

Abstract
NUCKS1 is a 27 kD vertebrate-specific protein, with a role in the DNA damage response. Here, we show that after 4 Gy total-body X-irradiation, Trp53+/− Nucks1+/− mice more rapidly developed tumors, particularly thymic lymphoma (TL), than Trp53+/− mice. TLs in both cohorts showed loss of heterozygosity (LOH) of the Trp53+ allele in essentially all cases. In contrast, LOH of the Nucks1+ allele was rare. Nucks1 expression correlated well with Nucks1 gene dosage in normal thymi, but was increased in the majority of TLs from Trp53+/− Nucks1+/− mice, suggesting that elevated Nucks1 message may be associated with progression towards malignancy in vivo. Trp53+/− Nucks1+/− mice frequently succumbed to CD4- CD8- TLs harboring translocations involving Igh but not Tcra/d, indicating TLs in Trp53+/− Nucks1+/− mice mostly originated prior to the double positive stage and at earlier lineage than TLs in Trp53+/- mice. Monoclonal rearrangements at Tcrb were more prevalent in TLs from Trp53+/− Nucks1+/− mice, as was infiltration of primary TL cells to distant organs (liver, kidney and spleen). We propose that, in the context of Trp53 deficiency, wild type levels of Nucks1 are required to suppress radiation-induced TL, likely through the role of the NUCKS1 protein in the DNA damage response.
Keywords: NUCKS1, double-strand break repair, ionizing radiation, thymic lymphoma, V(D)J recombination
INTRODUCTION
Ionizing radiation (IR) is an environmental carcinogen, and exposure to IR is associated with negative effects on health, such as reduced hematopoietic cell function and an elevated risk for cancer. These malignancies are considered to also result from the direct induction of mutations due to insufficient or imprecise repair of DNA damage after IR. IR induces a variety of DNA lesions, of which DNA double-strand breaks (DSBs) are considered to be the most detrimental [1]. To sense and repair DSBs, cells have evolved numerous highly efficient repair pathways, and the two main pathways for DSB repair in eukaryotes are classical non-homologous end joining (NHEJ) and homologous recombination (HR). Defects in either DNA repair pathway can cause genome instability and tumorigenesis [2, 3].
In addition to choosing the right DSB repair pathway, the capacity of cells to sense DNA damage and to signal to downstream effectors in the DNA damage response (DDR) network is crucial for genome stability and cancer avoidance. For example, when the Ataxia Telangiectasia Mutated Serine/Threonine-Protein Kinase (ATM), one of the key components of the DDR, is mutated, patients develop Ataxia-Telangiectasia (A-T), an autosomal recessive syndrome characterized by progressive neurodegeneration, radiosensitivity, immune dysfunction, cell cycle checkpoint defects and an increased predisposition to cancer [4]. ATM is one of the six members of the phosphoinositide 3-kinase-related protein kinase (PIKK) family that includes other DDR sensors such as AT and Rad3-related protein kinase (ATR) and DNA-dependent protein kinase catalytic subunit (DNA-PKcs).
More than 1,300 proteins are phosphorylated in response to DNA damage, as shown by the results from several studies using quantitative proteomics [5–9]. One such protein is Nuclear Casein Kinase and Cyclin-dependent Kinases Substrate 1 (NUCKS1), a nuclear and highly phosphorylated protein [10–12], which also is acetylated, methylated, ubiquitylated and formylated [13] (http://www.phosphosite.org/). Specifically, in phosphoproteomic screens, NUCKS1 Ser14 was identified as a substrate of either ATM or ATR after IR [5], and Ser54 and Ser181 were identified as ATM-dependent phosphorylation sites after neocarzinostatin [6]. In addition, our own results show that the DNA damage-induced phosphorylation of NUCKS1 at Ser54 is ATM-dependent and that it occurs in HeLa cells both after exposure to IR and after mitomycin C [14]. Collectively, the results from phosphoproteomic screens [5–9] and of our previous investigation [14] show that the NUCKS1 protein is an important new player in the DDR, although its precise functions still remain to be elucidated.
Albeit there are little functional cancer-related data on NUCKS1, several studies suggest that there are some links, particularly between NUCKS1/NUCKS1 expression and breast cancer [13, 15–17]. NUCKS1 also was identified as a colorectal cancer prognostic marker [18], as a biomarker for recurrence-free survival in cervical squamous cell carcinoma [19], as a risk factor for poor prognosis and recurrence in endometrial cancer [20], as an immunodiagnostic marker in hepatocellular carcinoma [21], and as aberrantly low expressed in adult T-cell leukemia-lymphoma [22] and in childhood acute lymphoblastic leukemia [23]. However, how expression of NUCKS1/NUCKS1 is linked to initiation and/or progression towards malignancy is currently unknown.
Using gene-specific knockdown of NUCKS1 in human cells, we have shown that NUCKS1 is a chromatin-associated protein with a novel role in the DDR and in HR, a DNA repair pathway critical for tumor suppression [14]. However, whether functional loss of NUCKS1 in mice would lead to an increased susceptibility to cancer had not been explored. Here we show that Trp53+/− Nucks1+/− mice more rapidly developed tumors than Trp53+/− mice after exposure to 4 Gy total-body X-irradiation (TBI). Notably, in Trp53+/− Nucks1+/− mice IR-induced thymic lymphomas (TLs) were more prevalent and arose at earlier lineage than in Trp53+/− mice, frequently with concomitant with an upregulated expression of Nucks1 transcript. We propose that, in the context of Trp53 deficiency, wild type levels of murine NUCKS1 are required to suppress radiation carcinogenesis, in line with an important role for NUCKS1 in the DDR.
RESULTS
Nucks1 shows haploinsufficiency for suppressing radiation carcinogenesis in mice
We tested for the consequences of total-body X-irradiation (TBI) of mice with partially inactivated Nucks1. We chose to do these experiments in the context of Trp53 heterozygosity to avoid Trp53 checkpoint activation in response to partial Nucks1 deficiency and/or IR exposure [24, 25]. To obtain Trp53+/− and Trp53+/−Nucks1+/− F2 littermates, a founder mouse heterozygous for Nucks1 (derived from strain: Nucks1Gt(XG374)Byg (BayGenomics)) and in a mixed genetic background, was backcrossed to mice heterozygous for Trp53 (strain: 129S1/SvImJ-Trp53tm1Tyj/J (Jackson Laboratory)). F1 mice on average were 25% C57BL/6, 62.5% 129S1/SvImJ and 12.5% CBA in genetic background. F1 mice heterozygous for both Nucks1 and Trp53 were further backcrossed to wild type 129S1/SvImJ mice to generate F2 mice that on average were 81.25% 129S1/SvImJ, 12.5% C57BL/6 and 6.25% CBA in genetic background (Supplementary Information; Supplementary Figure S1). F2 littermates were used for the experiments described below.
After 4 Gy TBI, all F2 Trp53+/− mice died within 45 weeks (Figure 1a). Tumor spectrum analysis revealed that Trp53+/− mice frequently developed TL (38.7%; Table 1). Trp53+/− mice also developed sarcoma and splenic lymphoma (Table 1). Tumor latency in F2 Trp53+/− Nucks1+/− mice was significantly reduced (P=0.018; Log-Rank test), and most mice died within 31 weeks (Figure 1a). The median survival times for Trp53+/− and Trp53+/− Nucks1+/− mice were 193 and 175 days, respectively. Tumor spectrum analysis revealed that the fraction of mice succumbing to TL was significantly higher in Trp53+/− Nucks1+/− mice (66.7%; Table 1; P=0.0398; Fisher's exact test). Trp53+/− Nucks1+/− mice also developed primary lung epithelial tumors that were not observed in Trp53+/− single heterozygous mice (Table 1; Supplementary Figure S2). When Kaplan-Meier survival curves for both cohorts were confined to mice with TL only (Figure 1b), a significant difference for TL-free survival between both cohorts was detected (P=0.004; Log-Rank test). Median survival times for succumbing to TL were 272 and 201 days for Trp53+/− and Trp53+/− Nucks1+/− mice, respectively.
Figure 1. Nucks1 deficiency promotes radiation carcinogenesis in mice.
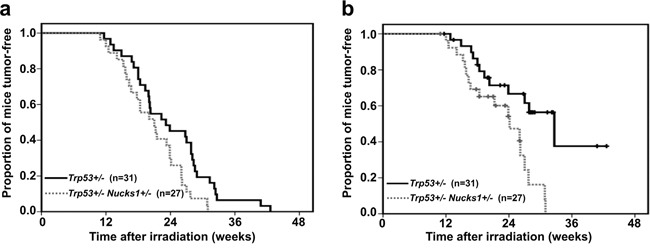
a. Kaplan-Meier survival curves for tumor-free survival of Trp53+/− Nucks1+/− and Trp53+/− mice after 4 Gy TBI (P=0.018; Log-Rank test). b. Kaplan-Meier survival curves for thymic lymphoma (TL)-free survival of Trp53+/− Nucks1+/− and Trp53+/− mice after 4 Gy TBI (P=0.004; Log-Rank test).
