40). α,β-Unsaturated aldehyde 535 reacted faster than ketone 534, but diketone 536 was the most rapid, reaching complete GSH adduct formation in less than 5 min. The diketone 536 also showed the largest increase in quantum yield (27-fold) upon addition of GSH, while ketone 534 and aldehyde 535 only resulted in a 1.7 or 2.7-fold increase in the quantum yield, respectively.
Scheme 40.

Coumarin Based Fluorogenic Probes and Second Order Rates Constants for GSH Addition
Fluorescent detection of covalent bond formation of quinazoline containing Michael acceptors to a protein has also been demonstrated.183 Quinazolines attached to Michael acceptors were incubated with a mutated tyrosine kinase c-Src (S345C), meant to mimic mutations in drug resistant EGFR. As in the fluorescence assays described above, this detection system relies on the increase in the fluorescence of quinazolines, such as 537-539, upon addition of a thiol to the Michael acceptor (Scheme 41). For irreversible inhibitors, this assay is simpler to perform and offers advantages over traditional activity-based assays because IC50 values may not be reliable for this class of compounds due to the time dependency of covalent adduct formation. This method was used to obtain kinact/Ki values for inhibition of S345C c-Src for compounds 537-539. Interestingly, IC50 values were measured after 90 min incubation with purified c-Src and correlated well with the Wilson-Kitz analysis of the inhibitors. Three representative compounds 537, 538, and 539 were determined to have time-dependent IC50 values of 240, 120, and 2 nM, respectively, for inhibition of mutated c-Src (S345C). The dimethylamino-acrylamide 537 (similar to afatinib (7)) had the lowest kinact/Ki value of 0.085 M−1s−1 compared to the unsubstituted acrylamide 538, 0.135 M−1s−1. The covalent binding efficiency of the vinyl sulfonamide 539 was approximately 10-fold higher than the acrylamides at 1.03 M−1s−1.183
Scheme 41.

Fluorescence of Quinazoline Michael Acceptors upon Covalent Modification of a Cysteine in c-Src
Avonto et al. developed a method using 1H NMR for measuring thiol reactivity by reacting a series of compounds with cysteamine in DMSO-d6.97 Costunolide (202), possessing an α-methylene-lactone and known to react with biological thiols, was used as a benchmark for measuring conjugate addition. Unactivated thiols dodecanethiol or NAC showed no reaction with 202 after 24 h in toluene, chloroform, MeCN, MeOH, or DMSO.97 However, conjugate addition of cysteamine, an activated thiol, to 202 occurs slowly in methanol and acetonitrile and instantaneously in DMSO. Several molecularly complex Michael acceptors were benchmarked against 202 and classified as irreversible, reversible, or nonreactive based on a Michael adduct formation and reversibility upon 10-fold dilution with CDCl3. Michael acceptors 200, 260, 543-548 formed thiol adducts with cysteamine, with cross-conjugated dienones 549 and 550 showing CDCl3 induced reversibility (Figure 40).97 Zerumbone (549) formed a bis-cysteamine adduct, but only cysteamine attached to C10 showed solvent induced reversibility; cysteamine addition to C6 was irreversible. Notably, cross-conjugated dienones α-santonin (551) and prednisone (552) gave no reaction with cysteamine (Figure 40).97 To further examine the cross-conjugated dienone reactivity, a series of zerumbone (549) analogs were examined in this assay. The C9-C10 cis alkene formed a single, irreversible adduct with cysteamine. Isomerizing the C6-C7 and C9-C10 alkenes showed the same reactivity as 549.184 When the C2-C3 alkene of 549 was epoxidized, the resulting analog showed the same thiol reactivity as 549; however, when the C2-C3 alkene was epoxidized and the C9-C10 alkene was isomerized, only cysteamine addition to C6 was observed. We propose that conformational changes induced by isomerization or epoxidation of alkenes in analogs of 549 resulted in changes to the thiol reactivity, reinforcing the sensitivity of Michael acceptors to steric and electronic affects. Other interesting reactivity properties of cysteamine were noted following studies with other compounds using this NMR assay. Namely, cysteamine reacted selectively with the α, β-unsaturated carbonyls and not with other reactive groups such as the epoxide of parthenolide (200) or peroxide of verlatorin (544).97 (+)-Piperitone (553) and (+)-verbenone (554), possessing a methyl substituent at the β or α positions of the enone gave no reaction with cysteamine while the structurally similar compound (R)-carvone (545) formed the corresponding thiol adduct (Figure 41).97 α,β-Unsaturated aldehydes citral (559) and periallaldehyde (560) reacted with cysteamine resulting in mixtures of 1,4-thiazepines 561 or 563 and bis adducts 562 or 564, respectively (Scheme 42).
Figure 41.

Compounds nonreactive toward cysteamine.
Scheme 42.

Reaction of α,β-Unsaturated Aldehydes with Cysteamine
ALARM NMR (a La assay to detect reactive molecules by nuclear magnetic resonance) is a an additional NMR method, developed by Abbot Laboratories for identifying compounds that can alkylate or oxidize cysteine residues.185 A fragment of the human La antigen protein (residues 100-324) affords high resolution HSQC NMR spectra and contains a reactive cysteine residue (Cys245) that upon undergoing chemical modification, changes and broadens the chemical shifts of the resonances assigned to Lys249, Lys294, and Lys296.185 219 commercial drugs were tested in the ALARM NMR assay, and seven of those compounds, which were known reactives, tested positive in this assay. The ALARM NMR assay was benchmarked to a fluorescent GSH assay that uses fluoroscein-5-maleimide.186 Only one of 28 compounds that were nonreactive by ALARM NMR showed reactivity in the fluorescent GSH assay; however, 10 of the 34 compounds that were reactive by ALARM NMR showed no reactivity with GSH in the fluorescent assay.185 This inconsistency was attributed to the ALARM NMR assay utilizing a thiol in a potentially more biologically relevant environment that may behave differently than a small molecule thiol like GSH. Another advantage of the La antigen is its stability to air oxidation, which can be a problem for small molecule thiols.
13C NMR chemical shifts have been used to measure the electrophilicity of a carbon, i.e., resonances shifted downfield correspond to higher electrophilicity. However, this is not always reliable. For example, methyl cinnamates (scaffold 565) with electron withdrawing substituents at the meta position of the aromatic ring resulted in an increase in the rate of GSH addition (Cl > Br > NO2 > H > OMe), but the chemical shifts of the β-carbon of the alkene was shifted slightly upfield relative to the unsubstituted methyl cinnamate; the resonances for the α-carbon of the alkene was shifted downfield as the rate of GSH addition increased, but no clear correlation was apparent (a correlation coefficient (R2) of 0.54 was obtained when the rate of GSH addition was plotted against the chemical shift of the α-carbons, Scheme 43).187 For para substituted methyl cinnamates the rate of GSH addition followed a similar pattern, both in terms of rates and chemical shift values, although p-NO2 methyl cinnamate was the fastest reacting and p-Br methyl cinnamate was slower to react than methyl cinnamate. The carbonyl carbon resonances were largely unaffected, changing by less than 1 ppm, for all methyl cinnamates in Scheme 43. ortho-Hydroxyl substitution of the aryl group resulted in an upfield shift of 3-6 ppm for the β-carbon of the alkene, but the rate of GSH addition increased significantly; this was attributed to phenol-assisted deprotonation of the thiol as it approached the β-carbon of the alkene (see compounds 228-233, Figure 26).188 These observations suggest that electron withdrawing groups can change the electron density of Michael acceptors in unexpected ways, where the net positive charge is increased at the α-position and decreased at the β-position. However, despite the unexpected resonance shifts of the α and β positions in the 13C NMR with electron withdrawing substituents, the rate of thiol addition still increased compared to unsubstituted methyl cinnamate.
Scheme 43.

Methyl Cinnamates in Order of Decreasing Rates of GSH Addition
Cusack et al. classified the chemical reactivity of α,β-unsaturated carbonyls in scaffolds 567, 568, and 569 using the 13C NMR chemical shift value of the β-carbon (marked with arrow, Figure 42).189 They found that the indolinopyrazolinones 567 (13C NMR 137-139 ppm) formed thiol adducts with β-mercaptoethanol; pyrrolopyrazolinones 568 (13C NMR 128-135 ppm) were reduced in the presence of high concentrations (5 mM) of NADPH (microsomal incubation), and benzothiazinones 569 (13C NMR 118-122 ppm) underwent photoisomerization (~10% isomerization by ambient light).
Figure 42.

Classification of scaffolds by 13C NMR chemical shift values.
9.2 Computational Measurements and Predictions
For N-arylacrylamides substituted with electron rich and electron poor aryl rings, it was shown that a reasonable correlation could be made between the rate of GSH addition and the chemical shifts of Hβ1 (R2 = 0.71) and Cβ (R2 = 0.80), even though only a small range of chemical shifts for all compounds was observed with Hβ1 falling between 5.65-5.86 ppm and Cβ falling between 125.6-128.7 ppm (Scheme 2).38 As predicted, downfield chemical shifts correlated with increased rates of reaction, confirming that NMR chemical shift values can be used to predict reactivity amongst a series of homologous substrates under the same reaction conditions. A good correlation between reaction rates and Hammett substituent constants was also observed. Additionally, transition state structures and activation parameters (activation energy, Ea, and kinetic reaction barrier, ΔG‡) were calculated at the IEFPCM-B3LYP/6-311 + G(d,p) level of theory using methanethiolate as a model nucleophile, and a reasonable correlation between experiment and theory was found (R2 = 0.83 for Ea vs KGSH and R2 = 0.75 for ΔG‡ vs KGSH).38 Another study was performed using a more diverse series of acrylamides possessing N-aryl, N-alkyl, and/or substituents at the α or β position of the acrylamide; transition state calculations of the kinetic reaction barrier, ΔG‡, gave good correlation to the measured half-lives (t1/2) for GSH addition to acrylamides with a correlation coefficient of 0.92 (Table 2).40 However, 13C NMR chemical shift values of either the α or β carbon of the alkene did not correlate to the measured reaction rates demonstrating that 13C NMR chemical shift values are not a reliable predictor of reactivity towards thiols when comparing structurally diverse substrates.
Transition state calculations offer one of the more reliable computational methods for predicting the reactivity of Michael acceptors. Transition state calculations of α,β-unsaturated esters and nitroalkenes were first used to explain the anti-selective addition of thiols to these Michael acceptors.190 Thomas and Kollman calculated that addition of methanethiolate was more exothermic (~10 kcal/mol) than addition of thiolate to acrolein, while for oxygen nucleophiles the addition of hydroxide was calculated to be more exothermic than the addition of methoxide.191 Kunakbaeva et al. used the semiempirical PM3 (parameterized Model 3) method to calculate the activation energies for the addition of β-mercaptopropionic acid to acrylamide, acrylonitrile, and methyl acrylate, as well as the addition of a series of thiols to acrylonitrile and found that the exponential term of the Arrhenius equation calculated from these activation energies correlated well with experimentally determined rates (R2 = 0.997 and 0.973); formation of the tetrahedral intermediate was found to be the rate determining step.192 It should be mentioned that other transition state calculations utilizing DFT methods provide support that favor alternative mechanisms other than 1,4-addition of a thiol to an α,β-unsaturated carbonyl to form an intermediate enolate or carbanion, including direct 1,2-alkene addition, concerted water mediated addition, and base catalyzed addition with ammonia or a histidine fragment.193 Recently, inaccuracies in the dipole moments and polarizabilities of Michael acceptors calculated from common DFT and ab initio methods have been reported.194 For example, calculations using Hartree-Fock methods overestimated dipole moments and underestimated polarizability, while DFT functionals gave the opposite results.195 CCSD, MP2, and hybrid DFT methods with the aug-cc-pVTV basis set could predict dipole moments and polarizabilities with reasonable accuracy, giving RMSD errors in the ranges of 0.12-0.13 D and 0.30-0.38 Å3, respectively.194 For hetero-Michael additions, energies calculated using DFT functionals, hybrid functionals, and range-separated hybrid functionals were compared to either a CCSD or a CBS-QB3 benchmark.195-196 Both CCSD and CBS-QB3 methods calculated the addition of the sulfide anion to the Michael acceptor to form an intermediate carbanion or enolate as the rate determining step. These studies showed that M06-2X, B2PLYP-D, and SCS-MP2 correlated within 1 kcal/mol of the CBS-QB3 energies,196 while M06, BNL, and ωB97x-D correlated within 2 kcal/mol of the CCSD(T)//MP2 energies.195 Other functionals such as B3LYP, MPW1PW91, B2LYP, MP2, PBE, PBE0, BLYP, TPSS, TPSSh, CAM-B3LYP, and LC-ωPBE, did not match the benchmark values.195-196 Using the CBS-QB3 enthalpies of reaction, it was found that an α-Me, β-Me, or β-Ph substituent reduced the exothermicity of the hetero-Michael addition by 1-3 kcal/mol compared to 3-buten-2-one and reactant destabilization, not steric hindrance, led to an increase in activation energies for substituted enones in the gas phase; this effect was larger in solution due to decreased solvation of the substituted enones.196
Other computational methods have been used to predict the reactivity of unsaturated carbonyl species. These parameters include the Fukui function, local softness, HOMO/LUMO energies and coefficients, electrophilicity indices, and related calculations. Thiols and thiolates are characterized as soft nucleophiles due to their polarizability, and the β carbon of the alkene of α,β-unsaturated carbonyls is a soft electrophile due to the charge delocalization of the conjugated system. Thus, the formation of carbon-sulfur covalent bonds via 1,4-addition of thiols to α,β-unsaturated carbonyls is a soft-soft interaction that is energetically favored.197 Molecular hardness and softness refer to a whole molecule, as does the electrophilicity index (ω), a measure of a compound's electrophilicity calculated from its chemical potential (μ) and hardness (η).197b, 198 The Fukui function, , is defined as a change in electron density of a region of a molecule due to a small change in the number of electrons.199 The electrophilic Fukui function, fk+, is used to calculate the propensity of nucleophilic attack at a particular atom of a molecule, and it has been used to predict the reactivity of Michael acceptors.200
Mondal et al. compared the use of atomic charges, electrophilic Fukui function (fk+), and local softnes (sk+), calculated using the Hartree-Fock model, three different basis sets, and Mulliken or Lowdin population analysis, for predicting the site of reactivity for α,β-unsaturated carbonyl compounds acrolein 570, methyl acrylate 571, cinnamaldehyde 572, methyl methacrylate 573, acryloyl chloride 574, and cinnamoyl chloride 575 (Figure 43).201 The carbonyl carbon always had the most positive atomic charge, although these compounds are known experimentally to react with thiols via 1,4- and not 1,2-addition. The condensed electrophilic Fukui function and local softness both had the largest values for the β-carbon, indicating preferential attack at this position by nucleophiles. The different basis sets and population analyses all indicated acrolein 570 as the most reactive with the largest Fukui function and local softness value, but these values, and thus the predicted order of reactivity, for the other compounds varied depending on which basis set and population analysis was used.201 Experimental RC50 values (vide infra) in the literature show that the reactivity order is acrolein 570 > methyl acrylate 571 > cinnamaldehyde 572 >> methyl methacrylate 573 (Figure 43).202 These experimental reactivities correlate with the calculated Fukui function and local softness for acrolein 570 being the most reactive, but the other compounds 571-575 do not correlate with the predicted reactivity. This indicates that the Fukui function and local softness can be good for predicting the site of reactivity but not the order of reactivity for different compounds.
Figure 43.

Relative experimental rates of GSH addition to α,β-unsaturated carbonyls.
The local electrophilicity index (ωk) is another computational parameter used to predict the reactivity of Michael acceptors using a combination of the global electrophilicity index and the Fukui function (fk+ω). Domingo et al. calculated the global electrophilicity indices for a set of α,β-unsaturated ketones, esters, anhydrides, nitriles, and nitro compounds and demonstrated that this index correctly predicts the β-carbon as the reactive site. The global electrophilicity index correlated linearly to the natural log of the experimental rate constants for addition of the β-mercaptoethanol anion to α-nitrostilbenes (R2 = 0.95) and gave better correlation to experimental rate constants, for thiol addition to unsaturated carbonyl compounds, than the Fukui function or local softness.203
LoPachin et al. calculated softness, electrophilicity indices, and HOMO and LUMO energies for N-ethyl maleimide 576, acrolein 570, methyl vinyl ketone 577, methyl acrylate 571, and acrylamide 578 (Table 10).204 These values were compared to the experimentally determined second order rate constants for the reaction of Michael acceptors 570-571, and 576-578 with NAC and with IC50 values for inhibition of dopamine uptake in striatal synaptosomes.204 Linear regression analysis showed that ELUMO and the global electrophilicity index (ω) correlated well with the rates of thiol addition (R2 = 0.93 and 0.87, respectively) but were only qualitatively related to inhibition of dopamine uptake (R2 = 0.75 and 0.64, respectively).
