

Abstract
Purpose
To evaluate consanguineous pedigrees from Pakistan with a clinical diagnosis of nonsyndromic congenital retinal nonattachment (NCRNA) and identify genes responsible for the disease as currently only one NCRNA gene is known (atonal basic helix-loop-helix transcription factor 7: ATOH7).
Methods
We implemented a three-step genotyping platform: single nucleotide polymorphism genotyping to identify loss of heterozygosity regions in patients, Retinal Information Network panel screening for mutations in currently known retinal genes. Negative patients were then subjected to whole exome sequencing.
Results
We evaluated 21 consanguineous NCRNA pedigrees and identified the causal mutations in known retinal genes in 13 out of our 21 families. We found mutations in ATOH7 in three families. Surprisingly, we then found mutations in familial exudative vitreoretinopathy (FEVR) genes; low-density lipoprotein receptor-related protein 5 mutations (six families), tetraspanin 12 mutations (two families), and NDP mutations (two families). Thus, 62% of the patients were successfully genotyped in our study with seven novel and six previously reported mutations in known retinal genes.
Conclusions
Although the clinical diagnosis of all children was NCRNA with severe congenital fibrotic retinal detachments, the molecular diagnosis determined that the disease process was in fact a very severe form of FEVR in 10 families. Because severe congenital retinal detachment has not been previously associated with all the FEVR genes, we have thus expanded the phenotypic spectrum of FEVR, a highly variable retinal detachment phenotype that has clinical overlap with NCRNA. We identified seven novel mutations. We also established for the first time genetic overlap between the Iranian and Pakistani populations. We identified eight NCRNA families that do not harbor mutations in any known retinal genes, suggesting novel causal genes in these families.
Keywords: ATOH7, LRP5, TSPAN12, NDP, FEVR, NCRNA, blindness, retinal detachments
Nonsyndromic congenital retinal nonattachment (NCRNA, OMIM #221900) is an autosomal recessive, devastating congenital form of retinal detachment leading to permanent blindness from birth. Children with NCRNA have congenital blindness caused by partial to complete retinal detachments. The main clinical findings are congenital insensitivity to light, nystagmus, microphthalmia, shallow anterior chamber (AC), and a massive fibrovascular mass behind the lens.1,2 Nonsyndromic congenital retinal nonattachment is reported to be clinically and genetically heterogeneous. The retina and blood vessels in one or both eyes display variable clinical and histologic anomalies. Systemic malformations associated with the disease have been shown in some individuals; however, no consistent association has been noticed.1
Autosomal recessive NCRNA is common in North Khorasan, Iran, among descendants of a Kurdish population. In this founder population, the locus was mapped to chromosome 10q21.1,3 Subsequently, a homozygous (hmz) 6523-bp deletion was found in atonal basic helix-loop-helix (BHLH) transcription factor 7 ([ATOH7] also known as Math5), in a cluster of three conserved noncoding elements that spans a remote cis regulatory element 20-kb upstream from ATOH7.1 Atonal BHLH transcription factor 7 encodes a member of the BHLH family of transcription factors that plays an important role in the development of retinal ganglion cell (RGC) and optic nerve formation.4,5 It was determined that the absence of RGCs creates neovascular growth of fetal blood vessels in the vitreous that results in early retinal detachment.1 Thus, the intrinsic mechanism of retinal cell fate determination is disrupted in NCRNA, which in turn leads to early onset retinal detachment and subsequent blindness.1
In addition to NCRNA, mild and partial acquired retinal detachments can occur in patients with familial exudative vitreoretinopathy (FEVR), a rare progressive genetic disorder affecting retinal blood vessel development.6 This disorder is characterized by partial vascularization of the peripheral retina and defective vascular differentiation.6 All modes of inheritance are seen in FEVR patients.6 The clinical presentation of FEVR patients is highly variable; most have mild disease, including neovascularization, exudation, dragged vessels and macula, retinal tears, and rhegmatogenous retinal detachments, which is not seen until the first or second decade of life.6,7 Usually, FEVR is an acquired condition (not congenital such as NCRNA). Some patients present with falciform retinal folds extending from the optic nerve to the temporal periphery, which sometimes comes in contact with the lens, causing leukocoria.8,9
Mutations in five genes have been reported to cause FEVR; NDP, a gene encoding Norrie disease protein (X-linked), FZD4, a gene encoding frizzled-4 (autosomal dominant and recessive), LRP5, a gene encoding low-density lipoprotein receptor-related protein 5 (autosomal dominant and recessive), TSPAN12, a gene encoding tetraspanin-12 (autosomal dominant and recessive), and ZNF408, a gene encoding zinc finger protein 408 (autosomal dominant).6,8,10–12 Individuals affected by mutations in the LRP5 gene often have reduced bone mineral density, osteopenia, and osteoporosis in addition to vision loss.6,8,13 Mutations of FEVR are associated with a wide range of phenotypes, ranging from mild retinal folds and avascular zones to later onset retinal detachments. Currently, FEVR mutations are not associated with congenital total retinal detachments and complete congenital blindness.
Because ATOH is currently the only gene associated with NCRNA, we aimed to identify the genetic causes of NCRNA in a Northern Pakistani population cohort, and at the same time validate a new gene discovery protocol established by our laboratories. In this study, we identified that mutations in genes associated previously with FEVR can result in the NCRNA phenotype. Our study suggests that although NCRNA and FEVR are classified as distinct diseases, a margin of overlap exists between the two. Thus, we expanded the phenotypic spectrum of FEVR. We also determined that a three-step gene discovery protocol established by our laboratories is highly effective for the identification of new genes and the elimination of previously known disease associated genes in Mendelian disorders.
