

Abstract
Objectives
Esophageal squamous cell carcinoma (ESCC) is one of the most aggressive malignancies owing to the high frequency of tumor recurrence. The identification of markers for early ESCC diagnosis and prediction of recurrence is expected to improve the long-term prognosis. Therefore, we searched for associations between tumor recurrence and cell-free DNA (cfDNA) mutations in blood plasma, which contains genetic markers for various cancer types.
Experimental Design
Genomic DNA from tumors and cfDNA from plasma were obtained from 13 patients undergoing treatment for newly diagnosed ESCC. Next-generation sequencing of cfDNA in plasma was performed to identify mutations in 53 cancer-related genes, in which recurrent mutations were previously detected in ESCC. cfDNA mutational profiles were compared before and after tumor resection in four patients. Furthermore, somatic mutations in serial plasma samples were monitored after treatment in four patients.
Results
We identified multiple concordant somatic mutations in cfDNA and primary tumor samples from 10 patients (83.3%) and in cfDNA and metastatic tumor samples from one patient (100%). Furthermore, the allele frequency of the concordant mutations in cfDNA changed concomitantly with tumor burden and increased approximately 6 months earlier than the detection of tumor recurrences by imaging tests in two patients. Conventional biomarkers, such as SCC and p53-Ab, did not reflect tumor recurrences.
Conclusions
The present multigene panel, which enabled the diagnosis of tumor recurrence with greater accuracy than did using standard tumor markers or imaging methods, is expected to greatly facilitate standard, postoperative follow-up monitoring in ESCC.
Keywords: esophageal squamous cell carcinoma, cell-free DNA, next-generation sequencing, tumor recurrence, somatic mutation
INTRODUCTION
Esophageal squamous cell carcinoma (ESCC), the most common subtype of esophageal cancer in East Asia, is one of the most aggressive malignancies owing to the high frequency of tumor recurrence [1, 2]. The identification of biomarkers for the accurate evaluation of tumor burden and early detection of tumor recurrence is crucial to ensure that effective therapy is administered in a timely manner [3].
Cell-free DNA (cfDNA) is present in the blood as small DNA fragments; these fragments can be released from dying tumor cells of not only primary tumors, but also metastatic tumors [4, 5]. Based on next-generation sequencing (NGS); droplet digital PCR; and beads, emulsion, amplification, and magnetics (BEAMing), cfDNA harbors genetic alterations associated with various malignancies [6–11]. Therefore, cfDNA, which may be obtained in a facile and non-invasive manner via “liquid biopsy,” is a potential source of diagnostic markers for the precise and early detection of ESCC.
In our previous study, we identified genes with recurrent mutations in Japanese ESCC patients by NGS [12]. Moreover, recent studies have provided important insights into the mutational landscape of ESCC and have identified recurrent mutations in driver genes [13–15]. The somatic mutations of such driver genes span large regions of the genome. Thus, it is difficult to examine mutations in these driver genes by PCR-based assays, and NGS analyses are more suitable for analyzing larger regions.
In this study, we constructed a panel of 53 previously identified driver genes in ESCC. We prospectively collected tumor and serial plasma samples and conducted NGS of the samples with the multigene panel. The aim of our study was to investigate the utility of NGS-based cfDNA analyses for the identification of clinically useful ESCC biomarkers.
RESULTS
Patient characteristics
The strategy adopted to assess the clinical utility of cfDNA in ESCC is shown in Figure 1A; 13 patients participated in our study. Genomic DNA from primary tumors (12 patients) or a recurrent tumor (one patient) and matched cfDNA samples were analyzed. cfDNAs from four patients (two with recurrence, two without) were obtained before and after surgery and monitored during follow-up. The clinical characteristics of 13 patients are shown in Table 1. We profiled 64 samples, including 12 primary tumor samples, 1 metastatic tumor sample, 38 plasma samples, and 13 matched normal tissue samples.
Figure 1.