Table 10.
Comparison of Calculated Electrophilicity Parameters to the Second Order Rate Constants of Thiol Addition and to the Inhibition of Dopamine Uptake
| Electrophile | ELUMO (eV) | ω (eV) | log k2 NAC | Uptake inhibition log IC50 |
|---|---|---|---|---|
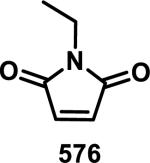
|
−2.36 | 4.73 | 2.17 | −4.33 |
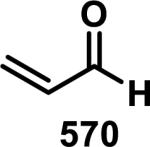
|
−1.70 | 3.57 | 0.332 | −4.28 |
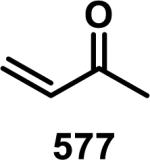
|
−1.33 | 3.00 | 0.037 | −3.48 |
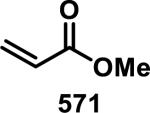
|
−1.01 | 2.76 | −1.893 | −0.336 |
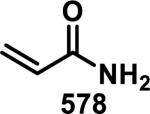
|
−0.69 | 2.30 | −3.651 | −0.359 |
Schüürmann and coworkers introduced two new parameters, energy weighted local electrophilicity (ωEr,s) and charge limited local electrophilicity (ωqr,s), which quantify the change in energy related to the gain or loss of electrons or charge, respectively.205 These new parameters take into account lower energy occupied and higher energy unoccupied molecular orbitals, instead of using only the HOMO and LUMO.205 Linear regression analysis of electrophilicity parameters with log kGSH gave R2 values of 0.84 to 0.91 for ωEr,s and ωqr,s for a series of 31 α,β-unsaturated ketones and esters.205 The benefit of these new local electrophilicity indices (ωEr,s and ωqr,s) is that they show less dependence on the population analysis used when compared to the Fukui function. The local charge limited electrophilicity index (ωq) was also used by Schwöbel et al. to predict the kinetic rate constants (kGSH) and RC50 values for a set of 66 α,β-unsaturated carbonyls.48c In this model, parameters for steric accessibility (Ω) of the α and β carbons were included with the charge limited local electrophilicity. Four models were tested, with the best having an R2 = 0.91 for the predicted vs. experimental rate constants. It was demonstrated that rate constants could be calculated from RC50 values and vice versa. Dividing compounds into separate classes such as aldehydes, ketones, and esters gave improved correlation coefficients.48c
10. Conclusions
To the best of our knowledge, this perspective has provided an exhaustive coverage of biologically relevant and molecularly complex α,β-unsaturated carbonyls that have demonstrated reactivity towards thiols. Thiol reactivity was presented as kinetic data, including rate constants and half-lives, as well as the yield and characterization of thiol adducts. The reversibility of thiol adducts was provided in terms of equilibrium or dissociation constants, crossover experiments, or adduct stability measurements. We have also highlighted the biological activity of α,β-unsaturated carbonyls through their cytotoxicity, inhibition of target proteins, and sites of covalent modification. Through comparisons of the rates of thiol addition to biological activities or toxicities, the effects of the electrophilicity of a Michael acceptor on its bioactivity were elucidated. Methods for analyzing the electrophilicity of α,β-unsaturated carbonyls through spectroscopy and computation provided a better understanding of the steric and electronic factors that influence hetero-Michael addition reactions.
The recent approval of several covalent drugs and the success of targeted covalent inhibitors for enhancing the ligand binding selectivity for structurally related proteins, increasing the binding affinity for proteins with shallow binding sites, and the ability to overcome drug resistance has led to a renewed interest in covalent drug design. When considering α,β-unsaturated carbonyls in the design of covalent inhibitors, it is important to understand the factors that govern their ability to form adducts with thiols, especially under biologically relevant conditions (pH = 6-8). Thiol adduct formation is one of the major pathways for covalent inhibition of proteins and is also a major pathway for covalent inhibitor deactivation when adducts are formed with free cellular thiols such as GSH. The ability to tune the electrophilicity of α,β-unsaturated carbonyls through careful selection of the type of carbonyl and selective modifications to the surrounding structure plays an important role in the future of covalent inhibitor design in order to maximize selectivity and efficacy for a desired protein. The factors that govern the reversibility of thiol adduct formation is another important consideration, since rapidly reversible covalent bonds offer a strong interaction that complements the typical noncovalent interactions used in the design of reversible inhibitors. This perspective has shown the effects of electronic and steric factors that influence the ability of Michael acceptors to form adducts with thiols and also presents methods used to measure the hetero-Michael addition of thiols to α,β-unsaturated carbonyls.
To briefly summarize, the rate of thiol addition to α,β-unsaturated carbonyls, under biologically relevant conditions, decreases in the order of enals > enones > α,β-unsaturated esters > acrylamides > α,β-unsaturated carboxylic acids. However, substituents at the α or β positions of a Michael acceptor can greatly influence the rate of thiol addition. Electron donating groups such as alkyl or electron rich aryl groups at the α or β position significantly reduce the rate of thiol addition relative to unsubstituted Michael acceptors. Electron withdrawing groups at the α position increase the rate of hetero-Michael addition, but also make the resulting adducts more prone to the reverse Michael reaction. The pH also has a significant impact on the rate of thiol adduct formation with more acidic conditions (pH < 7) slowing thiol adduct formation and basic conditions speeding it up. However, in some cases a pH > 9 promotes the reverse Michael reaction. We hope that this perspective has given readers a better understanding of the thiol reactivity of Michael acceptors and how this reactivity can be modulated by changing the structure and substituents on an α,β-unsaturated carbonyl. The information presented in this perspective demonstrates the potential of Michael acceptors as modulators of biological activity through examples of both natural products and synthetic compounds.
Acknowledgements
This work was supported by the NIH (R01-GM110129 to DAH and R01-GM054161 to KMB).
Abbreviations Used
- ABPP
activity based protein profiling
- ahpC
alkyl hydroperoxide reductase subunit C
- ALARM NMR
A La assay to detect reactive molecules by NMR
- ATM
ataxia telangiectasia mutated
- ATR
ataxia-telangiectasia and Rad3-related protein
- AurB
aurora B kinase
- Blk
B lymphocyte kinase
- Bmx
bone marrow kinase on chromosome X also known as ETK
- Btk
Bruton's tyrosine kinase
- CD
concentration of a compound that doubles the activity of NQO1
- CD117
cluster of differentiation molecule 117
- CDDP
1-p-Chlorophenyl-4,4-dimethyl-5-diethylamino-1-penten-3-one hydrobromide
- COMT
catechol-O-methyltransferase
- CRM
chromosome region maintenance
- c-Src
proto-oncogene tyrosine-protein kinase Src or cellular Src kinase
- CTD
C-terminal kinase domain
- CyPG
cyclopentenone prostaglandin
- DNA-PK
DNA-dependent protein kinase
- ERK
extracellular signal-related kinase
- FGFR
fibroblast growth factor receptor
- FLT3
FMS-like tyrosine kinase 3
- Gpx
glutathione peroxidase
- GR
glutathione reductase
- GSH
glutathione
- HCAN1
human calpain 1 protease
- HMAF
hydroxymethylacylfulvene
- HO-1
heme oxygenase
- HRV-3CP
human rhinovirus 3C protease
- HSF
heat shock transcription factor
- IκB
inhibitor of nuclear factor κ-B
- IKKβ
inhibitor of nuclear factor κ-B kinase subunit β
- JNK
c-jun NH2-terminal kinase
- JSP
JNK-stimulating phosphatase
- katG
catalase-peroxidase
- Keap1
Kelch-like ECH associated protein
- LMB
leptomycin B
- Lyn
Lck/Yes-related novel protein tyrosine kinase
- MelB
melampomagnolide B
- MgrA
global regulator in Staphylococcus aureus
- MNK
MAPK-interacting kinase
- Mps
monopolar spindle kinase
- NAC
N-acetyl cysteine
- Nek2
NIMA-related kinase 2
- NR-PKS
nonreducing fungal polyketide synthase
- NQO1
NADPH:quinone reductase
- NS5B
hepatitis C virus RNA polymerase
- PAINS
pan-assay interference compounds
- PDGFRα
platelet derived growth factor receptor α
- PIKK
PI3K-related kinase
- Plk
polo-like kinase
- PP
protein phosphatase
- PTL
parthenolide
- RAL
resorcylic acid lactone
- RSK
ribosomal s6 kinase
- S1P
sphingosine 1-phosphate receptor
- SarA
Staphylococcus aureus protein A
- SarR
Staphylococcus aureus protein R
- Skp2
S phase kinase-associated protein 2
- SL
sesquiterpene lactone
- Src
sarcoma
- SrtA
sortase A
- Syk
spleen tyrosine kinase
- TbCLK1
Trypanosoma brucei CDC2-like kinase
- thil
tRNA sulfurtransferase
- TNFα-PLAP
tumor necrosis factor phospholipase A2-activating protein
- TRPA
transient receptor potential cation channel
- Trx
thioredoxin
- TrxR
thioredoxin reductase
Biography
Paul Jackson received his B.S. degree from California University of Pennsylvania in 2011 and is currently pursuing his Ph.D. at the University of Pittsburgh under the supervision of Prof. Kay M. Brummond. His doctoral research is on the synthesis of tunable, electrophilic α-methylene-γ-lactam isosteres of γ-methylene-γ-lactone-containing guaianolide natural products by utilizing an allenic Pauson-Khand reaction of α-methylene-γ-lactam-tethered allene-ynes to generate the 5,7,5-tricyclic scaffold.
John C. Widen received his B.S. degree with honors in chemistry and a minor in biology from the University of Iowa in 2010. His undergraduate research was conducted with Christopher F. Pigge in the Department of Chemistry. After graduation, he worked for Penford Products Co. as a junior research chemist. In the summer of 2011 John matriculated to the University of Minnesota, Department of Medicinal Chemistry for graduate school and is currently a student in Daniel A. Harki's laboratory studying cysteine reactive probes.
Kay M. Brummond received her education and training as a synthetic chemist from the University of Nebraska-Lincoln, Pennsylvania State University, and the University of Rochester. She was a member of the faculty at West Virginia University and is currently Professor and Chair of the Chemistry Department at the University of Pittsburgh. Her research program focuses mainly on the development of new synthetic methods to expedite the preparation of, and to expand Nature's toolbox of biologically relevant compounds.
Daniel A. Harki obtained his B.A. in biology & chemistry from West Virginia University (1999) where he conducted undergraduate research with Kay Brummond on the synthesis of acylfulvene analogs. Dr. Harki then trained with Blake Peterson at The Pennsylvania State University on the design and synthesis of nucleosides for promoting antiviral lethal mutagenesis in collaboration with Craig Cameron's laboratory. Following completion of his Ph.D. in chemistry (2005), Dr. Harki began postdoctoral training with Peter Dervan at the California Institute of Technology (2005-2009) and worked on the solution phase synthesis of DNA-binding pyrrole-imidazole polyamides for applications including in vivo imaging by position emission tomography. In 2009, Dr. Harki started his independent career at the University of Minnesota where he is currently an Associate Professor of Medicinal Chemistry. The development of novel chemical probes of DNA-interactive proteins, such as APOBEC3 enzymes and transcription factors, are areas of research interest in the Harki laboratory.
Footnotes
The authors declare no competing financial interest.
References
- 1.Schöwobel JAH, Koleva YK, Enoch SJ, Bajot F, Hewitt M, Madden JC, Roberts DW, Schultz TW, Cronin MTD. Measurement and Estimation of Electrophilic Reactivity for Predictive Toxicology. Chem. Rev. 2011;111:2562–2596. doi: 10.1021/cr100098n. [DOI] [PubMed] [Google Scholar]
- 2.a Potashman MH, Duggan ME. Covalent Modifiers: An Orthogonal Ap proach to Drug Design. J. Med. Chem. 2009;52:1231–1246. doi: 10.1021/jm8008597. [DOI] [PubMed] [Google Scholar]; b Bauer RA. Covalent Inhibitors in Drug Discovery: From Accidental Discoveries to Avoided Liabilities and Designed Therapies. Drug Discov. Today. 2015;20:1061–1073. doi: 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Singh J, Petter RC, Baillie TA, Whitty A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Disc. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 4.a Kalgutkar AS, Dalvie DK. Drug Discovery for a New Generation of Covalent Drugs. Exp. Opin. Drug Disc. 2012;7:561–681. doi: 10.1517/17460441.2012.688744. [DOI] [PubMed] [Google Scholar]; b Johnson DS, Weerapana E, Cravatt BF. Strategies for Discovering and Derisking Covalent, Irreversible Enzyme Inhibitors. Future Med. Chem. 2010;2:949–964. doi: 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Parsons ZD, Gates KS. Redox Regulation of Protein Tyrosine Phosphatases: Methods for Kinetic Analysis of Covalent Enzyme Inactivation. In: Cadenas E, Packer L, editors. Methods in Enzymology Volume 528: Hydrogen Peroxide and Cell Signaling Part C. Vol. 528. Elsevier Science Publishing Co Inc; San Diego: 2013. pp. 130–154. [DOI] [PubMed] [Google Scholar]; b Kitz R, Wilson IB. Esters of Methanesulfonic Acid as Irreversible Inhibitors of Acetylcholinesterase. J. Biol. Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]; c Nagahara N, Sawada N, Nakagawa T. Affinity Labeling of a Catalytic Site, Cysteine 247, in Rat Mercaptopyruvate Sulfurtransfera se by Chloropyruvate as an Analog of a Substrate. Biochimie. 2004;86:723–729. doi: 10.1016/j.biochi.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Hagel M, Niu D, Martin TS, Sheets MP, Qiao L, Bernard H, Karp RM, Zhu Z, Labenski MT, Chaturvedi P, Nacht M, Westlin WF, Petter RC, Sing h J. Selective Irreversible Inhibition of a Protease by Targeting a Noncatalytic Cysteine. Nat. Chem. Biol. 2011;7:22–24. doi: 10.1038/nchembio.492. [DOI] [PubMed] [Google Scholar]
- 7.Grillot AL, Farmer LJ, Rao BG, Taylor WP, Weisberg IS, Jacobson IM, Perni RB, Kwong AD. Discovery and Development of Telaprevir. In: Kazmierski WM, editor. Antiviral Drugs: From Basic Discovery through Clinical Trials. Wiley; Hoboken, NJ: 2011. pp. 207–224. [Google Scholar]
- 8.Kohler J, Schuler M. Afatinib, Erlotinib and Gefitinib in the First-Line Therapy of EGFR Mu tation-Positive Lung Adenocarcinoma: A Review. Onkologie. 2013;36:510–518. doi: 10.1159/000354627. [DOI] [PubMed] [Google Scholar]
- 9.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, Sharma SV, Isselbacher KJ, Settleman J, Haber DA. Irreversible Inhibitors of the EGF Receptor May Circumvent Acquired Resistance to Gefitinib. PNAS. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solca F, Dahl G, Zoephel A, Bader G, Sa nderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol. Exp. Ther. 2012;343:342–50. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 11.a Michalczyk A, Klüter S, Rode HB, Simard JR, Grütter C, Rabiller M, Rauh D. Structural Insights into How Irreversible Inhibitors Can Overcome Drug Resistance in EGFR. Biorg. Med. Chem. 2008;16:3482–3488. doi: 10.1016/j.bmc.2008.02.053. [DOI] [PubMed] [Google Scholar]; b Miller VA, Hirsh V, Cadranel J, Chen Y-M, Park K, Kim S-W, Zhou C, Su W-C, Wang M, Sun Y, Heo DS, Crino L, Tan E-H, Chao T-Y, Shahidi M, Cong XJ, Lorence RM, Yang JC-H. Afatinib versus Placebo for Patients with Advanced, Metastatic Non-small-cell Lu ng Cancer After Failure of Erlotinib, Gefitinib, or Both, and One or Two Lines of Chemotherapy (LUX-Lung 1): a Phase 2b/3 Randomised Trial. The Lancet Oncology. 2012;13:528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 12.Engel J, Lategahn J, Rauh D. Hope and Disappointment: Covalent Inhib itors to Overcome Drug Resistance in Non-Small Cell Lung Cancer. ACS Med. Chem. Lett. 2016;7:2–5. doi: 10.1021/acsmedchemlett.5b00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a [8/8/2016];FDA Approves New Pill to Treat Certain Patients with Non-Small Cell Lung Cancer. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm472525.htm.; b Cross DAE, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MRV, Ward RA, Mellor MJ, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GHP, Cantarini M, Kim D-W, Ranson MR, Pao W. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, Papadimitrakopoulou V, Solomon BJ, Oxnard GR, Dziadziuszko R, Aisner DL, Doebele RC, Galasso C, Garon EB, Heist RS, Logan J, Neal JW, Mendenhall MA, Nichols S, Piotrowska Z, Wozniak AJ, Raponi M, Karlovich CA, Jaw-Tsai S, Isaacson J, Despain D, Matheny SL, Rolfe L, Allen AR, Camidge DR. Rociletinib in EFGR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015;372:1700–1709. doi: 10.1056/NEJMoa1413654. [DOI] [PubMed] [Google Scholar]; b Walter AO, Sjin RTT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, Martin TS, Karp R, Kalken D, Chaturvedi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Jaw-Tsai S, Raponi M, Dyke TV, Etter J, Weaver Z, Pao W, Singh J, Simmons AD, Harding TC, Allen A. Discovery of a Mutant-Selective Covalent Inhibitor of EGFR that Overcomes T790M-Mediated Resistance in NSCLC. Cancer Discov. 2013;3:1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, Barrett JC, Janne PA, Oxnard GR. Acquired EGFR C797S Mutation Med iates Resistance to AZD9291 in Non-small Cell Lung Cancer Harboring EGFR T790M. Nat. Med. 2015;21:560–562. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou W, Hur W, McDermott U, Dutt A, Xian W, Ficarro SB, Zhang H, Sharma SV, Brugge J, Meyerson M, Settleman J, Gray NS. A Structure Guided Approach to Creating Covalent FGFR Inhibitors. Chem. Biol. 2010;17:285–295. doi: 10.1016/j.chembiol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan Z, Scheerens H, Li S-J, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KCK, Grothaus PG, Jeffery DA, Spoerke JM, Honigberg LA, Young PR, Dalrymple SA, Palmer JT. Discovery of Selective Irreversible Inhibitors for Bruton's Tyrosine Kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 18.Lou Y, Owens TD, Kuglstatter A, Kondru RK, Goldstein DM. Bruton's Tyrosine Kinase Inhibitors: Approaches to Potent and Selective Inhibition, Preclinical and Clinical Evaluation for Inflammatory Diseases and B Cell Malignancies. J. Med. Chem. 2012;55:4539–4550. doi: 10.1021/jm300035p. [DOI] [PubMed] [Google Scholar]
- 19.a [8/8/16];FDA Approves Imbr uvica for Rare Blood Cancer. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm374761.htm.; b [8/8/16];FDA Approves Imbruvica to Treat Chronic Lymphocytic Leukemia. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm385764.htm.; c Rockoff JD, Loftus P. [9/8/16];AbbVie to Buy Pharmacyclics in $21 Billion Deal. http://www.wsj.com/articles/abbvie-to-buy-pharmacyclics-in-21-billion-deal-1425528086.
