

Summary
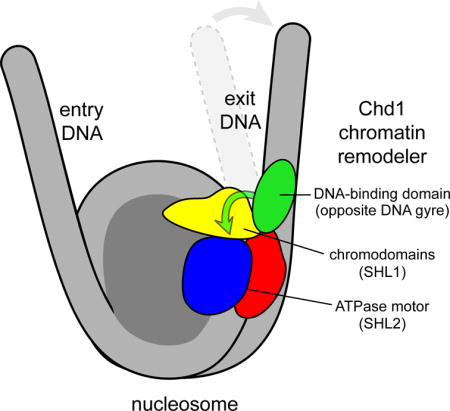
Chromatin remodelers use a helicase-like ATPase motor to reposition and reorganize nucleosomes along genomic DNA. Yet how the ATPase motor communicates with other remodeler domains in the context of the nucleosome has so far been elusive. Here we report for the Chd1 remodeler a unique organization of domains on the nucleosome that reveals direct domain-domain communication. Site-specific cross-linking shows that the chromodomains and ATPase motor bind to adjacent SHL1 and SHL2 sites, respectively, on nucleosomal DNA and pack against the DNA-binding domain on DNA exiting the nucleosome. This domain arrangement spans the two DNA gyres of the nucleosome and bridges both ends of a wrapped, ~90 bp nucleosomal loop of DNA, suggesting a means for nucleosome assembly. This architecture illustrates how Chd1 senses DNA outside the nucleosome core and provides a basis for nucleosome spacing and directional sliding away from transcription factor barriers.
Keywords: chromatin remodeling, nucleosome, ATPase motor, Snf2, CHD, ISWI
Graphical abstract
Introduction
Nucleosomes, the fundamental packaging unit of eukaryotic genomes, inherently restrict access to DNA (Kornberg and Lorch, 1999). Nucleosome reorganization, which is tightly coupled to transcriptional regulation, is driven by a diverse class of ATP-utilizing enzymes called chromatin remodelers (Ho and Crabtree, 2010). The Chd1 remodeler predominantly promotes transcriptional activation, and in mouse models Chd1 is essential for maintaining pluripotency of embryonic stem cells (Gaspar-Maia et al., 2009), as well as reprogramming endothelium into hematopoietic stem cells (Koh et al., 2015). Chd1 localizes to both gene promoters and coding regions and is believed to reassemble and reposition nucleosomes in the wake of RNA polymerase II (Lin et al., 2011; Pointner et al., 2012; Smolle et al., 2012). Like all chromatin remodelers, Chd1 possesses a conserved helicase-like ATPase motor responsible for translocating DNA past the histone core (Flaus et al., 2006). For each remodeler family, the ATPase motor is accompanied by unique auxiliary domains that are responsible for distinct remodeling outcomes. An area of intense interest is determining how these auxiliary domains coordinate with the ATPase motor to guide when and where the remodeler acts on nucleosome substrates.
Chd1 is named for N-terminal chromodomains, a central helicase-like ATPase motor, and a C-terminal DNA-binding domain (DBD; Figure 1A) (Delmas et al., 1993). One defining characteristic of Chd1 is its preference for directionally sliding mononucleosomes away from DNA ends (Stockdale et al., 2006). We previously showed that this directional sliding depends on the DBD (McKnight et al., 2011; Patel et al., 2013), yet how directionality is conferred has been unclear. Although the Chd1 DBD is not essential for nucleosome sliding, removal of this domain significantly impairs sliding activity. By fusing foreign binding domains to Chd1, we found that one role of the DBD is to tether the rest of the remodeler to nucleosome substrates (McKnight et al., 2011; Nodelman and Bowman, 2013; Patel et al., 2013). An important unanswered question, however, is how the Chd1 DBD might relay the availability of extranucleosomal DNA to the ATPase motor. Beyond simply tethering the remodeler to nucleosomes, interdomain communication could enable the remodeler to be more sensitive to DNA availability, yet evidence of such communication has so far been lacking.
Figure 1. The Chd1 DNA-binding domain preferentially binds DNA in a specific orientation and position at the edge of the nucleosome.
