

Abstract
Purpose
Mutations in the CYP1B1 gene are the most commonly identified genetic causes of primary infantile-onset glaucoma. Despite this disease association, the role of CYP1B1 in eye development and its in vivo substrate remain unknown. In the present study, we used zebrafish to elucidate the mechanism by which cyp1b1 regulates eye development.
Methods
Zebrafish eye and neural crest development were analyzed using live imaging of transgenic zebrafish embryos, in situ hybridization, immunostaining, TUNEL assay, and methylacrylate sections. Cyp1b1 and retinoic acid (RA) levels were genetically (morpholino oligonucleotide antisense and mRNA) and pharmacologically manipulated to examine gene function.
Results
Using zebrafish, we observed that cyp1b1 was expressed in a specific spatiotemporal pattern in the ocular fissures of the developing zebrafish retina and regulated fissure patency. Decreased Cyp1b1 resulted in the premature breakdown of laminin in the ventral fissure and altered subsequent neural crest migration into the anterior segment. In contrast, cyp1b1 overexpression inhibited cell survival in the ventral ocular fissure and prevented fissure closure via an RA-independent pathway. Cyp1b1 overexpression also inhibited the ocular expression of vsx2, pax6a, and pax6b and increased the extraocular expression of shha. Importantly, embryos injected with human wild-type but not mutant CYP1B1 mRNA also showed colobomas, demonstrating the evolutionary and functional conservation of gene function between species.
Conclusions
Cyp1b1 regulation of ocular fissure closure indirectly affects neural crest migration and development through an RA-independent pathway. These studies provide insight into the role of Cyp1b1 in eye development and further elucidate the pathogenesis of primary infantile-onset glaucoma.
Keywords: congenital glaucoma, neural crest, CYP1B1, retinoic acid, coloboma
In primary infantile-onset glaucoma (Online Mendelian Inheritance in Man [OMIM] 231300), elevated intraocular pressures resulting from the malformation of the trabecular meshwork lead to irreversible damage to the optic nerve, cornea, and sclera. Children with primary infantile-onset glaucoma often require multiple eye surgeries; however, eye pressures can be difficult to control, and many affected individuals are blind.1,2 Autosomal recessive mutations in the CYP1B1 gene are the most commonly identified genetic causes of primary infantile-onset glaucoma.3 Despite its association with human disease, the role of CYP1B1 in eye development remains unknown. Cyp1b1 knockout mice showed angle abnormalities involving Schlemm's canal, trabecular meshwork, cornea, and iris; yet these defects did not lead to elevations in intraocular pressure.4 Additional animal studies are required to understand the function of CYP1B1 during embryogenesis.
The CYP1B1 gene encodes a cytochrome p450 enzyme that regulates the in vitro synthesis of the essential morphogen retinoic acid (RA) through the conversion of retinol (vitamin A) into retinaldehyde and subsequently retinaldehyde into RA in a pathway independent of aldehyde dehydrogenases.5 Similar to other RA synthesis enzymes (e.g., raldh2 [aldh1a2] and raldh3 [aldh1a3]), cyp1b1 is expressed in the developing dorsal and ventral retina. In contrast, RA degradation enzymes, including cyp26a1, cyp26b1, and cyp26c1, are localized to the cranial and caudal retina.6–9 Together, these enzymes create RA gradients in the periocular mesenchyme centered on the dorsal and ventral axis of the eye.
We previously demonstrated that in zebrafish, RA produced by the developing retina regulates the formation of neural crest–derived structures in the eye and craniofacial region.10,11 The neural crest is a transient population of stem cells that contribute to many structures in the anterior segment, including the corneal stroma and endothelium, sclera, iris stroma, ciliary body stroma, and trabecular meshwork.12,13 The genetic or toxic disruption of ocular neural crest migration, proliferation, survival, and differentiation is responsible for many congenital eye diseases, including primary infantile-onset glaucoma, Axenfeld-Rieger syndrome (OMIM 180500), and Peters anomaly (OMIM 604229).14–16 Thus, investigating the molecular regulation of neural crest development is important for understanding the basis of congenital eye diseases.
In the present study, we demonstrated that cyp1b1 is expressed in the dorsal and ventral retina and retinal pigment epithelium, specifically in the ocular fissures, and maintains fissure patency, such that decreased expression caused premature closure, while overexpression resulted in colobomas. Although cyp1b1 regulated local RA levels, the effect on fissure closure was independent of RA. Cyp1b1 regulated the expression of vsx2, pax6, and shh, genes clinically associated with colobomas. Importantly, we demonstrated evolutionary and functional conservation between the human and zebrafish forms of cyp1b1. These studies better define the role of Cyp1b1 in eye development and, importantly, provide insight into the pathogenesis of primary infantile-onset glaucoma.
Materials and Methods
Animal Care and Animal Strains
Zebrafish (Danio rerio) were raised in a breeding colony under a 14-hour light/10-hour dark cycle as previously described.10,11,17,18 Embryos were maintained at 28.5°C and staged as previously described.19 The transgenic strains Tg(sox10:EGFP) and Tg(foxd3:GFP) were gifts from Thomas Schilling and Mary Halloran. The strains were crossed into the casper (roy−/−, nacre−/−) background as indicated.20–22 The Tg(RARE:mCHERRY) transgenic strain was generated using Gateway cloning techniques as previously described.23 Animal protocols were performed in accordance with the guidelines for the humane treatment of laboratory animals established by the University of Michigan Committee on the Use and Care of Animals (IACUC, protocol 10205) and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Morpholino Oligonucleotides and mRNA Injections
Translation blocking, 5-base pair mismatch, and standard control antisense morpholino oligonucleotides (MO; Gene Tools, LLC, Corvallis, OR, USA) directed against zebrafish Cyp1b1 (ATG and 5′ UTR targeted) and Raldh2 (ATG and 5′ UTR targeted) (Supplementary Table S1) were reconstituted in deionized water. Concentrations yielding consistent and reproducible phenotypes were determined for each MO. One-cell-stage embryos were injected with 1 to 2 nL MO at a concentration of 0.1 to 0.25 mM (2.1–2.4 ng/nL).
Two nonoverlapping translation blocking MO against Cyp1b1 were used to confirm the observed phenotype. The ATG-targeted MO against Cyp1b1 was previously verified for efficacy by confirming decreased protein expression, but this antibody is no longer available.24 No commercially available antibodies against human CYP1B1 with predicted cross-reactivity with zebrafish cyp1b1 showed consistent detection of the zebrafish protein by Western blot. The same ATG-targeted MO sequence and same range of concentrations as were used by Timme-Laragy et al.24 were used in these experiments. While the ATG-targeted Cyp1b1 MO is insensitive to the human mRNA coding sequence, there is overlap with the initial part of the zebrafish coding sequence. In contrast, the 5′ UTR-targeted Cyp1b1 MO is insensitive to both the zebrafish and human mRNA sequences (Supplementary Table S1). The 5′ UTR Cyp1b1 MO was used in cyp1b1 mRNA rescue experiments. In the current studies, both the ATG- and 5′ UTR-targeted Cyp1b1 MO showed similarity of phenotype.
Similarly, 2 nonoverlapping translation blocking Raldh2 MO were used to confirm the knockdown phenotype. The ATG-targeted Raldh2 MO, but not the 5′ UTR-targeted Raldh2 MO, overlapped with the zebrafish coding sequence (Supplementary Table S1). The 5′ UTR Raldh2 MO was used in raldh2 mRNA rescue experiments. Morpholino oligonucleotide knockdown (ATG or 5′ UTR) decreased Raldh2 protein while raldh2 mRNA overexpression increased Raldh2 protein detected by whole-mount immunostaining (Supplementary Figs. S1A–E).
The coding regions of zebrafish cyp1b1 and raldh2 genes and GFP were cloned into the pCS2p+ expression vector, and the sequences were verified. The human CYP1B1 coding region was cloned into the pCRII-TOPO expression vector. Site-directed mutagenesis using Pfu Turbo (QuikChange Site-Directed Mutagenesis Kit; Stratagene, La Jolla, CA, USA) and custom-designed primers were used to introduce point mutations (Supplementary Table S2). Capped mRNA was synthesized (mMessage mMachine kit; Ambion Biosystems, Foster City, CA, USA) and resuspended at 50 to 150 ng/μL in nuclease-free water. Approximately 1 to 2 nL (50–300 pg total RNA) of zebrafish cyp1b1 or raldh2 mRNA or human CYP1B1 (wild type or mutant) mRNA was coinjected with phenol red and GFP mRNA (50 ng/μL; 50–100 pg RNA) into one-cell-stage embryos.
For each experiment, ∼100 embryos were injected with MO and/or mRNA, and all experiments were replicated a minimum of three times. Embryos were counted, and phenotypes for developmental delay, abnormal craniofacial development, and coloboma were assessed at 72 and 96 hours post fertilization (hpf). The percentage of embryos with these phenotypes in each group was calculated. Morpholino- or mRNA-injected embryos were compared with control-injected embryos. Each experiment (of ∼100 embryos/group) was used as an experimental unit. The mean with standard error is shown in the text and tables. The data were statistically analyzed using ANOVA with post hoc Tukey pairwise tests, and P < 0.05 was considered statistically significant. Images shown are representative of all experiments.
Pharmacologic Treatments
All-trans retinoic acid (RA; Sigma-Aldrich Corp., St. Louis, MO, USA), diethylaminobenzaldehyde (DEAB; Sigma-Aldrich Corp.), and phenylthiourea (PTU; Sigma-Aldrich Corp.) were diluted in dimethylsulfoxide (DMSO; Sigma-Aldrich Corp.) at a 1000× final concentration. The pharmacologic treatments were administered in the embryo media at final concentrations of 25 to 50 nM RA, 10 μM DEAB, and 0.003% PTU. Treatments were initiated between 3 and 24 hpf as indicated. The medium was changed every 24 hours as indicated. Approximately 50 embryos were included for each condition, and all experiments were replicated a minimum of three times. Embryos were counted and phenotypes were assessed as described above. Images shown are representative of all experiments.