Table 1. Tumor types and frequencies in Trp53+/− and Trp53+/− Nucks1+/− mice.
| Tumor type1 | Tumor frequency (%) | |
|---|---|---|
| Trp53+/− | Trp53+/− Nucks1+/− | |
| Thymic lymphoma | 38.7 | 66.7 |
| Splenic lymphoma | 12.9 | 7.4 |
| Sarcoma | 16.7 | 22.2 |
| Lung tumor2 | - | 11.1 |
| Other tumors | 16.7 | 14.8 |
| Paralysis3 | 22.6 | 11.1 |
Only primary tumors, as examined by H&E or immunohistochemistry.
Epithelial-origin lung tumors were found in Trp53+/− Nucks1+/− mice only.
Mice were characterized as succumbing to paralysis when their limbs, usually their hind limbs, lost motility followed by severe weight loss.
TLs that arose from Trp53+/− and Trp53+/− Nucks1+/− mice showed LOH of the wild type Trp53 allele in 8/9 and 14/14 cases, respectively (Figure 2a). Only one TL (TL-3) from a Trp53+/− mouse maintained the Trp53 wild type allele, but showed greatly reduced expression of Trp53 mRNA (Figure 2b). In contrast, LOH of the Nucks1 wild type allele was rare and detected in one TL (TL-157; Trp53+/− Nucks1+/−) (Figure 2a). LOH of the mutant Nucks1 allele in TLs of Trp53+/− Nucks1+/− mice was also rare, and occurred in 1/14 cases only (TL-162; Figure 2a).
Figure 2. Nucks1 is haplo-insufficient in suppressing IR-induced TL.
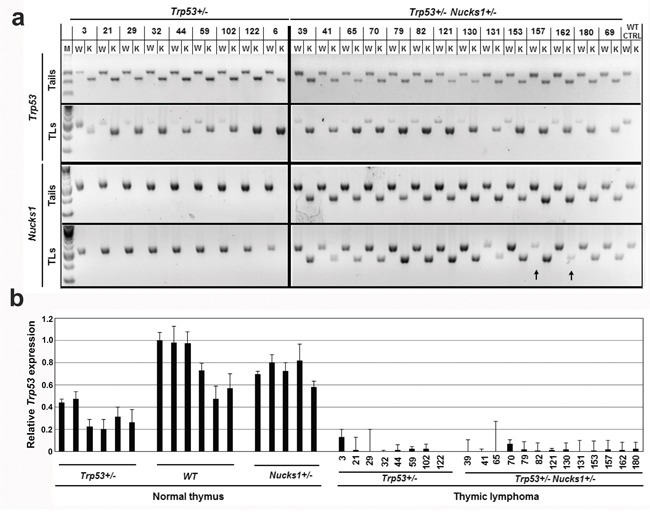
a. Agarose gels to show the results from semi-quantitative PCR analysis to detect LOH at Trp53 and Nucks1. Genomic DNAs from tail and TL of the same mouse were used as template. Arrows indicate LOH of the Nucks1 wild type and knockout alleles. M: molecular weight marker; W: wild type allele; K: knockout allele; WT CTRL: tail and thymus of a sham-irradiated 129S1/SvImJ mouse. b. Results from TaqMan qRT-PCR assays to assess Trp53 expression in normal thymi of Trp53+/−, wild type (WT) and Nucks1+/− mice, and in IR-induced TLs of Trp53+/− and Trp53+/−Nucks1+/− mice.
Activation of NOTCH1 by intragenic deletion of the Notch 1 exon 1 is common in lymphomas and irradiated thymi [26–28]. However, it was not clear whether deletion-based NOTCH1 activation also played a role in the radiation-induced TLs in Trp53-deficient mice, and if it was dependent on Nucks1 status. We found that Notch1 type 1 deletions were common in TLs from both cohorts, and that their frequencies were similar in TLs from Trp53+/− mice (8/9) and Trp53+/− Nucks1+/− mice (12/14; Supplementary Figure S3). These results suggest that, in the context of Trp53 deficiency and independent of Nucks1 status, deletion-based NOTCH1 activation is an important event during IR-induced T-cell lymphomagenesis.
Nucks1 expression is up-regulated in most TLs from Trp53+/− Nucks1+/− mice
We examined if Nucks1 mRNA levels were altered in TLs from Trp53+/− Nucks1+/− mice. In normal thymi, wild type Nucks1 mRNA expression correlated well with genotype in that ~40% Nucks1 mRNA was expressed in Nucks1+/− thymi compared to Nucks1+/+ thymi, and Trp53 heterozygosity did not affect Nucks1 mRNA expression (Figure 3a). Nucks1 mRNA expression was higher than in normal Nucks1+/+ thymi in 2/8 TLs (TL-3, TL-44) from Trp53+/− mice, but overall not significantly different between both groups (Figure 3a; P=0.703; Mann-Whitney test). The means of the dCTs for Nucks1 to Gapdh were 2.79 and 2.78 for normal wild type thymi and for TLs of Trp53+/− mice, respectively. Nucks1 expression in TLs from Trp53+/− mice was moderately decreased when compared to normal thymi from Trp53+/− mice (P=0.016; Mann-Whitney test). The means of the dCTs for Nucks1 to Gapdh were 2.61 and 2.78 for normal thymi and TLs of Trp53+/− mice, respectively. However, Nucks1 mRNA expression was significantly elevated in 9/13 TLs from Trp53+/− Nucks1+/− mice when compared to normal thymi of Nucks1+/− mice (P<0.0001; Mann-Whitney test; Figure 3a). The means of the dCTs for Nucks1 to Gapdh were 4.04 and 3.29 for normal thymi from Nucks1+/− mice and for TLs from Trp53+/− Nucks1+/− mice, respectively. These results suggest that the up-regulation of Nucks1 transcript is a frequent event in IR-induced TLs from Trp53+/− Nucks1+/− mice. Nucks1 expression in TLs from Trp53+/− mice compared to TLs from Trp53+/− Nucks1+/− mice was significantly different (P=0.0072; Mann-Whitney test), with mean dCTs for Nucks1 to Gapdh of 2.78 and 3.29, respectively.
Figure 3. Nucks1 mRNA expression in normal thymi and TLs.
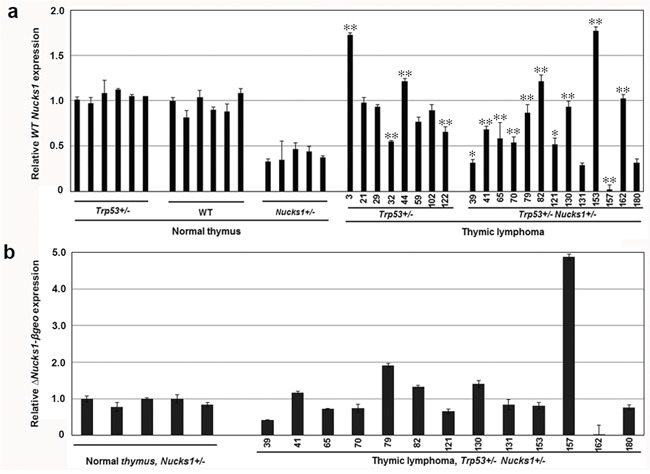
a. Results from TaqMan qRT-PCR assays to assess Nucks1 expression in normal thymi of Trp53+/−, wild type (WT) and Nucks1+/− mice, and in IR-induced TLs of Trp53+/− and Trp53+/−Nucks1+/− mice. *, P<0.05; **, P<0.01 (for Nucks1 expression in TL of Trp53+/− mice compared to normal thymus of Trp53+/− mice, and for Nucks1 expression in TL of Trp53+/− Nucks1+/− mice compared to normal thymus of Nucks1+/− mice). Note, that 9/13 TLs from Trp53+/− Nucks1+/− mice show significantly increased Nucks1, and 2/13 TLs from Trp53+/− Nucks1+/− mice show significantly decreased Nucks1. b. Results from TaqMan qRT-PCR assays to assess ΔNucks1-βgeo in normal thymi, and in IR-induced TLs of Trp53+/− Nucks1+/− mice.
We then examined the ΔNucks1-βgeo mRNA expression in normal thymi from Nucks1+/− mice and in TLs from Trp53+/− Nucks1+/− mice. In most TLs that arose from Trp53+/− Nucks1+/− mice, ΔNucks1-βgeo mRNA expression was not elevated when compared to normal thymi from Nucks1+/− mice (Figure 3b). TL-79 showed an ~2-fold increase in ΔNucks1-βgeo mRNA expression. TL-157 showed an ~5-fold increase in ΔNucks1-βgeo mRNA expression and TL-162 showed no ΔNucks1-βgeo mRNA expression, likely from gene conversion events involving gain and loss of the Nucks1 knockout allele, respectively (see Figure 2a). In support of these findings, Nucks1 mRNA expression was essentially zero in TL-157, but was ~2–fold increased in TL-162 when compared to normal Nucks1+/− thymi (Figure 3a).