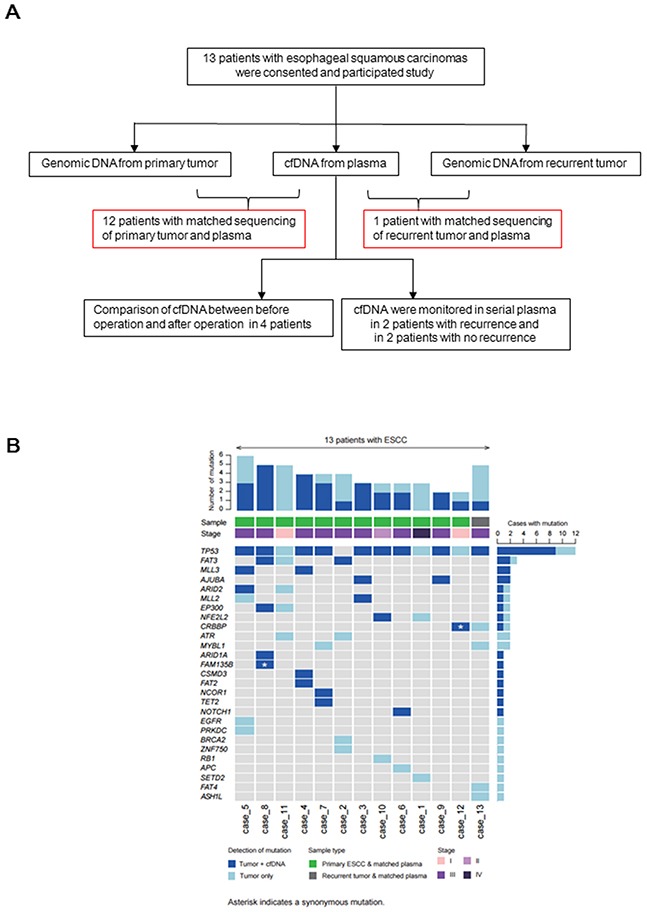
A. Study outline to assess the clinical utility of cfDNA in esophageal squamous cell carcinoma (ESCC); 13 patients participated in our study. Genomic DNA from primary tumors (12 patients) or a recurrent tumor (1 patient) and matched cfDNA samples were analyzed. Comparison of cfDNA before and after surgery in four patients; two patients with recurrence or no recurrence were monitored during follow-up. B. The number of somatic mutations in 53 genes in the tumor DNA and cfDNA samples from 13 patients (top), pathological (p)-stage and type of tumor sample (middle), and each mutated gene in the left column (bottom) are indicated.
Table 1. Characteristics of 13 patients in our study.
| Patient number | Age | Sex | Location | p-Stage | T | N | M | Differentiation | ly | v | Residual tumor after surgery | NAC | TP53 status in primary tumor | Recurrence | Tumor source |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 60 | Male | Lt | IV | 3 | 1 | 1 | Moderate | 1 | 0 | R0 | DCF | Mutant | − | Primary |
| 2 | 64 | Male | Mt | III A | 3 | 1 | 0 | Poor | 1 | 1 | R0 | DCF | Wild | − | Primary |
| 3 | 79 | Male | Lt | III A | 3 | 1 | 0 | Well | 1 | 1 | R0 | DCF | Mutant | − | Primary |
| 4 | 57 | Male | Lt | III C | 3 | 3 | 0 | Moderate | 1 | 1 | R0 | DCF | Mutant | + | Primary |
| 5 | 66 | Male | Lt | III B | 3 | 2 | 0 | Moderate | 1 | 1 | R0 | DCF | Mutant | − | Primary |
| 6 | 72 | Male | Ut | III A | 3 | 1 | 0 | Moderate | 1 | 0 | R0 | DCF | Mutant | + | Primary |
| 7 | 77 | Male | Mt | III B | 3 | 2 | 0 | Well | 1 | 1 | R0 | DCF | Mutant | − | Primary |
| 8 | 67 | Male | Ut Mt | III A | 3 | 1 | 0 | Moderate | 2 | 2 | R0 | − | Mutant | − | Primary |
| 9 | 67 | Male | Lt | III B | 3 | 2 | 0 | Moderate | 1 | 1 | R0 | DCF | Mutant | − | Primary |
| 10 | 48 | Male | Mt | II B | 2 | 1 | 0 | Moderate | 1 | 0 | R0 | DCF | Mutant | − | Primary |
| 11 | 53 | Female | Mt | I A | 1 | 0 | 0 | Well | 1 | 1 | R0 | − | Mutant | − | Primary |
| 12 | 64 | Male | Mt Lt | I A | 1 | 0 | 0 | Well | 0 | 0 | R0 | − | Mutant | − | Primary |
| 13 | 66 | Male | Mt | III C | 3 | 3 | 0 | Well | 2 | 1 | R0 | DCF | — | + | Recurrent |
NAC: Neoadjuvant chemotherapy
DCF: Docetaxel + Cisplatin + Fluorouracil
Concordant somatic mutations between tumor samples and plasma samples
We examined the mutational profiles of 53 genes in primary and metastatic tumor samples from 13 patients (Figure 1B and Supplementary Table S1) and identified 56 tumor-specific somatic mutations (mean = 4.3 mutations per patient), including genes with recurrent somatic alterations, e.g., TP53 (92.3%) and FAT3 (23.0%). The TP53H61L mutation was detected in only two patients. Next, we evaluated whether somatic mutations identified in tumor samples could be detected in the matched cfDNA samples. Twenty-nine somatic mutations detected in cfDNA samples were concordant with those in the primary or recurrent tumors (51.7%) (Figure 1B and Supplementary Table S1). We identified more than one concordant somatic mutation in cfDNA and primary tumor samples from 10 patients (83.3%) and in cfDNA and a recurrent tumor sample from one patient (100%). Among these, one concordant somatic mutation (CREBBP) was detected in a stage IA patient. Furthermore, to find more accurate biomarkers for identifying ESCC patients, we assessed the diagnostic utility of four genes (TP53, FAT3, MLL3, and AJUBA), mutations in which are the most recurrently detected. Mutations in these four genes were identified with 78.9% sensitivity, 100% specificity, and 92.3% accuracy in tumor and cfDNA samples from the patients (Table 2).