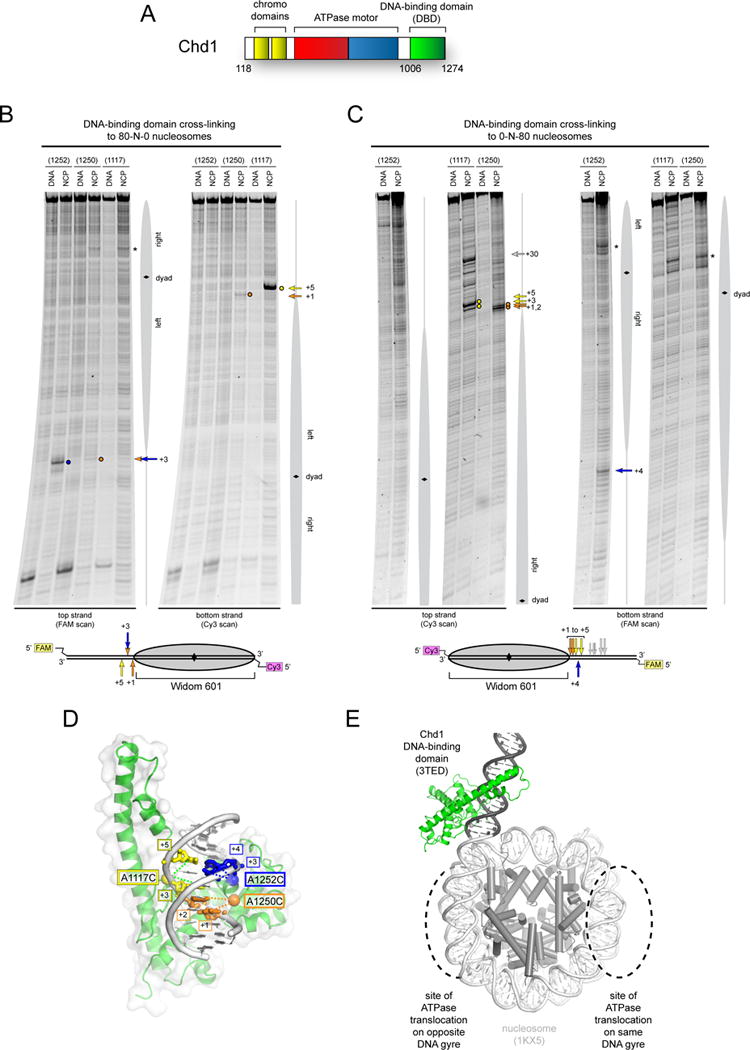
(A) Schematic of Chd1 domain architecture.
(B) and (C) Cross-linking of single cysteine variants within the Chd1 DNA-binding domain (DBD) to 80N0 and 0N80 nucleosomes or naked DNA in the presence of ADP∙BeF3. Lower schematics provide a summary of cross-linking positions, with the numbering referring to the distances from the nucleosome edge. Cross-linking sites reported here were observed in six or more independent reactions (n ≥ 6).
(D) Cross-links are consistent with a unique position and orientation of the DBD on DNA. The crystal structure of the Chd1 DBD bound to DNA (PDB code 3TED; (Sharma et al., 2011)) is shown, highlighting positions of cysteine substitutions as colored spheres and the corresponding cross-linked bases as colored sticks.
(E) A model of the Chd1 DBD at the edge of the nucleosome. Ideal B-form DNA (gray) was added to one end of the nucleosome crystal structure (1KX5, (Davey et al., 2002)), and the Chd1 DBD was positioned by docking DNA from the crystal structure (3TED) to match cross-linking as shown in (C).
See also Figure S1.
In this study, we report an unexpected organization of Chd1 domains on the nucleosome that explains how the ATPase motor can respond to DNA flanking the nucleosome. Through site specific cross-linking, we show that the DBD is stabilized specifically at the nucleosome edge in a manner that depends on the double chromodomains and ATPase motor (chromo-ATPase) portion of Chd1. Guided by site-specific cross-linking of the ATPase motor at the internal superhelical location 2 (SHL2) site, we identified a DNA-interacting loop of the chromodomains that places the chromodomains adjacent to the ATPase motor on the nucleosome. This location positions the chromodomains between the ATPase motor on one DNA gyre and the DBD bound to the opposite gyre, corresponding with exit-side DNA. Using streptavidin to site-selectively block the ATPase motor at one SHL2 site, we demonstrate physical coupling between the DBD and the chromo-ATPase across the DNA gyres of the nucleosome. Direct packing of the DBD against the chromo-ATPase is further supported by negative stain electron microscopy (EM), which suggests that stabilizing interactions with the DBD is coupled to DNA unpeeling from the nucleosome edge. We further show that the presence of DNA exiting the nucleosome slows Chd1 ATPase activity, and that disrupting the interface between the DBD and chromodomains abrogates this inhibition. This study reveals how interdomain communication on the nucleosome allows auxiliary remodeler domains to guide action of the ATPase motor.