Methods
This study was approved by the institutional review boards of the participating centers, according to the tenets of the Declaration of Helsinki. Informed consent was obtained from all patients at the initial visit in Pakistan at the El Shifa Trust Eye Hospital, where two of our clinical collaborators saw the babies (AK, SS) and another oversaw the DNA extractions (RQ); the consent form contained a paragraph about genetic analyses of retinal genes, including whole exome sequencing. Twenty-one NCRNA families (28 affected members) were recruited, phenotyped, and genotyped from Pakistan and included in this study. Entry criteria were as follows: the diagnosis of NCRNA was given to patients with the history of congenital blindness because of partial to complete retinal detachment from infancy, shallow anterior chamber, leukocoria, and abnormal B-scan. All children included in this study were born full term, free from uncomplicated pregnancies, and did not have any known medical issues.
All families were questioned for detailed, ocular, and visual histories, and pedigrees were drawn. Ophthalmic examinations were performed on all affected patients. Blood samples from all available NCRNA family members were collected at the Al Shifa Trust Eye Hospital Department of Pediatric Ophthalmology and Strabismus, Rawalpindi, Pakistan. Extraction of DNA from the NCRNA families and additional blood samples from 100 random unrelated healthy Pakistani control individuals were provided by the COMSATS Institute of Information Technology Department of Biosciences, Rawalpindi, Pakistan, for determination of allele frequency in the general Pakistani population.
DNA Extraction and Primer Design
Genomic DNA was extracted from peripheral blood leukocytes with an extraction kit (FlexiGene; QIAGEN, Hilden, Germany) and a blood kit (QIAamp DNA; QIAGEN), according to the instructions by the manufacturer. DNA quantity and quality was verified by spectrophotometer (NanoDrop 1000; Thermo Fisher Scientific, Wilmington, DE, USA). We used a PCR and Sanger sequencing approach to identify mutations in candidate genes. Primers were designed by Exon Primer (provided in the public domain by the Institute for Human Genetics, Technical University of Munich, Germany (http://ihg.gsf.de/ihg/ ExonPrimer.html/) and by Primer3 online program.14,15 To ensure the completeness and quality of the sequences and for detection of potential mutations located in splice sites, a minimal distance between primer and exon/intron boundary was selected of at least 60 bp when primers were designed. Sequences for the used primers are available upon request.
Step 1: Single Nucleotide Polymorphism (SNP) Microarrays
The genomes of 11 out of our 17 consanguineous NCRNA families were analyzed for homozygous chromosomal regions by using SNP homozygosity mapping (Infinium HD 660K; Illumina, Inc., San Diego, CA, USA), in accordance with the protocol provided by the manufacturer. Homozygous regions were visualized and identified with a commercial software package (Bead Studio pLink; Illumina, Inc.) and a freeware spreadsheet, ExcludeAR. Chromosomal segments were accepted as having significant homozygous regions if they contained ≥300 consecutive homozygous SNPs, as the likelihood that this would occur by chance is less than 1:100 with a particular focus to identified homozygous regions. All the known retinal genes (the retinal genes listed in Retinal Network information website) that resided in homozygous chromosomal segments were analyzed for mutations using Sanger sequencing.
Step 2: RetNet Known Retinal Disease Gene Panel Screening by Targeted Next Generation Sequencing (NGS)
Fourteen patients diagnosed with NCRNA were screened by our recently developed RetNet known retinal disease gene panel,16 which contains all the currently known retinal disease genes (163) by targeted NGS. The chip helped us to screen all retinal genes listed in the Retinal Information Network website and create a candidate retinal disease gene list for future study.
Step 3: Whole Exome Sequencing
We performed whole exome capture following the manufacturer's protocol (Illumina, Inc.). We used 2 μg of link-ligated DNA for the capture reaction. Exome capture (SureSelect V4; Agilent Technologies, Santa Clara, CA, USA) was performed according to the company's standard protocol. The amplified capture product was sequenced with commercial software (HiSeq 2000; Illumina, Inc.) to pinpoint the genetic variants responsible for the disease in those patients who did not carry mutations in known genes. Sequence capture was performed in accordance with the protocol of the manufacturer (Illumina, Inc.) in the sequencing platform of the McGill University and Genome Quebec Innovation Centre. Details of the protocol are described by Coussa et al.17
Mutation Analysis
Known retinal genes within the homozygous regions, retinal detachment genes shown in RetNet (Retinal Information Network, tables of genes and loci causing inherited retinal diseases), and all putative mutations found by RetNet panel screening were Sanger-sequenced. Complete sequencing of the candidate genes within the homozygous regions was done to exclude all possible mutations. If a patient had several homozygous regions carrying different known genes for the given disease, the screening of the genes was started from a gene located in the biggest homozygous segment.
Sanger Sequencing
Sanger sequencing was used to identify mutations in candidate genes. Primers and PCR conditions are available upon request. The assays were performed by the sequencing platform of the McGill University and Genome Quebec Innovation Centre using forward or reverse sequencing primers designed for PCR reactions that covered a designated exons and splice sites of the exons. The results were analyzed using an NGS software program (Sequencher; Gene Codes Corporation, Ann Arbor, MI, USA).
In Silico Mutational Analyses
For novel mutations found, we performed in silico analyses predicting the variants to be damaging by SIFT,18 Blosum62,19 and Polyphen-2.20 We then studied conservation of the residue in homologous proteins of lower animals and we verified that the variant has not been reported in the Exome Variant Server (NHLBI GO Exome Sequencing Project), dbSNP135, or 1000 Genome datasets. Finally, segregation of the mutations between family members was then performed and we excluded the newly found variant from normal controls that are culturally matched to the original family.
Results
In collaboration with a research group in Islamabad, Pakistan, we assembled 21 consanguineous pedigrees from Pakistan with an NCRNA diagnosis. Consanguineous families have homozygous regions, one of which contains the causal gene with homozygous mutations. This fact allows for a genetic strategy that is well established, called homozygosity mapping and inheritance by descent (IBD) studies. Our approach to finding the causal genes in NCRNA patients was to first identify all the significant homozygous regions in the probands (patients) and then look for overlap of all these regions with all affected members of that family, followed by searching for overlap with other families. This three-step process allows us to narrow and decrease the number of homozygous regions. The remaining overlapping regions can then be probed for candidate genes that are expressed in retina, using available databases.