Imaging
Embryos were analyzed under a M205FA combi-scope (Leica Microsystems CMS GmbH, Wetzlar, Germany). Representative images of at least four embryos in each group per experiment were obtained using brightfield DFC290 (Leica Microsystems CMS GmbH) and/or fluorescent ORCA-ER (Hamamatsu, Hamamatsu City, Japan) cameras. Sections were imaged under a DM6000B upright microscope (Leica Microsystems CMS GmbH) equipped with a DFC500 camera (Leica Microsystems CMS GmbH). The images were processed and analyzed using Adobe Photoshop (San Jose, CA, USA), LAS software (Leica Microsystems CMS GmbH), and/or AF6000 software (Leica Microsystems CMS GmbH).
Time-lapse imaging of Tg(foxd3:GFP) embryos was performed as previously described.23 Briefly, the embryos were imaged using a TCS SP5 MP multiphoton microscope (Leica Microsystems CMS GmbH) fitted with a Mai Tai DeepSee Ti-Sapphire laser (Spectra-Physics; Newport Corp., Irvine, CA, USA). The images were obtained every 20 minutes during the described time frame. Each time-lapse experiment was repeated three or four times. Images and movies included are representative of all experiments.
Immunostaining and TUNEL Assay
Staged zebrafish embryos were fixed in 4% paraformaldehyde overnight at 4°C. For sectioning, the embryos were cryoprotected in successive sucrose solutions, embedded in optical cutting temperature (OCT) compound, and subsequently sectioned at 10 μm.
Whole-mount immunostaining was performed as previously described using primary rabbit anti-laminin (1:200; Sigma-Aldrich Corp.), primary rabbit anti-Raldh2 (1:100; GeneTex, Irvine, CA, USA), and secondary goat anti-rabbit IgG conjugated to Cy3 or 488 (1:1000; Abcam, Cambridge, MA, USA).11 Section immunostaining for GFP was performed using mouse anti-GFP directly conjugated to FITC (1:100; EMD Millipore, Billerica, MA, USA).
The percentage of embryos with continuous laminin staining in the distal ocular fissure was calculated. Morpholino- or mRNA-injected embryos were compared with control-injected embryos. The experiments were repeated three times, and each experiment (of ∼20 embryos/group) was used as an experimental unit. The mean with standard error is shown in the text. The data were statistically analyzed using ANOVA with post hoc Tukey pairwise tests, and P < 0.05 was considered statistically significant. Images shown are representative of all experiments.
The terminal deoxynucleotidyl transferase (dUTP) nick-end labeling (TUNEL) assay was performed as previously described using standard protocols.11 Briefly, apoptotic cells were detected through the TdT-mediated incorporation of digoxigenin-labeled deoxyuridine triphosphate (Roche Life Sciences, Indianapolis, IN, USA). Sheep anti-digoxigenin conjugated to rhodamine (Roche Life Sciences) was used to detect the TUNEL signal. The sections were costained with 4′,6-diamidino-2-phenylindole (DAPI). For quantification, three to five consecutive sections through the equator of the lens of at least four embryos per group were counted. The total number of cell nuclei and TUNEL-positive cells in the eyes of each section were manually counted. The percentage of TUNEL-positive cells in the eyes of MO- or mRNA-injected embryos was compared with those of control-injected embryos. The data were statistically analyzed using ANOVA with post hoc Tukey pairwise tests, and P < 0.05 was considered statistically significant. Images shown are representative of all experiments.
In Situ Hybridization
In situ hybridization was performed through standard protocols using digoxigenin- or fluorescein-labeled RNA antisense probes.17,25 The cyp1b1 RNA probe was 1047 base pairs, the vsx1 RNA probe was 797 base pairs, the vsx2 RNA probe was 503 base pairs, the shha RNA probe was 911 base pairs, the shhb RNA probe was 876 base pairs, the pax6a RNA probe was 458 base pairs, and the pax6b RNA probe was 316 base pairs. All probes were hybridized at 56°C overnight. For colorimetric reactions and signal comparisons, the embryos were developed for equal amounts of time. Sense controls were also developed in parallel to ensure specific staining. The embryos were cryoprotected, embedded, and sectioned at 10 μm. The sections were washed in phosphate-buffered saline, coverslipped, and imaged as described above.
Western Blotting
Embryos were injected as described above, and total protein was isolated at 24 hpf. Twenty micrograms of protein was combined with sample buffer (10% glycerol, 62.5 mM Tris base, pH 6.8, 2.5% sodium dodecyl sulfate, 5% β-mercaptoethanol, and 0.002% bromophenol blue), boiled for 5 minutes, and loaded onto a 12% acrylamide gel (BioRad, Hercules, CA, USA). Protein was transferred to a nitrocellulose membrane (Life Technologies, Waltham, MA, USA), blocked in PBS/0.1% Tween–20/3% nonfat dry milk, and subsequently incubated with rabbit polyclonal anti-CYP1B1 (1:500, Ab157578, Ab78044; Abcam), mouse monoclonal anti-β-actin (1:1000, Ab8226; Abcam) overnight at 4°C. The membrane was washed in PBS/0.1% Tween-20 and subsequently incubated with antirabbit horseradish peroxidase (HRP)-conjugated secondary antibody (1:1000; Cell Signaling Technology, Danvers, MA, USA) or anti-mouse-HRP conjugated secondary antibody (1:1000, Cell Signaling Technology). Pierce ECL Western Blotting Substrate (Thermo Scientific, Waltham, MA, USA) was used for chemiluminescent detection. Commercially available antibodies (Abcam Ab157578 and Ab78044) against human CYP1B1 (∼61 kDa) did not show cross-reactivity with the zebrafish form (∼60 kDa) by Western blotting (see Fig. 7).
Figure 7.

Cyp1b1 differentially regulated vsx2, shh, and pax6. Whole-mount in situ hybridization at 24 hpf demonstrated that vsx2 expression in the eye decreased in response to cyp1b1 overexpression (D, D') compared with Cyp1b1 MO knockdown (C, C'), control-injected (B, B'), and uninjected (A, A'') embryos. Note the appearance of tissue bridging the two optic cups in the cyp1b1 mRNA-injected embryo, as demarcated by vsx2 (arrow). Shha at 24 hpf showed increased expression in the midline floor plate between the eyes in embryos injected with cyp1b1 mRNA (arrowheads, H, H') compared with Cyp1b1 MO knockdown (G, G'), control -injected (F, F'), and uninjected (E, D') embryos. Pax6a and pax6b expression in the retina at 48 hpf decreased in response to cypb1 overexpression (L, L') compared with Cyp1b1 knockdown (K, K'), control-injected (J, J'), and uninjected (I, I') embryos.
Methylacrylate Sections
Zebrafish embryos at 96 hpf were fixed in 2% paraformaldehyde/1.5% glutaraldehyde overnight at 4°C, followed by embedding in methylacrylate. The blocks were sectioned at 5 μm. The sections were stained with Lee's stain, coverslipped, and subsequently imaged as described above.
Results
Cyp1b1 Overexpression Inhibited Ocular Fissure Closure
Previously published studies have established an effective MO that blocks Cyp1b1 translation; however, these studies did not assess eye or craniofacial development.24 Because mutations in CYP1B1 are associated with primary infantile-onset glaucoma, reflecting abnormalities in the neural crest–derived iridocorneal angle, the Tg(sox10:EGFP) and Tg(foxd3:GFP) transgenic zebrafish strains in which GFP is expressed in different subpopulations of neural crest cells were used to analyze neural crest and eye development.
Two different translation blocking Cyp1b1 MOs (ATG or 5′ UTR) were used to determine the effects of knockdown on neural crest and eye development and verify consistency of phenotype. At 72 hpf, the most common phenotypes in Cyp1b1 ATG and 5′ UTR MO knockdown embryos included developmental delays of 12 to 24 hours and craniofacial abnormalities in 20.5 ± 12.1% and 26.8 ± 11.1% of embryos, respectively (Fig. 1A; Supplementary Table S3). This finding was not significantly different compared with the percentage of developmentally delayed and abnormal Cyp1b1 ATG mismatch MO injected (12.1 ± 9.8%, P = 0.56) or Cyp1b1 5′ UTR mismatch MO injected (21.0 ± 7.1%, P = 0.97). Further, there were no significant differences between mismatch control-injected embryos and uninjected (8.3% ± 6.3%, P = 0.98, ATG mismatch, and P = 0.10, 5′ UTR mismatch) embryos.
Figure 1.

Overexpression of cyp1b1 inhibited ocular fissure closure via an RA-independent pathway. The percentage of 72 hpf embryos with developmental delay and/or craniofacial abnormalities (A) due to Cyp1b1 MO (ATG or 5′ UTR) knockdown, cyp1b1 (mRNA) overexpression, Raldh2 MO knockdown, raldh2 (mRNA) overexpression compared to uninjected, mismatch (MM) MO-injected or gfp mRNA-injected controls. ([a] P < 0.0001; [b] P < 0.001; [c] P < 0.01; [d] P < 0.05). Live images of uninjected (B) and mismatch control-injected (C, G) Tg(sox10:EGFP) embryos showed that at 96 hpf, neural crest contributed to the pharyngeal arches (PA) and the ceratohyal (Ch) and Meckel's (Mk) cartilage of the jaw. Sections of 96 hpf Tg(foxd3:GFP) embryos showed that foxd3-positive neural crest cells contributed to the iris stroma in the dorsal (B', C', G', arrowhead) and ventral (double arrowhead) iridocorneal angle. In addition, differentiated retinal photoreceptors expressed foxd3, which was unrelated to neural crest cells. Injection of 5′ UTR Cyp1b1 MO had minimal effect on craniofacial neural crest (D) and overall eye (D') development. Overexpression of cyp1b1 through mRNA injection disrupted neural crest–derived Meckel's cartilage formation (E, K) and inhibited ocular fissure closure, resulting in prominent colobomas (closed arrows, E, E'). The retinal layers were properly differentiated, including the expression of foxd3. Injection of Cyp1b1 5′ UTR MO improved the ocular and neural crest phenotypes due to cyp1b1 mRNA (F, F'). As previously demonstrated, increasing (raldh2 mRNA, H, H') or decreasing (Raldh2 5′ UTR MO, I, I') RA inhibited pharyngeal arch development, disrupted ceratohyal and Meckel's cartilage formation, and decreased eye size. Injection of raldh2 mRNA rescued the ocular and neural crest phenotypes induced through Raldh2 5′ UTR MO knockdown (J, J'). Cyp1b1 MO improved eye size, retinal differentiation, and neural crest–derived pharyngeal arch and ceratohyal and Meckel's cartilage formation in embryos injected with raldh2 mRNA (K, K'). Knockdown of Raldh2 improved neural crest–derived pharyngeal arch and ceratohyal and Meckel's cartilage formation (L'), but did not rescue ocular fissure closure (arrows, L') in embryos injected with cyp1b1 mRNA.