To test if NUCKS1 protein levels change in wild type mice exposed to IR, five-week old wild type 129S1/SvImJ mice were either sham-irradiated or exposed to 4 Gy TBI, and thymi were harvested and fixed for immunohistochemistry (IHC) at 2 h, 4 h, 6 h, 24 h and 6 weeks post TBI. Cell death was observed within defined areas of the cortex as early as 4-6 h post treatment (Figure 4). Viable T-cells in the cortex overall expressed higher levels of NUCKS1 protein at 4-6 h post IR exposure than T-cells in sham-irradiated thymi, or in thymi that were recovered 6 weeks post IR exposure (Figure 4b). At 24 h post exposure, the vast majority of T-cells in the thymus cortex were dead, and viable T-cells with increased NUCKS1 were distributed mostly throughout the medulla (Figure 4a). These results suggest that NUCKS1 protein expression increases at early times post IR exposure in thymocytes of irradiated mice.
Figure 4. IR-induced cell death within the thymic cortex.
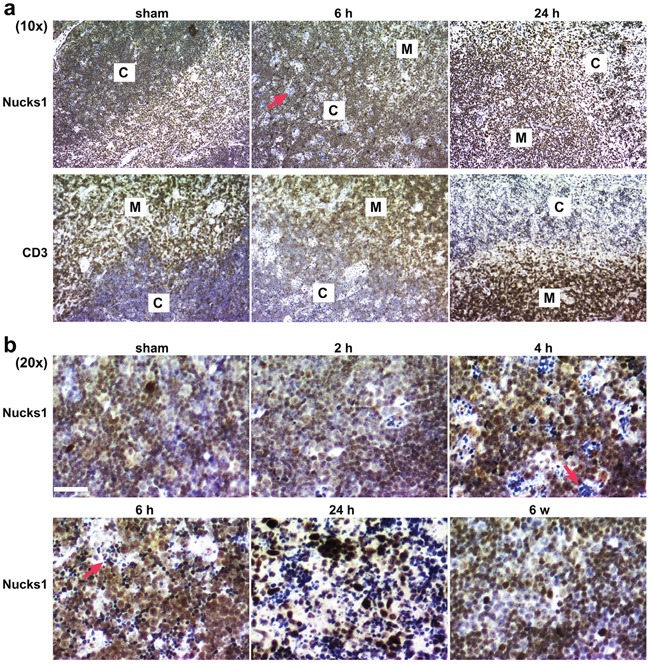
Five-week old wild type 129S1/SvImJ mice were either sham-treated or irradiated with 4 Gy TBI. CD3 and NUCKS1 expression was examined by IHC in FFPE sections of isolated thymi fixed at the times indicated. a. At 10× magnification, to show NUCKS1 and CD3 staining (brown) in both the cortex (C) and the medulla (M). b. At 20× magnification, to show NUCKS1 staining (brown) in the cortex. Red arrows: indicate apoptotic cells. Hematoxylin: nuclei. Scale bars: 200 μm.
Analyses of immunophenotypes and Tcrb clonality reveal differences in IR-induced TLs from Trp53+/− and Trp53+/− Nucks1+/− mice
We next analyzed the immunophenotypes of isolated TL cells from IR-induced TLs of Trp53+/− and Trp53+/− Nucks1+/− mice. CD3 expression for the majority of isolated TL cells from both cohorts was moderate, indicative of their immaturity (Figures 5a and 5b; Table 2). With the exception of TL-32, TL cells isolated from IR-induced TLs of Trp53+/− mice (8/9) were either CD4+ CD8+ double positive (DP) or CD8+ single positive (SP), or a mix of these two cell populations (Figure 5a; Table 2). TL-32 (from a Trp53+/− mouse) was the only rare case that showed a mixed population of CD4- CD8- double negative (DN) and uncommon CD4+ SP TL cells. In contrast, TLs of Trp53+/− Nucks1+/− double heterozygous mice frequently (5/8) contained CD4- CD8- DN TL cells (Figure 5b; Table 2). Among these, TL-180 and TL-121 were purely CD4- CD8- DN (Figure 5b), while TL-131, TL-82, TL-79 and TL-162 showed mixed populations of CD4- CD8- DN, CD4+ CD8+ DP and CD8+ SP TL cells (Figure 5b; Table 2).
Figure 5. Immunophenotyping analyses of TL cells isolated from IR-induced TLs of Trp53+/− and Trp53+/− Nucks1+/− mice.
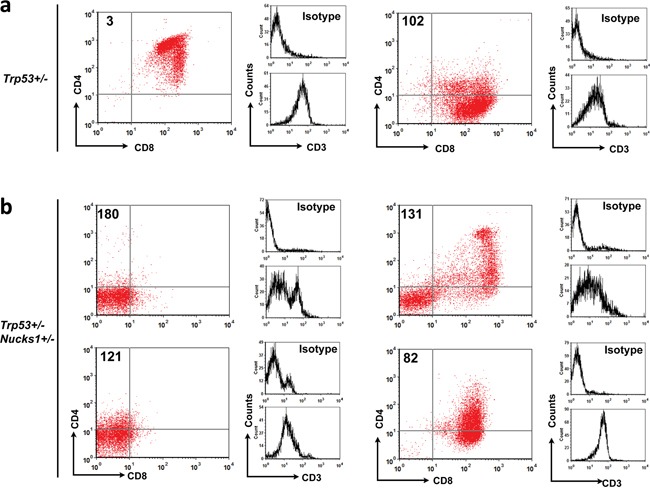
a. Primary TL cells from TLs of Trp53+/− mice were stained for CD4, CD8 and CD3 and analyzed by FACS. Representative 2-parameter dot plots showing that most primary TL cells from Trp53+/− mice were CD4+ CD8+ DP (TL-3) or CD8+ SP (TL-102) with moderate CD3 expression (see also Table 2). b. Same as in (a) for primary TL cells isolated from Trp53+/− Nucks1+/− mice. Note, that large compartments of CD4- CD8- DN TL cells were seen in TLs from Trp53+/− Nucks1+/− mice (TL-180, TL-121 and TL-131; see also Table 2).
Table 2. Summary: Immunophenotyping analyses of thymic lymphoma (TL) cells.
| Genotype | Mouse ID | TdT1 | CD32 | CD43 | CD83 | Tcra/d transloc. | Ightransloc. |
|---|---|---|---|---|---|---|---|
| Trp53+/− | 3 | + | + | + | + | N | Y |
| 29 | + | + | − and mod | + | n.d. | n.d. | |
| 32 | + | mod | − and + | − | Y | Y | |
| 44 | + | mod/+ | − and + | + | Y | Y | |
| 44, P154 | n.d. | mod | − | + | n.d. | n.d. | |
| 102 | + | mod | − | + | Y | N | |
| 102, P164 | n.d. | mod | − | + | n.d. | n.d. | |
| 122 | + | + and − | + | + | Y | Y | |
| Ly-1915 | + | + | −/mod | + | n.d. | n.d. | |
| Ly-2005 | + | − and + | − | + | n.d. | n.d. | |
| Ly-2335 | + | + | + and − | + | n.d. | n.d. | |
| Trp53+/− Nucks1+/− | 39 | + | + | mod/+ | + | N | N |
| 79 | + | mod | − | − and mod | N | Y | |
| 79, P154 | n.d. | − | − | − | n.d. | n.d. | |
| 82 | + | + | − and mod | + | N | N | |
| 82, P154 | n.d. | + | − | + | n.d. | n.d. | |
| 121 | + | mod | − | − | n.d. | n.d. | |
| 131 | + | +/mod | − and + | − and + | N | N | |
| 153 | + | +/mod | + | + | Y | N | |
| 162 | + | + | − | − and mod | N | Y | |
| 180 | + | − and + | − | − | N | Y | |
|
Trp53+/− Nucks1−/− |
573 | n.d. | + | − and + | − and + | n.d. | n.d. |
| 573, P174 | n.d. | + | − | − | n.d. | n.d. | |
| 576 | n.d. | + | − and + | − and + | n.d. | n.d. |
TdT was examined by IHC only.
CD3 immunophenotype was assessed by FACS using isolated primary TL cells, and by IHC using FFPE sections of TLs.
CD4 and CD8 were examined by FACS using isolated primary TL cells.
Established primary TL cell lines by sub-culturing: cells were split 1:10 ratio to maintain growth at a density ~5 × 10e5 cells/ml and counted as one passage.
These cell lines were derived from TLs of Trp53+/− 129S1/SvImJ mice irradiated with 4Gy TBI.
Definitions: “-”: negative; “mod”: positive, but with moderate intensity when compared to “+”; “+”: positive with good intensity; n.d.: not determined.
We also analyzed the immunophenotypes of TL cells isolated from two IR-induced TLs (TL-573 and TL-576) of Trp53+/− Nucks1−/− mice (further backcrossed to 129S1/SvImJ mice (i.e., F6 generation)). Both showed mixed populations of CD4- CD8- DN and CD4+ CD8+ DP TL cells (Table 2). Note, there was no difference in normal thymocyte maturation between sham-irradiated Trp53+/− and Trp53+/− Nucks1+/− mice (Supplementary Figure S4). Taken together, these results suggest that TLs in Trp53+/− Nucks1+/− mice originated from less mature thymocytes than TLs in Trp53+/− mice.