Results
The Chd1 DNA-binding domain prefers a specific position and orientation at the edge of the nucleosome
Despite sequence-independent interactions between the Chd1 DBD and DNA that would likely promote easy diffusion along DNA (Sharma et al., 2011), we hypothesized that communication with the rest of the remodeler would take place at a specific location with respect to the nucleosome core. To identify positions occupied by the DBD, we took a site-directed cross-linking approach that can report protein-DNA interactions at single-nucleotide resolution (Pendergrast et al., 1992). Using the crystal structure of the Chd1 DBD bound to DNA as a guide (Sharma et al., 2011), several amino acid positions were independently mutated to cysteine, labeled with the photoactivatable cross-linker azidophenacyl bromide (APB), and tested for cross-linking to nucleosomal DNA. These cysteines were introduced into a cysteine-free variant containing the chromodomains, ATPase motor, and DBD. To control for DNA sequence effects, cross-linking was carried out with both 80N0 and 0N80 nucleosome substrates, where N denotes the asymmetric Widom 601 positioning sequence (Lowary and Widom, 1998). In parallel, reactions were also carried out with the same DNA in the absence of histones to evaluate the specificity of cross-links for nucleosomes.
When Chd1 and nucleosomes were incubated in the presence of the ATP analog ADP∙BeF3, three single-cysteine substitutions gave specific and strong cross-links to nucleosome substrates: A1117C, A1250C, and H1252C (Figure 1B,C and Figure S1A). For both 80N0 and 0N80 nucleosome substrates, all cysteine-specific cross-links were made to extranucleosomal DNA. Cross-links were also detected 22–24 bp from the nucleosome dyad (Figure 1B,C, * mark); however, as these were also observed with a cys-free Chd1 variant (Figure S1B), APB labeling of Chd1 at a non-cysteine residue, likely on the ATPase motor (see below), was responsible for cross-linking to nucleosomal DNA. For the three single cysteine variants, cross-linking differed for the two sides of the nucleosome. The unique patterns highlight how cross-linking was influenced by DNA sequence, possibly reflecting differences in DBD binding or stability on the two sides of the nucleosome.
Interestingly, when the single cysteine substitutions are mapped onto the crystal structure, the relative locations of cross-links closest to the nucleosome edge suggests that a unique placement of the DBD on DNA could simultaneously satisfy most cross-links (Figure 1D). In agreement with this notion, aside from the A1250C variant on 80N0 nucleosomes, all cross-links were strand-specific, indicating a preferred orientation of the DBD relative to the nucleosome edge. The site and strand-specific cross-linking supports the preference of the DBD for the major groove as observed in the crystal structure (Sharma et al., 2011). To visualize how these cross-links define the relative positioning of the DBD on the nucleosome, we used idealized B-form DNA to extend the DNA duplex at one end of the 1.9 Å resolution nucleosome crystal structure (Davey et al., 2002). Aligning DNA from the Chd1 DBD-DNA crystal structure with DNA extending away from the nucleosome placed the DBD in the plane of the DNA superhelix, on the outermost edge of DNA facing away from the histone core (Figure 1E).
Cross-linking of the DNA-binding domain at the edge of the nucleosome requires the Chd1 chromo-ATPase
In addition to ADP∙BeF3, cross-linking was also performed in the presence of ADP and the nonhydrolyzable ATP analog AMP-PNP. On 80N0 nucleosomes, which reported on the left edge of the nucleosome, cross-linking was restricted to one or two positions whether ADP·BeF3 or AMP-PNP was present (Figure S2A). In contrast, the use of ADP·BeF3 versus AMP-PNP dramatically altered the distribution of cross-links to 0N80 nucleosomes for DBD variants A1117C and A1250C (Figure 2A,B and S2B). These distributed patterns on the right edge of the nucleosome were distinct for A1117C and A1250C variants, which we speculate may be due to the APB labeling at different places on the DNA-binding surface. For both variants, however, the presence of ADP·BeF3 always strengthened cross-links at the nucleosome edge. The influence of nucleotide state on DBD cross-linking suggested a physical connection between the position of the DBD and the nucleotide-bound conformation of the ATPase motor.
Figure 2. Cross-linking of the Chd1 DNA-binding at the nucleosome edge requires the chromo-ATPase.