At 96 hpf, MO knockdown of Cyp1b1 had a minimal effect on craniofacial neural crest development, including the pharyngeal arches (PA), ceratohyal cartilage (Ch), and Meckel's (Mk) cartilage (Fig. 1D; Supplementary Fig. S2B) compared with controls (Fig. 1C; Supplementary Fig. S2A) and uninjected (Fig. 1B) embryos. Although we previously showed that Cyp1b1 knockdown delayed eye development at 48 hpf,26 by 96 hpf, anterior segment and retinal development (Fig. 1D'; Supplementary Fig. S2B') had caught up to control-injected (Fig. 1C'; Supplementary Fig. S2A') and uninjected (Fig. 1B') embryos. This included foxd3-positive neural crest cells in the dorsal (arrowheads) and ventral (double arrowheads) iris stroma. In addition, foxd3 was expressed in differentiated retinal photoreceptors, which was unrelated to expression of this transcription factor in neural crest cells.
In contrast, the injection of cyp1b1 mRNA markedly affected craniofacial and eye development. At 72 hpf, a significant percentage (43.8 ± 12.1%, P < 0.0001) of cyp1b1 mRNA-injected embryos showed developmental delays and craniofacial abnormalities compared with gfp mRNA-injected (5.7 ± 4.9%) and uninjected embryos (Fig. 1A; Supplementary Table S3). Furthermore, at 72 hpf, 38.7 ± 8.5% (P = 0.0001) of cyp1b1 mRNA-injected embryos showed ocular fissure closure defects (delayed closure and coloboma), which were not observed in gfp-injected or uninjected embryos. At 96 hpf, cyp1b1 overexpression inhibited Mk cartilage development (Fig. 1E) and caused eye wall defects and colobomas (Figs. 1E, 1E', arrows). Although loss of the retinal pigment epithelial layer was observed in the area of the coloboma, the retinal layers were properly differentiated. In addition, foxd3-positive cells were observed in the neural crest–derived iris stroma (arrowheads and double arrowheads). The injection of Cyp1b1 ATG or 5′ UTR MO significantly decreased the percentage of embryos (20.9 ± 6.0%, P = 0.002 and 27.9 ± 4.8%, P = 0.03) with ocular and craniofacial defects due to cyp1b1 mRNA injection (Supplementary Table S3; Figs. 1A, 1F, 1F'). Although decreased cyp1b1 caused only mild developmental delays and abnormalities, the overexpression of cyp1b1 demonstrated that this gene plays a role in ocular fissure closure and cranial neural crest development.
Cyp1b1 Expression in the Ocular Fissure Was Regulated Through RA
The role of cyp1b1 in eye development has been hypothesized to involve RA, as cyp1b1 regulates RA synthesis in vitro.5 We determined whether cyp1b1 expression was regulated through alterations in RA levels. In situ hybridization revealed that cyp1b1 was expressed in the dorsal (arrowhead) and ventral (closed arrow) retina and retinal pigment epithelium at 24 (Figs. 2A, 2B, 2C) and 48 hpf (Figs. 2A', 2B', 2C') in untreated and DMSO-control treated embryos. Cyp1b1 expression was spatially and temporally correlated with the superior and inferior ocular fissures. Diethylaminobenzaldehyde is a pan-aldehyde dehydrogenase inhibitor that decreases endogenous RA synthesis mediated by raldh2 and raldh3. Treatment at 12 hpf with 10 μM DEAB did not alter cyp1b1 expression in the developing eye at 24 hpf (Fig. 2D), but did increase cyp1b1 expression in the ventral migrating neural crest (open arrowhead) compared with DMSO-treated control (Fig. 2C) and untreated embryos (Fig. 2A). At 48 hpf, DEAB increased cyp1b1 expression in the dorsal (arrowhead) and ventral (closed arrow) eye (Fig. 2D') compared with controls (Figs. 2A', 2C'). In contrast, treatment with exogenous RA (25 nM) at 12 hpf inhibited cyp1b1 expression in the eye at 24 (Fig. 2E) and 48 hpf (Fig. 2E'). Thus, cyp1b1 expression was affected by pharmacologic alterations in RA levels.
Figure 2.

Cyp1b1 expression in the developing eye was regulated through RA. Whole-mount in situ hybridization showed cyp1b1 expression along the dorsal (arrowheads)–ventral (arrows) axis in the developing eye at 24 and 48 hpf in uninjected (A, A') and 0.1% DMSO-treated (C, C') embryos. In sections, cyp1b1 localized to the retina and retinal pigment epithelium (B, B'). Treatment with 10 μM DEAB at 12 hpf increased cyp1b1 expression in the ventral neural crest, which subsequently forms the brachial arches (D, open arrowhead) at 24 hpf and the dorsal (arrowheads) and ventral (arrows) eye at 48 hpf (D'). Exogenous treatment with 25 nM all-trans RA at 12 hpf decreased cyp1b1 expression in the dorsal and ventral eye at 24 (E) and 48 hpf (E'). Live imaging of the Tg(RARE:mCherry) reporter line showed areas of high RA activity in the eye and olfactory placode (O) in uninjected (F) and control-injected (G) embryos. Within the eye (F', G'), RA activity was observed in the retinal pigment epithelium (arrows), retina (R), lens (L), and cornea (C). MO knockdown of Cyp1b1 (H, H') showed decreased RA activity in the medial retinal pigment epithelium (arrows) correlated with the area of cyp1b1 expression (B'). Overexpression of cyp1b1 through mRNA injection diffusely increased mCherry expression at 24 hpf throughout the craniofacial region (I). Increased RA activity in the eye was also observed in the medial retinal pigment epithelium (arrows) and ocular fissure (arrowhead).
To determine whether the converse was also true, a transgenic line (Tg[RARE:mCHERRY]), in which mCherry expression is driven by canonical RA response elements (RARE), was utilized. At 24 hpf, RA expression in the craniofacial region was greatest in the eye and the olfactory placode in uninjected and control-injected embryos (Figs. 2F, 2G; Supplementary Figs. S3A, S3B). At 48 hpf, within the eye, diffuse RA activity was observed throughout the retina, lens, and cornea (Fig. 3F'). Cyp1b1 MO knockdown did not show alterations in RA activity in the developing head at the doses tested (Fig. 2H; Supplementary Fig. S3C). In the eye, decreased RA activity was observed in the retinal pigment epithelium (Fig. 3H') in the area of cyp1b1 expression (Fig. 3B') compared with control-injected (Fig. 3G') and uninjected (Fig. 3F') embryos. In contrast, the overexpression of cyp1b1 by injecting mRNA at the one-cell stage caused a diffuse increase in mCherry expression throughout the craniofacial region (Fig. 3I; Supplementary Fig. S3D). Further, in the eye, increased RA activity was observed in the retinal pigment epithelium and retina (Fig. 3I', arrows) and in the region of the ocular fissure (arrowhead) compared with the eyes of control-injected (Fig. 3G') and uninjected (Fig. 3F') embryos. These studies suggest that cyp1b1 expression was regulated by RA, and the overexpression of cyp1b1 may alter local RA levels in and around the developing eye.
Figure 3.

Cyp1b1 and RA regulated cell survival in the developing eye. Cyp1b1 ATG (C) and 5′ UTR (E) MO knockdown did not significantly affect apoptosis in the eye at 24 hpf compared to uninjected (A) and control-injected (B) embryos. Cyp1b1 overexpression (E) increased apoptosis and the apoptotic cells were specifically localized to the inferior ocular fissure (arrows). Raldh2 5′ UTR (D') but not ATG (C') MO knockdown significantly decreased cell survival throughout the developing eye compared to uninjected (A) and control injected (B'). Similarly, overexpression of raldh2 (E') increased apoptosis in the eye. The percentage of apoptotic cells in 24 hpf developing eyes (F) of embryos injected with Cyp1b1 MO (ATG or 5′ UTR), cyp1b1 mRNA, Raldh2 (ATG or 5′ UTR) MO, raldh2 mRNA compared to uninjected and control injected (*P < 0.01).
Cyp1b1 Regulation of Ocular Fissure Closure Was Independent of RA
Since cyp1b1 expression was regulated through RA, we assessed whether cyp1b1 and raldh2 worked together to control eye and neural crest development. As previously demonstrated, raldh2 overexpression (Figs. 1H, 1H') and Raldh2 MO knockdown (Figs. 1I, 1I'; Supplementary Figs. S2C, S2C') significantly decreased eye size and inhibited neural crest–derived jaw and PA formation in 56.2 ± 17.7% (P < 0.002) and 80.4 ± 5.9% (P = 0.0001) of injected embryos, respectively (Fig. 1A; Supplementary Table S3).10 The coinjection of Raldh2 MO and raldh2 mRNA significantly decreased the percentage of embryos with craniofacial and ocular defects (20.5 ± 6.8%, P = 0.01), demonstrating the specificity of these reagents (Supplementary Table S3; Figs. 1A, 1H, 1H'). To determine whether the Cyp1b1-mediated regulation of neural crest and eye development reflects the regulation of RA synthesis, Cyp1b1 MO was coinjected with raldh2 mRNA. At 72 hpf, Cyp1b1 knockdown decreased the percentage of embryos with small eyes and craniofacial defects (31.4 ± 9.0%, P = 0.04, Fig. 1A; Supplementary Table S3) resulting from the overexpression of raldh2. At 96 hpf, Cyp1b1 knockdown with raldh2 overexpression improved eye size and craniofacial cartilage formation (Figs. 1K, 1K'; Supplementary Figs. S2D, S2D'). Cyp1b1 overexpression decreased the percentage of 72 hpf Raldh2 MO knockdown embryos with developmental delay, small, malformed eyes and craniofacial abnormalities (53.5 ± 7.7%, P = 0.0001, Fig. 1A; Supplementary Table S3). At 96 hpf, the overexpression of cyp1b1 improved eye size and jaw and PA development in Raldh2 MO knockdown embryos (Figs. 1L, 1L'; Supplementary Figs. S2E, S2E'). However, decreased RA through Raldh2 MO knockdown did not decrease the percentage of cyp1b1 mRNA-injected embryos with ocular fissure defects at 72 hpf (30.0 ± 8.7%, P = 0.17, Supplementary Table S3) and 96 hpf (Fig. 1L', Supplementary Fig. S2E').