PCR was used to test for Dβ-Jβ clonality at Tcrb. In Trp53+/− mice, 2/9 TLs (TL-6, TL-21) showed monoclonal expansion and 3/9 TLs (TL-29, TL-32 and TL-102) showed oligoclonal expansion (Figure 6a and 6b; Supplementary Table S1). As TL-29, TL-32 and TL-102 showed more than two predominant PCR products, likely these TLs originated from more than two cells. In contrast, 7/14 TLs from Trp53+/− Nucks1+/− mice showed clonal expansion (TL-41, TL-65, TL-79, TL-131, TL-153, TL-157 and TL-180), all of which showed just one or two predominant PCR products (Figure 6a).
Figure 6. Analyses of Dβ-Jβ clonality at Tcrb in IR-induced TLs of Trp53+/− and Trp53+/− Nucks1+/− mice.
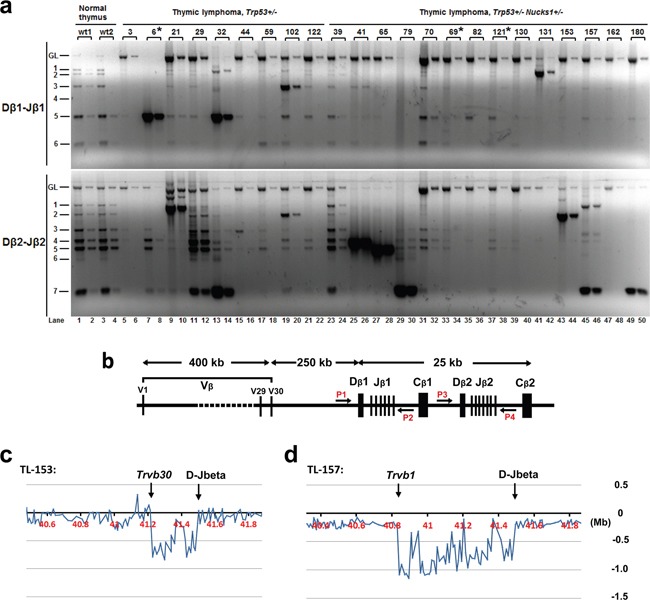
a. Representative agarose gels from semi-quantitative PCR analyses to assess Dβ1-Jβ1 and Dβ2-Jβ2 rearrangements at Tcrb using primer pairs P1 and P2 (Dβ1-Jβ1) and P3 and P4 (Dβ2-Jβ2), respectively, as show in (b). Fifty (odd lanes) and 5 ng (even lanes) of genomic DNA isolated from the thymi of normal wild type mice (wt1, wt2) and from IR-induced TLs of mice, with genotypes as indicated, were used. Asterisks: indicate TLs not analyzed by array-CGH. b. Schematic of the mouse Tcrb locus with relative genomic positions of the Vβ segments and Dβ-Jβ-Cβ clusters, and of PCR primers (P1, P2, P3 and P4). c, d. Tcrb rearrangements were also assessed by array-CGH analysis (representative expanded array-CGH profiles for TL-153 and TL-157 are shown). Note: For all TLs with genomic loss at Tcrb, the regions of loss spanned the Vβs and Dβ-Jβ clusters, indicative of Vβ to Jβ1 or Vβ to Jβ2 rearrangements. Minimal genomic loss was found in TL-153, as shown in c., which included Trvb30 segment, and maximal genomic loss was detected in TL-153, as shown in (d), which included the Trvb1 segment.
Clonality at Tcrb was confirmed by testing for Tcrb locus copy-number variations (CNVs) using array-CGH. Consistent with our results for Dβ-Jβ clonality, monoclonal Dβ-Jβ rearrangements (as detected by PCR) showed extreme loss within Tcrb by array-CGH (e.g., TL-21 (Trp53+/−), and TL-41, TL-65, TL-79, TL-131, TL-153, TL-157, TL-180 (all Trp53+/− Nucks1+/−); Figure 6c and 6d, Figure 7a and Supplementary Figure S5). Mild loss of Tcrb was detected in TL-29 (Trp53+/−; Figure 7a), and in TL-32 and TL-102 (Trp53+/−; data not shown), reflecting their oligoclonal properties. TL-82 and TL-162 (Trp53+/− Nucks1+/−) showed extreme loss within Tcrb by array-CGH (Supplementary Figure S5), but clonal expansion of Dβ-Jβ events was not detected (see Figure 6a). The reasons for this are unclear at this point. However, it is possible, that complex rearrangements within Tcrb may have occurred, not detectable by the primers used for Dβ1-Jβ1 and Dβ2-Jβ2 recombination events here.
Figure 7. Array-CGH analyses show CNVs in chromosomes 6, 14, 12 and 1 in TLs from Trp53+/− and Trp53+/− Nucks1+/− mice.
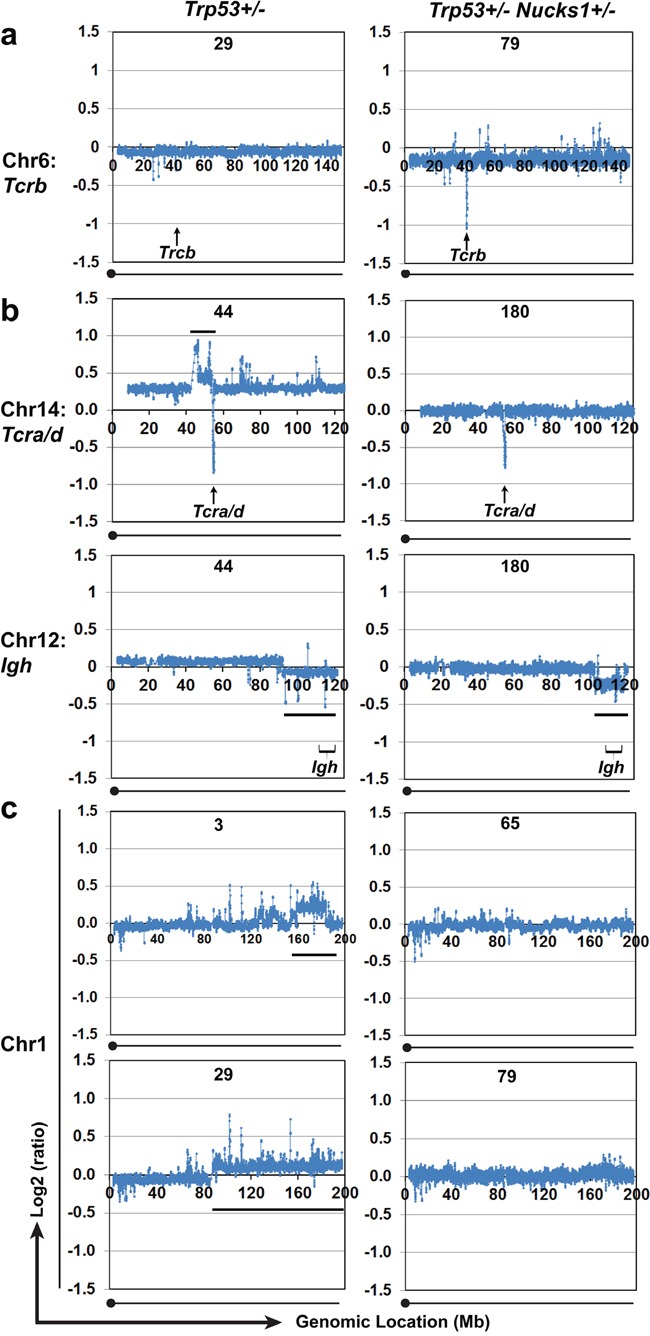
a. Array-CGH profiles of chromosome 6 to show differences in genomic loss at Tcrb between TLs from Trp53+/− (T-29) and Trp53+/− Nucks1+/− (TL-79) mice. Extensive genomic loss at Tcrb was detected in the majority (10/12) of TLs from Trp53+/− Nucks1+/− mice, but only in 1/8 TLs from Trp53+/− mice (TL-21; Supplementary Figure S5). b. Array-CGH profiles to show CNVs in chromosomes 14 (upper panels) and 12 (lower panels) in TLs from one Trp53+/− (TL-44) and one Trp53+/− Nucks1+/− (TL-180) mouse. Black horizontal lines indicate the regions of gain centromeric to Tcra/d (chromosome 14) and the telomeric regions of loss including Igh (chromosome 12). c. Array-CGH profiles of chromosome 1 to show differences in CNVs between TLs from Trp53+/− (TL-3, TL-29) and Trp53+/− Nucks1+/− (TL-65, TL-79) mice. Gain of the telomeric region of chromosome 1 was detected in ~50% of TLs from Trp53+/−, but never in TLs from Trp53+/− Nucks1+/− mice.