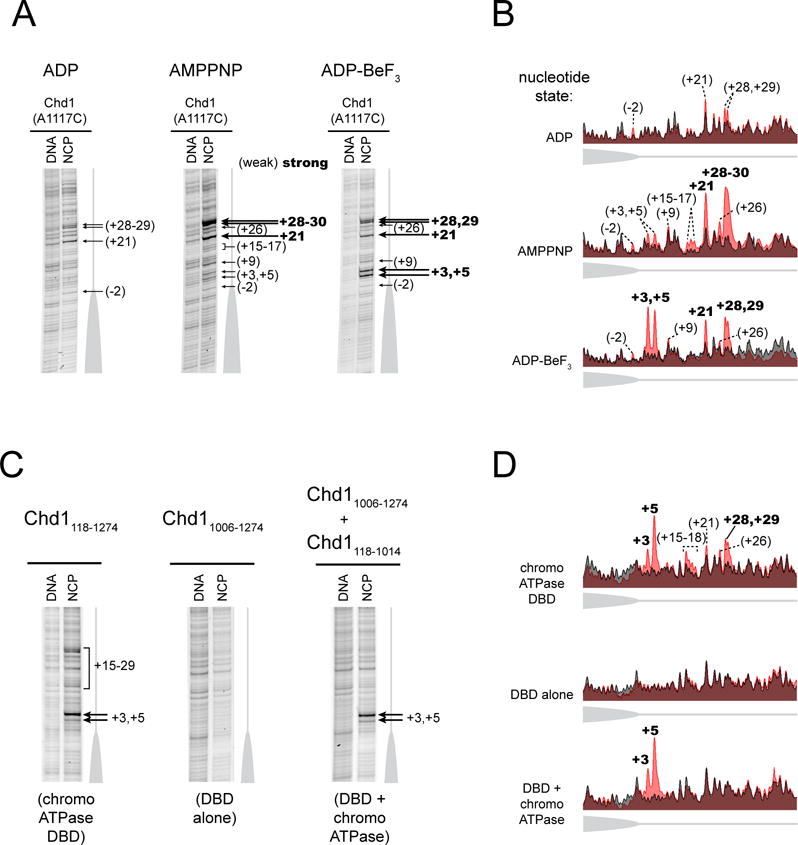
(A) The nucleotide-bound state of the remodeler biases the distribution of cross-links made by the DBD. Cross-linking reactions were carried out using 0N80 nucleosomes (n = 6).
(B) Intensity profiles of gel lanes shown in (A). Nucleosome cross-linking profiles (light red) are aligned with the background DNA-only cross-linking (gray).
(C) The chromo-ATPase is necessary for cross-linking of the DBD at the nucleosome edge and can facilitate cross-linking in trans. Cross-linking was carried out with the naturally connected chromo-ATPase-DBD (left), the isolated DBD (center), and the chromo-ATPase added to the DBD in trans (right) (n = 5).
(D) Intensity profiles of gel lanes shown in (C), as described in (B).
See also Figure S2.
The influence of nucleotide state on cross-linking could arise from the ATPase motor either assisting or inhibiting placement of the DBD on extranucleosomal DNA. The DBD alone (Chd11006–1274, A1117C variant) failed to yield specific cross-links (Figure 2C,D), indicating that the chromo-ATPase portion of Chd1 is required for stable positioning of the DBD. To see whether a direct interaction may be responsible for positioning the DBD, cross-linking was performed with the isolated DBD where the chromo-ATPase (Chd1118–1014) was added in trans. Remarkably, in ADP·BeF3 conditions, addition of the chromo-ATPase allowed cross-links closest to the nucleosome edge to be recovered (Figure 2C,D). These results strongly suggest that the closest cross-links arise from a direct interaction of the DBD with the Chd1 chromo-ATPase. In contrast, the cross-links observed farther from the nucleosome edge were not observed, suggesting that a covalent linkage with the rest of the remodeler is required. Thus, while more distant cross-links likely report a high local concentration of the DBD on flanking DNA due to tethering via the chromo-ATPase, the closest edge cross-links appear to reflect direct domain-domain communication.
The contribution of the DBD to nucleosome binding depends on the nucleotide-bound state of the ATPase motor
To examine how the ATPase motor and DBD of Chd1 contribute to nucleosome binding, we performed fluorescence-based binding experiments in the presence of either AMP-PNP or ADP·BeF3, using nucleosomes that possessed or lacked extranucleosomal DNA. With ADP·BeF3, Chd1 bound tightly to nucleosomes, regardless of whether extranucleosomal DNA was present (Figure 3, dark blue bars, Table S1). Since the observed affinities of 5 nM or stronger were near the upper limit of this binding assay, we increased KCl concentrations to 150 mM to weaken binding. This higher salt condition only modestly perturbed ADP·BeF3-dependent binding, with affinity decreasing to 12 ± 4 nM for nucleosomes lacking extranucleosomal DNA (0N0, light blue bars). In contrast, Chd1 binding was much more sensitive to both salt and extranucleosomal DNA in AMP-PNP conditions. While AMP-PNP conditions yielded tight binding to 0N63 nucleosomes in 100 mM KCl (K1/2 = 8.4 ± 1.6 nM), affinity was significantly weaker for 0N63 nucleosomes in 150 mM KCl (K1/2 = 160 ± 20 nM), which was similar to ~17-fold weaker binding observed for 0N0 nucleosomes in 100 mM KCl (K1/2 = 143 ± 10 nM, dark and light orange bars).
Figure 3. The DBD most strongly contributes to nucleosome binding under AMP-PNP conditions when flanking DNA is available.