Table 2. Calculation of sensitivity, specificity, and diagnostic accuracy of cfDNA analysis for 4 genes.
| Tumor | Sensitivity | Specificity | PPV | NPV | Accuracy | |||
|---|---|---|---|---|---|---|---|---|
| Mutant | Wild | (%) | (%) | (%) | (%) | (%) | ||
| TP53 | positive | 9 | 0 | 75 | 100 | 100 | 25 | 76.9 |
| cfDNA | negative | 3 | 1 | |||||
| FAT3 | positive | 2 | 0 | 66.6 | 100 | 100 | 90.9 | 92.3 |
| cfDNA | negative | 1 | 10 | |||||
| MLL3 | positive | 2 | 0 | 100 | 100 | 100 | 100 | 100 |
| cfDNA | negative | 0 | 11 | |||||
| AJUBA | positive | 2 | 0 | 100 | 100 | 100 | 100 | 100 |
| cfDNA | negative | 0 | 11 | |||||
| Total | positive | 15 | 0 | 78.9 | 100 | 100 | 89.1 | 92.3 |
| cfDNA | negative | 4 | 33 | |||||
Changes in the frequency of somatic mutations in cfDNA reflect changes in tumor burden
To elucidate whether somatic mutations in cfDNA were quantitatively correlated with tumor burden, we monitored somatic mutations in serial plasma samples (Supplementary Table S2). First, we compared the allele frequencies (AFs) of concordant mutations identified in pretreatment cfDNA samples with those of mutations in post-treatment cfDNA in four cases. In four patients, the AFs of all concordant somatic mutations decreased remarkably (Figure 2). Next, we investigated the association between tumor progression and AFs in cfDNA. In case 13, the AF of the TP53E207X mutation, which was concordant in recurrent tumor and matched cfDNA samples, remained extremely low until recurrence, at which point a 3.6% increase was observed (Figure 3). However, the concentration levels of cfDNA did not reflect either tumor reduction or tumor recurrence, which is consistent with previous findings [6]. These data suggested that concordant mutations in cfDNA change concomitantly with tumor burden.
Figure 2. Comparison of AFs of concordant mutations before and after treatment by targeted sequencing of cfDNA from four patients; changes in conventional biomarkers and AFs of the concordant mutations after neoadjuvant chemotherapy and surgery in case 2.
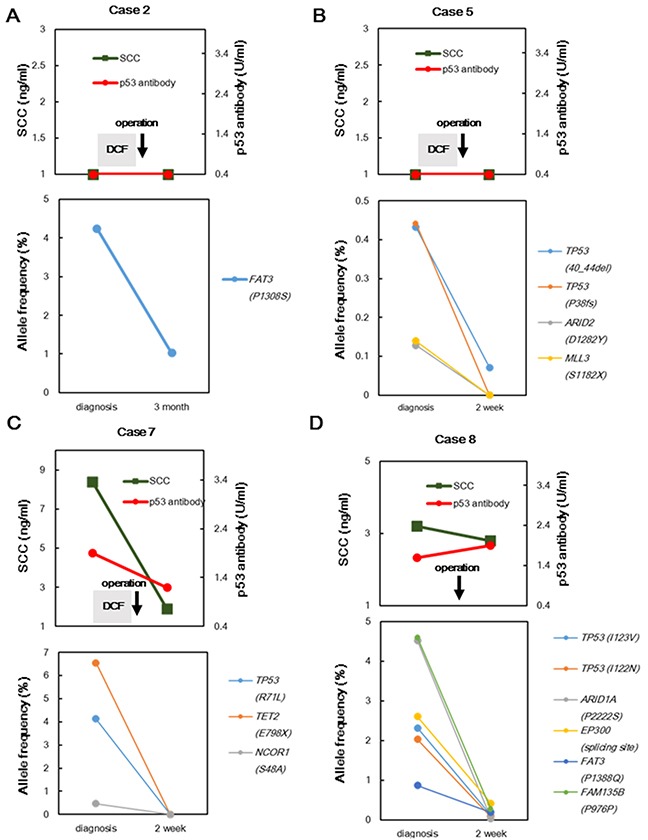
A. case 5 B. case 7 C. and case 8 D. SCC: squamous cell carcinoma-related antigen; DCF: docetaxel, cisplatin, and fluorouracil.
Figure 3. Comparison between before and after recurrence using targeted sequencing of cfDNA for case 13.