- 20.Woyach JA, Furman RR, Liu T-M, O zer HG, Zapatka M, Ruppert AS, Xue L, Li DH-H, Steggerda SM, Versele M, Dave SS, Zhang J, Yilmaz AS, Jaglowski SM, Blum KA, Lozanski A, Lozanski G, James DF, Barrientos JC, Lichter P, Stilgenbauer S, Buggy JJ, Chang BY, Johnson AJ, Byrd JC. Resistance Mechanisms for the Bruton's Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 2014;370:2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a D'Cruz OJ, Uckun FM. Novel Bruton's Tyrosine Ki nase Inhibitors Currently in Development. Onco Targets Ther. 2013;6:161–176. doi: 10.2147/OTT.S33732. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Eda H, Santo L, Cirstea DD, Yee AJ, Scullen TA, Nemani N, Mishima Y, Waterman PR, Arastu-Kapur S, Evans E, Singh J, Kirk CJ, Westlin WF, Raje NS. A Novel Bruton's Tyrosine Kinase Inhibitor CC-292 in Combination with the Proteasome Inhibitor Carfilzomib Impacts the Bone Microenvironment in a Multiple Myeloma Model with Resultant Antimyeloma Activity. Leukemia. 2014;28:1892–1901. doi: 10.1038/leu.2014.69. [DOI] [PubMed] [Google Scholar]
- 22.a Liu F, Zhang X, Weisberg E, Chen S, Hur W, Wu H, Zhao Z, Wang W, Mao M, Cai C, Simon NI, Sanda T, Wang J, Look AT, Griffin JD, Balk SP, Liu Q, Gray NS. Discovery of a Selective Irreversible BMX Inhibitor for P rostate Cancer. ACS Chem. Biol. 2013;8:1423–1428. doi: 10.1021/cb4000629. [DOI] [PubMed] [Google Scholar]; b Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, Look T, Mitsiades CS, Weng AP, Brown JR, Benes CH, Marto JA, Young RA, Gray NS. Targeting Transcription Regulation in Cancer with a Covalent CDK7 Inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, Laughlin JD, Park H, LoGrasso PV, Patricelli M, Nomanbhoy TK, Sorger PK, Alessi DR, Gray NS. Discovery of Potent and Selective C ovalent Inhibitors of JNK. Chem. Biol. 2012;19:140–154. doi: 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nacht M, Qiao L, Sheets MP, Martin TS, Labenski M, Mazdiyasni H, Karp R, Zhu Z, Chaturvedi P, Bhavsar D, Niu D, Westlin W, Petter RC, Medikonda AP, Singh J. Discovery of a Potent and Isoform-Selective Targeted Covalent Inhibitor of the Lipid Kinase PI3Kα. J. Med. Chem. 2013;56:712–721. doi: 10.1021/jm3008745. [DOI] [PubMed] [Google Scholar]
- 25.Wissner A, Floyd MB, Johnson BD, Fraser H, Ingalls C, Nittoli T, Dushin RG, Discafani C, Nilakanta n R, Marini J, Ravi M, Cheung K, Tan X, Musto S, Annable T, Siegel MM, Loganzo F. 2-(Quinazolin-4-ylamino)-[1,4]benzoquinones as Covalent-Binding, Irreversible Inhibitors of the Kinase Domain of Vascular Endothelial Growth Factor Recepto r-2. J. Med. Chem. 2005;48:7560–7581. doi: 10.1021/jm050559f. [DOI] [PubMed] [Google Scholar]
- 26.Gushwa NN, Kang S, Chen J, Taunton J. Selective Targeting of Distinct Active Site Nucleophiles by Irreversible Src-Family Kinase Inhibitors. J. Am. Chem. Soc. 2012;134:20214–20217. doi: 10.1021/ja310659j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a Kamps MP, Taylor SS, Sefton BM. Direct Evidence that Oncogenic Tyrosine Kinases and Cyclic AMP-Dependent Protein Kinase have Homologous ATP-Binding sites. Nature. 1984;310:589–592. doi: 10.1038/310589a0. [DOI] [PubMed] [Google Scholar]; b Pal PK, Wechter WJ, Colman RF. Affinity Labeling of the Inhi bitory DPNH Site of Bovine Liver Glutamate Dehydrogenase by 5'-Fluorosulfonylbenzoyl Adenosine. J. Biol. Chem. 1975;250:8140–8147. [PubMed] [Google Scholar]; c Zoller MJ, Taylor SS. Affinity Labeling of the Nucleotide Binding Site of the Catalytic Subunit of cAMP-dependent Protein Kinase Using p Fluorosulfonyl-[14-C]benzoyl 5'-Adenosine. J. Biol. Chem. 1979;254:8363–8368. [PubMed] [Google Scholar]; d Zoller MJ, Nelson NC, Taylor SS. Affinity Labeling of cAMP-dependent Protein Kinase with p-Fluorosulfonylbenzoyl Adenosine. J. Biol. Chem. 1981;256:10837–10842. [PubMed] [Google Scholar]; e Boettcher BR, Meister A. Covalent Modification of the Active Site of Carbamyl Phosphate Synthetase by 5'-p-Fluorosulfonylbenzoyladenosine. J. Biol. Chem. 1980;255:7129–7133. [PubMed] [Google Scholar]; f Kamoshenkova OM, Boiko VN. Reactio ns of 1,3,5-Tris(fluorosulfonyl)benzene with Some Nucleophilic Reagents. J. Fluorine Chem. 2010;131:248–253. [Google Scholar]
- 28.Henise JC, Taunton J. Irreversible Nek2 Kinase Inhibitors with Cellular Activity. J. Med. Chem. 2011;54:4133–4146. doi: 10.1021/jm200222m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wissner A, Fraser HL, Ingalls CL, Dushin RG, Floyd MB, Cheung K, Nittoli T, Ravi MR, Tan X, Loganzo F. Dual Irreversible Kinase Inhibitors: Quinazoline-Based Inhibitors Incorporating Two Independent Reactive Centers with each Targeting Differ ent Cysteine Residues in the Kinase Domains of EGFR and VEGFR-2. Biorg. Med. Chem. 2007;15:3635–3648. doi: 10.1016/j.bmc.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 30.a Garuti L, Roberti M, Bottegoni G. Irreversible Protein Kinase Inhibitors. Curr. Med. Chem. 2011;18:2981–2994. doi: 10.2174/092986711796391705. [DOI] [PubMed] [Google Scholar]; b Guterman L. Covalent Drugs Form Long-Lived Ties. Chem. Eng. News. 2011;89:19–26. [Google Scholar]
- 31.a Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Barf T, Kaptein A. Irreversible Protein Kinase Inhibitors: Balancing the Benefits and the Risks. J. Med. Chem. 2012;55:6243–6262. doi: 10.1021/jm3003203. [DOI] [PubMed] [Google Scholar]; c Mah R, Thomas JR, Shafer CM. Drug Discovery Considerations in the Development of Covalent Inhibitors. Bioorg. Med. Chem. Lett. 2014;24:33–39. doi: 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 32.a Schmidt TJ, Ak M, Mrowietz U. Reactivity of Dimethyl Fumarate and Methylhydrogen Fumarate Towards Glutathione and N-Acetyl-L-Cysteine--Preparation of S-Substituted Thiosuccinic Acid Esters. Biorg. Med. Chem. 2007;15:333–342. doi: 10.1016/j.bmc.2006.09.053. [DOI] [PubMed] [Google Scholar]; b [8/8/16];FDA Approves New Multiple Sclerosis Treatment: Tecfidera. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm345528.htm.
- 33.a Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frodin M, Taunton J. Reversible Targeting of Noncatalytic Cysteines with Chemically Tuned Electrophiles. Nat. Chem. Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Miller RM, Paavilainen VO, Krishnan S, Serafimova I, Taunton J. Electrophilic Fragment-Based Design of Reversible Covalent Kinase Inhibitors. J. Am. Chem. Soc. 2013;135:5298–5301. doi: 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Couch RD, Browning RG, Honda T, Gribble GW, Wright DL, Sporn MB, Anderson AC. Studies on t he Reactivity of CDDO, a Promising New Chemopreventive and Chemotherapeutic Agent: Implications for a Molecular Mechanism of Action. Bioorg. Med. Chem. 2005;15:2215–2219. doi: 10.1016/j.bmcl.2005.03.031. [DOI] [PubMed] [Google Scholar]; d Honda T, Honda Y, Favaloro FG, Gribble GW, Suh N, Place AE, Rendi MH, Sporn MB. A Novel Dicyanotriterpenoid, 2-Cyano-3,12-dioxooleana-1-9(11)-dien-28-onitrile, Active at Picomolar Concentrations for Inhibition of Nitric Oxide Production. Bioorg. Med. Chem. Lett. 2002;12:1027–1030. doi: 10.1016/s0960-894x(02)00105-1. [DOI] [PubMed] [Google Scholar]
- 34.a Adam GC, Sorensen EJ, Cravatt BF. Chemical Strategies for Functional Proteomics. Mol. Cell. Proteomics. 2002;1:781–790. doi: 10.1074/mcp.r200006-mcp200. [DOI] [PubMed] [Google Scholar]; b Speers AE, Cravatt BF. Chemical Strategies for Activity-Based Proteomics. ChemBioChem. 2004;5:41–47. doi: 10.1002/cbic.200300721. [DOI] [PubMed] [Google Scholar]; c Evans MJ, Cravatt BF. Mechanism-Based Profiling of Enzyme Families. Chem. Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]; d Kozarich JW. Activity-Based Proteomics: Enzyme Chemistry Redux. Curr. Opin. Chem. Biol. 2003;7:78–83. doi: 10.1016/s1367-5931(02)00013-3. [DOI] [PubMed] [Google Scholar]; e Jessani N, Cravatt BF. The Development and Applicatio n of Methods for Activity-Based Protein Profiling. Curr. Opin. Chem. Biol. 2004;8:54–59. doi: 10.1016/j.cbpa.2003.11.004. [DOI] [PubMed] [Google Scholar]; f Cravatt BF, Wright AT, Kozarich JW. Activity Based Protein Profiling: From Enzyme Chemistry to Proteomic Chemistry. Annu. Rev. Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]; g Nodwell MB, Sieber SA. Activity-Based Protein Profiling. Vol. 324. Springer-Verlag; Berlin: 2012. p. 165. [Google Scholar]; h Puri AW, Bogyo M. Using Small Molecules to Dissect Mechanisms of Microbial Pathogenesis. ACS Chem. Biol. 2009;4:603–616. doi: 10.1021/cb9001409. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Barglow KT, Cravatt BF. Activity-Based Protein Profiling for the Functional Annotation of Enzymes. Nat. Methods. 2007;4:822–827. doi: 10.1038/nmeth1092. [DOI] [PubMed] [Google Scholar]
- 35.a Kitagawa D, Yokota K, Gouda M, Narumi Y, Ohmoto H, Nishiwaki E, Akita K, Kirii Y. Activity-Based Kinase Profiling of Approved Tyrosine Kinase Inhibitors. Genes Cells. 2013;18:110–122. doi: 10.1111/gtc.12022. [DOI] [PubMed] [Google Scholar]; b Yang P-Y, Liu K, Ngai MH, Lear MJ, Wenk MR, Yao SQ. Activity-Based Proteome Profiling of Potential Cellular Targets of Orlistat-An FDA-Approved Drug with Anti-Tumor Activities. J. Am. Chem. Soc. 2010;132:656–666. doi: 10.1021/ja907716f. [DOI] [PubMed] [Google Scholar]; c Ngai MH, Yang P-Y, Liu K, Shen Y, Wenk MR, Yao SQ, Lear MJ. Click-Based Synthesis and Proteomic Profiling of Lipstatin Analogues. Chem. Commun. 2010;46:8335–8337. doi: 10.1039/c0cc01276a. [DOI] [PubMed] [Google Scholar]
- 36.Bruegger J, Haushalter B, Vagstad A, Shakya G, Mih N, Townsend CA, Burkart MD, Tsai S-C. Probing the Selectivity and Protein-Protein Interactions of a Nonreducing Fungal Polyketide Synthase Using Mechanism-Based Crosslinkers. Chem. Biol. 2013;20:1135–1146. doi: 10.1016/j.chembiol.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.a Wissner A, Overbeek E, Reich MF, Floyd MB, Johnson BD, Mamuya N, Rosfjord EC, Discafani C, Davis R, Shi X, Rabindran SK, Gruber BC, Ye F, Hallett WA, Nilakantan R, Shen R, Wang Y-F, Greenberger LM, Tsou H-R. Synthesis and Structure-Activity Relationships of 6,7-Disubstituted 4-Anilinoquinoline-3-carbonitriles. The Design of an Orally Active, Irreversible Inhibitor of the Tyrosine Kinase Activity of the Epider mal Growth Factor Receptor (EGFR) and the Human Epidermal Growth Factor Receptor-2 (HER-2). J. Med. Chem. 2003;46:49–63. doi: 10.1021/jm020241c. [DOI] [PubMed] [Google Scholar]; b Tsou H-R, Mamuya N, Johnson BD, Reich MF, Gruber BC, Ye F, Nilakantan R, Shen R, Discafani C, DeBlanc R, Davis R, Koehn FE, Greenberger LM, Wang Y-F, Wissner A. 6-Substituted-4-(3-bromophenylamino)quinazolines as Putative Irreversible Inhbitors of the Epidermal Growth Factor Receptor (EGFR) and Human Epidermal Growth Factor Receptor (HER-2) with Enhanced Antitumor Activity. J. Med. Chem. 2001;44:2719–2734. doi: 10.1021/jm0005555. [DOI] [PubMed] [Google Scholar]
- 38.Cee VJ, Volak LP, Chen Y, Bartberger MD, Tegley C, Arvedson T, McCarter J, Tasker AS, Fotsch C. Systematic Study of the Glutathione (GSH) Reactivity of N-Aryla crylamides: 1. Effects of Aryl Substitution. J. Med. Chem. 2015;58:9171–9178. doi: 10.1021/acs.jmedchem.5b01018. [DOI] [PubMed] [Google Scholar]
- 39.ln([electrophile]) = (−kpseudofirst)t + ln([electrophile])
- 40.Flanagan ME, Abramite JA, Anderson DP, Aulabaugh A, Dahal UP, Gilbert AM, Li C, Montg omery J, Oppenheimer SR, Ryder T, Schuff BP, Uccello DP, Walker GS, Wu Y, Brown MF, Chen JM, Hayward MM, Noe MC, Obach RS, Philippe L, Shanmugasundaram V, Shapiro MJ, Starr J, Stroh J, Che Y. Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J. Med. Chem. 2014;57:10072–10079. doi: 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- 41.a Drahl C, Cravatt BF, Sorenson EJ. Protein-Reactive Natural Products. Angew. Chem. Int. Ed. 2005;44:5788–5809. doi: 10.1002/anie.200500900. [DOI] [PubMed] [Google Scholar]; b Kondo F, Ikai Y, Oka H, Okumura M, Ishikawa N, Harada K-I, Matsuura K, Murata H, Suzuki M. Formation, Characterization, and Toxicity of the Glutathione and Cysteine Conjugates of Toxic Hepta peptide Microcystins. Chem. Res. Toxicol. 1992;5:591–596. doi: 10.1021/tx00029a002. [DOI] [PubMed] [Google Scholar]; c Runnegar M, Berndt N, Kong S-M, Lee EYC, Zhang L. In Vivo and In Vitro Binding of Microcystin to Protein Phosphatases 1 and 2A. Biochem. Biophys. Res. Commun. 1995;216:162–169. doi: 10.1006/bbrc.1995.2605. [DOI] [PubMed] [Google Scholar]; d Craig M, Luu HA, McCready TL, Holmes CFB, Williams D, Andersen RJ. Molecular Mechanisms Underlying the Interaction of Motuporin and Microcystins with Type-1 and Type-2A Protein Phosphatases. Biochem. Cell. 1996;74:569–578. doi: 10.1139/o96-061. [DOI] [PubMed] [Google Scholar]
- 42.Cl ement LL, Tsakos M, Schaffert ES, Scavenius C, Enghild JJ, Poulsen TB. The Amido-Pentadienoate-Functionality of the Rakicidins is a Thiol Reactive Electrophile - Development of a General Synthetic Strategy. Chem. Commun. 2015;51:12427–12430. doi: 10.1039/c5cc04500b. [DOI] [PubMed] [Google Scholar]