In a preliminary study of our cohort, we screened families for known NCRNA and retinal detachment genes (including FEVR genes) which resided in one of the top five homozygous segments documented by the SNP array. We hypothesized that the causal gene for NCRNA resides in one of the top five largest intervals of our identified homozygous regions in consanguineous families and a new causal gene underlying the disease may locate in the overlapping homozygous regions of different family members with the same disease phenotype.
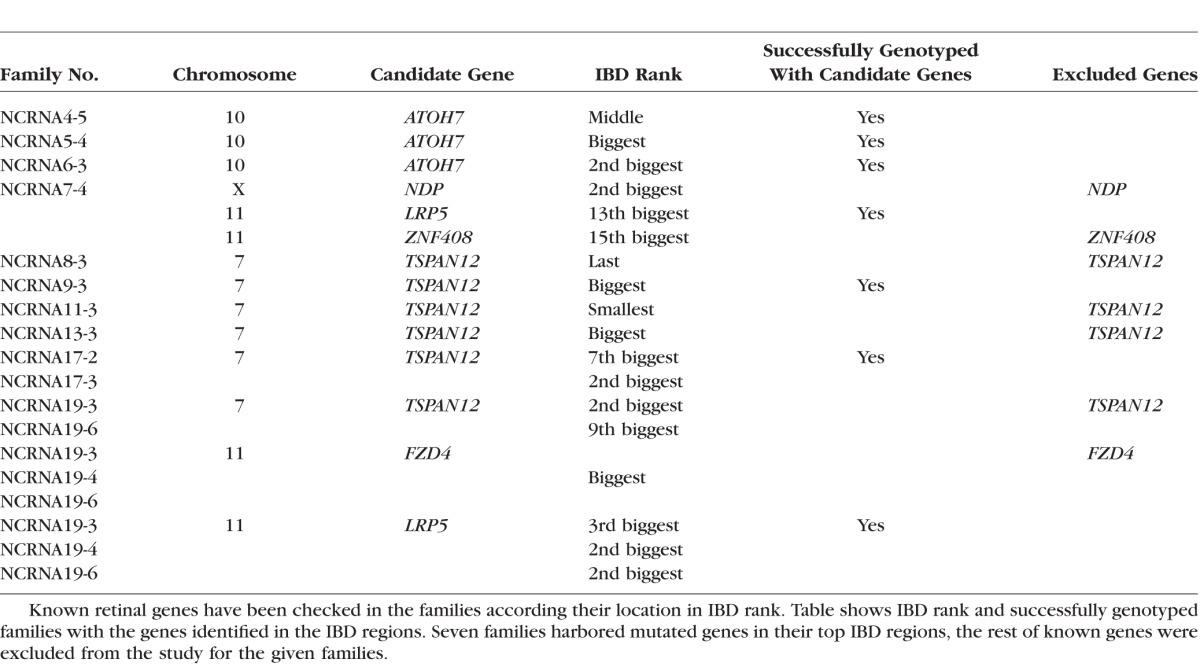
Analyzes of homozygous regions of the patients and direct sequencing of the candidate genes located in top five regions helped us to successfully genotype seven NCRNA families with known genes located in the regions of interest (Table 1).
Table 1.
Candidate Genes for NCRNA Families in Their IBD Regions
Clinical Findings
The affected individuals presented with the history of congenital blindness and presented with searching roving eye movements. Anterior segment examination showed small eyes. Some patients had cloudy corneas due to iridocorneal adhesion, shallow anterior chamber due to forward displacement of lens iris diaphragm, and irregular pupils secondary to posterior synechiae and leukocoria (white pupils); but there was no consistent elevation of intraocular pressure. Fundoscopy, when possible, revealed a dense fibrovasular mass behind the lens in each eye, originating from the optic nerve and attached behind the lens and peripheral fundus. In some cases, B-scan ultrasonography showed hyperechoic shadows involving the entire vitreous cavity. Other cases showed total retinal detachments and fibrous bands originating from optic nerve posteriorly and attached anteriorly behind the lens.
No other sensory or neurologic defects were detected in the affected individuals. Their auditory, olfactory, endocrine, and intellectual functions were intact. Together, these clinical findings suggest bilateral persistence of the fetal blood vessels, with complete congenital detachment of the neural retina and intravitreal vascular proliferation during infancy. The obligate heterozygotes are clinically normal. They have good vision, with no structural or functional eye pathology.
Known Genes for NCRNA; ATOH7.
In our 21 Pakistani NCRNA families, only three families had ATOH7 in their largest IBD regions (our mutation analysis confirmed this). Surprisingly, two of these families harbored the reported deletion in ATOH7, which is the well-known, common mutation in the founder population of North Khorasan, in Iran.1 Eight out of our 21 NCRNA families did not harbor mutations in ATOH7, nor in any other retinal detachment gene in their homozygous regions, suggesting that we have families with new gene/s for NCRNA. To be sure, we screened all our patients for ATOH7 exonic and promoter mutations by Sanger sequencing. Two families (NCRNA5 and NCRNA6) were found to have the well-known 6523-bp deletion located 20 kb upstream from ATOH7 (phenotypes of the patients are shown in Fig. 1). Segregation analyses for these families were conducted as described by Ghiasvand et al.1 (see Fig. 2). We also found a novel homozygous missense mutation p. Arg42Pro, altering a highly conserved arginine for a proline residue in ATOH7 in the third family (NCRNA4). This is the first missense mutation reported in ATOH7. In silico analyses predicts that this mutation is probably damaging (Table 2) and conservation analysis shows the significance of the residue (Fig. 3). All identified mutations are shown in Table 3.
Figure 1.