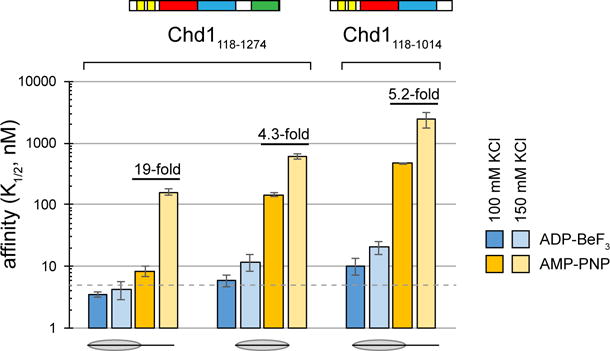
0N63 and 0N0 nucleosomes were used for AMP-PNP conditions, and 2N61 and 0N0 nucleosomes for ADP·BeF3 conditions. The dotted line marks 5 nM, which is near the limit for this assay. Error bars represent the standard deviations of three or more measurements.
See also Table S1.
To help further dissect domain contributions, binding experiments were also performed with a Chd1 protein lacking the DBD, Chd1118–1014. Although Chd1118–1014 binding was weaker overall, the binding profile was similar to Chd1 with 0N0 nucleosomes. In particular, in AMP-PNP, higher KCl decreased binding by ~4-fold (Chd1 with 0N0) or ~5-fold (Chd1118–1014 with 0N63), which contrasted with ~19-fold weaker binding observed for Chd1 with 0N63 nucleosomes (Figure 3, compare dark and light orange bars). These data are consistent with a greater reliance on the DBD engaging with extranucleosomal DNA when the ATPase motor is bound to AMP-PNP.
Both the ATPase motor and chromodomains of Chd1 contact nucleosomal DNA at specific sites
To better visualize how the DBD might communicate with the ATPase motor, we targeted the Chd1 ATPase motor with cys-directed cross-linking to reveal its positioning on the nucleosome. Previously, remodeling activities for several chromatin remodelers were shown to require intact DNA at the internal SHL2 site on the nucleosome, which is ~50 bp from the closest edge of the nucleosome (McKnight et al., 2011; Ranjan et al., 2015; Saha et al., 2005; Schwanbeck et al., 2004; Zofall et al., 2006). Cross-linking studies with ISWI and SWI/SNF remodelers demonstrated that the ATPase motor of the remodeler bound to SHL2 (Dang and Bartholomew, 2007; Dechassa et al., 2012); however, the photoactivatable cross-linkers in those studies originated from DNA and therefore did not provide sufficient information for uniquely orienting the protein domains on the nucleosome.
To design cysteine variants on the Chd1 ATPase motor, we turned to the crystal structures of Rad54 bound to duplex DNA (Dürr et al., 2005) and the Vasa DEAD-box RNA helicase bound to single-stranded RNA (Sengoku et al., 2006). Rad54 has a homologous fold to Chd1 and provided a model for how the first ATPase lobe engages with DNA. In this crystal structure, the second ATPase lobe of Rad54 was not in the expected arrangement for an active helicase motor, and therefore we used the domain organization described for Vasa, bound to an ATP analog and believed to represent an active state, to model the ATPase motor of Chd1 in a closed form on duplex DNA.
We introduced single cysteines into both ATPase lobes of Chd1 and tested for site-specific DNA cross-linking in ADP·BeF3. Using nucleosomes containing flanking DNA on both sides, we found nucleosome-specific cross-links for variants N459C and E493C on the first ATPase lobe and N650C, V721C and G743C on the second ATPase lobe (Figure 4A, Figure S3). These cross-links indicated unique placement of the ATPase motor at each SHL2 site of the nucleosome (Figure 4B) and agree with the expected 3′→5′ translocation along DNA away from the nucleosome dyad (Saha et al., 2005). As observed for the DBD, ATPase cross-links on the two sides of the nucleosome showed variation in placement and intensity, indicative of sequence bias. The strongest effect was observed for the N650C and G743C variants, which cross-linked to just the left and right sides of the 601 positioning sequence, respectively. Interestingly, N650 is located on a Snf2-specific insertion that sterically clashes with DNA when the ATPase is modeled in the closed form, suggesting that the path of DNA is likely altered in a closed, ATP-bound state (Hauk and Bowman, 2011).
Figure 4. The Chd1 chromodomains and ATPase motor sit in unique positions and orientations on nucleosomal DNA.