- 43.Lu L, Meehan MJ, Gu S, Chen Z, Zhang W, Zhang G, Liu L, Huang X, Dorrestein PC, Xu Y, Moore BS, Qian P-Y. Mechanism of Action of Thalassospiramides, A New Class of Calpain Inhibitors. Sci. Rep. 2015;5:1–8. doi: 10.1038/srep08783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ues ugi S, Fujisawa N, Yoshida J, Watanabe M, Dan S, Yamori T, Shiono Y, Kimura K.-i. Pyrrocidine A, a Metabolite of Endophytic Fungi, has a Potent Apoptosis-inducing Activity Against HL60 Cells through Caspase Activation via the Michael Addition. J. Antibiot. 2016;69:133–140. doi: 10.1038/ja.2015.103. [DOI] [PubMed] [Google Scholar]
- 45.Bohrisch J, Faltz H, Pätzel M, Liebscher J. Chiral 1,4-Diazepinones and 1,4-Thiazepinones by Diastereoselective Ring Chain Transformation of α,β-Unsaturated Lactones or Lactams. Tetrahedron. 1994;50:10701–10708. [Google Scholar]
- 46.Albrecht A, Albrecht L, Janecki T. Recent Advances in the Synthesis of α-Alkylidene-substituted δ-Lactones, γ-Lactams and δ-Lactams. Eur. J. Org. Chem. 2011:2747–2766. [Google Scholar]
- 47.a Arumugam V, Routledge A, Abell C, Balasubramanian S. Synthesis of 2-Oxindole Derivatives via the Intramolecular Heck Reaction on Solid Support. Tet. Lett. 1997;38:6473–6476. [Google Scholar]; b Kornet MJ. Synthesis and Anticonvulsant Activity of 1-Aryl-3-Methylene-2-Pyrrolidinone Adducts. J. Heterocycl. Chem. 1985;22:129–130. [Google Scholar]; c Pettigrew NE, Brush EJ, Colman RF. 3-Methyleneoxindole: An Affinity Label of Glutathione S-Transferase pi Which Targets Tryptophan 38. Biochem. 2001;40:7549–7558. doi: 10.1021/bi002840w. [DOI] [PubMed] [Google Scholar]; d Wieland T, Unger O. Synthese Einiger Methoxy-oxindole Und-indoline. Chem. Ber. 1963;96:253–259. [Google Scholar]; e Klutchko S, Hoefle ML, Smith RD, Essenburg AD, Parker RB, Nemeth VL, Ryan MJ, Dugan DH, Kaplan HR. Synthesis and Angio tensin-Converting Enzyme Inhibitory Activity of 3-(Mercaptomethyl)-2-Oxo-1-Pyrrolidineacetic Acids and 3-(Mercaptomethyl)-2-Oxo-1-Piperidineacetic Acids. J. Med. Chem. 1981;24:104–109. doi: 10.1021/jm00133a021. [DOI] [PubMed] [Google Scholar]
- 48.a Böhme A, Thaens D, Schramm F, Paschke A, Schüürmann G. Thiol Reactivity and Its Impact on the Ciliate Toxicity of α,β-Unsaturated Aldehydes, Ketones, and Esters. Chem. Res. Toxicol. 2010;23:1905–1912. doi: 10.1021/tx100226n. [DOI] [PubMed] [Google Scholar]; b Böhme A, Thaens D, Paschke A, Schüürmann G. Kinetic Glutathione Chemoassay To Quantify Thiol R eactivity of Organic Electrophiles -- Application to α, β-Unsaturated Ketones, Acrylates, and Propiolates. Chem. Res. Toxicol. 2009;22:742–750. doi: 10.1021/tx800492x. [DOI] [PubMed] [Google Scholar]; c Schwöbel JAH, Wondrousch D, Koleva YK, Madden JC, Cronin MTD, Schüürmann G. Predictio n of Michael-Type Acceptor Reactivity Toward Glutathione. Chem. Res. Toxicol. 2010;23:1576–1585. doi: 10.1021/tx100172x. [DOI] [PubMed] [Google Scholar]
- 49.Fukami T, Yokoi T. The Emerging Role of Human Esterases. Drug Metab. Pharmacokinet. 2012;27:466–477. doi: 10.2133/dmpk.dmpk-12-rv-042. [DOI] [PubMed] [Google Scholar]
- 50.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Prodrugs: Design and Clinical Applications. Nat Rev Drug Discov. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 51.Matthews DA, Dragovich PS, Webber SE, Fuhrman SA, Patick AK, Zalman LS, Hendrickson TF, Love RA, Prins TJ, Marakovits JT, Zhou R, Tikhe J, Ford CE, Meador JW, Ferre RA, Brown EL, Binford SL, Brothers MA, DeLisle DM, Worland ST. Structure-Assisted Design of Mechanism-Based Irreversible Inhibitors of Hu man Rhinovirus 3C Protease with Potent Antiviral Activity Against Multiple Rhinovirus Serotypes. PNAS. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.a Zhang X, Song Z, Qin B, Zhang X, Chen L, Hu Y, Yuan Z. Rupintrivir is a Promising Candidate for Treating Sever e Cases of Enterovirus-71 Infection: Evaluation of Antiviral Efficacy in a Murine Infection Model. Antiviral Res. 2013;97:264–269. doi: 10.1016/j.antiviral.2012.12.029. [DOI] [PubMed] [Google Scholar]; b Rocha-Pereira J, Nascimento MSJ, Ma Q, Hilgenfeld R, Neyts J, Jochmans D. The Enterovirus Protease Inhib itor Rupintrivir Exerts Cross-Genotypic Anti-Norovirus Activity and Clears Cells from the Norovirus Replicon. Antimicrob. Agents Chemother. 2014;58:4675–4681. doi: 10.1128/AAC.02546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rosell NRR, Mokhlesi L, Milton NE, Sweeney TR, Zunszain PA, Curry S, Leatherbarrow RJ. Design and Synthesis of Irreversible Inhibitors of Foot-and-Mouth Disease Virus 3C Protease. Bioorg. Med. Chem. Lett. 2014;24:490–494. doi: 10.1016/j.bmcl.2013.12.045. [DOI] [PubMed] [Google Scholar]
- 53.Dragovich PS, Webber SE, Babine RE, Fuhrman SA, Patick AK, Matthews DA, Lee CA, Reich SH, Prins TJ, Markovits JT, Littlefield ES, Zhou R, Tikhe J, Ford CE, Wallace MB, Meador JW, Ferre RA, Brown EL, Binford SL, Harr JEV, DeLisle DM, Worland ST. Structure-Based Desi gn, Synthesis, and Biological Evaluation of Irreversible Human Rhinovirus 3C Protease Inhibitors. I. Michael Acceptor Structure-Activity Studies. J. Med. Chem. 1998;41:2806–2818. doi: 10.1021/jm980068d. [DOI] [PubMed] [Google Scholar]
- 54.Webber SE, Okano K, Little TL, Reich SH, Xin Y, Fuhrman SA, Matthews DA, Love RA, Hendrickson TF, Patick AK, Meador JW, Ferre RA, Brown EL, Ford CE, Bindford SL, Worland ST. Tripeptide Aldehyde Inhibitors of Human Rhinovirus 3C Protease: Design, Synthesis, Biological Evaluation, and Cocrystal Structure of P1 Glutamine Isosteric Replacements. J. Med. Chem. 1998;41:2786–2805. doi: 10.1021/jm980071x. [DOI] [PubMed] [Google Scholar]
- 55.Kathman SG, Xu Z, Statsyuk AV. A Fragment-Based Method to Discover Irrreversible Covalent Inhibitors of Cysteine Proteases. J. Med. Chem. 2014;57:4969–4974. doi: 10.1021/jm500345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kathman SG, Statsyuk AV. Covalent Tethering of Fragments for Covalent Probe Discovery. MedChemComm. 2016;7:576–585. doi: 10.1039/c5md00518c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nonoo RH, Armstrong A, Mann DJ. Kinetic Template-Guided Tethering of Fragments. ChemMedC hem. 2012;7:2082–2086. doi: 10.1002/cmdc.201200404. [DOI] [PubMed] [Google Scholar]
- 58.Kalesse M, Christmann M. The Chemistry and Biology of the Leptomycin Family. Synthesis. 2002;8:981–1003. [Google Scholar]
- 59.Bonazzi S, Eidam O, Guettinger S, Wach J-Y, Zemp I, Kutay U, Gademann K. Anguinomycins and Derivative s: Total Syntheses, Modeling, and Biological Evaluation of the Inhibition of Nucleocytoplasmic Transport. J. Am. Chem. Soc. 2010;132:1432–1442. doi: 10.1021/ja9097093. [DOI] [PubMed] [Google Scholar]
- 60.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B Inactivates CRM1/Exportin 1 by Covalent Modification at a Cysteine Residue in the Central Conserved Region. PNAS. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kudo N, Wolff B, Sekimoto T, Schreiner EP, Yoneda Y, Yanagida M, Horinouchi S, Yoshida M. Leptomycin B Inhibition of Signal-Mediated Nuclear Export by Direct Binding to CRM1. Exp. Cell Res. 1998;242:540–547. doi: 10.1006/excr.1998.4136. [DOI] [PubMed] [Google Scholar]
- 62.Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, MacMillan J, Chook YM. Nuclear Export Inhibition Through Covalent Co njugation and Hydrolysis of Leptomycin B by CRM1. PNAS. 2013;110:1303–1308. doi: 10.1073/pnas.1217203110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ando R, Amano Y, Nakamura H, Arai N, Kuwajima I. Design, Synthesis, and Evaluation of Novel Kazusamycin A Derivatives as Potent Antitumor Agents. Bioorg. Med. Chem. L ett. 2006;16:3315–3318. doi: 10.1016/j.bmcl.2006.03.056. [DOI] [PubMed] [Google Scholar]
- 64.Mandal PK, Maiti GH, Misra AK. Catalyst-Free Efficient Synthesis of 3-Thio-2-deoxysugar Derivatives in Water. J. Carbohydr. Chem. 2008;27:238–257. [Google Scholar]
- 65.Ikeda H, Kaneko E, Okuzawa S, Takahashi D, Toshima K. Chemical and Biological Evaluation of Unusual Sugars, α-Aculosides, as Novel Michael Acceptors. Org. Biomol. Chem. 2014;12:8832–8835. doi: 10.1039/c4ob01877j. [DOI] [PubMed] [Google Scholar]
- 66.Bartmann W, Beck G, Granzer E, Jendralla H, Kerekjarto BV, Wess G. Convenient Two-Step Stereospecific Hydroxy-Substitution with Retention in β-Hydroxy-δ-Lactones. Tet. Lett. 1986;27:4709–4712. [Google Scholar]
- 67.a Chen J-L, You Z-W, Qing F-L. Total Synthesis of γ-Trifluoromethylated Analogs of Goniothalamin and their Derivatives. J. Fluorine Chem. 2013;155:143–150. [Google Scholar]; b Wolberg M, Dassen BHN, Schurmann M, Jennewein S, Wubbolts MG, Schoemaker HE, Mink D. Large-Scale Synthesis of New Pyranoid Building Blocks Based on Aldolase-Catalysed Carbon-Carbon Bond Formation. Adv. Synth. Catal. 2008;350:1751–1759. [Google Scholar]; c Urbanski MJ, Chen RH, Demarest KT, Gunnet J, Look R, Ericson E, Murray WV, Rybczynski PJ, Zhang X. 2,5-Disubstituted 3,4-Dihydro-2H-Benzo[b][1,4]thiazepines as Potent and Selective V2 Arginine Vasopressin Rece ptor Antagonists. Bioorg. Med. Chem. Lett. 2003;13:4031–4034. doi: 10.1016/j.bmcl.2003.08.051. [DOI] [PubMed] [Google Scholar]
- 68.Calvet S, Courillon C, Malacria M. Diastereoselective Thiol Conjugate Addition on δ-Alkylated-γ-Silylated-α,β-Unsaturated-δ-Lactones. Arkivoc. 2002:189–195. [Google Scholar]
- 69.a Fitton AO, Frost JR, Houghton PG, Suschitzky H. Reactions of Formylchromone Derivatives. Part 2. Addition Reactions of 3-(Aryliminomethyl)chromones. J. Chem. Soc., Perkin Trans. 1. 1979:1691–1694. [Google Scholar]; b Fitton AO, Houghton PG, Suschitzky H. Reactions of Formylchomone Derivatives; 3. A Facile Synthesis of Fused Benzopyrano-Benzothiazepinones,-Benzoxazepinones, and-Benzodiazepinones. Synthesis. 1979:337–339. [Google Scholar]; c Ahmad-Junan SA, Walkington AJ, Whiting DA. Aryloxymethyl Radical Cyclizations Mimic king Biological C-C Bond Formation to Methoxy Groups. J. Chem. Soc., Perkin Trans. 1. 1992:2313–2320. [Google Scholar]
- 70.Mustafa A, Kamel M, Allam MA, Harhash AHE-S, Hassan AEAA. Reactions with Mercaptans. III. Action of Aromatic Thiols on Coumarins, 4-Styryl-Coumarins and 2-Styrylchomones. J. Am. Chem. Soc. 1956;78:5011–5016. [Google Scholar]
- 71.a Song HY, Ngai MH, Song ZY, MacAry PA, Hobley J, Lear MJ. Practical Synthesis of Maleimides and Coumarin-Linked Probes for Protein and Antibody La belling via Reduction of Native Disulfides. Org. Biomol. Chem. 2009;7:3400–3406. doi: 10.1039/b904060a. [DOI] [PubMed] [Google Scholar]; b Chung CC, Ohwaki K, Schneeweis JE, Stec E, Varnerin JP, Goudreau PN, Chang A, Cassaday J, Yang L, Yamakawa T, Kornienko O, Hodder P, Inglese J, Ferrer M, Strulovici B, Kusunoki J, Tota MR, Takagi T. A Fluorescence-Based Thiol Quantification Assay for Ultra-High-Throughput Screening for Inhibitors of Coenzyme A Production. Assay Drug Dev. Technol. 2008;6:361–374. doi: 10.1089/adt.2007.105. [DOI] [PubMed] [Google Scholar]; c Zuo Q-P, Li B, Pei Q, Li Z, Liu S-K. A Highly Selective Fluorescent Probe for Detection of Biological Samples Thiol and Its Application in Living Cells. J. Fluoresc. 2010;20:1307–1313. doi: 10.1007/s10895-010-0673-6. [DOI] [PubMed] [Google Scholar]; d Roubinet B, Renard P-Y, Romieu A. New Insights into the W ater-Solubilization of Thiol-Sensitive Fluorogenic Probes Based on Long-Wavelength 7-Hydroxycoumarin Scaffolds. Dyes Pigm. 2014;110:270–284. [Google Scholar]; e Jung HS, Han JH, Pradhan T, Kim S, Lee SW, Sessler JL, Kim TW, Kang C, Kim JS. A Cysteine-Selective Fluorescent Probe for the Cellular Detection of Cysteine. Biomaterials. 2012;33:945–953. doi: 10.1016/j.biomaterials.2011.10.040. [DOI] [PubMed] [Google Scholar]; f Lim S-Y, Lee S, Park SB, Kim H-J. Highly Selective Fluorescence Turn-on Probe for Glutathione. Tet. Lett. 2011;52:3902–3904. [Google Scholar]; g Ha H-J, Yoon D-H, Park S, Kim H-J. Fluorescence Turn-on Probe for Biothiols: Intramolecular Hydrogen Bonding Effect on the Michael Reaction. Tetrahedron. 2011;67:7759–7762. [Google Scholar]
- 72.Jones JB, Middleton HW. Studies Related to the Mechanism of Action of Cardiac Glycosides. On the Reactions of Thiols with Unsaturated Lactones and on the Structure of the Compound Previously Reported as Ouabagenin. The Preparation and Properties of Authentic Ouabagenin. Can. J. Chem. 1970;48:3819–3826. [Google Scholar]
- 73.Kupchan SM, Giacobbe TJ, Krull IS, Thomas AM, Eakin MA, Fessler DC. Reaction of Endocyclic α,β-Unsaturated-γ-lactones with Thiols. J. Org. Chem. 1970;35:3539–3543. [Google Scholar]
- 74.Rao YS. Chemistry of Butenolides. Chem. Rev. 1964;64:353–388. [Google Scholar]