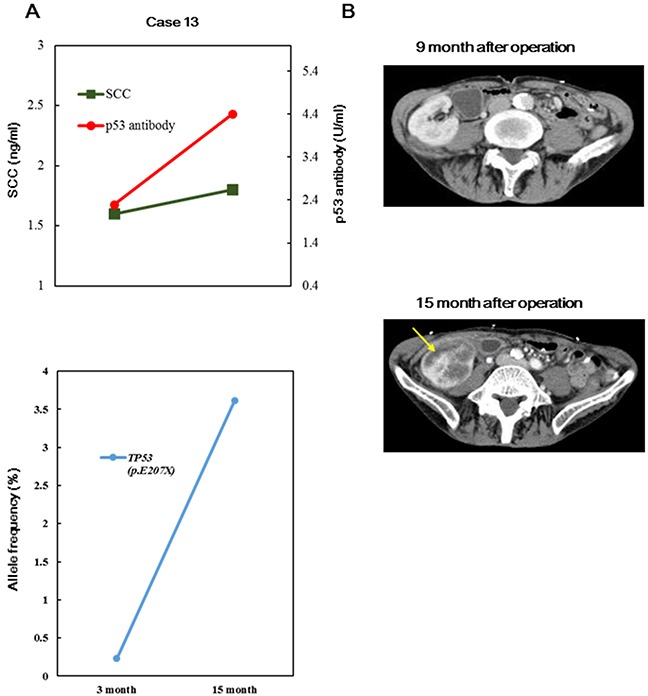
A, B. Changes in conventional biomarkers, AFs of concordant mutations, and computed tomography (CT) findings before and after recurrence are indicated. Yellow allows indicate the recurrence of tumors in the kidney. SCC: squamous cell carcinoma-related antigen; DCF: docetaxel, cisplatin, and fluorouracil
Monitoring somatic mutations in cfDNA for postoperative follow-up in ESCC
Next, we assessed whether cfDNA could provide clinically meaningful follow-up information for ESCC. We tracked serial samples after treatment in four patients. During the follow-up period, two patients (cases 4 and 6) had tumor recurrences, whereas two patients (cases 1 and 9) showed no recurrences. In case 4, SCC and p53-Ab were negative during the entire follow-up period and all concordant mutations were absent after treatment; however, the AFs of all concordant mutations increased 9 months before recurrence was detected by imaging tests (Figures 4A and 4B). Furthermore, in case 6, the AF of the TP53R141C mutation decreased, and NOTCHW327C was not detected after neoadjuvant chemotherapy and surgery (Figures 4C and 4D). Intriguingly, although the NOTCHW327C mutation remained absent during follow-up, the AF of TP53R141C gradually increased 6 months before hepatic metastasis was detected. SCC and p53-Ab levels remained within the normal range during the follow-up period. However, different results were observed in the two patients with no tumor recurrence. In one patient with non-recurrence (case 9), the concordant mutations did not increase in frequency during the follow-up period (Figure 5A). In the other patient with non-recurrence (case 1), somatic mutations derived from the primary tumor were also absent in serial plasma during the follow-up period (Figure 5B).
Figure 4.
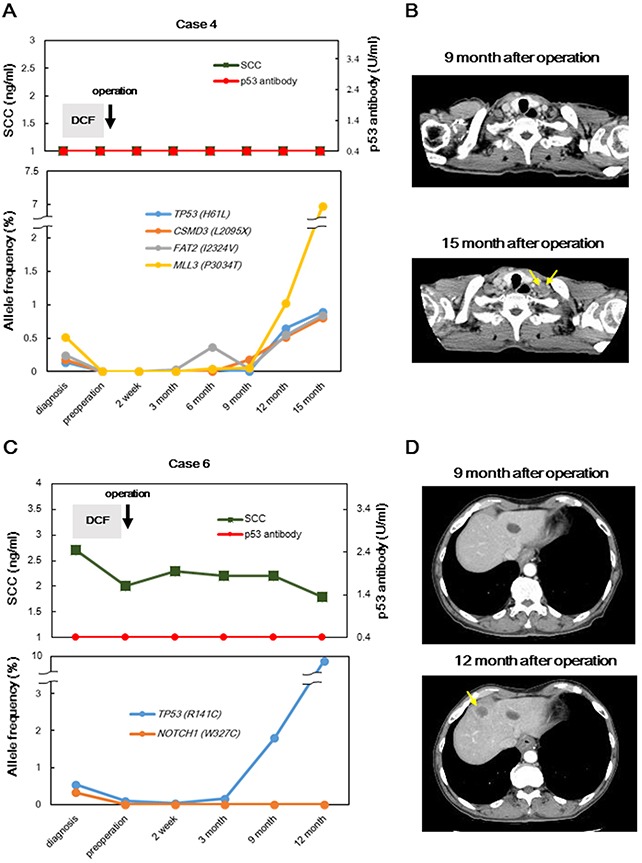
Disease monitoring in two patients with recurrences, from diagnosis to tumor recurrence, by targeted sequencing of cfDNA samples; changes in conventional biomarkers and AFs of concordant mutations and CT findings before and after recurrence in case 4 A, B. and case 6 C, D. are indicated. Yellow allows indicate recurrent tumors. AFs of non-concordant mutations were not detected in serial plasma samples in four patients. SCC: squamous cell carcinoma-related antigen; DCF: docetaxel, cisplatin, and fluorouracil.
Figure 5.