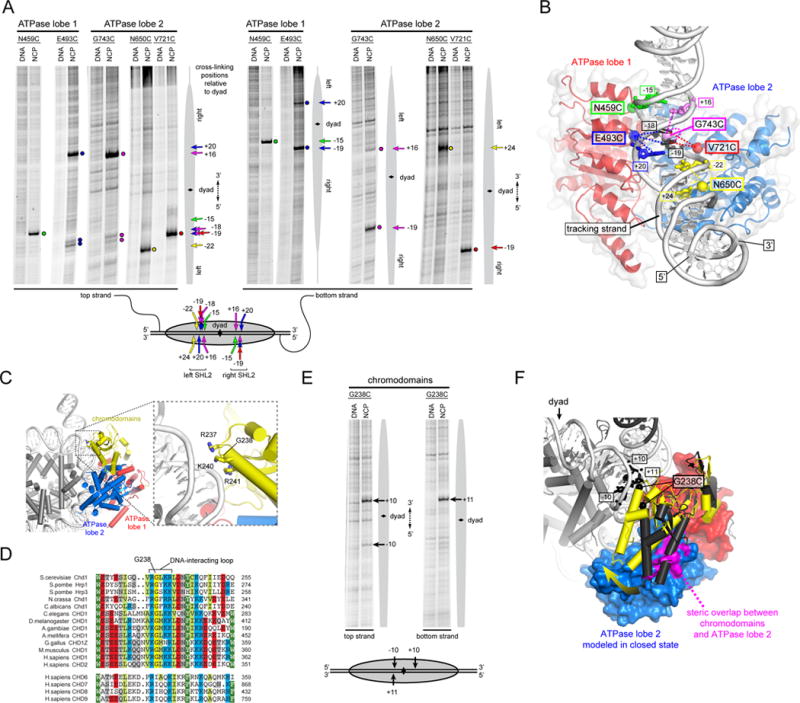
(A) Single-cysteine variants in both ATPase lobes of Chd1 form specific cross-links to nucleosomal DNA. Cross-linking reactions shown were performed in the presence of AMP-PNP (E493C) or ADP·BeF3 (all others) with either 10N70 nucleosomes (N459C, E493C) or 29N19 nucleosomes (N650C, V721C, G743C). The lower schematic summarizes the cross-linking positions on the nucleosome, with the numbering representing the distances from the dyad in either the 5′ direction (negative) or 3′ direction (positive). (n ≥ 4)
(B) A model for the Chd1 ATPase motor in a closed, nucleotide-bound state, docked onto the nucleosome crystal structure (1KX5; (Davey et al., 2002)) to match cysteine-substituted positions (spheres) with cross-linked nucleotides (sticks). This orientation shows a view from the histone-interacting side of one SHL2 site, with the histones not shown for clarity.
(C) Docking the chromo-ATPase crystal structure onto the nucleosome suggests a potential DNA-interacting loop on the chromodomains. The same docking of the ATPase motor is shown as in (B), but from a view showing the face of the nucleosome disk, with the chromodomains positioned relative to the first ATPase lobe as in the 3MWY structure.
(D) Sequence alignment shows high conservation of the predicted DNA-interacting loop of the chromodomains. The upper sequences show Chd1 orthologs, whereas the lower group is from the related CHD6-9 clade. Although the CHD6-9 group does not maintain the highly conserved Gly in the middle of this loop, additional basic residues on either side of this loop segment are conserved.
(E) The Chd1 chromodomains specifically contact nucleosomal DNA. Cross-linking reactions with the G238C variant were carried out in ADP·BeF3 with 29N19 nucleosomes (n = 4).
(F) A predicted shift of chromodomains on the nucleosome. The position of the chromodomains from the crystal structure (3MWY, charcoal) sterically overlaps with the second ATPase lobe in the modeled closed state (magenta). A shift of the chromodomains by ~23° (yellow) relieves steric clash and places G238 close to cross-linking sites on nucleosomal DNA.
See also Figures S3 and S4.
Docking the ATPase motor of Chd1 on the nucleosome unexpectedly suggested a potential DNA-interacting surface on the chromodomains (Figure 4C). We previously solved a crystal structure of the chromo-ATPase fragment of Chd1, which showed an inhibitory placement of the chromodomains against the DNA-binding surface of the second ATPase lobe (Hauk et al., 2010). Using the site-specific cross-links of the ATPase motor, docking the chromo-ATPase crystal structure onto nucleosomal DNA positioned a loop on chromodomain 1 containing three basic residues close to the DNA backbone. This loop is highly conserved among Chd1 orthologs, and also shows a conserved basic character in the related but distinct CHD subfamily that encompasses CHD6, CHD7, CHD8, and CHD9 (Figure 4D). To determine whether this loop directly contacts DNA, we tested cross-linking of chromodomain variants R237C and G238C. The G238C variant gave strong cross-links to nucleosomal DNA at SHL1, ~10 bp from the dyad (Figure 4E,F and S3). Compared to the other cross-linking sites, which were separated from target nucleotides by ~7–11 Å as expected for the 11 Å reach of APB (Pendergrast et al., 1992), docking the chromo-ATPase structure via the ATPase cross-links placed the Cα of G238 ~14 and ~17 Å away from the strong +11 and +10 cross-linking sites, respectively. This larger separation suggests that on the nucleosome, the chromodomains are in a similar orientation yet shifted away from the ATPase motor relative to the crystal structure. Consistent with this idea, the crystallographic positioning of the chromodomains sterically clashed with the second ATPase lobe when modeled in the closed, active state (Figure 4F, magenta). This clash can be relieved by a ~23° rigid-body rotation of the chromodomains away from ATPase lobe 2, which also repositions G238 to within cross-linking distance of SHL1. The cross-linking of this DNA-interacting loop therefore demonstrates that the Chd1 chromodomains remain in close proximity to the ATPase motor, even after engagement of both ATPase lobes with nucleosomal DNA.