Trp53+/− Nucks1+/− mice succumb to IR-induced TLs with clonal translocations involving Igh but not Tcra/d
We asked whether both cohorts of mice would harbor distinct clonal translocations, and if those would involve the Tcra/d and Igh loci [29, 30]. To do so, we analyzed array-CGH plots for translocation patterns involving Tcra/d and Igh. In Trp53+/− mice, typical patterns of centromeric amplification coupled with Tcra/d loss were seen in 3/8 TLs (TL-44 (Figure 7b), and in TL-32 and TL-102, and an atypical pattern with gain telomeric to Tcra/d was observed in TL-122 (Supplementary Figure S6A)). These results show that around half of the TLs from Trp53+/− mice harbored clonal translocations involving Tcra/d. In contrast, TLs from Trp53+/− Nucks1+/− mice, which contained mixed CD4- CD8- DN TL cell populations (TL-79, TL-131 and TL-162) or were solely CD4- CD8- DN (TL-121 and TL-180), clonal translocations involving the Tcra/d were rare and detected in TL-65 and TL-153 only (Supplementary Figure S6A). Interestingly, TL cells isolated from TL-153 were CD4+ CD8+ DP without any CD4- CD8- DN TL cells (Table 2). TL-65, unfortunately, was unavailable for surface staining due to poor cell viability after isolation.
We next examined Igh-involving translocations in TLs from both Trp53+/− and Trp53+/− Nucks1+/− mice. In Trp53+/− mice, loss of the region telomeric to Igh was detected in TL-44 (Figure 7b), and in TL-3, TL-32 and TL-122 (Supplementary Figure S6B). In Trp53+/− Nucks1+/− mice, loss of the region telomeric to Igh was detected in TL-180 (Figure 7b) and in TL-65, TL-79, TL-157 and TL-162 (Supplementary Figure S6B), suggesting that translocations involving the Igh locus occurred in both cohorts with similar frequencies.
Array-based CGH analysis shows that CNVs in TLs from Trp53+/− and Trp53+/− Nucks1+/− mice are both specific and overlapping
Array-based CGH analysis was used to examine genome-wide CNVs in TLs from Trp53+/− and Trp53+/− Nucks1+/− mice. The overall patterns of genome-wide CNVs were similar between TLs from both cohorts, including changes at Igh and Tcrg (Figure 7b, Supplementary Figure S6B; data not shown). In addition, chromosome regions that contain the Ikaros [31, 32] and Pten genes [33, 34] were lost frequently in TLs from both Trp53+/− and Trp53+/− Nucks1+/− mice (data not shown). CNVs, other than at Tcra/d and Tcrb, that were more specific to one but not the other cohort were also detected. For example, gains in the telomeric arm of chromosome 1, reported to correlate tightly with cancer occurrence [35], were observed in ~50% of the TLs from Trp53+/− mice (TL-3, TL-29 (Figure 7c), TL-32 and TL-102 (Supplementary Figure S6C)). However, none of the TLs from Trp53+/− Nucks1+/− mice showed gain in the telomeric arm of chromosome 1 (Figure 7c; data not shown).
TLs from Trp53+/− Nucks1+/− mice show increased frequency of infiltration
In 1/10 of the Trp53+/− mice, TL cells (i.e., TL-122) were found to infiltrate the liver (Figure 8; Supplementary Table S2). In contrast, in 4/14 Trp53+/− Nucks1+/− mice TL cells were found to infiltrate distant organs, including kidney (TL-65 and TL-162), spleen (TL-157 and TL-131), and liver (TL-162) (Figure 8; Supplementary Table S2). To confirm these observations, additional Trp53+/− and Trp53+/− Nucks1+/− mice (further backcrossed to 129S1/SvImJ mice (F6 generation, with an average genetic background of > 99% 129S1/SvImJ)) were exposed to 4 Gy TBI. At the time the mice became moribund from TLs, mice were euthanized, assessed for the occurrence of TL and for infiltrating TL cells to distant organs. Similar to the observations for the F2 generation, increased infiltration of TL cells to distant organs was detected in Trp53+/− Nucks1+/− mice (4/13; liver, kidney and spleen) compared to Trp53+/− mice (1/10; liver only; Supplementary Table S2).
Figure 8. TLs from Trp53+/− Nucks1+/− mice infiltrate distant organs more frequently than TLs from Trp53+/− mice.
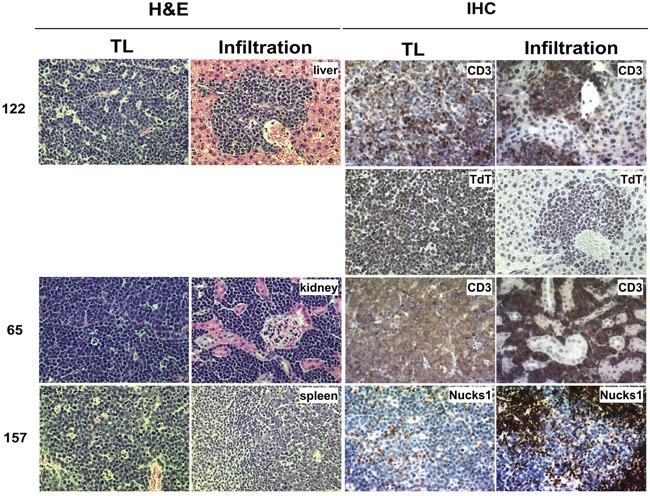
TL cells infiltrating target organs were surrounded by blood vessels and were discriminated from the target tissue based on H&E staining (left). Further evidence of infiltration was obtained by IHC using anti-CD3 (TL-122 and TL-65), anti-TdT (TL-122) and anti-NUCKS1 (TL-157) antibodies. TL-122: Trp53+/−; TL-65 and TL-157: Trp53+/− Nucks1+/−.
DISCUSSION
Environmental factors, including exposure to IR, are recognized as exogenous risk factors that induce genetic changes to drive carcinogenesis. Mice harbor many single-nucleotide polymorphisms and CNVs similar to those observed in humans [36, 37], and mouse models are powerful tools for the identification of alleles associated with susceptibility or resistance to carcinogenesis.
The effects of Nucks1 inactivation on carcinogenesis in mice had not been investigated. In human cells, however, NUCKS1 deficiency after siRNA-mediated knockdown impairs DNA damage signaling, DSB repair and genome stability [14], suggesting that this protein may function as a tumor suppressor. Here, we investigated the phenotypic consequences of partial Nucks1 deficiency and the susceptibility of these mice to radiation carcinogenesis. As the constitutive inactivation of HR genes in mice frequently leads to embryonic lethality [24, 25, 38–40], we chose to conduct our studies in a Nucks1+/− Trp53+/− double heterozygous context. Using a Trp53+/− mouse model that is prone to radiation-induced TL [32, 34, 41, 42], we found that heterozygous Nucks1 synergistically accelerated radiation lymphomagenesis and also led to other changes in the associated tumor spectrum. Importantly, IR-induced TLs that arose in double heterozygous Nucks1+/− Trp53+/− mice developed at earlier lineage stage and more frequently led to the infiltration of distant organs than TLs in single heterozygous Trp53+/− mice. We speculate that, in our mouse model, Trp53 loss itself (as observed in both cohorts to essentially full extent) does not suffice to fully drive invasive migration, in accord with what has been reported by others [43, 44]. Our findings for Trp53+/− mice also are consistent with our unpublished results (J.-H. Mao) that show that IR-induced TLs from Trp53+/− mice rarely infiltrate distant organs (e.g., liver, kidney and spleen). In contrast, TLs from Nucks1+/− Trp53+/− mice show a much greater propensity to infiltrate distant organs, and it will be important in the future to investigate the additional molecular determinants involved in this difference.
Many tumors contain chromosomal translocations and deletions (resulting from mis-repaired or unrepaired DSBs) leading to the activation of oncogenes and/or inactivation of tumor suppressor genes [45]. As such, DSBs likely play a major role in driving malignant transformation. Notably, NUCKS1 deficiency in human cells has been linked to a DSB repair defect [14], and inactivation of NHEJ and HR factors in mice leads to increased tumor burden, frequently resulting from immature T-cell lymphomas [30, 46, 47]. Defective repair of IR-induced DSBs can lead to TLs associated with translocations, deletions and CNVs [30, 48–51], similar to the findings reported here. Specifically, we show that TLs in Trp53+/− mice predominantly exhibited CNVs indicative of chromosomal translocations at Tcra/d, as reported previously [52], whereas TLs in Trp53+/− Nucks1+/− mice predominantly exhibited CNVs indicative of chromosomal translocations at Igh. Combined with the observation that the majority of TLs from Trp53+/− mice were CD4+ CD8+ DP or CD8+ SP, we suggest that IR-induced TLs in Trp53+/− mice largely originated at the DP lineage stage, the stage during which Tcra/d recombination occurs. In contrast, in Trp53+/− Nucks1+/− mice, 5 of the 8 analyzed TLs contained mixed CD4- CD8- DN TL cell populations or were purely CD4- CD8- DN, suggesting that a large proportion of TLs in Trp53+/− Nucks1+/− mice originated earlier than the DP lineage stage. Among the TLs that contained CD4- CD8- DN TL cell populations or that were purely CD4- CD8- DN, clonal translocations involving Tcra/d were not detected. Interestingly, the presence of CD4- CD8- DN cells detected in this study in TLs from Trp53+/− Nucks1+/− mice is similar to that reported in TLs derived from mice with somatic mutation of Trp53 in hematopoietic stem cells [53], and could potentially be related to the disruption of the T cell checkpoint pathways through PD-1 expression [54].