This finding was further confirmed using a DEAB dose–response curve (1–5 μM) in combination with cyp1b1 overexpression (Supplementary Fig. S4; Supplementary Table S4). Treatment of GFP mRNA-injected embryos with 1 μM DEAB (at 3 hpf) did not significantly affect ocular fissure closure (28.0 ± 7.0%, P = 0.10) compared with untreated GFP-injected controls (3.3 ± 1.0%). However, treatment with 2 or 5 μM DEAB significantly increased the percentage of embryos (38.0 ± 9.4% [P = 0.008] and 55.2 ± 4.6% [P < 0.0001], respectively) with ocular closure defects at 72 hpf. At 96 hpf in GFP-injected control embryos, increasing concentrations of DEAB (Supplementary Figs. S4E, S4G, S4I) altered PA and Ch and Mk cartilage formation, decreased eye size, and delayed ocular fissure closure (arrowhead) compared with untreated (Supplementary Fig. S4A) and DMSO-control treated (Supplementary Fig. S4C) embryos. Treatment with DEAB did not improve or rescue the ocular fissure defects in cyp1b1 mRNA-injected embryos, but rather significantly increased the percentage of embryos with defects at each concentration (82.2 ± 13.4% at 1 μM DEAB [P = 0.0007], 84.5 ± 14.4% at 2 μM DEAB [P = 0.0003], and 100% at 5 μM DEAB [P < 0.0001]). Cyp1b1 mRNA-injected embryos treated with 1, 2, or 5 μM DEAB showed open ocular fissures (arrowheads), ocular wall defects (arrows), small eyes, and jaw and PA defects (Supplementary Figs. S4F, S4H, S4J). Concentrations of DEAB higher than 5 μM were lethal in both GFP- and cyp1b1 mRNA-injected embryos (data not shown). Thus, the effect of cyp1b1 overexpression on ocular fissure closure was not mediated through RA.
Next, the effect of cyp1b1 on cell survival in the developing eye, particularly in the ocular fissure, was assessed. At 24 hpf, TUNEL assay of the eyes of embryos injected with cyp1b1 mRNA (5.9 ± 2.8%, Figs. 3E, 3F) showed a statistically significant (P = 0.005) 2-fold increase in apoptosis compared with uninjected (2.6 ± 1.1%, Fig. 2A) and control-injected embryos (1.8 ± 0.8% Fig. 3B). Many of the apoptotic cells were localized to the inferior ocular fissure (arrows). In contrast, injection of Cyp1b1 ATG (2.1 ± 1.0%, Fig. 2C) or 5′ UTR MO (3.6 ± 1.2%, Fig. 3D) did not significantly affect apoptosis. Raldh2 overexpression (8.2 ± 4.1%, Fig. 3E') significantly (P = 0.003) increased the percentage of apoptotic cells in the developing eye compared to uninjected (Fig. 3A) or control-injected embryos (2.9 ± 0.7%, Fig. 3B'). Injection of Raldh2 5′ UTR MO (8.7 ± 2.1%, Fig. 3D'), but not ATG MO (6.1 ± 1.7%, Fig. 3C'), significantly (P = 0.005) increased apoptosis. However in both the raldh2 overexpression and Raldh2 knockdown embryos, apoptotic cells were distributed throughout the eye and were not specifically localized to the ocular fissure. Thus, Cyp1b1 increased apoptosis in the ventral ocular fissure and regulated fissure closure via an RA-independent pathway.
Cyp1b1 Maintained the Patency of the Inferior Ocular Fissure
Ocular fissure closure is a complex process, requiring optic vesicle evagination, spatial specification along the proximal–distal and ventral–dorsal axes, breakdown of the basement membrane that demarcates the fissure, and fusion of the two sides (reviewed in Refs. 27, 28). The temporal and spatial expression of cyp1b1 in the ocular fissure and the colobomatous defects resulting from cyp1b1 overexpression suggested that cyp1b1 plays a role in fissure closure. We used whole-mount laminin staining to examine basement membrane integrity in the ocular fissure as a measure for the timing of ocular fissure closure. In uninjected (Figs. 4A–A''') and control-injected (Figs. 4B–B''') embryos, laminin staining was observed in the distal ocular fissure (arrows) between 36 and 54 hpf. In uninjected embryos, laminin staining was found in a continuous line indicating presence of the basement membrane between the two sides of the ocular fissure in 100% of 36 and 42 hpf embryos, 93.4 ± 2.9% of 48 hpf embryos, and 79.1 ± 11.7% of 54 hpf embryos (Fig. 4F). Similarly, control-injected embryos showed laminin staining in 100% of 36 hpf embryos, 99.3 ± 1.7% of 42 hpf embryos, 92.1 ± 8.8% of 48 hpf embryos, and 79.8 ± 8.8% of 54 hpf embryos, (Fig. 4G). Injection of Cyp1b1 ATG (Figs. 4C–C''') or 5′ UTR (Figs. 4D–D''') MO caused early breakdown of the basement membrane in the ocular fissure. At 42 hpf, injection of Cyp1b1 ATG or 5′ UTR MO did not significantly decrease the percentage of embryos with continuous laminin staining in the fissure (90.8 ± 8.0% and 80.4 ± 17.2%, respectively). However, injection of Cyp1b1 ATG or 5′ UTR MO significantly decreased the percentage of embryos with continuous laminin staining in the ocular fissure at 48 hpf (52.2 ± 15.0%, P = 0.002, and 59.3 ± 12.2%, P = 0.001) and 54 hpf (33.7 ± 10.5%, P = 0.0005, and 30.1 ± 18.3%, P = 0.0002). Cyp1b1 overexpression (Figs. 4E', 4E''') showed laminin in the fissure in 100% of 36, 42, and 48 hpf and 93.9 ± 5.4% of 54 hpf embryos.
Figure 4.

Alterations in the basement membrane integrity as a result of decreased or increased Cyp1b1. Whole-mount laminin staining showed that in uninjected (A–A''') and control-injected (B–B''') embryos, a continuous basement membrane (arrows) was evident in the distal ocular fissure at 36, 42, 48, and 54 hpf. Cyp1b1 ATG (C–C''') and 5′ UTR (D–D''') MO knockdown showed discontinuous staining of laminin at 48 (C'', D'') and 54 hpf (C''', D'''), suggesting premature basement membrane breakdown. In cyp1b1 mRNA-injected embryos, laminin staining remained prominent within the fissure at all time points (E–E'''). The percentage of embryos with continuous laminin staining in the distal ocular fissure at 36, 42, 48, and 54 hpf (G), (*P < 0.001).
The neural crest migrates into the anterior segment of the eye, contributing to the cornea, iris, and iridocorneal angle. Because cyp1b1 was expressed in the retina and retinal pigment epithelium and alterations in cyp1b1 affected fissure closure, we used time-lapse imaging to investigate whether cyp1b1 affected neural crest migration into the developing eye. Between 30 and 60 hpf, foxd3-positive neural crest cells [Tg(foxd3:GFP)] entered the anterior segment of the eye (Figs. 5A–J). Foxd3-positive cells migrated between the surface ectoderm and optic cup, with the majority of cells localized to the dorsal–posterior quadrant (Figs. 5E–H, dorsal denoted with “d”, ventral denoted with “v”, anterior denoted with “a” and posterior denoted with “p”). Additional cells travelled adjacent to and through the ocular fissure (Figs. 5A–F; white arrowhead denotes migrating neural crest, red dashed line demarcates ocular fissure). At 60 hpf, foxd3-positive cells coalesced and organized to completely encircle the lens (Figs. 5I, 5J, closed arrows), indicating closure of the fissure. In addition, at 60 hpf, foxd3 was also expressed in the photoreceptors (Figs. 5H–J, open arrowhead). Cyp1b1 knockdown altered foxd3-positive cell migration between 30 and 60 hpf (Figs. 5A'–J'). Foxd3-positive cells travelled between the surface ectoderm and optic cup in the dorsal quadrants (Figs. 5A'–E'), but poorly migrated in specific ventral quadrants (denoted “v”) adjacent to and through the ocular fissure (demarcated by red dashed line). Foxd3-positive cells encircled the lens earlier than in control embryos (Figs. 5D'–H' versus Figs. 5D–H), suggesting premature closure of the fissure. At 60 hpf, fewer foxd3-positive cells were observed in the eyes of Cyp1b1 MO knockdown embryos (Fig. 5J'). In embryos in which cyp1b1 was overexpressed, ocular neural crest migration was less organized, as foxd3-positive cells were located throughout the posterior half of the eye (Figs. 5A''–G''). Further, the ocular fissure (demarcated by red dashed lines) remained open, preventing the continuous coalescence of neural crest cells around the lens (Figs. 5H''–J'', open arrows). Thus, the mis-expression of cyp1b1 altered ocular neural crest migration and subsequent iris stromal organization.
Figure 5.