- 75.Trost BM, Aponick A. Palladium-Catalyzed Asymmetric Allylic Alkylation of meso- and dl-1,2-Divinylethy lene Carbonate. J. Am. Chem. Soc. 2006;128:3931–3933. doi: 10.1021/ja0578348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.a Calderon A, Font J, Ortuno RM. Studies on Structurally Simple α,β-Butenolides. IV. Behaviour of Protoanemonin as Electrophile Towards Alcohols and Thiols. Tetrahedron. 1984;40:3787–3794. [Google Scholar]; b Camps P, Cardellach J, Corbera J, Font J, Oruno RM, Ponsati O. Studies on Structurally Simple α,β-Butenolides. III. Behaviour of (-)-(S)-δ-Heterosubstituted γ-Methyl-α,β-Butenolides Towards Nucleophiles. Protoanemonin as Intermediate in Elimination-Addition Mechanism. Tetrahedron. 1983;39:395–400. [Google Scholar]
- 77.Hakimelahi GH, Moosavi-Movahedi AA, Sambaiah T, Zhu J-L, Ethiraj KS, Pasdar M, Hakimelahi S. Reactions of Purines-Containing Butenolides with L-Cysteine or N-Acetyl-L-C ysteine as Model Biological Nucleophiles: a Potent Mechanism-Based Inhibitor of Ribonucleotide Reductase Caused Apoptosis in Breast Carcinoma MCF7 Cells. Eur. J. Med. Chem. 2002;37:207–217. doi: 10.1016/s0223-5234(02)01333-8. [DOI] [PubMed] [Google Scholar]
- 78.Kitson RR, Millemaggi A, Taylor RJ. The Renaissance of alpha-Methylene-gamma-Butyrolactones: New Synthetic Approaches. Angew. Chem. Int. Ed. 2009;48:9426–9451. doi: 10.1002/anie.200903108. [DOI] [PubMed] [Google Scholar]
- 79.a Polo LM, Castro CM, Cruzado MC, Collino CJG, Cuello-Carrión FD, Ciocca DR, Giordano OS, Ferrari M, López LA. 11,13-Dihydro-dehydroleucodine, a Derivative of Dehydroleucodine with an Inactivated Alkylating Function Conserves the Anti-proliferative Activity in G2 but does not Cause Cytotoxicity. Eur. J. Pharmacol. 2007;556:19–26. doi: 10.1016/j.ejphar.2006.10.049. [DOI] [PubMed] [Google Scholar]; b Beekman AC, Woerdenbag HJ, Uden W, Pras N, Konings AW, Wikstrom HV, Schmidt TJ. Structure-Cytotoxicity Relationships of Some Helenanolide-Type Sesquiterpene Lactones. J. Nat. Prod. 1997;60:252–257. doi: 10.1021/np960517h. [DOI] [PubMed] [Google Scholar]; c Csuk R, Heinold A, Siewert B, Schwarz S, Barthel A, Kluge R, Strohl D. Synthesis and Biological Evaluation of Antitumor-Active Arglabin Derivatives. Arch Pharm (Weinheim) 2012;345:215–222. doi: 10.1002/ardp.201100065. [DOI] [PubMed] [Google Scholar]
- 80.a Woods JR, Mo HP, Bieberich AA, Alavanja T, Colby DA. Amino-Derivatives of the Sesquiterpene Lactone Class of Natural Products as Prodrugs. MedChemComm. 2013;4:27–33. [Google Scholar]; b Amslinger S. The Tunable Functionality of alpha,beta-Unsaturated Carbonyl Compounds Enables Their Differential Application in Biological Systems. ChemMedChem. 2010;5:351–356. doi: 10.1002/cmdc.200900499. [DOI] [PubMed] [Google Scholar]; c Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N. What Made Sesquiterpene Lactones Reach Cancer Clinical Trials? Drug Discov. Today. 2010;15:668–678. doi: 10.1016/j.drudis.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 81.Kupchan SM, Fessler DC, Eakin MA, Giacobbe TJ. Reactions of alpha Methylene Lactone Tumor Inhibitors with Model Biological Nucleophiles. Science. 1970;168:376–378. doi: 10.1126/science.168.3929.376. [DOI] [PubMed] [Google Scholar]
- 82.Hall IH, Lee K-H, Mar EC, Starnes CO, Waddell TG. Antitumor Agents 21. A Proposed Mechanism for Inhibition of Ca ncer Growth by Tenulin and Helenalin and Related Cyclopentenones. J. Med. Chem. 1977;20:333–337. doi: 10.1021/jm00213a003. [DOI] [PubMed] [Google Scholar]
- 83.Schmidt TJ. Helenanolide-Type Sesquiterpene Lactones-III. Rates and Stereochemistry in the Reaction of Helenalin and Related Helenanolides with Sulfhy dryl Containing Biomolecules. Biorg. Med. Chem. 1997;5:645–653. doi: 10.1016/s0968-0896(97)00003-5. [DOI] [PubMed] [Google Scholar]
- 84.Schmidt TJ, LyB G, Pahl HL, Merfort I. Helenanolide Type Sesquiterpene Lactones. Part 5. The Role of Glutathione Addition Under Physiological Conditions. Bioorg. Med. Chem. 1999;7:2849–2855. doi: 10.1016/s0968-0896(99)00234-5. [DOI] [PubMed] [Google Scholar]
- 85.a Khazir J, Singh PP, Reddy DM, Hyder I, Shafi S, Sawant SD, Chashoo G, Mahajan A, Alam MS, Saxena AK, Arvinda S, Gupta BD, Kumar HM. Synthesis and Anticancer Activity of Novel Spiro-Isoxazolin e and Spiro-Isoxazolidine Derivatives of alpha-Santonin. Eur. J. Med. Chem. 2013;63:279–289. doi: 10.1016/j.ejmech.2013.01.003. [DOI] [PubMed] [Google Scholar]; b Vuckovic I, Vujisic L, Klaas CA, Merfort I, Milosavljevic S. NF-kappa B DNA Binding Activity of Sesquiterpene Lactones from Anthemis Arvensis and Anthemis Cotula. Nat. Prod. Res. 2011;25:800–805. doi: 10.1080/14786410902941402. [DOI] [PubMed] [Google Scholar]; c Torres F, Quintana J, Cabrera J, Loro JF, Leon F, Bermejo J, Estevez F. Induction of G2-M Phase Arrest and Apoptosis by alpha-Methylene-gamma-Butyrolactones in Human Leukemia Cells. Can cer Lett. 2008;269:139–147. doi: 10.1016/j.canlet.2008.04.028. [DOI] [PubMed] [Google Scholar]; d Nasim S, Crooks PA. Antileukemic Activity of Aminoparthenolide Analogs. Bioorg. Med. Chem. Lett. 2008;18:3870–3873. doi: 10.1016/j.bmcl.2008.06.050. [DOI] [PubMed] [Google Scholar]; e Cateni F, Zilic J, Zacchigna M, Bonivento P, Frausin F, Scarcia V. Synthesis and Biol ogical Properties of New alpha-Methylene-gamma-Butyrolactones and alpha,beta-Unsaturated delta-Lactones. Eur. J. Med. Chem. 2006;41:192–200. doi: 10.1016/j.ejmech.2005.10.009. [DOI] [PubMed] [Google Scholar]; f Hughes MA, McFadden JM, Townsend CA. New alpha-Methylene-gamma-Butyrolactones with Antimycobacteri al Properties. Bioorg. Med. Chem. Lett. 2005;15:3857–3859. doi: 10.1016/j.bmcl.2005.05.119. [DOI] [PubMed] [Google Scholar]; g Kempema AM, Widen JC, Hexum JK, Andrews TE, Wang D, Rathe SK, Meece FA, Noble KE, Sachs Z, Largaespada DA, Harki DA. Synthesis and Antileukemic Activiti es of C1 – C10-Modified Parthenolide Analogues. Biorg. Med. Chem. 2015;23:4737–4745. doi: 10.1016/j.bmc.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Simonsen H, Weitzel C, Christensen S. Guaianolide Sesquiterpenoids: Pharmacology and Biosynthesis. In: Ramawat KG, Mérillon J-M, editors. Natural Products. Spri nger Berlin Heidelberg; 2013. pp. 3069–3098. [Google Scholar]
- 87.a Canova S, Lépine R, Thys A, Baron A, Roche D. Synthesis and Biological Properties of Macrolactam Analogs of the Natural Product Macrolide (-)-A26771B. Bioorg. Med. Chem. Lett. 2011;21:4768–4772. doi: 10.1016/j.bmcl.2011.06.073. [DOI] [PubMed] [Google Scholar]; b Janecki T, Blaszczyk E, Studzian K, Janecka A, Krajewska U, Rozalski M. Novel Synthesis, Cytotoxic Evaluation, and Structure-Activity Relationship Studies of a Series of alpha-Alkylidene-gamma-Lactones and Lactams. J. Med. Chem. 2005;48:3516–3521. doi: 10.1021/jm048970a. [DOI] [PubMed] [Google Scholar]
- 88.a Kunzmann MH, Staub I, Bottcher T, Sieber SA. Protein Reactivity of Natural Product-Derived gamma-Butyrolactones. Biochem. 2011;50:910–916. doi: 10.1021/bi101858g. [DOI] [PubMed] [Google Scholar]; b Kunzmann MH, Bach NC, Bauer B, Sieber SA. alpha-Methylene-gamma-Buty rolactones Attenuate Staphylococcus Aureus Virulence by Inhibition of Transcriptional Regulation. Chem. Sci. 2014;5:1158–1167. [Google Scholar]; c Pei SS, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye HB, Liesveld JL, O'Dwyer KM, Li Z, Shi L, Greninger P, Settleman J, Benes C, Hagen FK, Munger J, Crooks PA, Becker MW, Jordan CT. Targeting Aberrant Glutathione Metabolism to Eradicate Human Acute Myelogenous Leukemia Cells. J. Biol. Chem. 2013;288:33542–33558. doi: 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.a Kwok BH, Koh B, Ndubuisi MI, Elofsson M, Crews CM. The Anti-Inflammatory Natural Product Parthenolide from the Medicinal Herb Feverfew Directly Binds to and Inhibits IkappaB Kinase. Chem Biol. 2001;8:759–766. doi: 10.1016/s1074-5521(01)00049-7. [DOI] [PubMed] [Google Scholar]; b Wells SM, Widen JC, Harki DA, Brummond KM. Alkyne Ligation Handles: Propargylation of Hydroxyl, Sulfhydryl, Amino, and Carboxyl Groups via the Nicholas Reaction. Org. Lett. 2016;18:4566–4569. doi: 10.1021/acs.orglett.6b02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nasim S, Pei SS, Hagen FK, Jordan CT, Crooks PA. Melampomagnolide B: A New Antileukemic Sesquiterpene. Biorg. Med. Chem. 2011;19:1515–1519. doi: 10.1016/j.bmc.2010.12.045. [DOI] [PubMed] [Google Scholar]
- 91.a Kunzmann MH, Sieber SA. Target Analysis of α-Alkylidene-β-Butyrolactones in Uropathogenic E. Coli. Mol. BioSyst. 2012;8:3061–3067. doi: 10.1039/c2mb25313e. [DOI] [PubMed] [Google Scholar]; b Förster S, Helmchen G. Stereoselective Synthesis of a Lactam Analogue of Brefeldin C. SYNLETT. 2008;6:831–836. [Google Scholar]
- 92.Dinkova-Kostova AT, Massiah MA, Boz ak RE, Hicks RJ, Talalay P. Potency of Michael Reaction Acceptors as Inducers of Enzymes that Protect Against Carcinogenesis Depends on their Reactivity with Sulfhydryl Groups. PNAS. 2001;98:3404–3409. doi: 10.1073/pnas.051632198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dimmock JR, Smith LM. The Reactio n of Some Nuclear Substituted Acyclic Conjugated Styryl Ketones and Related Mannich Bases with Ethanethiol. Can. J. Chem. 1980;58:984–991. [Google Scholar]
- 94.Mutus B, Wagner JD, Talpas CJ, Dimmock JR, Philips OA, Reid RS. 1-p-Chlorophenyl-4,4-dimethyl-5-dimethylamino-1-penten-3-one Hydrobromide, a Sulfhydryl-Specific Compound Which Reacts Irreversibly with Protein Thiols but Reversibly with Small Molecular Weight Thiols. Anal. Biochem. 1989;177:237–243. doi: 10.1016/0003-2697(89)90045-6. [DOI] [PubMed] [Google Scholar]
- 95.Al-Rifai N, Rucker H, Amslinger S. Opening or Closing the Lock? When Reactivity is the Key to Biological Activity. Chem. Eur. J. 2013;19:15384–15395. doi: 10.1002/chem.201302117. [DOI] [PubMed] [Google Scholar]
- 96.Amslinger S, Al-Rifai N, Winter K, Wormann K, Scholz R, Baumeister P, Wild M. Reactivity Assessment of Chalcones by a Kine tic Thiol Assay. Org. Biomol. Chem. 2013;11:549–554. doi: 10.1039/c2ob27163j. [DOI] [PubMed] [Google Scholar]
- 97.Avonto C, Taglialatela-Scafati O, Pollastro F, Minassi A, Marzo VD, Petrocellis LD, Appendino G. An NMR Spectroscopic Method to Identify and Classify Thiol-Trapping Agents: Revival of Michael Acceptors for Drug Discovery. Angew. Chem. Int. Ed. 2011;50:467–471. doi: 10.1002/anie.201005959. [DOI] [PubMed] [Google Scholar]
- 98.Awasthi S, Pandya U, Singhal SS, Lin JT, Thiviyanathan V, Seifert WE, Awasthi YC, Ansari GAS. Curcumin-Glutathione Interactions and the Role o f Human Glutathione S-Transferase P1-1. Chem. Biol. Interact. 2000;128:19–38. doi: 10.1016/s0009-2797(00)00185-x. [DOI] [PubMed] [Google Scholar]
- 99.Usta M, Wortelboer HM, Vervoort J, Boersma MG, Rietjens IMCM, Bladeren P. J. v., Cnubben NHP. Human Glutathione S-Transferase-Mediated Glutathione Conjugation of Curcumin and Efflux of These Conjugates in Caco-2 Cells. Chem. Res. Toxicol. 2007;20:1895–1902. doi: 10.1021/tx7002245. [DOI] [PubMed] [Google Scholar]
- 100.Sun A, Lu YJ, Hu H, Shoji M, Liotta DC, Snyder JP. Curcumin Analog Cytotoxicity Against Breast Cancer Cells: Exploitatio n of a Redox-dependent Mechanism. Bioorg. Med. Chem. Lett. 2009;19:6627–6631. doi: 10.1016/j.bmcl.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.a Brase S, Glaser F, Kramer CS, Lindner S, Linsenmeier AM, Masters K-S, Meister AC, Ruff BM, Zhong S. The Chemistry of Mycotoxins. 1 ed. Vol. 97. Sprin ger-Verlag Wein; Heidelberg, New York, Dordrecht, London: 2013. p. 300. [PubMed] [Google Scholar]; b Winssinger N, Barluenga S. Chemistry and Biology of Resorcylic Acid Lactones. Chem. Commun. 2007:22–36. doi: 10.1039/b610344h. [DOI] [PubMed] [Google Scholar]; c Ayers S, Graf TN, Adcock AF, Kroll DJ, Matthew S, de Blanco EJC, Shen Q, Swanson SM, Wani MC, Pearce CJ, Oberlies NH. Resorcylic Acid Lactones with Cytotoxic and NF-kappa B Inhibitory Activities and Their Structure-Activity Relationships. J. Nat. Prod. 2011;74:1126–1131. doi: 10.1021/np200062x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wei L, Wu J, Li G, Shi N. Cis-Enone Resorcylic Acid Lactones (RALs) as Irreversible Protein Kinase Inhibitors. Curr. Pharm. Des. 2012;18:1186–1198. doi: 10.2174/138161212799436395. [DOI] [PubMed] [Google Scholar]
- 103.Nishino M, Choy JW, Gushwa NN, Oses-Prieto JA, Koupparis K, Burlingame AL, Renslo AR, McKerrow JH, Taunton J. Hypothemycin, a Fungal Natural Product, Identifies Therapeutic Targets in Trypanosoma Brucei. eLife. 2013;2:1–15. doi: 10.7554/eLife.00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jogireddy R, Barluenga S, Winssinger N. Molecular Editing of Kinase-Targeting Resorc ylic Acid Lactones (RAL): Fluoroenone RAL. ChemMedChem. 2010;5:670–673. doi: 10.1002/cmdc.201000047. [DOI] [PubMed] [Google Scholar]
- 105.Schirmer A, Kennedy J, Murli S, Reid R, Santi DV. Targeted Covalent Inactivation of Protein Kinases by Resorcylic Acid Lactone Polyketides. PNAS. 2006;103:4234–4239. doi: 10.1073/pnas.0600445103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xu J, Chen A, Joy J, Xavier VJ, Ong EHQ, Hill J, Chai CLL. Rational Design of Resorcylic Acid Lactone Analogues as Covalent MNK1/2 Kinase Inhibitors by Tuning the Reactivity of an Enamide Michael Acceptor. ChemMedChem. 2013;8:1483–1494. doi: 10.1002/cmdc.201300231. [DOI] [PubMed] [Google Scholar]