Our results suggest that ~ 50% of TLs in Trp53+/− Nucks1+/− mice originated from one or two cells, while TLs in Trp53+/− mice largely appear to have originated from several pre-cancerous cells. Moreover, TLs in Trp53+/− Nucks1+/− mice frequently developed from less mature thymocytes than those in Trp53+/− mice. The heterogeneous expression of CD4 and CD8 in TL-79 and TL-131 from two double heterozygous mice suggests that these TLs originated from single thymocytes and accumulated subsequent changes during tumor progression.
It has been well established that IR almost exclusively induces LOH of the Trp53 wild type allele in TLs derived from Trp53+/− mice [41, 55], in line with the results presented here. However, the exact course of events taken during TL development for loss of the wild type Trp53 allele to occur is unclear. Notably, LOH can be caused by several mechanisms, including mitotic recombination, mitotic non-disjunction and also by multi-locus deletion events. We speculate that partial NUCKS1 deficiency may lead to the accelerated loss of the Trp53 wild type allele in TLs from Trp53+/− Nucks1+/− mice, that compared to TLs from Trp53+/− mice arise at an earlier stage during T-cell development (i.e. show increased DN T-cell populations), predominate in Tcrb mono- and bi-clonality, and show relatively higher fractions of Igh- than of Tcra/d-associated translocation events, suggestive of their overall dependency on fewer genetic changes for tumor formation. Interestingly, Trp53+/− Nucks1+/− mice also developed primary lung epithelial tumors that were never observed in Trp53+/− single heterozygous mice, in accord with a previous report investigating Trp53+/− mice only [52]. In contrast, loss of the remaining Nucks1 wild type allele was rare in Trp53+/− Nucks1+/− mice, suggesting linkage of murine Nucks1 to essential loci on chromosome 1.
NUCKS1 expression is reduced in adult T-cell leukemia-lymphoma and in childhood acute lymphoblastic leukemia [22, 23]. However, the human NUCKS1 gene also belongs to a group of co-expressed genes located on chromosomal region 1q32.1 that is amplified in some breast cancers [16, 17, 56, 57], and in other cancers [18, 19, 21, 23, 35, 58, 59]. Interestingly, in mouse lung adenocarcinoma, amplification of a similar region on mouse chromosome 1, which results in elevated expression of several genes including Nucks1, correlates with tumor invasiveness [58]. It is interesting to note that we find increased expression (relative to normal thymi from Nucks1+/− mice) of the wild type Nucks1 transcript in 9/13 of the TLs isolated from Trp53+/− Nucks1+/− mice. We speculate that, within a subset of IR-induced TLs in Trp53+/− Nucks1+/− mice, increased expression of Nucks1/NUCKS1 may be associated with a selective advantage during tumor initiation and development, enabling early neoplastic cells to overcome replication stress during lymphomagenesis (Figure 9), thereby limiting DSBs resulting from collapsed replication forks [60, 61]. Of note, NUCKS1 plays an important role in mitigating replication stress in human cells, as we [14] and others [21] have shown. It is also possible that, in TLs from Trp53+/− Nucks1+/− mice, Nucks1 expression is upregulated as part of the activated DDR network during tumorigenesis, as described previously for clinical specimens from different stages of human tumors [62].
Figure 9. Model to explain the role of NUCKS1 in tumor initiation and progression.
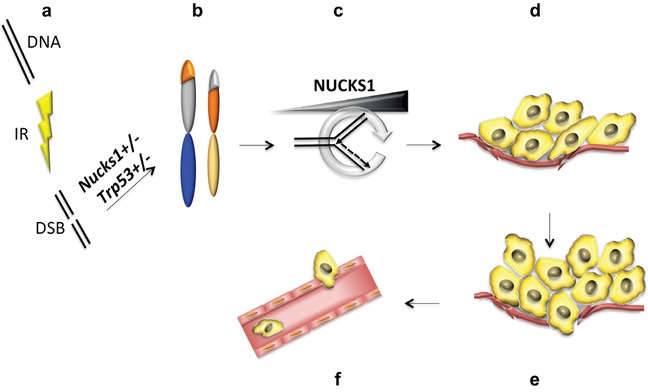
a, b. Partial deficiency of NUCKS1 leads to a defect in DNA double-strand break (DSB) repair and to genome rearrangements (i.e., translocations) after ionizing radiation (IR) exposure. c. Tumor initiating cells experience increased replication stress, as described previously [60–62], which potentially may be ameliorated by up-regulation of NUCKS1 expression. d. High levels of NUCKS1 provide a selective advantage and promote TL growth, and e. invasion of the tumor border leading to f. intravasation of TL cells into the circulatory system.
We have shown that there is a direct link between Nucks1 status and radiation carcinogenesis in mice. The results of our investigation are relevant to many published reports [16–19, 21–23, 35, 56–59] that indirectly have described an association between NUCKS1/NUCKS1 and several human cancer types. In a different genetic background, Nucks1−/− mice with wild type Trp53 were reported to exhibit decreased insulin signaling and increased body weight/fat mass along with impaired glucose tolerance and reduced insulin sensitivity [63], related to the role of NUCKS1 in the hypothalamus [64]. These studies demonstrate that NUCKS1 can function as a tissue-specific transcriptional regulator of the insulin receptor, critical for insulin signaling and consequent peripheral metabolic activities. Interestingly, the DDR intersects the insulin-IGF1-PI3K-AKT pathway at many points [5]. While Qiu et al. [63, 64] did not expose their Nucks1−/− mice to DNA damaging agents and did not investigate the direct phenotypic consequences of persistent or mis-repaired DNA damage in these mice, persistent and repetitive DNA damage has been proposed to alter insulin-IGF1 signaling, thereby contributing to diabetes and other age-associated metabolic disorders [5]. In future investigations it therefore will be important to further dissect the role of the NUCKS1 protein in metabolic syndrome and in cancer avoidance to improve both diagnosis and targeted therapy of these prevalent ailments in humans.
MATERIALS AND METHODS
Irradiations
Five week-old F2 Trp53+/− and Trp53+/− Nucks1+/− littermates (Supplementary Materials and Methods) were exposed to 4 Gy TBI using a 360 kVp X-ray machine (Precision X-ray Inc., North Branford, CT, USA). Necropsies were performed when mice appeared morbid, or at the end of the study (45 weeks post TBI). The Kaplan-Meier method was used to compare the tumor development between different genotypes. The study was carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Array based-comparative genomic hybridization (array-CGH)
Array-CGH experiments were performed on two-color Agilent SurePrint G3 Mouse CGH Microarrays (4×180k; Ambry Genetics, Aliso Viejo, CA, USA). Microsoft Excel-based programs (XY Scatter) were used for ratio data normalization and profile presentation.
Immunohistochemistry
Formalin-fixed paraffin embedded (FFPE) tumor tissues were used to generate 0.5 μm tissue sections (Mouse Pathology Core, UCSF, San Francisco, CA, USA). Sections were heated to 60°C for 30 min and deparaffinized using xylene. Heat-induced epitope retrieval was carried out using Antigen Retrieval Solution (Dako, Carpinteria, CA, USA). Sections were blocked with egg white solution (2/200 ml H2O) and 5% non-fat dried milk powder in PBST (PBS with 0.1% Tween-20) for 20 min at room temperature. Horseradish peroxidase (HRP) activity was quenched by incubating in 0.3% H2O2 diluted in 0.1% NaN3/PBS for 20 min at room temperature. Antibody incubations were carried out as described (VECTASTAIN; Vector Laboratories, Burlingame, CA, USA) using ImmPACT DAB peroxidase substrate (Vector Laboratories). Sections were then counterstained with hematoxylin, dehydrated and mounted with Permount mounting medium (Fisher Scientific, Waltham, MA, USA). Primary antibodies used were: anti-CD3e (Abcam, Cambridge, MA, USA; ab5690), anti-TdT (Abcam; ab85148), anti-NUCKS1 (Bethyl Laboratories, Montgomery, TX, USA; IHC-00303) and anti-β-galactosidase (Life Technologies, Carlsbad, CA, USA; AB_10055437). Secondary antibodies were biotinylated horse anti-mouse or anti-rabbit IgG (Vector Laboratories).
Immunophenotyping of primary TL cells by flow cytometry
Primary TL cells were isolated from dissected murine TLs and cultured in RPMI-1640 medium with 10% FBS and 1% antibiotics/antimycotics. Freshly isolated or sequentially expanded TL cells were used for cell surface marker staining. The following fluorochrome-conjugated primary antibodies (BD Biosciences, San Jose, CA, USA) were used: PE-Cy7-CD3e, PE-CD4, and APC-CD8a. A FACSCalibur flow cytometer (BD Biosciences) with CellQuest Software (BD Biosciences) was used for data acquisition. Live cells were gated based on forward and side scatter. Data analyses were done using FCS Express 4 software package (De Novo Software, Los Angeles, CA, USA).