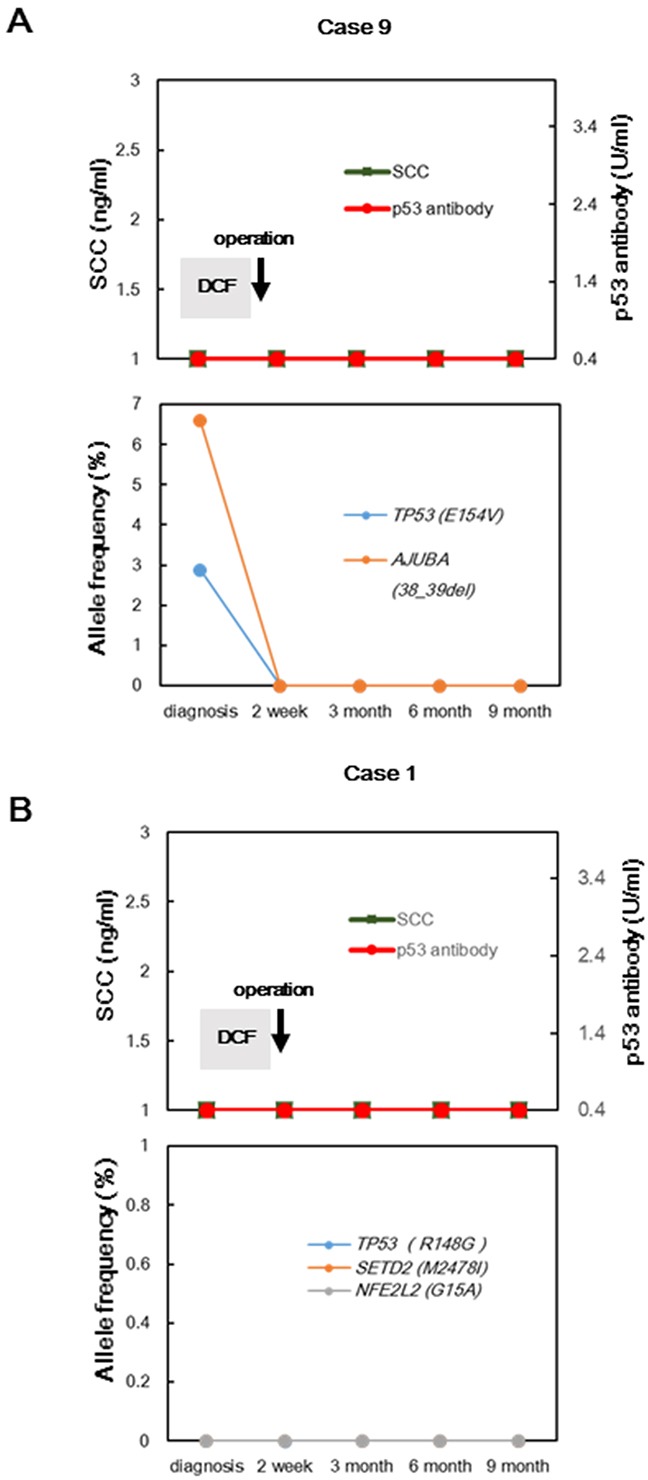
A. Disease monitoring in two patients with no recurrence during follow-up using targeted sequencing of cfDNA; changes in conventional biomarkers and AFs of concordant mutations for case 9; AFs of non-concordant mutations were not detected in serial plasma. B. Changes in conventional biomarkers and AFs of mutations detected only in primary tumor samples from case 1.
DISCUSSION
In the current study, we applied NGS with a multigene panel to detect mutations in plasma samples in ESCC. Recent exome studies in ESCC have identified mutations in driver genes such as TP53, FAT3, MLL3, and AJUBA. Furthermore, plasma contains a very small quantity of cfDNA, which makes it difficult to detect mutant fractions emerging from malignant tissues. Therefore, we amplified cfDNA samples for NGS and focused on 53 genes; this approach enabled deeper sequencing coverage. In our cohort, 55 somatic mutations, but not TP53H61L, in tumor samples were detected in a single individual, indicating that NGS is adequate to examine mutational profiles in ESCC. This is consistent with the results of several recent studies using cfDNA [6, 10], suggesting that NGS with a gene panel can be used to effectively identify tumor-derived somatic mutations in various cancer types, using plasma samples.
In contrast to the study of other malignancies such as colorectal cancer, we found a clinical advantage in sequencing cfDNA from patients with ESCC to identify mutations in target genes. We previously observed diverse mutations in driver genes in colorectal cancer among patients with distinctive inter-tumor heterogeneity [16]. Therefore, it is difficult to establish a genetic panel for patients with colorectal cancer that is broadly informative. However, almost all patients with ESCC had mutations of one gene out of the top four genes (Table 2). We attained deep sequence coverage, which enabled us to accurately measure the tumor burden; therefore, using cfDNA is appropriate for ESCC cases.