- 107.Napolitano C, Palwai VR, Eriksson LA, Murphy PV. Synthesis, Kinase Activity and Molecular Modeling of a Resorcylic Acid Lactone Incorporating an Amide and a trans-Enone in the Macrocycle. Tetrahedron. 2012;68:5533–5540. [Google Scholar]
- 108.Xu J, Ong EHQ, Hill J, Chen A, Chai CLL. Design, Synthesis and Biological Evaluation of FLT3 Covalent Inhibitors with a Resorcylic Acid Core. Bioorg. Med. Chem. 2014;22:6625–6637. doi: 10.1016/j.bmc.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 109.a Garzón B, Oeste CL, Díez-Dacal B, Pérez-Sala D. Proteomic Studies on Protein Modification by Cyclopentenone Prostaglandins: Expanding our View on Electrophile Actions. J. Proteomics. 2011;74:2243–2263. doi: 10.1016/j.jprot.2011.03.028. [DOI] [PubMed] [Google Scholar]; b Díez-Dacal B, Pérez-Sala D. A-Class Prostaglandins: Early Findings and New Perspectives for Overcoming Tumor Chemoresistance. Cancer Lett. 2012;320:150–157. doi: 10.1016/j.canlet.2012.03.003. [DOI] [PubMed] [Google Scholar]; c Straus DS, Glass CK. Cyclopentenone Prostaglandins: New Insights on Biological Activities and Cellular Targets. Med. Res. Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- 110.Grudzinskas CV, We iss MJ. Prostaglandins and Congeners IV. The Synthesis of Certain 11-Substituted Derivatives of 11-Deoxyprostaglandin E2 and F2α from 15-O-Acetylprostaglandin A2 Methyl Ester. Tet. Lett. 1973;14:141–144. [Google Scholar]
- 111.a Cagen LM, Fales HM, Pisano JJ. Formation of Glutathione Conjugates of Prostaglandin A1 in Human Red Blood Cells. J. Biol. Chem. 1976;251:6550–6554. [PubMed] [Google Scholar]; b Ham EA, Oien HG, Ulm EH, Kuehl FA. The Reaction of PGA with Sulfhydryl Groups: A Component in the Binding of A-Type Prostaglandins to Proteins. Prostaglandins. 1975;10:217–229. doi: 10.1016/0090-6980(75)90041-6. [DOI] [PubMed] [Google Scholar]
- 112.Atsmon J, Sweetman BJ, Baertschi SW, Harris TM, Roberts LJ. Formation of Thiol Conjugates of 9-deoxy-Δ9,Δ12-Prostaglandin D2 and Δ12-Prostaglandin D2. Biochem. 1990;29:3760–3765. doi: 10.1021/bi00467a023. [DOI] [PubMed] [Google Scholar]
- 113.Atsmon J, Freeman ML, Meredith MJ, Sweetman BJ, Roberts LJ. Conjugation of 9-Deoxy-Δ9,Δ12-prostaglandin D2 with Intracellular Glutathione and Enhancement of Its Antiproliferative Activity by Glutathione Depletion. Cancer Res. 1990;50:1879–1885. [PubMed] [Google Scholar]
- 114.Suzuki M, Mori M, Niwa T, Hirata R, Furuta K, Ishikawa T, Noyori R. Chemical Implications for Antitumor and Antiviral Prostaglandins: Reaction of D7-Prostaglandin A1 and Prostaglandin A1 Methyl Esters with Thiols. J. Am. Chem. Soc. 1997;119:2376–2385. [Google Scholar]
- 115.Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, Eguchi M, Wada Y, Kumag ai Y, Yamamoto M. The Antioxidant Defense System Keap1-Nrf2 Comprises a Multiple Sensing Mechanism for Responding to a Wide Range of Chemical Compounds. Mol. Cell. Biol. 2009;29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gayarre J, Stamatakis K, Renedo M, Pérez-Sala D. Dif ferential Selectivity of Protein Modification by the Cyclopentenone Prostaglandins PGA1 and 15-Deoxy-Δ12,14-PGJ2: Role of Glutathione. FEBS Lett. 2005;579:5803–5808. doi: 10.1016/j.febslet.2005.09.069. [DOI] [PubMed] [Google Scholar]
- 117.Sánchez-Gómez FJ, Gayarre J, Avellano MI, Pérez-Sala D. Direct Evidence for the Covalent Modification of Glutathione-S-Transferase P1-1 by Electrophilic Prostaglandins: Impl ications for Enzyme Inactivation and Cell Survival. Biochem. Biophys. 2006;457:150–159. doi: 10.1016/j.abb.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 118.Sánchez-Gómez FJ, Díez-Dacal B, Pajares MA, Llorca O, Pérez-Sala D. Cyclopentenone Prostaglandins with Dienone Structure Promote Cross-Linking of th e Chemoresistance-Inducing Enzyme Glutathione Transferase P1-1. Mol. Pharmacol. 2010;78:723–733. doi: 10.1124/mol.110.065391. [DOI] [PubMed] [Google Scholar]
- 119.Oeste CL, Díez-Dacal B, Bray F, Lacoba M. G. d., Torre B. G. d. l., Andreu D, Ruiz-Sánchez AJ, Pérez-Inestrosa E, García-Dominguez CA, Rojas JM, Pérez-Sala D. The C-Terminus of H-Ras as a Target for the Covalent Binding of Reactive Compounds Modulating Ras-Dependent Pathways. PLoS ONE. 2011;6:e15866. doi: 10.1371/journal.pone.0015866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tanaka H, Kitade M, Iwashima M, Iguchi K, Takahashi T. Effective Irreversible Alkylating Reagents Based on the Structure of Clavulones. Bioorg. Med. Chem. Lett. 2004;14:837–840. doi: 10.1016/j.bmcl.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 121.Bickley J, Ciucci A, Evans P, Roberts SM, Ross N, Santoro MG. Reactions of some Cyclopentenones with Selected Cysteine D erivatives and Biological Activities of the Product Thioethers. Biorg. Med. Chem. 2004;12:3221–3227. doi: 10.1016/j.bmc.2004.03.061. [DOI] [PubMed] [Google Scholar]
- 122.Danishefsky S, Kahn M. Regiospecific Michael Reactions to an Enedione. Tet. Lett. 1981;22:485–488. [Google Scholar]
- 123.Siwapinyoyos T, Thebtaranonth Y. Reg iospecific Michael Additions to α-Methylene Cyclopentenones. Tet. Lett. 1984;25:353–356. [Google Scholar]
- 124.Castelli V. v. A., Bernardi F, Cort AD, Mandolini L, Rossi I, Schiaffino L. Rates and Equilibria of the Michael-Type Addition of Benzenethiol to 2-Cyclopenten-1-ones. J. Org. Chem. 1999;64:8122–8126. doi: 10.1021/jo9906882. [DOI] [PubMed] [Google Scholar]
- 125.Kakiuchi K, Ue M, Takeda M, Tadaki T, Kato Y, Nagashima T, Tobe Y, Koike H, Ida N, Odaira Y. Antiproliferating Polyquinanes. V. Di-and Triquinanes Involving α-Methylene or α-Alkylidene Cyclopentanone, Cyclopen tenone, and γ-Lactone Systems. Chem. Pharm. Bull. 1987;35:617–631. doi: 10.1248/cpb.35.617. [DOI] [PubMed] [Google Scholar]
- 126.a Sun H-D, Huang S-X, Han Q-B. Diterpenoids from Isodon Species and their Biological Activities. Nat. Prod. Rep. 2006;23:673–698. doi: 10.1039/b604174d. [DOI] [PubMed] [Google Scholar]; b Wang L, Li D, Wang C, Zhang Y, Xu J. Recent Progress in the Development of Natural ent-Kaurane Diterpenoids with Anti-tumor Activity. Mini. Rev. Med. Chem. 2011;11:910–919. doi: 10.2174/138955711796575416. [DOI] [PubMed] [Google Scholar]; c Fujita E, Node M. Progress in the Chemistry of Organic Natural Products. Vol. 46. Springer-Verlag; Vienna: 1984. [DOI] [PubMed] [Google Scholar]
- 127.a Lee J-H, Koo TH, Hwang BY, Lee JJ. Kaurane Diterpene, Kamebakaurin, Inhibits NF-κB by Directly Targeting the DNA-binding Activity of p50 and Blocks the Expression of Antiapoptotic NF-κB Target Genes. J. Biol. Chem. 2002;277:18411–18420. doi: 10.1074/jbc.M201368200. [DOI] [PubMed] [Google Scholar]; b Leung C-H, Grill SP, Lam W, Gao W, Sun H-D, Cheng Y-C. Eriocalyxin B Inhibits Nuclear Factor-κB Activation by Interfering with the Binding of Both p65 and p50 to the Response Element in a Noncompetitive Manner. Mol. Pharmacol. 2006;70:1946–1955. doi: 10.1124/mol.106.028480. [DOI] [PubMed] [Google Scholar]
- 128.a Liao Y-J, Bai H-Y, Li Z-H, Zou J, Chen J-W, Zheng F, Zhang J-X, Mai S-J, Zeng M-S, Sun H-D, Pu J-X, Xie D. Longikaurin A, a Natural ent-Kaurane, Induces G2/M Phase Arrest via Downregulation of Skp2 and Apoptosis Induction Through ROS/JNK/c-Jun Pathway in Hepatocellular Carcinoma Cells. Cell Death Di s. 2014;5:e1137. doi: 10.1038/cddis.2014.66. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Piaz FD, Cotugno R, Lepore L, Vassallo A, Malafronte N, Lauro G, Bifulco G, Belisario MA, Tommasi ND. Chemical Proteomics Reveals HSP70 1A as a Target for the Anticancer Diterpene Oridonin in Jurkat Cells. J. P roteomics. 2013;82:14–26. doi: 10.1016/j.jprot.2013.01.030. [DOI] [PubMed] [Google Scholar]
- 129.Kubo I, Taniguchi M, Satomura Y, Kubota T. Antibacterial Activity and Chemical Structure of Diterpenoids. Agric. Biol. Chem. 1974;38:1261–1262. [Google Scholar]
- 130.Fujita E, Nagao Y, Keneko K, Nakazawa S, Kuroda H. The An titumor and Antibacterial Activity of the Isodon Diterpenoids. Chem. Pharm. Bull. 1976;24:2118–2127. doi: 10.1248/cpb.24.2118. [DOI] [PubMed] [Google Scholar]
- 131.a Murayama C, Nagao Y, Sano S, Ochiai M, Fuji K, Fujita E, Mori T. Effect of Oridonin, a Rabdosia Diterpenoid, on Radiosensitization with Misonidazole. Experientia. 1987;43:1221–1223. doi: 10.1007/BF01945533. [DOI] [PubMed] [Google Scholar]; b Kuo L-M, Kuo C-Y, Hung M-F, Shen J-J, Hwang T-L. Intracellular Glutathione Depletion by Oridonin Leads to Apoptosis in Hepatic Stellate Cells. Molecules. 2014;19:3327–3344. doi: 10.3390/molecules19033327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Piaz FD, Nigro P, Braca A, Tommasi ND, Belisario MA. 13-Hydroxy-15-oxo-zoapatlin, an ent-Kaurane Diterpene, Induces Apoptosis in Human Leukemia Cells, Affecting Thiol-mediated Redox Regulation. Free Radical Biol. Med. 2007;43:1409–1422. doi: 10.1016/j.freeradbiomed.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 133.Hexu m JK, Tello-Aburto R, Struntz NB, Harned AM, Harki D. Bicyclic Cyclohexenones as Inhibitors of NF-κB Signaling. ACS Med. Chem. Lett. 2012;3:459–464. doi: 10.1021/ml300034a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.a Magnus P, Lewis RT. Synthetic Studies on the Esperamicin/Calicheamicin Antit umor Antibiotics. Conjugate Addition of Thiol to Initiate 1,4-Diyl Formation. Tet. Lett. 1989;30:1905–1906. [Google Scholar]; b Myers AG, Cohen SB, Kwon BM. A Study of the Reaction of Calicheamicin γ1 with Glutathione in the Presence of Double-Stranded DNA. J. Am. Chem. Soc. 1994;116:1255–1271. [Google Scholar]
- 135.a Kumar KJS, Yang H-L, Tsai Y-C, Hung P-C, Chang S-H, Lo H-W, Shen P-C, Chen S-C, Wang H-M, Wang S-Y, Chou C-W, Hseu Y-C. Lucidone Protects Human Skin Keratinocytes Against Free Radical-induced Oxidative Damage and Inflammation Through the Up-regulation of HO-1/Nrf2 Antioxidant Genes and Down-regulation of NF-κB Signaling Pathway. Food Chem. Toxicol. 2013;59:55–66. doi: 10.1016/j.fct.2013.04.055. [DOI] [PubMed] [Google Scholar]; b Chen W-C, Wang S-Y, Chiu C-C, Tseng C-K, Lin C-K, Wang H-C, Lee J-C. Lucidone Suppresses Hepatitis C Virus Replication by Nrf2-Mediated Heme Oxygenase-1 Induction. Antimicro b. Agents Chemother. 2013;57:1180–1191. doi: 10.1128/AAC.02053-12. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kumar KJS, Liao J-W, Xiao J-H, Vani MG, Wang S-Y. Hepatoprotective Effect of Lucidone Against Alcohol-induced Oxidative Stress in Human Hepatic HepG2 Cells Through the Up-regulation of HO-1/Nrf-2 Antioxidant Genes. Toxicol. In Vitro. 2012;26:700–708. doi: 10.1016/j.tiv.2012.03.012. [DOI] [PubMed] [Google Scholar]; d Kumar KJS, Hsieh HW, Wang S-Y. Anti-inflammatory Effect of Lucidone in Mice via Inhibition of NF-κB/MAP Kinase Pathway. Int. Immunopharmacol. 2010;10:385–392. doi: 10.1016/j.intimp.2009.12.013. [DOI] [PubMed] [Google Scholar]; e Kumar KJS, Wang S-Y. Lucidone Inhibits iNOS and COX-2 Expression in LPS-Induced RAW 264.7 Murine Macrophage Cells via NF-κB and MAPKs Signaling Pathways. Planta Med. 2009;75:494–500. doi: 10.1055/s-0029-1185309. [DOI] [PubMed] [Google Scholar]
- 136.a Aoyama Y, Konoike T, Kanda A, Naya N, Nakajima M. Total Synthesis of Human Chymase Inhibitor Methyllinderone and Structure-Activity Relationships of its Derivatives. Bioorg. Med. Chem. Lett. 2001;11:1695–1697. doi: 10.1016/s0960-894x(01)00265-7. [DOI] [PubMed] [Google Scholar]; b Paes-Gonçalves H, Facundo VA, Santos DMF, Silva AGC, Ballico LJ, Lima DKS, Stábeli RG, Silva-Jardim I. The Leishmanicidal Activity of a Cyclopentenedione Derivative Isolated from the Roots of a Native Amazonian Pepper (Piper Carniconnectivu m). Braz. J. Pharmacog. 2012;22:1018–1023. [Google Scholar]; c Oh H-M, Choi S-K, Lee JM, Lee S-K, Kim H-Y, Han DC, Kim H-M, Son K-H, Kwon B-M. Cyclopentenediones, Inhibitors of Farnesyl Protein Transferase and Anti-tumor Compounds Isolated fr om the Fruit of Lindera Erythrocarpa Makino. Biorg. Med. Chem. 2005;13:6182–6187. doi: 10.1016/j.bmc.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 137.Capkova K, Hixon MS, Pellett S, Barbieri JT, Johnson EA, Janda KD. Benzylidene Cyclopentenediones: First Irreversible Inhibitors Against Botulinum Neurotoxin A's Zinc Endopeptidase. Bioorg. Med. Chem. Lett. 2010;20:206–208. doi: 10.1016/j.bmcl.2009.10.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Qian J, Klomsiri C, Wright MW, King SB, Tsang AW, Poole LB, Furdui CM. Simple Synthesis of 1,3-Cyclopentanedione Derived Probes for Labeling Sulfenic Acid Proteins. Chem. Commun. 2011;47:9203–9205. doi: 10.1039/c1cc12127h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Healy J, Ekkerman S, Pliotas C, Richard M, Bartlett W, Grayer SC, Morris GM, Miller S, Booth IR, Conway SJ, Rasmussen T. Understanding the Structural Requirements for Activators of the Kef Bacterial Potassium Efflux System. Biochem. 2014;53:1982–1992. doi: 10.1021/bi5001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Billington S, Mann J, Quazi P, Alexander R, Eaton MAW, Miller K, Millican A. The Synthesis of Novel Bifunctional Linker Molecules. Tetrahedron. 1991;47:5231–5236. [Google Scholar]