Chd1 domains communicate across the DNA gyres of the nucleosome
DNA cross-linking of the chromodomains, ATPase motor, and DBD suggests a direct means of domain-domain communication. On the nucleosome crystal structure flanked by idealized DNA, docking Chd1 domains shows a large (>50 Å) separation between the chromo-ATPase and the DBD on the same DNA gyre, whereas the chromo-ATPase appears positioned to easily make direct contact with the DBD on the opposite DNA gyre (Figure 5A). To investigate the prediction that the DBD contacts the chromo-ATPase on the opposite gyre, we tested whether loss of flanking DNA on one side of the nucleosome altered cross-linking of the ATPase motor to SHL2. Using single cysteine variants of the ATPase motor, cross-linking reactions with 0N80 and 80N0 nucleosomes showed a systematic influence of flanking DNA on SHL2 cross-linking, with stronger cross-linking often correlating with flanking DNA on the opposite gyre (Figure S4). Although several variants yielded strong cross-links to the right SHL2 regardless of the flanking DNA, G743C was notably stronger on the 80N0 compared to 0N80 nucleosomes, highlighting a connection to DNA on the opposite side. Similarly, the variants N459C, V721C, and N650C cross-linked markedly more strongly to the left SHL2 with 0N80 compared with 80N0 nucleosomes. With differing effects on the two sides of the 601 positioning sequence, these experiments again highlight the sensitivity of cross-linking to DNA sequence. It is important to note that cross-linking efficiency may not simply report on DNA binding/occupancy but could reflect changes in local positioning or conformation. Given the location of the DBD on flanking DNA shown by cross-linking (Figure 1E), these correlations between flanking DNA and ATPase cross-linking support the idea that the presence of the DBD can affect the conformation or organization of the ATPase motor when bound to the opposite DNA gyre.
Figure 5. The Chd1 chromo-ATPase and DNA-binding domain interact across the gyres of the nucleosome.
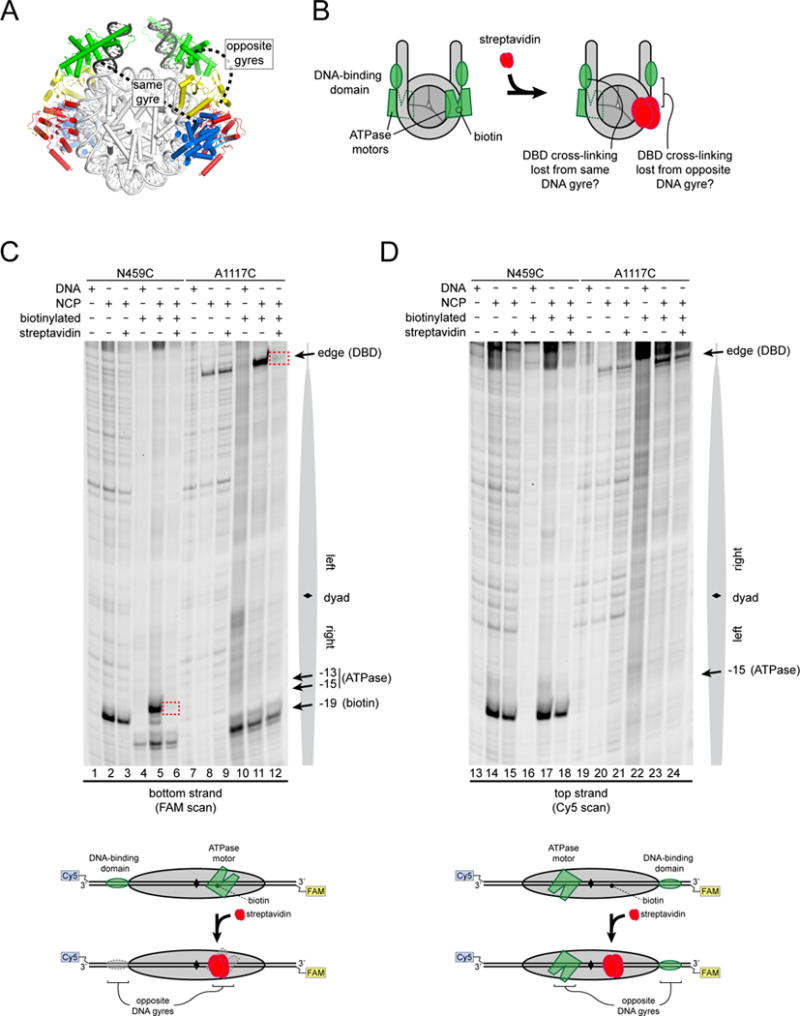
(A) Model of Chd1 domains docked on the nucleosome crystal structure (1KX5, (Davey et al., 2002)) based on cross-linking.