PCR detection of Tcrb DJ rearrangements
Tcrb gene DJ rearrangements were analyzed as described [65]. Five and 50 ng of genomic DNA were used for semi-quantitative PCR to detect Tcrb D-J recombination. Primer sequences were as listed in Supplementary Table S1.
Statistical analyses
Comparisons between different groups of mice for tumor-free survival were made using a log-rank test (SPSS Statistics, IBM Software). Comparisons of Nucks1 expression between groups of mice were made using the Kruskal-Wallis test (SPSS Statistics, IBM Software) and Mann-Whitney test (Prism GraphPad). Otherwise, Fisher's exact or Student's t tests were used, as stated, to determine significance (Prism GraphPad).
For further Materials and Methods see Supplementary Information.
SUPPLEMENTARY MATERIALS FIGURES AND TABLES
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by National Institutes of Health grants R01ES021454 and R01CA116481, a U.S. Department of Energy LBNL LDRD grant, a grant from the Norwegian Cancer Society A02122/005, and by the Low Dose Scientific Focus Area, Office of Biological and Environmental Research, U.S. Department of Energy under Contract No. DE AC02-05CH11231.
REFERENCES
- 1.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 2.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu T, Huang J. Quality control of homologous recombination. Cell Mol Life Sci. 2014;71:3779–3797. doi: 10.1007/s00018-014-1649-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA repair. 2004;3:1187–1196. doi: 10.1016/j.dnarep.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 5.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 6.Bensimon A, Schmidt A, Ziv Y, Elkon R, Wang SY, Chen DJ, Aebersold R, Shiloh Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci Signal. 2010;3:rs3. doi: 10.1126/scisignal.2001034. [DOI] [PubMed] [Google Scholar]
- 7.Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, Andersen JS. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics. 2010;9:1314–1323. doi: 10.1074/mcp.M900616-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, Choudhary C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Molecular cell. 2012;46:212–225. doi: 10.1016/j.molcel.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elia AE, Boardman AP, Wang DC, Huttlin EL, Everley RA, Dephoure N, Zhou C, Koren I, Gygi SP, Elledge SJ. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Molecular cell. 2015;59:867–881. doi: 10.1016/j.molcel.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grundt K, Skjeldal L, Anthonsen HW, Skauge T, Huitfeldt HS, Ostvold AC. A putative DNA-binding domain in the NUCKS protein. Arch Biochem Biophys. 2002;407:168–175. doi: 10.1016/s0003-9861(02)00513-1. [DOI] [PubMed] [Google Scholar]
- 11.Grundt K, Haga IV, Aleporou-Marinou V, Drosos Y, Wanvik B, Ostvold AC. Characterisation of the NUCKS gene on human chromosome 1q32. 1 and the presence of a homologous gene in different species. Biochem Biophys Res Commun. 2004;323:796–801. doi: 10.1016/j.bbrc.2004.08.153. [DOI] [PubMed] [Google Scholar]
- 12.Grundt K, Haga IV, Huitfeldt HS, Ostvold AC. Identification and characterization of two putative nuclear localization signals (NLS) in the DNA-binding protein NUCKS. Biochim Biophys Acta. 2007;1773:1398–1406. doi: 10.1016/j.bbamcr.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 13.Wisniewski JR, Zougman A, Kruger S, Ziolkowski P, Pudelko M, Bebenek M, Mann M. Constitutive and dynamic phosphorylation and acetylation sites on NUCKS, a hypermodified nuclear protein, studied by quantitative proteomics. Proteins. 2008;73:710–718. doi: 10.1002/prot.22104. [DOI] [PubMed] [Google Scholar]
- 14.Parplys AC, Zhao W, Sharma N, Groesser T, Liang F, Maranon DG, Leung SG, Grundt K, Dray E, Idate R, Ostvold AC, Schild D, Sung P, Wiese C. NUCKS1 is a novel RAD51AP1 paralog important for homologous recombination and genome stability. Nucleic Acids Res. 2015;43:9817–9834. doi: 10.1093/nar/gkv859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Pan J, Li JL, Lee JH, Tunkey C, Saraf K, Garbe JC, Whitley MZ, Jelinsky SA, Stampfer MR, Haney SA. Transcriptional changes associated with breast cancer occur as normal human mammary epithelial cells overcome senescence barriers and become immortalized. Mol Cancer. 2007;6:7. doi: 10.1186/1476-4598-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drosos Y, Kouloukoussa M, Ostvold AC, Grundt K, Goutas N, Vlachodimitropoulos D, Havaki S, Kollia P, Kittas C, Marinos E, Aleporou-Marinou V. NUCKS overexpression in breast cancer. Cancer cell international. 2009;9:19. doi: 10.1186/1475-2867-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Symonowicz K, Dus-Szachniewicz K, Wozniak M, Murawski M, Kolodziej P, Osiecka B, Jurczyszyn K, Ziolkowski P. Immunohistochemical study of nuclear ubiquitous casein and cyclin-dependent kinase substrate 1 in invasive breast carcinoma of no special type. Experimental and therapeutic medicine. 2014;8:1039–1046. doi: 10.3892/etm.2014.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kikuchi A, Ishikawa T, Mogushi K, Ishiguro M, Iida S, Mizushima H, Uetake H, Tanaka H, Sugihara K. Identification of NUCKS1 as a colorectal cancer prognostic marker through integrated expression and copy number analysis. Int J Cancer. 2013;132:2295–2302. doi: 10.1002/ijc.27911. [DOI] [PubMed] [Google Scholar]
- 19.Gu L, Xia B, Zhong L, Ma Y, Liu L, Yang L, Lou G. NUCKS1 overexpression is a novel biomarker for recurrence-free survival in cervical squamous cell carcinoma. Tumour biology. 2014;35:7831–7836. doi: 10.1007/s13277-014-2035-5. [DOI] [PubMed] [Google Scholar]
- 20.Liu T, Tan S, Xu Y, Meng F, Yang C, Lou G. Increased NUCKS expression is a risk factor for poor prognosis and recurrence in endometrial cancer. Am J Cancer Res. 2015;5:3659–3667. [PMC free article] [PubMed] [Google Scholar]
- 21.Cheong JY, Kim YB, Woo JH, Kim DK, Yeo M, Yang SJ, Yang KS, Soon SK, Wang HJ, Kim BW, Park JH, Cho SW. Identification of NUCKS1 as a putative oncogene and immunodiagnostic marker of hepatocellular carcinoma. Gene. 2016 doi: 10.1016/j.gene.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Alizadeh AA, Bohen SP, Lossos C, Martinez-Climent JA, Ramos JC, Cubedo-Gil E, Harrington WJ, Jr, Lossos IS. Expression profiles of adult T-cell leukemia-lymphoma and associations with clinical responses to zidovudine and interferon alpha. Leuk Lymphoma. 2010;51:1200–1216. doi: 10.3109/10428191003728628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akbari Moqadam F, Boer JM, Lange-Turenhout EA, Pieters R, den Boer ML. Altered expression of miR-24, miR-126 and miR-365 does not affect viability of childhood TCF3-rearranged leukemia cells. Leukemia. 2014;28:1008–1014. doi: 10.1038/leu.2013.308. [DOI] [PubMed] [Google Scholar]
- 24.Smiraldo PG, Gruver AM, Osborn JC, Pittman DL. Extensive chromosomal instability in Rad51d-deficient mouse cells. Cancer Res. 2005;65:2089–2096. doi: 10.1158/0008-5472.CAN-04-2079. [DOI] [PubMed] [Google Scholar]
- 25.Kuznetsov SG, Haines DC, Martin BK, Sharan SK. Loss of Rad51c leads to embryonic lethality and modulation of Trp53-dependent tumorigenesis in mice. Cancer Res. 2009;69:863–872. doi: 10.1158/0008-5472.CAN-08-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsuji H, Ishii-Ohba H, Ukai H, Katsube T, Ogiu T. Radiation-induced deletions in the 5′ end region of Notch1 lead to the formation of truncated proteins and are involved in the development of mouse thymic lymphomas. Carcinogenesis. 2003;24:1257–1268. doi: 10.1093/carcin/bgg071. [DOI] [PubMed] [Google Scholar]
- 27.Tsuji H, Ishii-Ohba H, Katsube T, Ukai H, Aizawa S, Doi M, Hioki K, Ogiu T. Involvement of illegitimate V(D)J recombination or microhomology-mediated nonhomologous end-joining in the formation of intragenic deletions of the Notch1 gene in mouse thymic lymphomas. Cancer Res. 2004;64:8882–8890. doi: 10.1158/0008-5472.CAN-03-1163. [DOI] [PubMed] [Google Scholar]