- 141.a LoPachin RM, Gavin T. Molecular Mechanisms of Aldehyde Toxicity: A Chemical Perspective. Chem. Res. Toxicol. 2014;27:1081–1091. doi: 10.1021/tx5001046. [DOI] [PMC free article] [PubMed] [Google Scholar]; b O'Brien PJ, Siraki AG, Shangari N. Aldehyde Sources, Metabolism, Molecular Toxicity Mec hanisms, and Possible Effects on Human Health. Crit. Rev. Toxicol. 2005;35:609–662. doi: 10.1080/10408440591002183. [DOI] [PubMed] [Google Scholar]; c Zhu Q, Sun Z, Jiang Y, Chen F, Wang M. Acrolein Scavengers: Reactivity, Mechanism and Impact on Health. Mol. Nutr. Food Res. 2011;55:1375–1390. doi: 10.1002/mnfr.201100149. [DOI] [PubMed] [Google Scholar]; d Witz G. Biological Interactions of α,β-Unsaturated Aldehydes. Free Radical Biol. Med. 1989;7:333–349. doi: 10.1016/0891-5849(89)90137-8. [DOI] [PubMed] [Google Scholar]
- 142.Esterbauer H, Zollner H, Scholz N. Reaction of Glutathione with Conjugated Carbonyls. Z. Naturforsch., C: Biosci. 1975;30c:466–473. doi: 10.1515/znc-1975-7-808. [DOI] [PubMed] [Google Scholar]
- 143.Esterbauer H, Ertl A, Scholz N. The Reaction of Cysteine With α,β-Unsaturated Aldehydes. Tetrahedron. 1976;32:285–289. [Google Scholar]
- 144.a Cai J, Bhatnagar A, Pierce WM. Protein Modification by Acrolein: Formation and Stability of Cysteine Adducts. Chem. Res. Toxicol. 2009;22:708–716. doi: 10.1021/tx800465m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Randall MJ, Hristova M, Vliet A. v. d. Protein Alkylation by the α,β-Unsaturated Aldehyde Acrolein. A Reversible Mechanism of Electrophile Signaling? FEBS Lett. 2013;587:3804–3814. doi: 10.1016/j.febslet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chan K, Poon R, O'Brien PJ. Application of Structure-a ctivity Relationships to Investigate the Molecular Mechanisms of Hepatocyte Toxicity and Electrophilic Reactivity of α,β-Unsaturated Aldehydes. J. Appl. Toxicol. 2008;28:1027–1039. doi: 10.1002/jat.1369. [DOI] [PubMed] [Google Scholar]
- 146.Vidal N, Cavaille JP, Graziani F, Robin M, Ouari O, Piet ri S, Stocker P. High Throughput Assay for Evaluation of Reactive Carbonyl Scavenging Capacity. Redox Biol. 2014;2:590–598. doi: 10.1016/j.redox.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pritchard RB, Lough CE, Currie DJ, Holmes HL. Equilibrium Reactions of n-Butanethiol with some Conjugated H eteroenoid Compounds. Can. J. Chem. 1968;46:775–781. [Google Scholar]
- 148.Krishnan S, Miller RM, Tian B, Mullins RD, Jacobson MP, Taunton J. Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. C hem. Soc. 2014;136:12624–12630. doi: 10.1021/ja505194w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.a Liby K, Yore MM, Roebuck BD, Baumgartner KJ, Honda T, Sundararajan C, Yoshizawa H, Gribble GW, Williams CR, Risingsong R, Royce DB, Dinkova-Kostova AT, Stephenson KK, Egner PA, Yates MS, Groopman JD, Kensler TW, Sporn MB. A Novel Acetylenic Tricyclic bis-(Cyano Enone) Potently Induces Phase 2 Cytoprotective Pathways and Blocks Liver Carcinogenesis Induced by Aflatoxin. Cancer Res. 2008;68:6727–6733. doi: 10.1158/0008-5472.CAN-08-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dinkova-Kostova AT, Talalay P, Sharkey J, Zhang Y, Holtzclaw WD, Wang XJ, David E, Schiavoni KH, Finlayson S, Mierke DF, Honda T. An Exceptionally Potent Inducer of Cytoprotective Enzymes: Elucidation of the Structural Featu res That Determine Inducer Potency and Reactivity with Keap1. J. Biol. Chem. 2010;285:33747–33755. doi: 10.1074/jbc.M110.163485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zheng S, Laxmi YRS, David E, Dinkova-Kostova AT, Shiavoni KH, Ren Y, Zheng Y, Trevino I, Bumeister R, Ojima I, Wigley WC, Bliska JB, Mierke DF, Honda T. Synthesis, Chemical Reactivity as Michael Acceptors, and Biological Potency of Monocyclic Cyanoenones, Novel and Highly Potent Anti-inflammatory and Cytoprotective Agents. J. Med. Chem. 2012;55:4837–4846. doi: 10.1021/jm3003922. [DOI] [PubMed] [Google Scholar]
- 151.Segura-Aguilar J, Paris I, Muñoz P, Ferrari E, Zecca L, Zucca FA. Protective and Toxic Roles of Dopamine in Parkinson's Disease. J. Neurochem. 2014;129:898–915. doi: 10.1111/jnc.12686. [DOI] [PubMed] [Google Scholar]
- 152.Baur JA, Sinclair DA. Therapeutic Potential of Resveratrol: The In Vivo Evidence. Nat. Rev. Drug Disc. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 153.a Ito S, Kato T, Fujita K. Covalent Binding of Catechols to Proteins through the Sulphydryl-Group. Biochem. Pharmacol. 1988;37:1707–1710. doi: 10.1016/0006-2952(88)90432-7. [DOI] [PubMed] [Google Scholar]; b Cavalieri E, Rogan E, Chakravarti D. Methods Enzymol. Vol. 382. Academic Press; San Diego: 2004. The Role of Endogenous Catechol Quinones in the Initiation of Cancer and Neurodegenerative Diseases. pp. 293–319. [DOI] [PubMed] [Google Scholar]
- 154.Mbiya W, Chipinda I, Siegel PD, Mhike M, Simoyi RH. Substituent Ef fects on the Reactivity of Benzoquinone Derivatives with Thiols. Chem. Res. Toxicol. 2013;26:112–123. doi: 10.1021/tx300417z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tanasova M, Sturla SJ. Chemistry and Biology of Acylfulvenes: Sesquiterpene-Derived Antitumor Agents. Chem. Rev. 2012;112:3578–3610. doi: 10.1021/cr2001367. [DOI] [PubMed] [Google Scholar]
- 156.a Liu X, Sturla SJ. Profiling Patterns of Glutathione Reductase Inhibition by the Natural Product Illudin S and its Acylfulvene Analogues. Mol. BioSyst. 2009;5:1013–1024. doi: 10.1039/b904720d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu X, Pietsch KE, Sturla SJ. Susceptibility of the Antioxidant Selenoenzymes Thioredoxin Reductase and Glutathione Peroxidase to Alkylation-Mediated Inhibition by Anticancer Acylfulvenes. Chem. Res. Toxicol. 2011;24:726–736. doi: 10.1021/tx2000152. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gong J, Vaidyanathan VG, Yu X, Kensler TW, Peterson LA, Sturla SJ. Dep urinating Acylfulvene-DNA Adducts: Characterizing Cellular Chemical Reactions of a Selective Antitumor Agent. J. Am. Chem. Soc. 2007;129:2101–2111. doi: 10.1021/ja0665951. [DOI] [PubMed] [Google Scholar]
- 157.Fang J, Lu J, Holmgren A. Thioredoxin Reductase is Irreversibly Modified by Curcumin: A Novel Mo lecular Mechanism for its Anticancer Activity. J. Biol. Chem. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- 158.a McMorris TC, Kelner MJ, Wang W, Moon S, Taetle R. On the Mechanism of Toxicity of Illudins: The Role of Glutathione. Chem. Res. Toxicol. 1990;3:574–579. doi: 10.1021/tx00018a013. [DOI] [PubMed] [Google Scholar]; b Tanaka K, Inoue T, Tezuka Y, Kikuchi T. Michael-Type Addition of Illudin S, a Toxic Substance from Lampteromyces Japonicus, with Cysteine and Cysteine-Containing Peptides In Vitro. Chem. Pharm. Bull. 1996;44:273–279. doi: 10.1248/cpb.44.273. [DOI] [PubMed] [Google Scholar]; c McMorris TC, Kelner MJ, Wang W, Estes LA, Montoya MA, Taetle R. Structure-Activity Relationships of Illudins: Analogs with Improved Therapeutic Index. J. Org. Chem. 1992;57:6876–6883. [Google Scholar]; d McMorris TC, Kelner MJ, Wang W, Diaz MA, Estes LA, Taetle R. Acylfulvenes, a New Class of Potent Antitumor Agents. Experientia. 1996;52:75–80. doi: 10.1007/BF01922420. [DOI] [PubMed] [Google Scholar]; e McMorris TC, Yu J, Estes LA, Kelner MJ. Reaction of Antitumor Hydroxymethylacylfulvene (HMAF) with Thiols. Tetrahedron. 1997;53:14579–14590. [Google Scholar]
- 159.Dick RA, Yu X, Kensler TW. NADPH Alkenal/One Oxidoreductase Activity Determines Sensitivity of Cancer Cells to the Chemotherapeutic Alkylating Agent Irofulven. Clincal Cancer Research. 2004;10:1492–1499. doi: 10.1158/1078-0432.ccr-03-0162. [DOI] [PubMed] [Google Scholar]
- 160.a Shen Y-C, Cheng Y-B, Lin Y-C, Guh J-H, Teng C-M, Ko C-L. New Prostanoids with Cytotoxic Activity from Taiwanese Octocoral Clavularia Viridis. J. Nat. Prod. 2004;67:542–546. doi: 10.1021/np030435a. [DOI] [PubMed] [Google Scholar]; b Perry NB, Blunt JW, Munro MHG. Cytotoxic Pigments from New Zealand Spon ges of the Genus Latrunculia : Discorhabdins A, B and C. Tetrahedron. 1988;44:1727–1734. [Google Scholar]; c Teeyapant R, Woerdenbag HJ, Kreis P, Hacker J, Wray V, Witte L, Proksch P. Antibiotic and Cytotoxic Activity of Brominated Compounds from the Marine Sponge. Verongia Aerophoba Z. Naturforsch., C: Biosci. 1993;48:939–945. doi: 10.1515/znc-1993-11-1218. [DOI] [PubMed] [Google Scholar]; d Geroni C, Marchini S, Cozzi P, Galliera E, Ragg E, Colombo T, Battaglia R, Howard M, D'Incalci M, Broggini M. Brostallicin, a Novel Anticancer Agent Whose Act ivity is Enhanced upon Binding to Glutathione. Cancer Res. 2002;62:2332–2336. [PubMed] [Google Scholar]; e Verbitski SM, Mullally JE, Fitzpatrick FA, Ireland CM. Punaglandins, Chlorinated Prostaglandins, Function as Potent Michael Receptors to Inhibit Ubiquitin Is opeptidase Activity. J. Med. Chem. 2004;47:2062–2070. doi: 10.1021/jm030448l. [DOI] [PubMed] [Google Scholar]
- 161.Fekry MI, Price NE, Zang H, Huang C, Harmata M, Brown P, Daniels JS, Gates KS. Thiol-Activated DNA Damage by α-Bromo-2-cyclopentenone. Chem. Res. Toxicol. 2011;24:217–228. doi: 10.1021/tx100282b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Farina F, Maestro MC, Martin MR, Martin MV, Sánchez F, Soria ML. Pseudoesters and Derivatives. XXIII. Reaction of 3-Bromo-5-methoxyfuran-2(5H)-one with Nucleophiles. F ormation of Cyclopropane Derivatives. Tetrahedron. 1986;42:3715–3722. [Google Scholar]
- 163.Tomasic T, Masic LP. Rhodanine as a Privileged Scaffold in Drug Discovery. Curr. Med. Chem. 2009;16:1596–1629. doi: 10.2174/092986709788186200. [DOI] [PubMed] [Google Scholar]
- 164.a Baell J, Walters MA. Chemical Con Artists Foil D rug Discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]; b Baell J. Observations on Sreening-based Research and Some Concerning Trends in the Literature. Future Med. Chem. 2010;2:1529–1546. doi: 10.4155/fmc.10.237. [DOI] [PubMed] [Google Scholar]; c Baell JB, Holloway GA. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 165.Tomasic T, Masic LP. Rhodanine as a Scaffold in Drug Discovery: A Critical Review of its Biological Activities and Mechanisms of Target Modulation. Expert Opin. Drug Discov. 2012;7:549–560. doi: 10.1517/17460441.2012.688743. [DOI] [PubMed] [Google Scholar]
- 166.Mendgen T, Seuer C, Klein CD. Privileged Scaffolds or Promiscuous Binders: A Comparative Study on Rhodanines and Related Heterocycles in Medicinal Chemistry. J. M ed. Chem. 2012;55:743–753. doi: 10.1021/jm201243p. [DOI] [PubMed] [Google Scholar]
- 167.a Ramirez MA, Borja NL. Epalrestat: An Aldose Reductase Inhibitor of the Treatment of Diabetic Neuropathy. Pharmacotherapy. 2008;28:646–655. doi: 10.1592/phco.28.5.646. [DOI] [PubMed] [Google Scholar]; b Hotta N, Akanuma Y, Kawamori R, Matsuoka K, Oka Y, Shiciri M, Toyota T, Nakashima M, Yoshimura I, Sakamoto N, Shigeta Y. Long-term Clinical Effects of Epalrestat, an Aldose Reductase Inhibitor, on Diabetic Peripheral Neuropathy: The 3-year, Multicenter, Comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetes Care. 2006;29:1538–1544. doi: 10.2337/dc05-2370. [DOI] [PubMed] [Google Scholar]
- 168.Powers JP, Piper DE, Li Y, Mayorga V, Anzola J, Chen JM, Jaen JC, Lee G, Liu J, Peterson MG, Tonn GR, Ye Q, Walker NPC, Wang Z. SAR and Mode of Action o f Novel Non-Nucleoside Inhibitors of Hepatitis C NS5B RNA Polymerase. J. Med. Chem. 2006;49:1034–1046. doi: 10.1021/jm050859x. [DOI] [PubMed] [Google Scholar]
- 169.Cutshall NS, O'Day C, Prezhdo M. Rhodanine Derivatives as Inhibitors of JSP-1. Bioorg. Med. Chem. Lett. 2005;15:3374–3379. doi: 10.1016/j.bmcl.2005.05.034. [DOI] [PubMed] [Google Scholar]
- 170.a Suree N, Yi SW, Thieu W, Marohn M, Damoiseaux R, Chan A, Jung ME, Clubb RT. Discovery and Structure-activity Relationship Analysis of Staphylococcus Aureus Sortase A Inhibitors. Biorg. Med. Chem. 2009;17:7174–7185. doi: 10.1016/j.bmc.2009.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Piali L, Froidevaux S, Hess P, Nayler O, Bolli MH, Schlosser E, Kohl C, Steiner B, Clozel M. The Selective Sphingosine 1-Phosphate Receptor 1 Agonist Ponesimod Protects against Lymphocyte-Mediated Tissue Inflammation. J. Pharmacol. Exp. Ther. 2011;337:547–556. doi: 10.1124/jpet.110.176487. [DOI] [PubMed] [Google Scholar]