We identified somatic mutations derived not only from primary tumors, but also from recurrent tumors, in cfDNA with high sensitivity and accuracy, suggesting that the genetic profiles of cfDNA can accurately reflect the status of tumors. Moreover, our analysis demonstrated that the AFs of concordant mutations in serial plasma samples are useful not only for evaluating the tumor status, but also for predicting tumor recurrence in ESCC. In particular, we demonstrated an increased frequency of concordant somatic mutations in cfDNA 6 months earlier than tumor recurrences were detected based on imaging tests in two patients. Similarly, Sausen et al. reported that mutations in cfDNA in pancreatic adenocarcinoma could be detected approximately 6 months before the detection of recurrences by imaging tests, which supported the utility of cfDNA analyses in ESCC in predicting tumor recurrences [11]. These data suggested that somatic mutations in cfDNA may reflect minor tumors that are not detectable by imaging tests and, accordingly, may provide a basis for the development of a diagnostic tool in ESCC. In this study, 1 concordant somatic mutation in CREBBP was detected in a stage IA patient. CREBBP mutations have been reported both in various solid cancers and in hematologic malignancies. In ESCC, CREBBP mutations and deletions of have been recurrently detected, and CREBBP acetyltransferase activities may be tumor suppressive [12–20]. CREBBP encodes a highly conserved and ubiquitously expressed nuclear phosphoprotein that belongs to the KAT3 family of histone/protein lysine acetyltransferases. CREBBP, together with the closely related protein EP300, also function as transcription factors involved in multiple signaling and developmental pathways by modifying lysine residues on both histone and non-histone nuclear proteins [21–24]. We propose that CREBBP might serve a fundamental function in ESCC tumorigenesis and that mutant CREBBP alleles might be frequently released into the bloodstream, as observed in the case of mutated TP53.
The AF of mutations in cfDNA was more strongly associated with tumor burden than conventional biomarkers of ESCC. Interestingly, in the two patients with tumor recurrence, the p53-Ab levels did not increase during recurrence, which was not consistent with the AF of the TP53 mutation. Although p53-Ab is a circulating antibody against the mutated p53 protein in serum, its sensitivity is remarkably low according to a meta-analysis [25, 26]. In recent studies, it was demonstrated that ESCC is associated with various hotspot mutations in TP53 [12–15]. In general, the p53-Ab is applied to treat tumors expressing mutated TP53 protein. The mutated TP53 gene produces alternative epitopes in the variant TP53 proteins. In contrast, the limited epitopes recognized by the TP53-Ab cannot distinguish the variety of esophageal cancer cells with diverse TP53 gene mutations. These results suggest that the AFs of somatic mutations in cfDNA accurately reflect tumor status and may be superior to standard biomarkers currently used in ESCC.
Several mutations were detected in tumor samples, but not in cfDNA, and such mutations were absent in serial plasma samples from all patients. Furthermore, in case 6, although the TP53R141C mutation gradually increased before tumor recurrence was detected, the NOTCHW327C mutation remained absent during follow-up. We inferred that these results may reflect tumor heterogeneity. Heterogeneity exists within tumors and is an important factor underlying low therapeutic efficacy [27]. Tumors evolve by processes of branched evolution via the acquisition of genetic and epigenetic alterations, leading to the formation of various clones of cancer cells, including genetically distinct subclones, which contribute to tumor heterogeneity. Mutations detected in tumor samples, but not in cfDNA, may be derived from subclones of tumor samples and may rarely affect the formation of metastatic lesions. Thus, considering that tumor heterogeneity has an influence on tumor-derived mutations in cfDNA, it is important to investigate several driver genes, including TP53, related to ESCC in cfDNA. In addition, in case 1, we did not detect concordant somatic mutations despite the fact that the patient had stage IV ESCC. There is no definitive answer for this issue; however, we speculate that the outcome depends on the diversity of tumors in primary cases. According to the cancer-evolution model [16], greater diversity of mutations in primary tumors is observed in cancers in stage IV patients than in cancers from stage I–III patients. Therefore, the detectability of mutations in cfDNA derived from primary tumors might be reduced in patients with stage IV ESCC, compared to that in other patients. In addition, it is necessary to increase the accuracy of technologies used for cfDNA analysis.
In conclusion, although additional studies analyzing a larger number of samples are required, our findings suggest that NGS using a multigene panel is an effective method for detecting somatic mutations in plasma cfDNA. Our data support the use of cfDNA in clinical assessments of the tumor burden and suggest that cfDNA analysis may help predict tumor recurrence in ESCC.
MATERIALS AND METHODS
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the authors' institutional review board.
Patient selection and sample collection
The study included 13 patients who were undergoing treatment for newly diagnosed ESCC between September 2013 and August 2015. The patients provided informed consent, and the study was approved by the Department of Gastroenterological Surgery (Osaka University) and the Department of Surgery (Kyushu University Beppu Hospital). All patients underwent tumor biopsy through an upper endoscopy. The diagnosis of primary ESCC was confirmed by histologic review. In 12 patients, tumor samples by biopsy were obtained from patients before they underwent neoadjuvant chemotherapy. For germline controls, adjacent esophageal normal tissue was obtained. In one patient (case 13), a biopsy sample was not collected because the primary tumor was too small. No patients received adjuvant chemotherapy following radical surgery. Plasma samples were routinely collected at the time of initial diagnosis, at the time of surgery in the operation room, at postoperative day 14, and at follow-up. p53 antibody (p53-Ab) and squamous cell carcinoma (SCC)-related antigen were measured simultaneously as conventional biomarkers [25, 28]. In three patients, tumor recurrences were identified by computed tomography tests. In 1 patient (case 13), the sample was collected from a recurrent tumor.