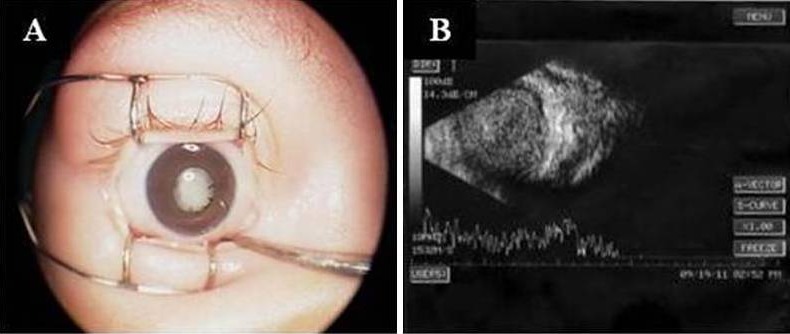
Anterior segment and B scan of NCRNA5-4. Family NCRNA5 was found to have the well-known 6523-bp deletion located 20 kb upstream from ATOH7. Anterior segment with leukocoria (A) and disorganized B scan showing retrolental mass with total vitreous condensation (B) caused by the variation in ATOH7 gene in NCRNA5-4.
Figure 2.
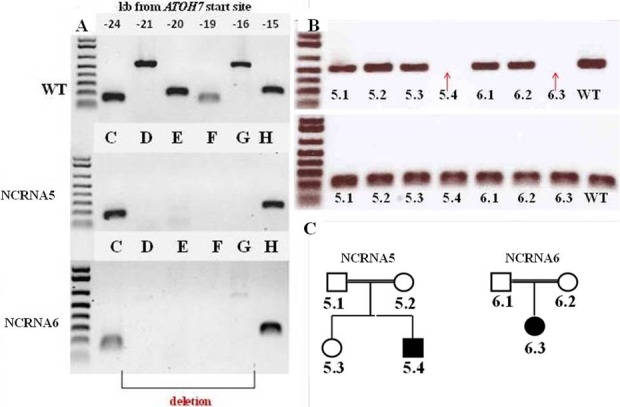
Polymerase chain reaction–based genotyping of homozygous 6523-kb deletion in 5′ATOH7 in NCRNA5 and NCRNA6 families. (A) A polymerase chain reaction–based deletion confirmation in the NCRNA5 and NCRNA6 families conducted as described by Ghiasvand et al.1 Letters (C, D, E, F, G, H) represent six adjacent amplicons in an NCRNA subject that amplify conserved noncoding elements ∼20 kb upstream of ATOH7 transcription start site. As it can be seen from the figure, C and H amplicons are present in both wild-type (WT) and mutant subjects, while the rest of the amplicons are missing in NCRNA5 and NCRNA6 family members. (B) Cosegregation results between family members using primers designed by Ghiasvand et al.1 In the first panel, primers for G and H amplicons are pooled, whereas in the second panel, only primers for H amplicons were used to verify if deletion of the area is present in the members of the family. Red arrows show deletion in affected family members compared with a WT control band. (C) Pedigrees of NCRNA5 and NCRNA6 families.
Table 2.
Summary of Analysis Done by Bioinformatics Tools for Novel Mutations
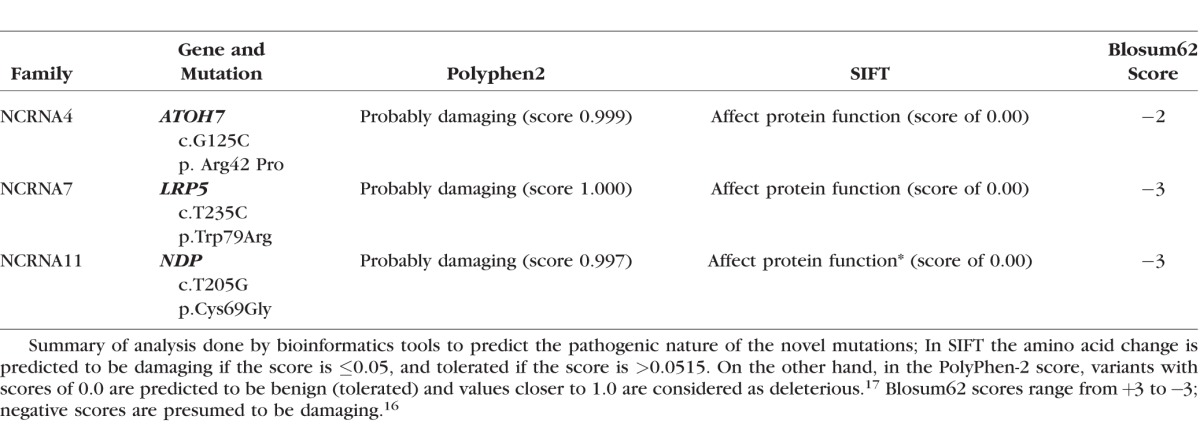
Figure 3.

Mutation conservation analysis of ATOH7. We found a novel homozygous missense mutation p. Arg42Pro, altering a highly conserved arginine for a proline residue in ATOH7 in the NCRNA4 family; this is the first missense mutation reported in ATOH7. In silico analyses predicts that this mutation is probably damaging (Table 2) and conservation analysis shows the significance of the residue.
Table 3.
Summary of Successfully Genotyped NCRNA Families
Mutations in LRP5.
The gene LRP5 encodes a transmembrane low-density lipoprotein receptor and plays a key role in eye development, in skeletal homeostasis, and many bone density–related diseases (LRP5, OMIM #603506).
Although the LRP5 gene is known for mild retinal detachments and folds and was located in IBD regions of the patients, FEVR was not considered as the primary diagnosis since these patients presented with congenital blindness. However, LRP5 was the only attractive retinal candidate gene in the region, and Sanger sequencing identified the mutations in the gene.
We found five homozygous and one heterozygous (htz) mutations in the FEVR gene LRP5; namely, two members of each NCRNA2 (NCRNA2-5 and NCRNA2-6) and NCRNA3 (NCRNA3-4 and NCRNA3-7) families were carrying the reported p.Gly610Arg and p.Asp434Asn homozygous mutations, respectively. A member from a third family (NCRNA7-4) had a novel homozygous p.Trp79Arg mutation. We then identified the reported homozygous p.Gly1401Asp mutation in three members of a single family (NCRNA19-3, 19-4, and 19-6). Panel screening with RetNet then identified htz and hmz LRP5 mutations in an additional two families (NCRNA16 and NCRNA20, respectively); NCRNA16 has the previously reported p.Arg142Gln change and the NCRNA20 family carries a novel p.Leu541Pro alteration. Cosegregation of these mutations in the family members could not been checked.