- 171.Kokel D, Cheung CYJ, Mills R, Coutinho-Budd J, Huang L, Setola V, Sprague J, Jin S, Jin YN, Huang X-P, Bruni G, Woolf CJ, Roth BL, Hamblin MR, Zylka MJ, Milan DJ, Peterson RT. Photochemical Ac tivation of TRPA1 Channels in Neurons and Animals. Nat. Chem. Biol. 2013;9:257–263. doi: 10.1038/nchembio.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Carlson EE, May JF, Kiessling LL. Chemical Probes of UDP-Galactopyranose Mutase. Chem. Biol. 2006;13:825–837. doi: 10.1016/j.chembiol.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 173.a Mustafa A, Asker W, Sobhy M. E. E.-d. On the Reactivity of the Exocyclic Double Bond in 5-Arylidene-3-aryl-2,4-thiazolidinediones; Their Reaction with Diazoalkanes, p-Thiocresol and Piperidine. J. Am. Chem. Soc. 1960;82:2597–2601. [Google Scholar]; b Nagase H. Studies on Fungicides. XXIV. Reactio n of 5-Methoxycarbonyl-methylidene-2-thioxo(or oxo)-4-thiazolidones with o-Aminobenzenethiol and other Thiols. Chem. Pharm. Bull. 1974;22:42–49. doi: 10.1248/cpb.22.42. [DOI] [PubMed] [Google Scholar]
- 174.a Norman BH, Shih C, Toth JE, RAy JE, Dodge JA, Johnson DW, Rutherford PG, Schultz RM, Worzalla JF. Studies on the Mechanism of Phosphatidylinositol 3-Kinase Inhibition by Wortmannin and Related Analogs. J. Med. Chem. 1996;39:1106–1111. doi: 10.1021/jm950619p. [DOI] [PubMed] [Google Scholar]; b Wipf P, Minion DJ, Halter RJ, Berggren MI, Ho CB, Chiang GG, Kirkpatrick L, Abraham R, Powis G. Synthesis and Biological Evaluation of Synthetic Viridins Derived from C(20)-Heteroalkylation of the Steroidal PI-3-Kinase Inhibitor Wortmannin. Org. Biomol. Chem. 2004;2:1911–1920. doi: 10.1039/b405431h. [DOI] [PubMed] [Google Scholar]
- 175.a Wymann MP, Bulgare lli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, Panayotou G. Wortmannin Inactivates Phosphoinositide 3-Kinase by Covalent Modification of Lys-802, a Residue Involved in the Phosphate Transfer Reaction. Mol. Cell. Biol. 1996;16:1722–1733. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Structural Determinants of Phosphoinositide 3-Kinase Inhibition by Wortmannin, LY294002, Quercetin, Myricetin, and Staurosporine. Mol. Cell. 2000;6:909–919. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- 176.a Yee M.-c., Fas SC, Stohlmeyer MM, Wandless TJ, Cimprich KA. A Cell-permeable, Activity-based Probe for Protein and Lipid Kinases. J. Biol. Chem. 2005;280:29053–29059. doi: 10.1074/jbc.M504730200. [DOI] [PubMed] [Google Scholar]; b Liu Y, Shreder KR, Gai W, Corral S, Ferris DK, Rosenblum JS. Wortmannin, a Widely Used Phosphoinositide 3-Kinase Inhibitor, also Potently Inhibits Mammalian Polo-like Kinase. Chem. Biol. 2005;12:99–107. doi: 10.1016/j.chembiol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 177.a Bowles DW, Senzer N, Hausman D, Peterson S, Vo A, Walker L, Cohen RB, Jimeno A. A Multicenter Phase 1 Study of PX-866 and Cetuximab in Patients with Metastic Colorectal Carcinoma or Recurrent/Metastatic Squamous Cell Carcinoma of the Head and Neck. Invest. New Drugs. 2014;32:1197–1203. doi: 10.1007/s10637-014-0124-3. [DOI] [PubMed] [Google Scholar]; b Jimeno A, Bauman JE, Weissman C, Adkins D, Schnadig I, Beauregard P, Bowles DW, Spira A, Levy B, Seetharamu N, Hausman D, Walker L, Rudin CM, Shirai K. A Randomized, Phase 2 Trial of Docetaxel with or without PX-866, an Irreversible Oral Phosphatidylinositol 3-Kinase Inhibitor, in Patients with Relapsed or Metastatic Head and Neck Squamous Cell Cancer. Oral Oncol. 2015;51:383–388. doi: 10.1016/j.oraloncology.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yuan H, Barnes KR, Weissleder R, Cantley L, Josephson L. Coval ent Reactions of Wortmannin under Physiological Conditions. Chem. Biol. 2007;14:321–328. doi: 10.1016/j.chembiol.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 179.a Huntington KM, Yi T, Wei Y, Pei D. Synthesis and Antibacterial Activity of Peptide Deformylase Inhibitors. Biochem. 2000;39:4543–4551. doi: 10.1021/bi992452y. [DOI] [PubMed] [Google Scholar]; b Lee HS, Kim DH. Synthesis and Evaluation of α,β-Disubstituted-3-mercaptopropanoic Acids as Inhibitors for Carboxypeptidase A and Implications with Respect to Enzyme Inhibitor Design. Biorg. Med. Chem. 2003;11:4695–4691. doi: 10.1016/j.bmc.2003.08.017. [DOI] [PubMed] [Google Scholar]; c Gao S, Tzeng T, Sastry MNV, Chu C-M, Liu J-T, Lin C, Yao C-F. Iodine Catalyzed Conjugate Addition of Mercaptans to α,β-Unsaturated Carboxylic Acids Under Solvent-free Conditions. Tet. Lett. 2006;47:1889–1893. [Google Scholar]; d Gao F, Tseng C, Tsai CH, Yao C-F. Fluori de Ion-catalyzed Conjugate Addition for Easy Synthesis of 3-Sulfanylpropionic Acid from Thiol and α,β-Unsaturated Carboxylic Acid. Tetrahedron. 2008;64:1955–1961. [Google Scholar]
- 180.Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS. The Proto-oncometabolite Fumarate Binds Glutathione to Amplify ROS-Dependent Signaling. Mol. Cell. 2013;51:236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dai C-G, Du X-J, Song Q-H. Acid-Activatable Michael-Type Fluorescent Pr obes for Thiols and for Labeling Lysosomes in Live Cells. J. Org. Chem. 2015;80:12088–12099. doi: 10.1021/acs.joc.5b02041. [DOI] [PubMed] [Google Scholar]
- 182.Kim G-J, Yoon D-H, Yun M-Y, Kwon H, Ha H-J, Kim H-J. Ratiometric Fluorescence Probes Based on a Michael Acceptor Type of Coumarin and their Application for the Multichannel Imaging of In Vivo Glutathione. RSC Advances. 2014;4:18731–18736. [Google Scholar]
- 183.Kluter S, Simard JR, Rode HB, Grutter C, Pawar V, Raaijmakers HC, Barf TA, Rabiller M, van Otterlo WA, Rauh D. Characteriz ation of Irreversible Kinase Inhibitors by Directly Detecting Covalent Bond Formation: A Tool for Dissecting Kinase Drug Resistance. ChemBioChem. 2010;11:2557–2566. doi: 10.1002/cbic.201000352. [DOI] [PubMed] [Google Scholar]
- 184.Appendino G, Minassi A, Collado JA, Pollastro F, Chianese G, Taglialatela-Scafati O, Ayyari M, Garcia V, Muñoz E. The Thia-Michael Reactivity of Zerumbone and Related Cross-Conjugated Dienones: Disentangling Stoichiometry, Regiochemistry, and Addition Mode with an NMR-Spectroscopy-Based Cysteamine Assay. Eur. J. Org. Che m. 2015:3721–3726. [Google Scholar]
- 185.Huth JR, Mendoza R, Olejniczak ET, Johnson RW, Cothron DA, Liu Y, Lerner CG, Chen J, Hajduk PJ. ALARM NMR: A Rapid and Robust Experimental Method to Detect Reactive False Positives in Biochemical Screens. J. Am. Chem. Soc. 2005;127:217–224. doi: 10.1021/ja0455547. [DOI] [PubMed] [Google Scholar]
- 186.Epps DE, Taylor BM. A Competitive Fluorescence Assay to Measure the Reactivity of Compounds. Anal. Biochem. 2001;295:101–106. doi: 10.1006/abio.2001.5173. [DOI] [PubMed] [Google Scholar]
- 187.Spencer SR, Xue L, Klenz EM, Talalay P. The Potency of Indu cers of NAD(P)H: (Quinone-acceptor) Oxidoreductase Parallels their Efficiency as Substrates for Glutathione Transferases. Biochem. J. 1991;273:711–717. doi: 10.1042/bj2730711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dinkova-Kostova AT, Abeygunawardana C, Talalay P. Chemoprotective Properties of Phenylprop enoids, Bis(benzylidene)cycloalkanones, and Related Michael Reaction Acceptors: Correlation of Potencies as Phase 2 Enzyme Inducers and Radical Scavengers. J. Med. Chem. 1998;41:5287–5296. doi: 10.1021/jm980424s. [DOI] [PubMed] [Google Scholar]
- 189.Cusack KP, Arnold LD, Barberis CE, Chen H, Eri csson AM, Gaza-Bulseco GS, Gordon TD, Grinnell CM, Harsch A, Pellegrini M, Tarcsa E. A 13C NMR Approach to Categorizing Potential Limitations of α,β-Unsaturated Carbonyl Systems in Drug-like Molecules. Bioorg. Med. Chem. Lett. 2004;14:5503–5507. doi: 10.1016/j.bmcl.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 190.a Miyata O, Shinada T, Ninomiya I, Naito T, Date T, Okamura K, Inagaki S. Stereospecific Nucleophilic Addition Reactions to Olefins. Addition of Thiols to α,β-Unsaturated Carboxylic Acid Derivatives. J. Org. Chem. 1991;56:6556–6564. [Google Scholar]; b Kamimura A, Sasatani H, Hashimoto T, Kawai T, Hori K, Ono N. Anti-Selective Michael Addition of Thiols and their Analogues to Nitro Olefins. J. Org. Chem. 1990;55:2437–2442. [Google Scholar]
- 191.Thomas BE, Kollman PA. An ab Initio Molecular Orbital Study of the First Step of the Catalytic Mechanism of Thymidylate Synthase: The Michael Addition of Sulfur and Oxygen Nucleophiles. J. Org. Chem. 1995;60:8375–8381. [Google Scholar]
- 192.Kunakbaeva Z, Carrasco R, Rozas I. An Approximation to the Mechanism of Inhibition of Cysteine Proteases: Nucleophilic Sulphur Addition to Michael Acceptor Type Compounds. J. Mol. Struc-Theochem. 2003;626:209–216. [Google Scholar]
- 193.a Mulliner D, Wondrousch D, Schüürmann G. Predicting Michael-Acceptor Reactivity and Toxicity Through Quantum Chemical Transition-State Calculations. Org. Biomol. Chem. 2011;9:8400–8412. doi: 10.1039/c1ob06065a. [DOI] [PubMed] [Google Scholar]; b Pereira SR, Vasconcelos VM, Antunes A. Computational Study of the Covalent Bonding of Microcystins to Cysteine Residues-a Reaction Involved in the Inhibition of the PPP Family of Protein Phosphatases. FEBS Journal. 2013;280:674–680. doi: 10.1111/j.1742-4658.2011.08454.x. [DOI] [PubMed] [Google Scholar]; c Paasche A, Schiller M, Schirmeister T, Engels B. Mechanistic Study of the Reaction of Thiol-Containing Enzymes with α,β-Unsaturated Carbonyl Substrates by Computation and Chemoassays. ChemMedChem. 2010;5:869–880. doi: 10.1002/cmdc.201000020. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Carlqvist P, Svedendahl M, Branneby C, Hult K, Brinck T, Berglund P. Exploring the Active-site of a Rationally Redesigned Lipase for Catalysis of Michael-type Additions. ChemBioChem. 2005;6:331–336. doi: 10.1002/cbic.200400213. [DOI] [PubMed] [Google Scholar]
- 194.Hickey AL, Rowley CN. Benchmarking Quantum Chemical Methods for the Calculation of Molecular Dipole Moments and Polarizabilities. J. Phys. Chem. A. 2014;118:3678–3687. doi: 10.1021/jp502475e. [DOI] [PubMed] [Google Scholar]
- 195.Smith JM, Alahmadi YJ, Rowley CN. Range-Separated DFT Functionals are Necessary to Model Thio-Michael Additions. J. Chem. Theory Comput. 2013;9:4860–4865. doi: 10.1021/ct400773k. [DOI] [PubMed] [Google Scholar]
- 196.Krenske EH, Petter RC, Zhu Z, Houk KN. Transition States and Energetics of Nucleophilic Additions of Thiols to Substituted α,β-Unsaturated Ketones: Substituent Effects Involve Enone Stabilization, Product Branching, and Solvation. J. Org. Chem. 2011;76:5074–5081. doi: 10.1021/jo200761w. [DOI] [PubMed] [Google Scholar]
- 197.a Parr RG, Pearson RG. Absolute Hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983;105:7512–7516. [Google Scholar]; b Meza R, Gordillo B, Galvan M. Local HSAB Principle in the Conjugate Addition of p-Substituted Thiophenols to Cyclohexenone. Int. J. Quantum Chem. 2005;104:29–37. [Google Scholar]; c Pearson RG. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963;85:3533–3530. [Google Scholar]; d Pearson RG. Hard and Soft Acids and Bases, HSAB, Part 1: Fundamental Principles. J. Chem. Educ. 1968;45:581–587. [Google Scholar]; e Pearson RG. Hard and Soft Acids and Bases, HSAB, Part II: Underlying Theories. J. Chem. Educ. 1968;45:643–648. [Google Scholar]
- 198.Parr RG, Szentpaly L. v., Liu S. Electrophilicity Index. J. Am. Chem. Soc. 1999;121:1922–1924. [Google Scholar]
- 199.Ayers P, Yang W, Bartolot ti LJ. Chemical Reactivity Theory: A Density Functional View. Vol. 1. CRC Press; Boca Raton, FL: 2009. pp. 255–267. [Google Scholar]
- 200.a Parr RG, Yang W. Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc. 1984;106:4049–4050. [Google Scholar]; b Lee C, Yang W, Parr RG. Local Softness and Chemical Reactivity in the Molecules CO, SCN−, and H2CO. J. Mol. Struc-Theochem. 1988;163:305–313. [Google Scholar]
- 201.Mondal P, Hazarika KK, Deka RC. Reactivity of α,β-Unsaturated Carbonyl Compounds Towards Nucleophilic Addition Reactions: A Local Hard-Soft Acid-Base Approach. PhysChemComm. 2003;6:24–27. [Google Scholar]
- 202.a Yarbrough JW, Schultz TW. Abiotic Sulfhydryl Reactivity: A Predictor of Aquatic Toxicity for Carbonyl-Containing α,β-Unsat urated Compounds. Chem. Res. Toxicol. 2007;20:558–562. doi: 10.1021/tx600344a. [DOI] [PubMed] [Google Scholar]; b Schultz TW, Rogers K, Aptula AO. Read-across to Rank Skin Sensitization Potential: Subcategories for the Michael Acceptor Domain. Contact Derm. 2009;60:21–31. doi: 10.1111/j.1600-0536.2008.01473.x. [DOI] [PubMed] [Google Scholar]
- 203.Domingo L, Perez P, Contreras R. Reactivity of the Carbon-Carbon Double Bond Towards Nucleophilic Additions. A DFT Analysis. Tetrahedron. 2004;60:6585–6591. [Google Scholar]
- 204.LoPachin RM, Gavin T, Geohagen BC, Das S. Neurotoxic Mechanisms of Electrophilic Type-2 Alkenes: Soft-Soft Interactions Described by Quantum Mechanical Parameters. Toxicol. Sci. 2007;98:561–570. doi: 10.1093/toxsci/kfm127. [DOI] [PubMed] [Google Scholar]
- 205.Wondrousch D, Böhme A, Thaens D, Ost N, Schüürmann G. Local Electrophilicity Predicts the Toxicity-Relevant Reactivity of Michael Acceptors. J. Phys. Chem. Lett. 2010;1:1605–1610. [Google Scholar]