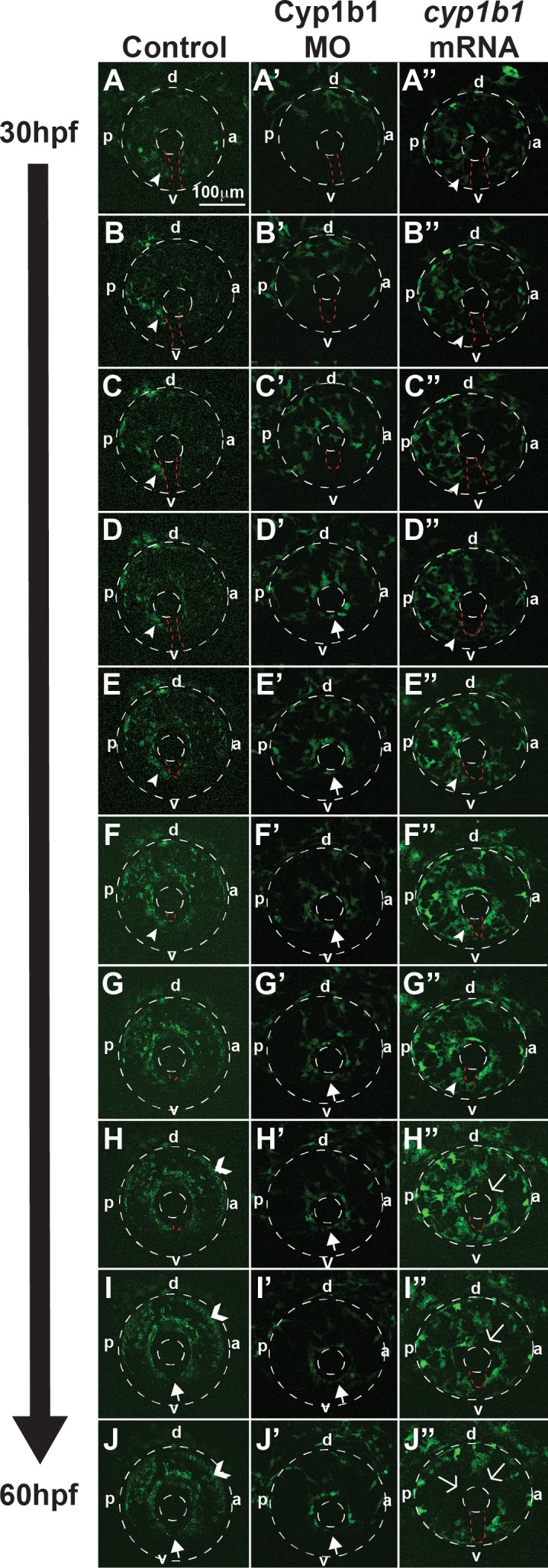
Cyp1b1 regulates ocular neural crest migration. Time-lapse imaging of Tg(foxd3:EGFP) control embryos from 30 to 60 hpf (A–J) showed that foxd3-positive cells entered into the anterior chamber between the surface ectoderm and optic cup, with more cells localized to the dorsal (d)–posterior (p) quadrant (E–H) compared with the anterior (a) and ventral (v) areas. In addition, foxd3-positive cells migrated adjacent to and through the ocular fissure (A–F, white arrowhead denotes migrating neural crest, red dashed line demarcates ocular fissure). At 60 hpf, foxd3-positive cells completely encircled the lens (I, J, closed arrows), indicating the closure of the fissure. In addition, at 60 hpf, foxd3 was also expressed in photoreceptors (H–J, open arrowhead). In Cyp1b1 MO knockdown embryos, Foxd3-positive cells travelled between the surface ectoderm and optic cup in the dorsal quadrants (A'–E'), but there were few cells adjacent to and in the ocular fissure. Foxd3-positive cells encircled the lens prior to controls (D'–H' versus D–H), indicating the premature closure of the fissure. At 60 hpf, there were fewer foxd3-positive cells in the eye (J'). In embryos overexpressing cyp1b1, ocular neural crest migration was less organized, as foxd3-positive cells were located throughout the posterior half of the eye (A''–G''). Further, the ocular fissure remained open and prevented the continuous coalescence of neural crest cells around the lens (H''–J'', open arrows), resulting in fewer neural crest cells at 60 hpf (J'').
Tyrosinase Did Not Modify the Effect of Cyp1b1 in Zebrafish Eye Development
Previously published studies in mice have demonstrated that tyrosinase modifies the effect of Cyp1b1 knockout; however, the specific interactions between these two enzymes in neural crest and anterior segment development remain unknown.4 This relationship was further investigated in zebrafish embryos. In 24 hpf wild-type zebrafish embryos, the tyrosinase gene (tyr) was expressed in the migrating neural crest and in the retinal pigment epithelium in uninjected and control-injected embryos (Figs. 6A, 6B). At 48 hpf, tyr expression (Figs. 6A', 6A'', 6B', 6B'') followed the skin pigmentation pattern and was also observed in the cranial neural crest (arrowheads), retinal pigment epithelium, migrating neural crest cells in the ocular fissure (closed arrows), and iris stroma (open arrows). At 48 hpf, Cyp1b1 knockdown decreased tyr expression in the cranial neural crest (arrowhead) and ocular fissure (Figs. 6C, 6C'). Overexpression of cyp1b1 did not alter tyr expression (Figs. 6D, 6D', 6D'') at 24 and 48 hpf. Mutant mice deficient for both Tyr and Cyp1b1 genes showed a worse anterior segment phenotype. We utilized pharmacologic and genetic strategies to decrease tyrosinase activity. Phenylthiourea is often used in zebrafish embryos to inhibit tyrosinase and subsequent pigmentation, but can cause developmental delays and abnormalities when administered prior to 22 hpf or at concentrations higher than 0.003%.29 To inhibit tyrosinase activity prior to tyr and cyp1b1 expression, embryos were treated with 0.003% PTU between 10 and 12 hpf. Treatment of uninjected embryos with PTU did not significantly increase the percentage of embryos with developmental delays and craniofacial abnormalities (17.6 ± 7.0%, P = 0.99) compared with untreated/uninjected embryos. Compared with PTU-treated uninjected embryos, PTU did not cause a significant increase in developmental delays or abnormalities in control-injected (26.3 ± 3.5%, P = 0.28) or Cyp1b1 MO-injected embryos (29.0 ± 7.5%, P = 0.85). In addition, we utilized the casper mutant strain, which has decreased tyr expression in neural crest–derived cells. Injection of Cyp1b1 MO in casper embryos did not significantly increase the percentage of embryos with craniofacial abnormalities or developmental delays (25.0 ± 11.0%) compared to control-injected (19.4 ± 12.8%, P = 0.71) and uninjected casper embryos (8.4 ± 10.4 %). Hence, in zebrafish, alterations in tyrosinase expression and activity did not alter the Cyp1b1 effect on eye development.
Figure 6.
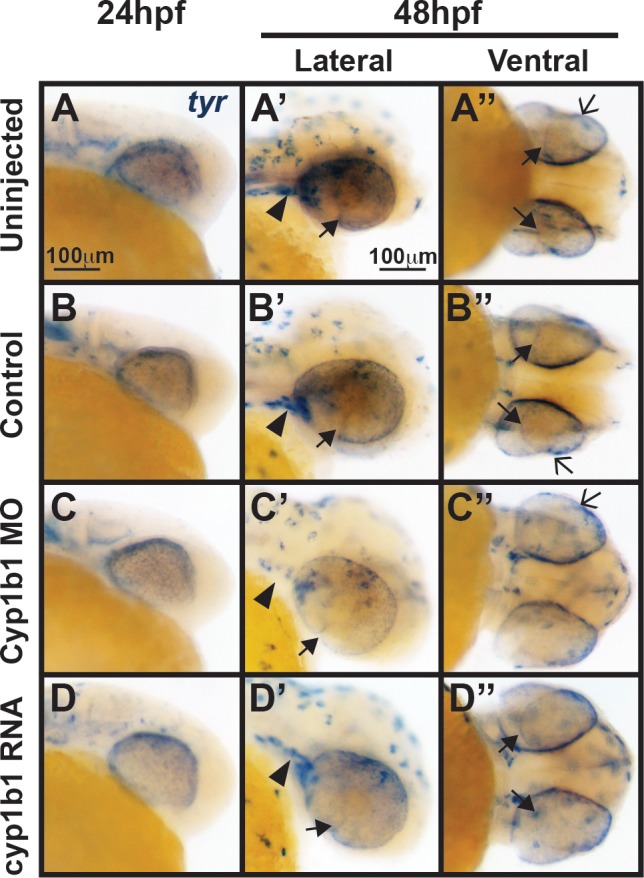
Tyr expression is not influenced by alterations in cyp1b1 expression. In situ hybridization demonstrated that at 24 hpf, tyr was expressed in the migrating neural crest and retinal pigment epithelium in uninjected (A) and control-injected (B) embryos. At 48 hpf, tyr expression (A', A'', B', B'') followed the skin pigmentation pattern and was also observed in the cranial neural crest (arrowheads), retinal pigment epithelium, migrating neural crest cells in the ocular fissure (closed arrows), and iris stroma (open arrows). Knockdown of cyp1b1 did not alter tyr expression at 24 hpf, but did decrease tyr expression in the cranial neural crest (arrowhead) and ocular fissure (C, C') at 48 hpf. Overexpression of cyp1b1 did not alter tyr expression (D, D', D'') at 24 and 48 hpf.
Cyp1b1 Differentially Regulated Genes Associated With Colobomas
Because Cyp1b1 regulation of ocular fissure closure was not mediated through RA, genes clinically associated with colobomas were investigated as downstream targets. In situ hybridization at 24 hpf demonstrated that cyp1b1 overexpression inhibited the expression of the homeobox transcription factor vsx2 in the developing eye (Figs. 7D, 7D') compared with Cyp1b1 MO knockdown (Figs. 7C, 7C'), control-injected (Figs. 7B, 7B'), and uninjected (Figs. 7A, 7A') embryos. Further, in situ hybridization revealed the presence of vsx2-expressing tissue bridging the area between the optic cups (Fig. 7D', arrow) in response to cyp1b1 overexpression. In contrast, cyp1b1 overexpression increased shha expression in the floor plate between the developing eyes (Figs. 7H, 7H', arrowheads) compared with Cyp1b1 MO knockdown (Figs. 7G, 7G), control-injected (Figs. 7F, 7F'), and uninjected (Figs. 7E, 7E') embryos. Alterations in cyp1b1 levels did not affect the expression of vsx1 and shhb (data not shown) at 24 hpf. Although the expression of pax6a and pax6b was not affected at 24 hpf (data not shown), the expression of these two pax6 genes was decreased in the retina in response to cyp1b1 overexpression at 48 hpf (Figs. 7L, 7L') compared with Cyp1b1 MO knockdown (Figs. 7K, 7K'), control-injected (Figs. 7J, 7J'), and uninjected (Figs. 7I, 7I') embryos. Thus, cyp1b1 overexpression altered the expression of key signaling molecules involved in eye development and ocular fissure closure.