A summary of the LRP5 mutations is given in Table 3. Since the p.Gly1401Asp mutation in LRP5 found in the NCRNA19 family members has previously been reported to cause osteoporosis-pseudoglioma,10 we checked bone mineral density (BMD) in patient NCRNA19-4 and the result confirmed that the patient has reduced BMD and osteoporosis. The rest of the families were not able to come back for the given test.
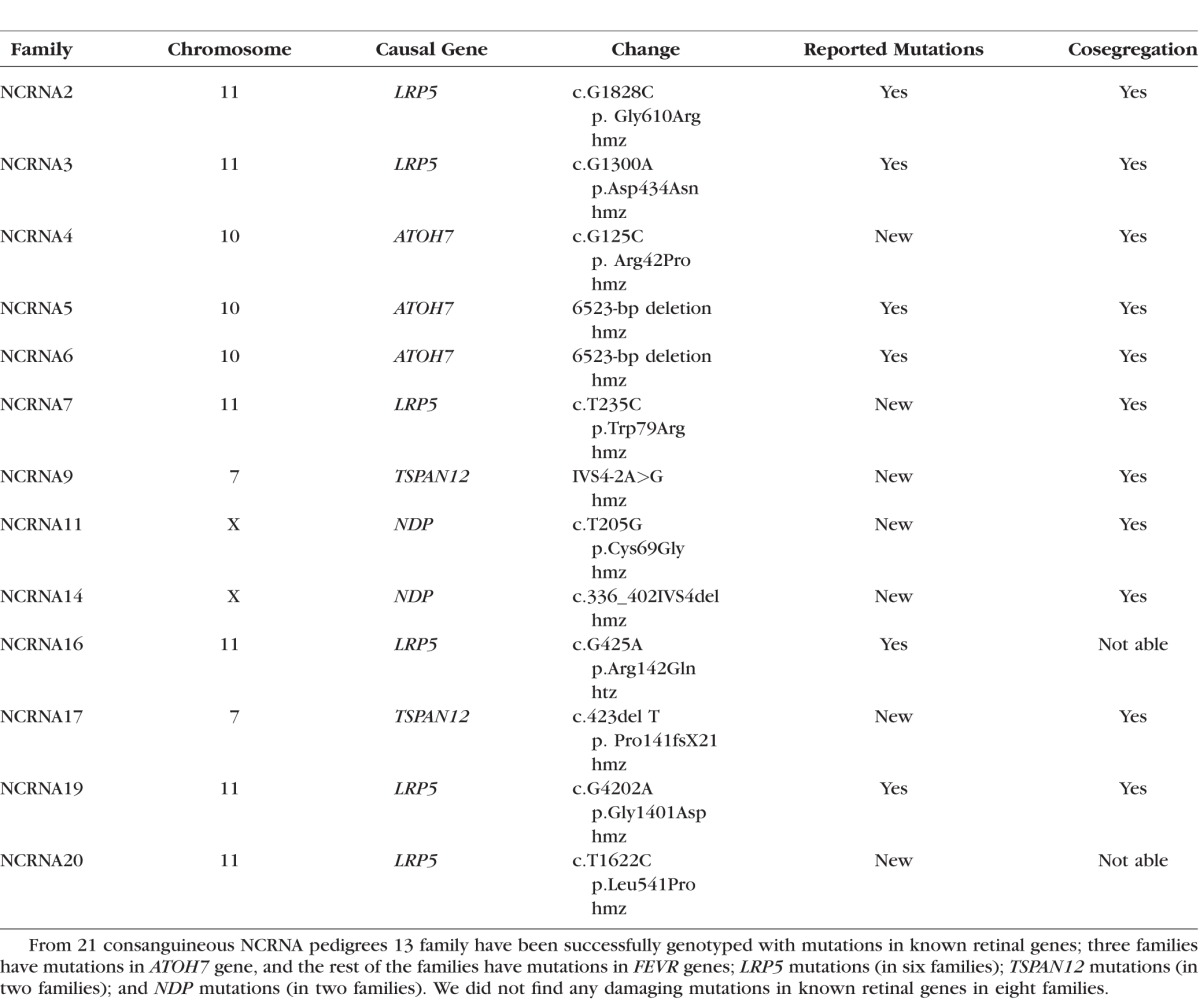
The eye phenotype in our study consisted of congenital blindness with congenital retinal detachments and corneal opacity, posterior synechia, shallow AC, due to anteriorly displaced lens and iris, leukocoria, retrolenticular fibrovascular plaque, retinal folds (in some patients), and complete retinal detachment, unlike the current phenotypes associated with LRP5 type FEVR (Fig. 4). The altered residues are all evolutionarily conserved (Fig. 5). We then documented delayed growth and skeletal abnormalities in some patients in these families (Fig. 4). Pedigrees for successfully genotyped NCRNA2, NCRNA3, NCRNA7 and NCRNA19 family positive for homozygous LRP5 mutations are shown in Figure 6.
Figure 4.
Eye phenotype for NCRNA2, NCRNA3, NCRNA7 and NCRNA19 families and skeletal abnormalities found in a NCRNA19 family member. (A) The fundus photo shows the fibrovascular fold arising from the optic nerve and extending to the retrolental area in the temporal region in NCRNA2-5; family NCRNA2 has been identified to carry a homozygous p. Gly610Arg alteration in LRP5 gene. (B) Fundus photograph of NCRNA3-4 show hemorrhage retinal fold with a fibrovascular mass behind the lens which is a typical presentation of NCRNA1 caused by p.Asp434Asn mutation in LRP5 gene in NCRNA3-4 proband. (C, D) Anterior segment and B-scan for NCRNA7-4. (C) Total leukocoria. (D) B-scan shows atrophy of the detached retina. (E) A member of NCRNA19 family with p. Gly1401Asp mutation showing dense corneal opacity. (F) Skeletal abnormalities (NCRNA19-4).