- 28.Koch U, Radtke F. Notch in T-ALL: new players in a complex disease. Trends Immunol. 2011;32:434–442. doi: 10.1016/j.it.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Zha S, Bassing CH, Sanda T, Brush JW, Patel H, Goff PH, Murphy MM, Tepsuporn S, Gatti RA, Look AT, Alt FW. ATM-deficient thymic lymphoma is associated with aberrant tcrd rearrangement and gene amplification. The Journal of experimental medicine. 2010;207:1369–1380. doi: 10.1084/jem.20100285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deriano L, Chaumeil J, Coussens M, Multani A, Chou Y, Alekseyenko AV, Chang S, Skok JA, Roth DB. The RAG2 C terminus suppresses genomic instability and lymphomagenesis. Nature. 2011;471:119–123. doi: 10.1038/nature09755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimada Y, Nishimura M, Kakinuma S, Okumoto M, Shiroishi T, Clifton KH, Wakana S. Radiation-associated loss of heterozygosity at the Znfn1a1 (Ikaros) locus on chromosome 11 in murine thymic lymphomas. Radiat Res. 2000;154:293–300. doi: 10.1667/0033-7587(2000)154[0293:raloha]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Kubota T, Yoshikai Y, Tamura Y, Mishima Y, Aoyagi Y, Niwa O, Kominami R. Comparison of properties of spontaneous and radiation-induced mouse thymic lymphomas: role of Trp53 and radiation. Radiat Res. 2005;163:159–164. doi: 10.1667/rr3303. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi Y, Takabatake T, Kakinuma S, Amasaki Y, Nishimura M, Imaoka T, Yamauchi K, Shang Y, Miyoshi-Imamura T, Nogawa H, Kobayashi Y, Shimada Y. Complicated biallelic inactivation of Pten in radiation-induced mouse thymic lymphomas. Mutation research. 2010;686:30–38. doi: 10.1016/j.mrfmmm.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Mao JH, Wu D, Perez-Losada J, Nagase H, DelRosario R, Balmain A. Genetic interactions between Pten and p53 in radiation-induced lymphoma development. Oncogene. 2003;22:8379–8385. doi: 10.1038/sj.onc.1207083. [DOI] [PubMed] [Google Scholar]
- 35.Corson TW, Huang A, Tsao MS, Gallie BL. KIF14 is a candidate oncogene in the 1q minimal region of genomic gain in multiple cancers. Oncogene. 2005;24:4741–4753. doi: 10.1038/sj.onc.1208641. [DOI] [PubMed] [Google Scholar]
- 36.Cho EK, Tchinda J, Freeman JL, Chung YJ, Cai WW, Lee C. Array-based comparative genomic hybridization and copy number variation in cancer research. Cytogenet Genome Res. 2006;115:262–272. doi: 10.1159/000095923. [DOI] [PubMed] [Google Scholar]
- 37.Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, Pethiyagoda CL, Stuve LL, Johnson FM, Daly MJ, Wade CM, Cox DR. A sequence-based variation map of 8. 27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- 38.Bouwman P, Drost R, Klijn C, Pieterse M, van der Gulden H, Song JY, Szuhai K, Jonkers J. Loss of p53 partially rescues embryonic development of Palb2 knockout mice but does not foster haploinsufficiency of Palb2 in tumour suppression. J Pathol. 2011;224:10–21. doi: 10.1002/path.2861. [DOI] [PubMed] [Google Scholar]
- 39.Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A. Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes & development. 1997;11:1226–1241. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- 40.Bennett LM, McAllister KA, Blackshear PE, Malphurs J, Goulding G, Collins NK, Ward T, Bunch DO, Eddy EM, Davis BJ, Wiseman RW. BRCA2-null embryonic survival is prolonged on the BALB/c genetic background. Mol Carcinog. 2000;28:174–183. doi: 10.1002/1098-2744(200007)28:3<174::aid-mc6>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 41.Jen KY, Song IY, Banta KL, Wu D, Mao JH, Balmain A. Sequential mutations in Notch1, Fbxw7, and Tp53 in radiation-induced mouse thymic lymphomas. Blood. 2012;119:805–809. doi: 10.1182/blood-2011-01-327619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mao JH, Wu D, DelRosario R, Castellanos A, Balmain A, Perez-Losada J. Atm heterozygosity does not increase tumor susceptibility to ionizing radiation alone or in a p53 heterozygous background. Oncogene. 2008;27:6596–6600. doi: 10.1038/onc.2008.280. [DOI] [PubMed] [Google Scholar]
- 43.Attardi LD, Jacks T. The role of p53 in tumour suppression: lessons from mouse models. Cell Mol Life Sci. 1999;55:48–63. doi: 10.1007/s000180050269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller PA, Vousden KH, Norman JC. p53 and its mutants in tumor cell migration and invasion. J Cell Biol. 2011;192:209–218. doi: 10.1083/jcb.201009059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Javadekar SM, Raghavan SC. Snaps and mends: DNA breaks and chromosomal translocations. FEBS J. 2015;282:2627–2645. doi: 10.1111/febs.13311. [DOI] [PubMed] [Google Scholar]
- 46.Dasika GK, Lin SC, Zhao S, Sung P, Tomkinson A, Lee EY. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene. 1999;18:7883–7899. doi: 10.1038/sj.onc.1203283. [DOI] [PubMed] [Google Scholar]
- 47.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 48.Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- 49.Jacobs C, Huang Y, Masud T, Lu W, Westfield G, Giblin W, Sekiguchi JM. A hypomorphic Artemis human disease allele causes aberrant chromosomal rearrangements and tumorigenesis. Hum Mol Genet. 2011;20:806–819. doi: 10.1093/hmg/ddq524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bi X, Feng D, Korczeniewska J, Alper N, Hu G, Barnes BJ. Deletion of Irf5 protects hematopoietic stem cells from DNA damage-induced apoptosis and suppresses gamma-irradiation-induced thymic lymphomagenesis. Oncogene. 2014;33:3288–3297. doi: 10.1038/onc.2013.295. [DOI] [PubMed] [Google Scholar]
- 51.Prochazkova J, Loizou JI. Programmed DNA breaks in lymphoid cells: repair mechanisms and consequences in human disease. Immunology. 2016;147:11–20. doi: 10.1111/imm.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–779. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- 53.DeMicco A, Yang-Iott K, Bassing CH. Somatic inactivation of Tp53 in hematopoietic stem cells or thymocytes predisposes mice to thymic lymphomas with clonal translocations. Cell cycle (Georgetown, Tex) 2013;12:3307–3316. doi: 10.4161/cc.26299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chikuma S. Basics of PD-1 in self-tolerance, infection, and cancer immunity. Int J Clin Oncol. 2016;21:448–455. doi: 10.1007/s10147-016-0958-0. [DOI] [PubMed] [Google Scholar]
- 55.Kemp CJ, Wheldon T, Balmain A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet. 1994;8:66–69. doi: 10.1038/ng0994-66. [DOI] [PubMed] [Google Scholar]
- 56.Naylor TL, Greshock J, Wang Y, Colligon T, Yu QC, Clemmer V, Zaks TZ, Weber BL. High resolution genomic analysis of sporadic breast cancer using array-based comparative genomic hybridization. Breast Cancer Res. 2005;7:R1186–1198. doi: 10.1186/bcr1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soliman NA, Zineldeen DH, El-Khadrawy OH. Effect of NUCKS-1 overexpression on cytokine profiling in obese women with breast cancer. Asian Pac J Cancer Prev. 2014;15:837–845. doi: 10.7314/apjcp.2014.15.2.837. [DOI] [PubMed] [Google Scholar]
- 58.Sargent LM, Ensell MX, Ostvold AC, Baldwin KT, Kashon ML, Lowry DT, Senft JR, Jefferson AM, Johnson RC, Li Z, Tyson FL, Reynolds SH. Chromosomal changes in high- and low-invasive mouse lung adenocarcinoma cell strains derived from early passage mouse lung adenocarcinoma cell strains. Toxicol Appl Pharmacol. 2008;233:81–91. doi: 10.1016/j.taap.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 59.Yang M, Wang X, Zhao Q, Liu T, Yao G, Chen W, Li Z, Huang X, Zhang Y. Combined evaluation of the expression of NUCKS and Ki-67 proteins as independent prognostic factors for patients with gastric adenocarcinoma. Tumour biology. 2014;35:7505–7512. doi: 10.1007/s13277-014-1880-6. [DOI] [PubMed] [Google Scholar]
- 60.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 61.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 62.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 63.Qiu B, Shi X, Wong ET, Lim J, Bezzi M, Low D, Zhou Q, Akincilar SC, Lakshmanan M, Swa HL, Tham JM, Gunaratne J, Cheng KK, Hong W, Lam KS, Ikawa M, et al. NUCKS is a positive transcriptional regulator of insulin signaling. Cell reports. 2014;7:1876–1886. doi: 10.1016/j.celrep.2014.05.030. [DOI] [PubMed] [Google Scholar]
- 64.Qiu B, Shi X, Zhou Q, Chen HS, Lim J, Han W, Tergaonkar V. Hypothalamic NUCKS regulates peripheral glucose homoeostasis. Biochem J. 2015;469:391–398. doi: 10.1042/BJ20150450. [DOI] [PubMed] [Google Scholar]
- 65.Wang X, Xiao G, Zhang Y, Wen X, Gao X, Okada S, Liu X. Regulation of Tcrb recombination ordering by c-Fos-dependent RAG deposition. Nature immunology. 2008;9:794–801. doi: 10.1038/ni.1614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.