Human CYP1B1 and Zebrafish cyp1b1 Genes Are Evolutionarily and Functionally Conserved
Mutations in CYP1B1 disrupt the development of the neural crest–derived trabecular meshwork and outflow channels and are the most commonly identified genetic causes of primary infantile-onset glaucoma.3 To investigate whether Cyp1b1 function is conserved between humans and zebrafish, human CYP1B1 mRNA was injected into zebrafish embryos at the one-cell stage. Western blotting verified human CYP1B1 protein expression in 24 hpf mRNA-injected embryos (Fig. 8G). Similar to the effects observed with the zebrafish gene, the overexpression of human CYP1B1 caused large colobomas in 71.8 ± 17.9% of embryos (P = 0.0003, Supplementary Table S5), reflecting the defective closure of the ocular fissure (Figs. 8B, 8B') compared to controls (Figs. 8A, 8A'). Further, similar to the overexpression of zebrafish cyp1b1, human CYP1B1 overexpression disrupted neural crest–derived jaw (Mk and Ch cartilage) and PA formation. The ATG MO-mediated knockdown of endogenous zebrafish Cyp1b1 showed improvement of craniofacial defects, but did not statistically significantly decrease the percentage of embryos with colobomas and craniofacial defects (36.4 ± 17.0, P = 0.06) due to human CYP1B1 overexpression (Figs. 8C, 8C').
Figure 8.
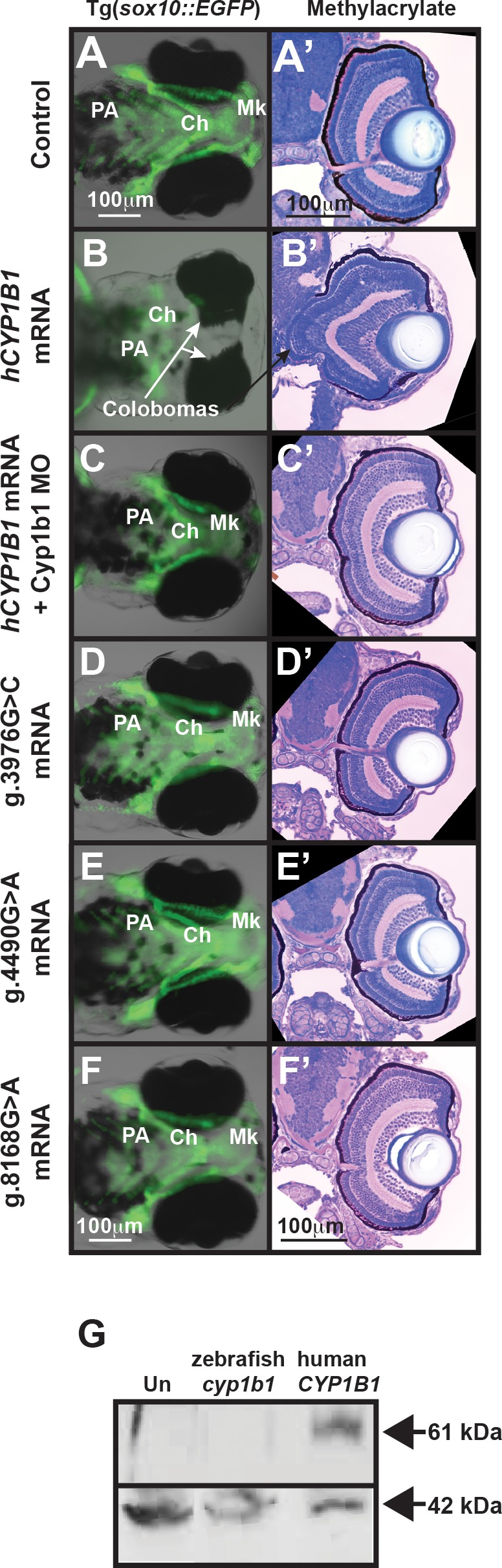
Human CYP1B1 and zebrafish cyp1b1 genes are evolutionarily and functionally conserved. Live images and methylacrylate sections of 96 hpf Tg(sox10:EGFP) embryos demonstrated that the injection of human CYP1B1 mRNA disrupted neural crest–derived jaw and pharyngeal arch formation and inhibited ocular fissure closure (B, B') compared with controls (A, A'). The knockdown of endogenous Cyp1b1 restored fissure closure (C') and partially rescued the jaw and pharyngeal arch defects resulting from the injection of human CYP1B1 mRNA (C). The injection of human CYP1B1 mRNA containing clinically relevant g.3976G>C (D, D'), g.4490G>A (E, E'), or g.8168G>A (F, F') mutations did not disrupt eye or neural crest development. Western blot analysis (G) showed expression of human CYP1B1 protein (61 kDa) in embryos injected with human CYP1B1 mRNA sequence, but not in uninjected embryos or embryos injected with zebrafish cyp1b1 mRNA. β-actin (42 kDa) was used as a loading control.
The genetic analysis of patients affected with primary infantile-onset glaucoma showed that disease-causing mutations typically occur in the second or third exon of the CYP1B1 gene.30 Using site-directed mutagenesis, three mutations (genomic DNA nomenclature based on GenBank U56438) were introduced into the coding region of human CYP1B1, and the mRNAs for each of these mutants were transcribed and injected into one-cell embryos (Supplementary Table S2). The g.3976G>C mutation results in a tryptophan to cysteine (W57C) amino acid substitution that disrupts the hinge region connecting the N-terminal membrane-bound domain to the cytosolic portion of CYP1B1.3,31 The g.4490G>A mutation results in a glutamic acid to lysine (E229K) amino acid substitution that alters the ionic bonds necessary for substrate binding.3,32–35 The g.8168G>A mutation leads to an arginine to glutamine (R444Q) amino acid substitution that destabilizes the heme-binding domain.36 Unlike wild-type human CYP1B1 mRNA, injecting g.3976G>C, g.4490G>A, or g.8168G>A mutant CYP1B1 mRNA into one-cell-stage zebrafish embryos did not significantly affect eye size, neural crest development, or ocular fissure closure (Supplementary Table S5; Figs. 8D, 8D', 8E, 8E', 8F, 8F') compared with control embryos. Thus, these studies demonstrated the evolutionary conservation of CYP1B1 gene function between humans and zebrafish.
Discussion
In primary infantile-onset glaucoma, mutations in the CYP1B1 gene lead to increased intraocular pressure and subsequent vision loss resulting from damage to the optic nerve, cornea, and sclera.1–3 However, the mechanism by which CYP1B1 regulates eye development has not been defined, and the in vivo substrate for this cytochrome p450 enzyme during embryogenesis has not previously been identified. In the present study, we used a zebrafish model to improve our understanding of the pathophysiology of primary infantile-onset glaucoma.
CYP1B1 mutations associated with primary infantile-onset glaucoma have primarily been detected in the coding region, comprising the second and third exons of this gene.3,37 The CYP1B1 protein comprises an N-terminal membrane-binding domain, a short “hinge” region, and a large C-terminal cytosolic domain. Disease-causing point mutations frequently affect highly conserved regions in the cytosolic portion, including the heme-binding region, substrate-binding region, and the substrate access channel. For example, the g.4490G>A (E229K) mutation, one of the most commonly identified point mutations associated with primary infantile-onset glaucoma, destabilizes ionic interactions between helices in the substrate-binding domain, resulting in decreased protein activity.3,30 In the present study, the overexpression of either the zebrafish cyp1b1 or human CYP1B1 yielded a similar phenotype: colobomatous defects and abnormal jaw and PA formation. However, the injection of human mRNA containing clinically significant point mutations had less effect on eye and craniofacial development. The effects of the injection of wild type were rescued after knocking down endogenous Cyp1b1, demonstrating functional conservation between human and zebrafish forms of this gene and indicating that zebrafish can be used as a model for studying the in vivo function of CYP1B1 in eye development.
In zebrafish, Cyp1b1 plays a role in metabolizing polycyclic aromatic hydrocarbons, such as 2,3,7,8-tetrachlorodibenzodioxin. Cyp1b1 transcription is directly regulated via exposure to exogenous toxins through the activation of the aryl hydrocarbon receptor.24,38 However, this regulatory pathway is not activated until 72 hpf, indicating that early cyp1b1 expression in the developing eye is regulated through developmental pathways, and this enzyme has an endogenous substrate. In vitro studies have shown that Cyp1b1 catalyzes both enzymatic steps in the conversion of retinol into RA.5 This mechanism represents an alternative pathway from the well-characterized canonical pathway, which uses alcohol dehydrogenases to convert retinol into retinal and retinaldehyde dehydrogenases to convert retinal into RA. In mice, chickens, and zebrafish, the developing eye expresses retinaldehyde dehydrogenases in the dorsal and ventral retina. The local synthesis of RA is counteracted by degradation enzymes (Cyp26a1, Cyp26b1, and Cyp26c1) expressed in the cranial and caudal retina.6–9 The specific expression of synthesis and degradation enzymes generates RA gradients, where the highest concentrations of RA are localized along the dorsal–ventral axis of the developing eye. Retinoic acid is critical for both craniofacial and ocular neural crest development. We previously demonstrated that decreased or increased RA levels disrupt neural crest contributions to the cornea, iris, jaw, and PA in zebrafish.17 The present study demonstrated that cyp1b1 expression was regulated by alterations in RA levels and the overexpression of cyp1b1 increased RA levels throughout developing zebrafish embryos. Further, the increase in RA as a result of cyp1b1 overexpression decreased the microphthalmia induced through RA deficiency (Raldh2 MO). Cyp1b1 knockdown had minimal effect on RA expression and eye development. However, decreased Cyp1b1 expression improved the effects of increased RA as a result of the overexpression of raldh2. Taken together, cyp1b1 may regulate RA synthesis in vivo through a secondary pathway that can be either up- or downregulated when the primary aldehyde dehydrogenase pathway is genetically or pharmacologically altered; however, the primary endogenous role of cyp1b1 in eye development seems to be independent of RA.