Figure 5.

Mutation conservation analysis for LRP5. We identified a novel homozygous p.Trp79Arg mutation in family NCRNA7. Conservation analysis showed that the altered residue is evolutionarily conserved in many species.
Figure 6.
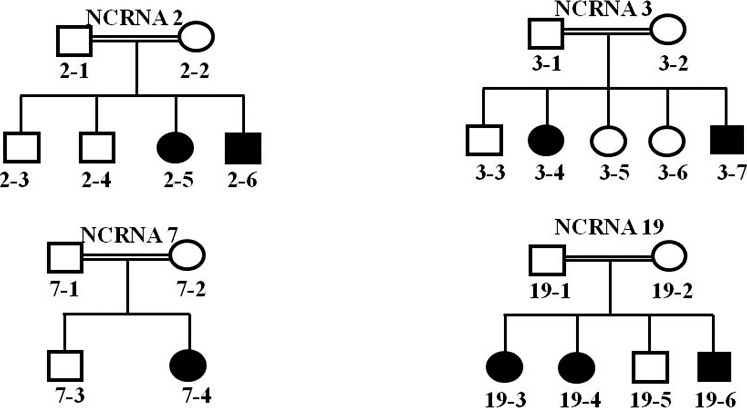
Successfully genotyped NCRNA2, NCRNA3, NCRNA7 and NCRNA19 family pedigrees positive for homozygous LRP5 mutations. Four families carrying mutation in LRP5 gene were consanguineous.
To confirm the molecular diagnosis, we performed NGS in these patients and excluded other possible mutations. It is important to note that this is the first report relating LRP5 mutations with the NCRNA phenotype.
Mutations in TSPAN12.
The gene TSPAN12 encodes a cell-surface protein that is characterized by the presence of four hydrophobic domains and is predicted to mediate signal transduction events that play a role in the regulation of cell development, activation, growth and motility (TSPAN12, OMIM#613138). Tetraspanin-12 is currently known to cause autosomal dominant and recessive FEVR,21–23 but not autosomal recessive NCRNA, and it is not known to cause severe retinal detachments.
We identified two new TSPAN12 mutations for the first time in NCRNA. We confirmed a novel homozygous splice site mutation, IVS4-2A>G and a novel homozygous frameshift mutation, p. Pro141fsX21 in TSPAN12 in two of our families (NCRNA9 and NCRNA17; Table 3). The Berkeley Drosophila Genome Project–splice site predictor analysis tool showed that the IVS4-2 A>G mutation found in NCRNA9 deletes the acceptor site. Cosegregation (the tendency for closely linked genes and genetic markers to segregate together) analysis showed perfect cosegregation of both mutations in the families. We revisited the phenotypes in those patients and found leukocoria, corneal opacities, a shallow AC, posterior synechiae, and total retinal detachment (Fig. 7). Six families had TSPAN12 as a candidate gene in their significant homozygous regions (Table 1), but only NCRNA9 and NCRNA17 had mutations in this gene. Pedigrees of the families positive for TSPAN12 mutations are shown in Figure 8.
Figure 7.

Phenotypes of patients with TSPAN12 mutations. (A, C) Anterior segment photographs show shallow AC, posterior synechiae, white pupils. (B) Total vitreous condensation. (D) Retrolental mass and atrophic retina with thin atrophic band attachment to optic nerve, confirming severe disease (red arrow shows site of the optic nerve).
Figure 8.
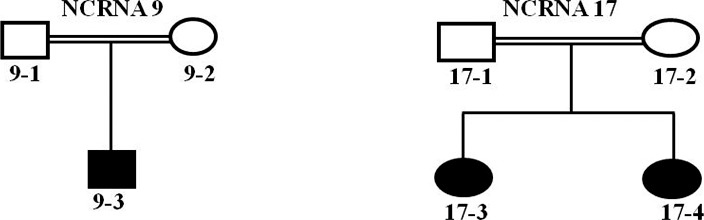
Pedigrees of patients with TSPAN12 mutations. Pedigrees of families carrying mutations in TSPAN12 gene.
Thus, for the first time, we found recessive mutations in TSPAN12 in clinically diagnosed NCRNA patients.
Mutations in NDP.
The gene NDP encodes a protein that plays a key role in retinal vascularization and activates the Wnt signaling pathway with the help of its coreceptors, FZD4 and LRP5. The gene is also thought to be implicated in neural cell differentiation and proliferation, as well as neuroectodermal cell-cell interaction (NDP, OMIM#300658). Mutations in NDP are known to cause X-linked exudative vitreoretinopathy.24
With the help of the RetNet panel and WES, we found NDP mutations in two families (NCRNA11 and NCRNA14). A novel homozygous 244-bp deletion in NDP, which starts from the end of the third exon, deleting 67-bp from exon three and 177-bp for 3′ UTR, was identified in one of our families. Another novel mutation, p. Cys69Gly, was identified in a second family (NCRNA11, Table 3). This novel mutation is predicted to be probably damaging by an online polymorphism phenotyping tool, PolyPhen,17 with a score of 0.997 (Table 2) and the Cys residue is evolutionally highly conserved (Fig. 9). Both the deletion and the missense mutations cosegregate within the families. Segregation of the mutation between the NCRNA11 family members is shown in Figure 10.
Figure 9.

Mutation conservation analysis for NDP variation. A novel mutation, p. Cys69Gly was identified in NCRNA11 family. This novel mutation is predicted to be probably damaging with a score of 0.997 (PolyPhen, Table 2) and the Cys residue is evolutionally highly conserved.
Figure 10.