Sample processing and DNA extraction
Plasma samples obtained from patients were collected in EDTA tubes. Plasma was centrifuged at 2500 × g for 10 min, added to microcentrifuge tubes, and further centrifuged at 16,000 × g for 10 min to remove debris. DNA was extracted from plasma with the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany), per the manufacturer's protocol. DNA from tumor samples and the corresponding normal samples were extracted using the QIAamp DNA Mini Kit (Qiagen), following the manufacturer's protocol.
Next-generation sequencing library construction
Indexed Illumina NGS libraries were prepared from plasma, tumor, and germline DNA. Plasma DNA was used for library construction without additional fragmentation. Tumor and germline genomic DNA were sheared before library construction with a Covaris S2 instrument (Woburn, MA, USA) to obtain 200-bp fragments. The NGS libraries of plasma DNA were constructed using the KAPA Hyper Prep Kit (Kapa Biosystems, Wilmington, MA, USA), following the manufacturer's protocols. A sequence library was prepared using a combination of the KAPA Hyper Prep Kit (Kapa Biosystems) and the SureSelect Target Enrichment System (Agilent Technologies, Santa Clara, CA, USA). End repair and A-tailing reactions were performed in 60-μL reaction volumes. The mixtures were incubated at 20°C for 30 minutes and 65°C for 30 minutes. Adapter ligation was performed using 110 μL and samples were incubated at 16°C for 16 hours using Agilent SureSelect Adapter. After post-ligation cleanup, the ligated fragments were amplified in 50 μL containing 2× KAPA HiFi HotStart ReadyMix and 10× KAPA Library Amplification Primer Mix. The following cycling protocol was used: 98°C for 45 s; 14–16 cycles (depending on the input DNA mass) of 98°C for 15 s, 65°C for 30 s, and 72°C for 30 s; and 1 cycle of 72°C for 5 min. Library purity, library concentration, and fragment length were determined using a 2100 Bioanalyzer (Agilent).
Targeted massively parallel sequencing
Tumor, germline, and plasma DNA extracted from the samples of ESCC patients were captured using SureSelectXT Custom 1Kb-499kb, 16 (Agilent Technology), following the manufacturer's instructions. A panel of 53 genes was designed; recurrent mutations were previously identified in these genes, which are listed as potential driver genes in the Catalogue of Somatic Mutations in Cancer [12–15, 29] (Supplementary Table S3). The captured DNA was sequenced using the HiSeq2000 to generate paired-end (75–100 bp) reads for each sample. Targeted deep sequencing was performed for all samples using the multigene panel for a mean sequencing depth of 3810×.
Mutation calling for genomic DNA from primary tumor and metastatic tumor samples
The sequence data for genomic DNA from tumor samples were processed using an in-house pipeline (http://genomon.hgc.jp/exome/). The sequencing reads were aligned to the NCBI Human Reference Genome Build 37 hg19 with BWA version 0.5.10 using default parameters (http://bio-bwa.sourceforge.net/). Mutation calling was conducted using the following parameters: (i) mapping quality score ≥ 25, (ii) base quality score ≥ 15, (iii) mismatched bases ≤ 5, (iv) both tumor and normal depths ≥ 100, (v) variant allele frequencies in tumor samples ≥ 0.05, (vi) variant allele frequencies in normal samples ≤ 0.05, and (vii) Fisher's exact test P-values < 0.05.
Identification of mutations in plasma DNA
The sequence data for cfDNA from plasma samples were aligned to NCBI Human Reference Genome Build 37 hg19, following the same methods for genomic DNA obtained from tumors. Mutation calling was performed only at the positions with mutations detected in genomic DNA from tumor samples. The following parameters were used: (i) mapping quality score ≥ 25, (ii) base quality score ≥ 15, (iii) mismatched bases ≤ 5, (iv) cfDNA depth ≥ 100, (v) variant allele frequencies for cfDNA samples ≥ 0.0005, (vi) numbers of reads supporting mutation in cfDNA ≥ 2, and (vii) Fisher's exact test P-values < 0.05.
SUPPLEMENTARY TABLES
Acknowledgments
This research was performed using computational resources of the K computer provided by the RIKEN Advanced Institute for Computational Science through the HPCI System Research project (Project ID: hp140230). Computational time was also provided by the Supercomputer System, Human Genome Center, Institute of Medical Science, and University of Tokyo. We would like to thank Ms. Fusano Todokoro, Ms. Kiyomi Imamura, Ms. Terumi Horiuchi, and Ms. Yuni Ishikawa for providing technical assistance with next-generation sequencing. In addition, we would like to thank Ms. M. Kasagi, T. Ms. Kohno, Ms. K. Oda, Ms. Aoyagi, and Ms. T Kawano for providing assistance with molecular biology procedures.