A key function of cyp1b1 may be the regulation of ocular fissure closure. Cyp1b1 expression in the developing eye was localized to the retina and retinal pigment epithelium within and immediately surrounding the ocular fissures. Although the inferior ocular fissure is well defined, the closure of the superior fissure prior to its ventral counterpart in zebrafish is less well understood (Stach T, et al. IOVS 2013;54:ARVO E-Abstract 3362). Cypb1 expression was correlated with the patency of ocular fissures, as cypb1 expression is highest between 24 and 36 hpf, after which expression becomes attenuated and is undetectable at 60 hpf. This finding suggests a role for cyp1b1 in maintaining the patency of ocular fissures. Ocular fissure closure is a complex process that requires proper optic vesicle evagination from the forebrain, specific spatial patterning of the optic cup along the dorsal–ventral and proximal–distal axes, breakdown of the basement membrane enveloping the anterior and posterior edges, and tissue fusion.27,28,39,40 In zebrafish embryos, the ventral fissure closure is initiated in the middle of the optic cup and proceeds anteriorly and posteriorly between 36 and 72 hpf.41 In the present study, the premature breakdown of laminin in the basement membrane was observed in Cyp1b1 MO knockdown embryos, suggesting premature ocular fissure closure compared with control MO-injected and uninjected embryos. Time-lapse imaging further revealed that the premature closure of the fissure disrupted neural crest migration adjacent to and through the ocular fissure into the anterior segment. In zebrafish embryos, premature fissure closure did not seem to affect neural crest contributions to the cornea and iris at 96 hpf; thus, we propose that this mechanism may account for the specific disruption of the neural crest–derived trabecular meshwork in primary infantile-onset glaucoma. In humans, neural crest cells travel in three separate waves, with the first wave generating the components of the cornea, the second wave contributing to the iris, and the third wave forming the trabecular meshwork and angle structures. While neural crest cells destined for the cornea and iris enter the anterior segment, the early closure of the ocular fissure may prevent neural crest specifically destined for the trabecular meshwork from migrating into the anterior segment. Differences in the effects on the neural crest in cyp1b1 mutant zebrafish, mice, and chickens may reflect species differences in the number of waves of neural crest migration (e.g., two in chickens and one in mice) and anatomic differences in aqueous outflow pathways.42,43 Taken together, the regulation of ocular fissure patency may explain the mechanism by which cyp1b1 affects neural crest contributions to the anterior segment of the eye.
Interestingly, overexpression of cyp1b1 resulted in large colobomas. CYP1B1 mutations have not been reported in cases of inferior colobomas, but this gene is not included in the typical genetic screens used in clinical testing. Notably, a CYP1B1 mutation was identified in a patient with a rare superior coloboma (Stach T, et al. IOVS 2013;54:ARVO E-Abstract 3362). However, whether this variant is a gain-of-function mutation remains unknown, indicating that additional studies are required to further identify and assess mutations in human CYP1B1 in cases of colobomas.
Given the timing of the changes in the expression of shh, pax6, and vsx2 and the increased apoptosis in the inferior ocular fissure, we hypothesized that cyp1b1 overexpression had an early effect on the spatial patterning of the optic cup. In humans, heterozygous mutations in SHH are associated with holoprosencephaly and a spectrum of eye malformations, including anophthalmia, microphthalmia, and colobomas.44–46 Shh is expressed in the floor plate of the diencephalon and is required for patterning the optic cup through the specific spatial expression of transcription factors.47 Shh regulates Vax2 and Pax2 in the optic stalk and ventral neural retina to maintain the expression of these transcription factors within the more proximal structures of the eye.48,49 Mice with Vax2 or Pax2 mutations show the inhibition of basement membrane breakdown in the fissure, resulting in colobomas. In contrast, Shh inhibits Pax6, restricting its expression to the distal optic cup, which is critical for lens induction from the surface ectoderm.50,51 Human mutations in PAX6 are most commonly associated with aniridia (OMIM 706108), an autosomal dominant congenital eye disease characterized by cataract, limbal stem cell deficiency, glaucoma, and hypoplasia of the iris, optic nerve, and fovea. While large disruptions in gene function resulting from frameshift or nonsense mutations lead to these widespread ocular defects,52,53 rare missense PAX6 mutations have been associated with colobomas.54 In mice, Pax6 regulates other genes associated with colobomas, such as Vsx2 (Chx10), Maf1, and Six3.55–57 The current studies showed that in zebrafish, cyp1b1 overexpression increased shha gene expression in the midline floor plate, while decreasing pax6a, pax6b, and vsx2 gene expression in the developing retina. Notably, RA is also a key regulator of ocular fissure closure, as both increased and decreased RA result in colobomas in mice and zebrafish.58–61 Exposure to exogenous RA inhibits Shh expression in the midline floor plate.62 We observed that the overexpression of cyp1b1 not only increased RA levels throughout the developing head and eye, but also increased shha expression. Furthermore, genetically or pharmacologically decreasing endogenous RA synthesis to compensate for cyp1b1 overexpression did not rescue the observed colobomatous defects. It is unclear whether these signaling pathways are affected only in the overexpression state or represent normal endogenous targets of cyp1b1. We did not observe alterations in the levels of shh, pax6, or vsx2 with decreased Cyp1b1, suggesting that there are additional targets of Cyp1b1 that regulate fissure patency. Taken together, the results of the present study provide evidence that the overexpression of cyp1b1 regulates ocular fissure closure through an RA-independent mechanism involving shh and pax6.
In summary, we investigated the mechanisms by which Cyp1b1 regulates eye development. The functions of the zebrafish and human forms of the CYP1B1 gene were evolutionarily conserved. Cyp1b1 plays a primary role in regulating ocular fissure closure, which indirectly regulates neural crest migration, and this mechanism was independent of RA. These results improve the current understanding of the complex signals that regulate eye development and, importantly, provide insight into the molecular pathogenesis of primary infantile-onset glaucoma.
Supplementary Material
Acknowledgments
The authors thank Elisa Schley, Mitchell Gillett, and the Kellogg Eye Center Morphology and Imaging Module for technical assistance.
Supported by grants from the National Eye Institute of the National Institutes of Health (K08EY022912-01), Vision Research Core (P30 EY007003), a Research to Prevent Blindness Career Development Award, and an Edward Mallinckrodt Jr. Foundation Grant.
Disclosure: A.L. Williams, None; J. Eason, None; B. Chawla, None; B.L. Bohnsack, Alcon Research Institute (F)
References
- 1. Aponte EP,, Diehl N,, Mohney BG. Inicidence and clinical characteristics of childhood glaucoma: a population-based study. Arch Ophthalmol. 2010; 128: 478–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papadopoulos M,, Cable N,, Rahi J,, Khaw PT; BIG Eye Study Investigators. The British Infantile and Childhood Glaucoma (BIG) Eye Study. Invest Ophthalmol Vis Sci. 2007; 48: 100–106. [DOI] [PubMed] [Google Scholar]
- 3. Li N,, Zhou Y,, Du L,, Wei M,, Chen X. Overview of cytochrome p450 1B1 gene mutations in patients with primary congenital glaucoma. Exp Eye Res. 2011; 93: 572–579. [DOI] [PubMed] [Google Scholar]
- 4. Libby RT,, Smiths RS,, Savinova OV,, et al. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science. 2003; 299: 1578–1581. [DOI] [PubMed] [Google Scholar]
- 5. Chambers D,, Wilson L,, Maden M,, Lumsden A. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development. 2007; 134: 1369–1383. [DOI] [PubMed] [Google Scholar]
- 6. McCaffery P,, Wagner E,, O'Neil J,, Petkovich M,, Drager UC. Dorsal and ventral retinal territories defined by retinoic acid synthesis, break-down and nuclear receptor expression. Mech Dev. 1999; 82: 119–130. [DOI] [PubMed] [Google Scholar]
- 7. Grandel H,, Brand M. Zebrafish limb development is triggered by a retinoic acid signal during gastrulation. Dev Dyn. 2011; 240: 1116–1126. [DOI] [PubMed] [Google Scholar]
- 8. Cvekl A,, Wang W-L. Retinoic acid signaling in mammalian eye development. Exp Eye Res. 2009; 89: 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aulehla A,, Pourquie O. Signaling gradients during paraxial mesoderm development. Cold Spring Harb Perspect Biol. 2010; 2: a000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bohnsack BL,, Kasprick D,, Kish PE,, Goldman D,, Kahana A. A zebrafish model of Axenfeld-Rieger Syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Invest Ophthalmol Vis Sci. 2012; 53: 7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bohnsack BL,, Kahana A. Thyroid hormone and retinoic acid interact to regulate zebrafish craniofacial neural crest development. Dev Biol. 2013; 373: 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trainor PA. Specification of neural crest cell formation and migration in mouse embryos. Semin Cell Dev Biol. 2005; 16: 683–693. [DOI] [PubMed] [Google Scholar]
- 13. Gage PJ,, Rhoades W,, Prucka SK,, Hjalt T. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci. 2005; 46: 4200–4208. [DOI] [PubMed] [Google Scholar]