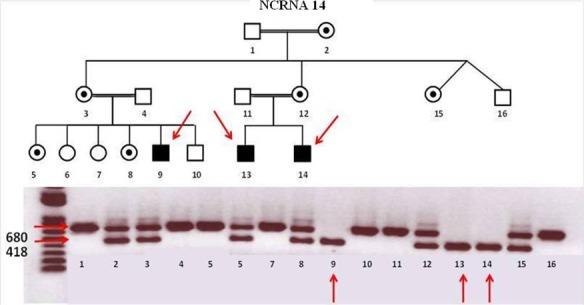
Family NCRNA14 pedigree and cosegregation result for NDP gene deletion. A novel homozygous 244-bp deletion in NDP gene (gene deletion: 43808839–43809101) was identified in NCRNA14 family. Mutation cosegregates within the family members. Red arrows show the affected members of the family. Family members with double bands (correspond to the sizes 418 and 680, WT and mutant bands, respectively) are heterozygous female carriers in the pedigree.
Phenotypes of the patients with NDP mutations is consistent with the phenotype of previously described NCRNA families in Iran1 and also similar to the phenotypes of families carrying mutations in ATOH7, LRP5, and TSPAN12 genes described in this study; bilateral corneal opacities, shallow AC, posterior synechiae, retrolenticular fibrovascular plaque, and complete congenital retinal detachment.
Genetic causes of NCRNA in the remaining eight families were not identified after extensive SNP, WES, and RetNet panel testing and are still under investigations for novel candidate NCRNA genes.
Discussion
Nonsyndromic congenital retinal nonattachment (OMIM #221900) is a rare genetic disorder defined by congenital retinal detachments and children are completely blind at birth. Often, the retinas are found to be fibrotic and appear as a white mass behind the crystalline lens.1–3 The goal of this study was to dissect an inbred cohort of clinically diagnosed NCRNA patients, to determine the genetic causes of NCRNA, in the hope of getting a molecular or genetic entry point into adult retinal detachments, a much more common condition, without any known molecular determinants.
In this study, 28 Pakistani blind children from 17 consanguineous and 4 nonconsanguineous families with the diagnosis of NCRNA were analyzed with variety of gene-identifying techniques, including SNP microarrays searching for homozygous regions. All the patients were from Northern Pakistan where consanguineous marriages are very prevalent. As expected, the majority of the patients in our cohort carried significant homozygous regions in their genomes as a result of consanguineous marriages commonly practiced in the region.
In 7 (41%) of 17 consanguineous families, homozygosity mapping successfully identified the region and subsequently a disease-causing mutation in the only currently known NCRNA gene or other retinal disease genes. It is important to note that the mutated gene was in the largest or second largest homozygous region in 6 of the 11 families (Table 1) successfully genotyped using homozygosity mapping. In accordance with our previous study,25 the data obtained in this current study suggest that the ranking of the IBD regions (continuous regions over which two haplotypes are identical by descent) can be very helpful to identify which homozygous segment contains the possible underlying causal and mutant gene. In the rest of the families, the known NCRNA and retinal disease genes located in significant homozygous regions were excluded from the study. The additional six successfully genotyped families were identified by RetNet panel screening (Table 3).
Although the clinical diagnosis and strict entry criteria of all children was NCRNA, in eight families, the molecular diagnosis determined that the disease process was in fact a new and severe form of FEVR. Familial exudative vitreoretinopathy is a rare disease and usually characterized by a much milder, acquired phenotype characterized by retinal folds, inferotemporal dragging of the optic disc and macula, increased vessels in the equatorial region, and a peripheral avascular zone.26 Familial exudative vitreoretinopathy was first described by Criswick and Schepens in 196927; it is currently not known to be associated with the severe developmental and congenital retinal detachment in babies and children. Therefore, we have expanded the phenotypic spectrum of FEVR to a severe retinal detachment phenotype that clinically overlaps with NCRNA. In addition, we identified the identical large deletion (6523 bp) in the ATOH7 gene found in the Kurdish founder population of Northern Iranian with a high incidence of NCRNA,1 suggesting genetic overlap between the Iranian and Pakistani populations. Therefore, we suggest that this founder mutation in ATOH7 gene links the two populations.
Our molecular genetics study of clinically evaluated and diagnosed patients with NCRNA revealed five important patterns. First, our work illustrates that NCRNA and FEVR overlap clinically; FEVR can be so severe that it can mimic NCRNA. Therefore, we show for the first time that the NCRNA phenotype can be caused by FEVR mutations. Second, we found the ATOH7 deletion links the Pakistani population to the Iranian population. Possible explanations for this could be a common ancestor for both populations or substantial interbreeding by the patients' ancestors who all lived in the same general large geographical area. Third, we have found seven novel mutations: (1) a hmz c.125G>C, p.Arg42Pro mutation in ATOH7, (2) a hmz c.235T>C, p.Trp79Arg and (3) a hmz c.1622T>C, p.Leu541Pro mutation in LRP5, (4) a hmz splice site mutation IVS4-2 A>G and (5) a hmz frameshift mutation c.423del T, p. Pro141fsX21 in TSPAN12, and (6) a hmz c.205T>G, p.Cys69Gly and (7) a hmz 244-bp c.336_402IVS4del deletion in NDP. These families were informed about their results and were genetically counseled about family planning and future treatment options. Fourth, we confirmed that recessive mutations in TSPAN12 can cause much more severe FEVR phenotype than the previously known dominant mutations related to the autosomal dominant FEVR in accordance with a previous study.18 A summary of the successfully genotyped NCRNA patients are given in Table 3. In addition, we used our newly developed RetNet known retinal disease gene panel, which for the first time contains all the currently known retinal disease genes by targeted NGS which allowed us to screen all the genes listed in Retinal Information Network website and create candidate retinal disease gene list for future study, in case if they are involved in new unknown mechanisms.
Conclusions
Although the clinical diagnosis of all children was NCRNA, the molecular diagnosis determined that the disease process was in fact a severe new form of FEVR in 13 families. However, the phenotype was severe congenital retinal detachment, previously not known to be associated with FEVR mutations. Therefore, we have expanded the phenotypic spectrum of FEVR, and we also show that the disease spectrum is much wider than previously established. Out of 21 Pakistani families with clinically diagnosed NCRNA, 10 families were discovered to have mutations in three FEVR genes: LRP5 (six families), TSPAN12 (two families), and NDP (two families), and three families were found to have mutations in ATOH7. Thus, eight NCRNA families in our cohort do not carry mutations in any known gene, which suggests that ATOH7 is not the only gene to cause NCRNA in children. These NCRNA families likely have new causal genes. We show that SNP studies—followed by RetNet, WES, and Sanger confirmation—is a powerful protocol to identify the genetic cause of NCRNA. Finally, we established a genetic link between the Iranian and Pakistani populations, through a common founder deletion in ATOH7.