Footnotes
CONFLICTS OF INTEREST
None of the authors have any potential conflicts of interest.
FINANCIAL SUPPORT
Funding was provided by Grants-in-Aid for Scientific Research (grant numbers 26461980, 26670608, 26861003, 15H04921, 15H05791, 15K10168, and 15K10170) and the Funding Program for Next Generation World-Leading Researchers (LS094).
REFERENCES
- 1.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 2.Shimada H, Kitabayashi H, Nabeya Y, Okazumi S, Matsubara H, Funami Y, Miyazawa Y, Shiratori T, Uno T, Itoh H, Ochiai T. Treatment response and prognosis of patients after recurrence of esophageal cancer. Surgery. 2003;133:24–31. doi: 10.1067/msy.2003.31. [DOI] [PubMed] [Google Scholar]
- 3.Abate E, DeMeester SR, Zehetner J, Oezcelik A, Ayazi S, Costales J, Banki F, Lipham JC, Hagen JA, DeMeester TR. Recurrence after esophagectomy for adenocarcinoma: defining optimal follow-up intervals and testing. J Am Coll Surg. 2010;210:428–435. doi: 10.1016/j.jamcollsurg.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 5.Murtaza M, Dawson SJ, Pogrebniak K, Rueda OM, Provenzano E, Grant J, Chin SF, Tsui DW, Marass F, Gale D, Ali HR, Shah P, Contente-Cuomo T, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015;6:8760. doi: 10.1038/ncomms9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW, Jr, Alizadeh AA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M, Hendifar AE, Wang Z, Atreya CE, Kelley RK, Van Loon K, Ko AH, Tempero MA, et al. Cell-free DNA next-generation sequencing in pancreatobiliary carcinomas. Cancer Discov. 2015;5:1040–1048. doi: 10.1158/2159-8290.CD-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 10.Takai E, Totoki Y, Nakamura H, Morizane C, Nara S, Hama N, Suzuki M, Furukawa E, Kato M, Hayashi H, Kohno T, Ueno H, Shimada K, et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci Rep. 2015;5:18425. doi: 10.1038/srep18425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, Hruban CA, Scharpf R, White JR, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;6:7686. doi: 10.1038/ncomms8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawada G, Niida A, Uchi R, Hirata H, Shimamura H, Suzuki Y, Shiraishi Y, Chiba K, Imoto S, Takahashi Y, Iwaya T, Sudo T, Hayashi T, et al. The genomic landscape of Japanese esophageal squamous cell carcinoma. Gastroenterology. 2016;150:1171–1182. doi: 10.1053/j.gastro.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 13.Song Y, Li L, Ou Y, Gao Z, Li E, Li X, Zhang W, Wang J, Xu L, Zhou Y, Ma X, Liu L, Zhao Z, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–95. doi: 10.1038/nature13176. [DOI] [PubMed] [Google Scholar]
- 14.Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, Garg M, Liu LZ, Yang H, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46:467–473. doi: 10.1038/ng.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, Zhao YD, Sun J, Zhou CC, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 16.Uchi R, Takahashi Y, Niida A, Shimamura T, Hirata H, Sugimachi K, Sawada G, Iwaya T, Kurashige J, Shinden Y, Iguchi T, Eguchi H, Chiba K, et al. Integrated multiregional analysis proposing a new model of colorectal cancer evolution. PLoS Genet. 2016;12:e1005778. doi: 10.1371/journal.pgen.1005778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, Heatley SL, Holmfeldt L, Collins-Underwood JR, Ma J, Buetow KH, Pui CH, Baker SD, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–239. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, Rossi D, Chadburn A, Murty VV, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, He M, Li Z, Sun X, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kishimoto M, Kohno T, Okudela K, Otsuka A, Sasaki H, Tanabe C, Sakiyama T, Hirama C, Kitabayashi I, Minna JD, Takenoshita S, Yokota J. Mutations and deletions of the CBP gene in human lung cancer. Clin Cancer Res. 2005;11:512–519. [PubMed] [Google Scholar]
- 21.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 22.Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145–1155. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 23.Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 24.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 25.Angelopoulou K, Diamandis EP, Sutherland DJ, Kellen JA, Bunting PS. Prevalence of serum antibodies against the p53 tumor suppressor gene protein in various cancers. Int J Cancer. 1994;58:480–487. doi: 10.1002/ijc.2910580404. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Xv Z, Wu X, Li K. Potential diagnostic value of serum p53 antibody for detecting esophageal cancer: a meta-analysis. PLoS One. 2012;7:e52896. doi: 10.1371/journal.pone.0052896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;28:141. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Shimada H, Nabeya Y, Okazumi S, Matsubara H, Shiratori T, Gunji Y, Kobayashi S, Hayashi H, Ochiai T. Prediction of survival with squamous cell carcinoma antigen in patients with resectable esophageal squamous cell carcinoma. Surgery. 2003;133:486–494. doi: 10.1067/msy.2003.139. [DOI] [PubMed] [Google Scholar]
- 29.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.