- 14. Strungaru MH,, Dinu I,, Walter MA. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci. 2007; 48: 228–237. [DOI] [PubMed] [Google Scholar]
- 15. Harissi-Dagher M,, Colby K. Anterior segment dysgenesis: Peters anomaly and sclerocornea. Int Ophthalmol Clin. 2008; 48: 35–42. [DOI] [PubMed] [Google Scholar]
- 16. Vincent A,, Billingsley G,, Priston M,, et al. Phenotypic heterogeneity of CYP1B1: mutations in a patient with Peters Anomaly. J Med Genet. 2001; 36: 152–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bohnsack BL,, Gallina D,, Thompson H,, et al. Development of extraocular muscles require early signals from periocular neural crest and the developing eye. Arch Ophthalmol. 2011; 129: 1030–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bohnsack BL,, Kahana A. Phenothiourea sensitizes zebrafish cranial neural crest and extraocular muscle developmnt to changes in retinoic acid and insulin-like growth factor signaling. PLoS One. 2011; 6: e22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kimmel CB,, Ballard WW,, Kimmel SR,, Ullmann BB,, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995; 203: 253–310. [DOI] [PubMed] [Google Scholar]
- 20. Curran K,, Raible DW,, Lister JA. Foxd3 controls melanophore specification in the zebrafish neual crest by regulation of Mitf. Dev Biol. 2009; 332: 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dutton K,, Dutton JR,, Pauliny A,, Kelsh RN. A morpholino phenocopy of the colourless mutant. Genesis. 2001; 30: 188–189. [DOI] [PubMed] [Google Scholar]
- 22. Dutton KA,, Pauliny A,, Lopes SS,, et al. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development. 2001; 128: 4113–4125. [DOI] [PubMed] [Google Scholar]
- 23. Chawla B,, Schley E,, Williams AL,, Bohnsack BL. Retinoic acid and pitx2 regulate early neural crest survival and migration in craniofacial and ocular development. Birth Defects Res B Dev Reprod Toxicol. 2016; 107: 126–135. [DOI] [PubMed] [Google Scholar]
- 24. Timme-Laragy AR,, Noyes PD,, Buhler DR,, Di Giulio RT. CYP1B1 knockdown does not alter synergistic developmental toxicity of polycyclic aromatic hydrocarbons in zebrafish (Danio rerio). Mar Environ Res. 2008; 66: 85–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barthel LK,, Raymond PA. In situ hybridization studies of retinal neurons. Methods Enzymol. 2000; 316: 579–590. [DOI] [PubMed] [Google Scholar]
- 26. Williams AL,, Bohnsack BL. Neural crest derivatives in ocular development: discerning the eye of the storm. Birth Defects Res C Embryo Today. 2015; 105: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gregory-Evans CY,, Williams MJ,, Halford S,, Gregory-Evans K. Ocular coloboma: a reassessment in the age of molecular neuroscience. J Med Genet. 2004; 41: 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gongal PA,, French CR,, Waskiewicz AJ. Aberrant forebrain signaling during early development underlies the generation of holoprosencephaly and coloboma. Biochim Biophys Acta. 2011; 1912: 390–401. [DOI] [PubMed] [Google Scholar]
- 29. Bohnsack BL,, Gallina D,, Kahana A. Phenothiourea sensitizes zebrafish cranial neural crest and extraocular muscle developmnt to changes in retinoic acid and insulin-like growth factor signaling. PLoS One. 2011; 6: e22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Achary MS,, Reddy AB,, Chakrabarti S,, et al. Disease-causing mutations in proteins: structural analysis of the CYP1B1 mutations causing primary congenital glaucoma in humans. Biophys J. 2006; 91: 4329–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stoilov I,, Akarsu AN,, Alozie I,, et al. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P450 1B1. Am J Hum Genet. 1998; 62: 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Acharya M,, Huang L,, Fleisch VC,, Allison WT,, Walter MA. A complex regulatory network of transcription factors critical for ocular development and disease. Hum Mol Genet. 2011; 20: 1610–1624. [DOI] [PubMed] [Google Scholar]
- 33. Badeeb OM,, Micheal S,, Koenekoop RK,, den Hollander AI,, Hedrawi MT. CYP1B1 mutations in patients with primary congenital glaucoma from Saudia Arabia. BMC Med Genet. 2014; 15: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chavarria-Soley G,, Sticht H,, Akillu E,, et al. Mutations in CYP1B1 cause primary congenital glaucoma by reduction of either activity or abundance of the enzyme. Hum Mutat. 2008; 29: 1147–1153. [DOI] [PubMed] [Google Scholar]
- 35. Hollander DA,, Sarfarazi M,, Stoilov I,, et al. Genotype and phenotype correlations in congenital glaucoma: CYP1B1 mutations, goniodysgenesis, and clinical characteristics. Am J Ophthalmol. 2006; 142: 993–1004. [DOI] [PubMed] [Google Scholar]
- 36. Mashima Y,, Suzuki Y,, Sergeev Y,, et al. Novel cytochrome P4501B1 (CYP1B1) gene mutations in Japanese patients with primary congenital glaucoma. Invest Ophthalmol Vis Sci. 2001; 42: 2211–2216. [PubMed] [Google Scholar]
- 37. Lloret-Vilaspasa F,, Jensen HJ,, de Roos K,, et al. Retinoid signalling is required for information transfer from mesoderm to neuroectoderm during gastrulation. Int J Dev Biol. 2010; 54: 599–608. [DOI] [PubMed] [Google Scholar]
- 38. Yin H-C,, Tseng H-P,, Chung H-Y,, et al. Influence of TCDD on zebrafish CYP1B1 transcription during development. Toxicol Sci. 2008; 103: 158–168. [DOI] [PubMed] [Google Scholar]
- 39. Matsushima D,, Heavner W,, Pevny LH. Combinatorial regulation of optic cup progenitor cell fate by SOX2 and PAX6. Development. 2011; 138: 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reza HM,, Takahashi Y,, Yasuda K. Stage-dependent expression of Pax6 in optic vesicle/cup regulates patterning genes through signaling molecules. Differentiation. 2007; 75: 726–736. [DOI] [PubMed] [Google Scholar]
- 41. Pillai-Kastoori L,, Wen W,, Wilson SG,, et al. Sox11 is required to maintain proper levels of hedgehog signaling during vertebrate ocular morphogenesis. PLoS Genet. 2014; 10: e1004491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hay ED,, Revel JP. Fine structure of the developing avian cornea. Monogr Dev Biol. 1969; 1: 1–144. [PubMed] [Google Scholar]
- 43. Pei YF,, Rhodin JA. The prenatal development of the mouse eye. Anat Rec. 1970; 168: 105–125. [DOI] [PubMed] [Google Scholar]
- 44. Schimmenti LA,, de la Cruz J,, Lewis RA,, et al. Novel mutation in sonic hedgehog in non-syndrome colobomatous microphthalmia. Am J Med Genet. 2003; 116A: 215–221. [DOI] [PubMed] [Google Scholar]
- 45. Geng X,, Speirs C,, Lagutin O,, et al. Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev Cell. 2008; 15: 236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nanni L,, Ming JE,, Bocian M,, et al. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet. 1999; 8: 2479–2488. [DOI] [PubMed] [Google Scholar]
- 47. Ingham PW,, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet. 2006; 7: 841–850. [DOI] [PubMed] [Google Scholar]
- 48. Barbieri AM,, Broccoli V,, Bovolenta P,, et al. Vax2 inactivation in mouse determines alteration of the eye dosal-ventral axis, misrouting of the optic fibres and eye coloboma. Development. 2002; 129: 805–813. [DOI] [PubMed] [Google Scholar]
- 49. Torres M,, Gomez-Pardo E,, Gruss P. Pax2 contributes to inner ear patterning and optic nerve trajectory. Development. 1996; 122: 3381–3391. [DOI] [PubMed] [Google Scholar]
- 50. Macdonald R,, Barth KA,, Xu Q,, Holder N,, Mikkola I,, Wilson SW. Midline signalling is required for Pax gene regulation and patterning of the eyes. Development. 1995; 121: 3267–3278. [DOI] [PubMed] [Google Scholar]
- 51. Canto-Soler MV,, Adler R. Optic cup and lens development requires Pax6 expression in the early optic vesicle during a narrow time window. Dev Biol. 2006; 294: 119–132. [DOI] [PubMed] [Google Scholar]
- 52. Hingorani M,, Hanson I,, van Heyningen V. Aniridia. Eur J Hum Genet. 2012; 20: 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee H,, Khan R,, O'Keefe M. Aniridia: current pathology and management. Acta Ophthalmol. 2008; 86: 708–715. [DOI] [PubMed] [Google Scholar]
- 54. Azuma N,, Yamaguchi Y,, Handa H,, et al. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet. 2003; 72: 1565–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ferda-Percin E,, Ploder LA,, Yu JJ,, et al. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet. 2000; 25: 397–401. [DOI] [PubMed] [Google Scholar]
- 56. Jamieson RV,, Perveen R,, Kerr B,, et al. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet. 2002; 11: 33–42. [DOI] [PubMed] [Google Scholar]
- 57. Wallis EC,, Rosessler E,, Hehr U,, et al. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999; 22: 196–198. [DOI] [PubMed] [Google Scholar]
- 58. Stull DL,, Wikler KC. Retinoid-dependent gene expression regulates early morphological events in the development of the murine retina. J Comp Neurol. 2000; 417: 289–298. [DOI] [PubMed] [Google Scholar]
- 59. Marsh-Armstrong N,, McCaffery P,, Gilbert W,, Dowling JE,, Drager UC. Retinoic acid is necessary for development of the ventral retina in zebrafish. Proc Natl Acad Sci U S A. 1994; 91: 7286–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Molotkov A,, Molotkova N,, Duester G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development. 2006; 133: 1901–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. See AW,, Clagett-Dame M. The temporal requirement for vitamin A in the developing eye: mechanism of action in optic fissure closure and new roles for the vitamin in regulating cell proliferation and adhesion in the embryonic retina. Dev Biol. 2009; 325: 94–105. [DOI] [PubMed] [Google Scholar]
- 62. Helms JA,, Kim CH,, Hu D,, Minkoff R,, Thaller C,, Eichele G. Sonic hedgehog participates in craniofacial morphogenesis and is down-regulated by teratogenic doses of retinoic acid. Dev Biol. 1997; 187: 25–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.