Acknowledgments
The authors thank the families and patients who participated in this study.
Supported by Canadian Institutes for Health Research (CIHR); the FRSQ; Reseau Vision; The MCH Foundation; The Eyeball Foundation; grants from the Retinal Research Foundation; Foundation Fighting Blindness (BR-GE-0613-0618-BCM); the National Eye Institute (R01EY022356, R01EY018571); and a scholarship awarded by the Republic of Azerbaijan (VK). RKK was financially supported by The Foundation Fighting Blindness (FFB Canada).
This work is part of the thesis for a master of science at McGill University awarded to VK.
Disclosure: V. Keser, None; A. Khan, None; S. Siddiqui, None; I. Lopez, None; H. Ren, None; R. Qamar, None; J. Nadaf, None; J. Majewski, None; R. Chen, None; R.K. Koenekoop, None
References
- 1. Ghiasvand NM,, Rudolph DD,, Mashayekhi M,, Brzezinski JA, IV,, Goldman D,, Glaser T. Deletion of a remote enhancer near ATOH7 disrupts retinal neurogenesis, causing NCRNA disease. Nat Neurosci. 2011; 14: 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Phillips CI,, Stokoe NL. Congenital hereditary bilateral nonattachment of retina: a sibship of two males. J Pediatr Ophthalmol Strabismus. 1979; 16: 358–363. [DOI] [PubMed] [Google Scholar]
- 3. Ghiasvand NM,, Shirzad E,, Naghavi M,, Vaez Mahdavi MR. High incidence of autosomal recessive nonsyndromal congenital retinal nonattachment (NCRNA) in an Iranian founding population. Am J Med Genet. 1998; 78: 226–232. [PubMed] [Google Scholar]
- 4. Brown NL,, Patel S,, Brzezinski J,, Glaser T. Math5 is required for retinal ganglion cell and optic nerve formation. Development. 2001; 28: 2497–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang SW,, Kim BS,, Ding K,, et al. Requirement for Math5 in the development of retinal ganglion cells. Genes Dev. 2001; 15: 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gilmour DF. Familial exudative vitreoretinopathy and related retinopathies. Eye (Lond). 2015; 29: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pendergast SD,, Trese MT. Familial exudative vitreoretinopathy. Results of surgical management. Ophthalmology. 1998; 105: 1015–1023. [DOI] [PubMed] [Google Scholar]
- 8. Warden SM,, Andreoli CM,, Mukai S. The Wnt signaling pathway in familial exudative vitreoretinopathy and Norrie disease. Semin Ophthalmol. 2007; 22: 211–217. [DOI] [PubMed] [Google Scholar]
- 9. Warburg, M. Heterogeneity of congenital retinal non-attachment, falciform folds and retinal dysplasia: a guide to genetic counseling. Hum Hered. 1976; 26: 137–148. [DOI] [PubMed] [Google Scholar]
- 10. Robitaille J,, MacDonald ML,, Kaykas A,, et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat Genet. 2002; 32: 326–330. [DOI] [PubMed] [Google Scholar]
- 11. Toomes C,, Bottomley HM,, Jackson RM,, et al. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am J Hum Genet. 2004; 74: 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiao X,, Ventruto V,, Trese MT,, Shastry BS,, Hejtmancik JF. Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am J Hum Genet. 2004; 75: 878–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ai M, Heeger S, Bartels CF, Schelling DK; Osteoporosis-Pseudoglioma Collaborative Group. . Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am J Hum Genet. 2005; 77: 741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Untergasser A,, Cutcutache I,, Koressaar T,, et al. Primer3- new capabilities and interfaces. Nucleic Acids Res. 2012: 40;e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koressaar T,, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007; 23: 1289–1291. [DOI] [PubMed] [Google Scholar]
- 16. Wang F,, Wang H,, Tuan HF,, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014; 133: 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coussa RG,, Otto EA,, Gee HY,, et al. WDR19: an ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clin Genet. 2013; 84: 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar P,, Henikoff S,, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 19. Henikoff S,, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992; 89: 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adzhubei IA,, Schmidt S,, Peshkin L,, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010: 248–249. [DOI] [PMC free article] [PubMed]
- 21. Poulter JA,, Davidson AE,, Ali M,, et al. Recessive mutations in TSPAN12 cause retinal dysplasia and severe familial exudative vitreoretinopathy (FEVR). Invest Ophthalmol Vis Sci. 2012; 53: 2873–2879. [DOI] [PubMed] [Google Scholar]
- 22. Poulter JA,, Ali M,, Gilmour DF,, et al. Mutations in TSPAN12 cause autosomal-dominant familial exudative vitreoretinopathy. Am J Hum Genet. 2010; 86: 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nikopoulos K,, Gilissen C,, Hoischen A,, et al. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. Am J Hum Genet. 2010; 86: 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen ZY,, Battinelli EM,, Fielder A,, et al. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat Genet. 1993; 5: 180–183. [DOI] [PubMed] [Google Scholar]
- 25. den Hollander AI,, Lopez I,, Yzer S,, et al. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Vis Sci. 2007; 48: 5690–5698. [DOI] [PubMed] [Google Scholar]
- 26. Boonstra FN,, van Nouhuys CE,, Schuil J,, et al. Clinical and molecular evaluation of probands and family members with familial exudative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2009; 50: 4379–4385. [DOI] [PubMed] [Google Scholar]
- 27. Criswick VG,, Schepens CL. Familial exudative vitreoretinopathy. Am J Ophthalmol. 1969; 68: 578–594. [DOI] [PubMed] [Google Scholar]