(B) Experimental design for determining which Chd1 domains interact on the nucleosome. Targeting streptavidin via biotin should block the ATPase motor at only one SHL2 site. The streptavidin block should reveal which DBD depends on this ATPase motor.
(C) Blocking the ATPase motor at one SHL2 site disrupts cross-linking of the DBD on the opposite DNA gyre of the nucleosome. Chd1 was added to 29N19 nucleosomes at a 1:1 molar ratio. Dotted red boxes highlight cross-links that are specifically lost upon addition of streptavidin to biotinylated nucleosomes. Schematic below illustrates the relative positioning of domains detected by cross-linking.
(D) The ATPase motor remaining on the nucleosome stabilizes the DBD on the opposite DNA gyre. The same cross-linking reactions shown in (B) but following cross-linking to the other (non-biotinylated) DNA strand (n = 4).
See also Figure S5.
To further test the model that the Chd1 chromo-ATPase interacts with the DBD on the opposite DNA gyre of the nucleosome, we generated a nucleosome containing a biotin moiety on the right SHL2 site, 19 nt from the dyad. This biotin position is in the middle of the ATPase binding site, and was designed to selectively block SHL2 binding upon addition of streptavidin, without interfering with the DBD at the nucleosome edge (Figure 5B). Since the chromo-ATPase is required for the DBD to cross-link at the edge of the nucleosome, disruption of ATPase binding at one SHL2 site should likewise prevent cross-linking of the DBD at one edge position. The side of the nucleosome that loses or maintains cross-linking from the DBD in the presence of streptavidin should therefore reveal whether communication occurs through the same or opposite gyres of the nucleosome.
To control for potential interference from the streptavidin alone, parallel reactions were performed using a non-biotinylated nucleosome. As shown in Figure 5C, the N459C variant, which reports on ATPase binding, still yielded a specific cross-link to the biotinylated DNA strand at SHL2, though the cross-linked position was shifted relative to the non-biotinylated control. Upon addition of streptavidin, N459C cross-linking to the non-biotinylated nucleosome was unaffected, whereas cross-linking to the biotinylated DNA was blocked at the targeted SHL2 site (compare lanes 3, 5 and 6). Cross-linking of N459C was not impeded at the other SHL2 site (Figure 5D, compare lanes 17 and 18), confirming that this strategy site-specifically blocks association of the ATPase motor. To evaluate the impact of the streptavidin block on the DBD, cross-linking was performed using the A1117C variant. The A1117C variant cross-linked to the strand with 5′→3′ polarity moving away from the nucleosome edge, which corresponds to same strand cross-linked by N459C on the opposite gyre SHL2 site. Importantly, A1117C yielded the same cross-linking patterns observed for N459C: edge cross-linking opposite the non-biotinylated SHL2 site was unaffected (Figure 5D, lanes 23 and 24), whereas cross-linking opposite the biotinylated SHL2 site was lost (Figure 5C, lanes 11 and 12). Analogous experiments were carried out using other cysteine variants and showed similar results: cross-linking of G743C (ATPase) and G238C (chromodomains) were strongly reduced on the right but not the left side in a streptavidin-dependent manner, whereas H1252C (DBD) was blocked on the left (opposite gyre) but not right (same gyre) side, where the biotin moiety was located (Figure S5). Taken together, these experiments demonstrate that the DBD and chromo-ATPase of Chd1 communicate across the DNA gyres of the nucleosome.
Domain-domain interactions across DNA gyres are dynamic yet significantly populated
While the biotin-streptavidin experiments confirmed that the DBD communicates with the chromo-ATPase on the opposite DNA gyre of the nucleosome, it was unclear how stable such domain-domain contacts might be, since transient interactions could be overrepresented with cross-linking. To visualize the predominant domain organization(s) of Chd1 on the nucleosome, we collected negative stain EM data for Chd1118–1274 and Chd1118–1014, which possessed and lacked the DBD, respectively. Class averages obtained in ADP·BeF3 showed a strongly preferred orientation of Chd1118–1274-nucleosome complexes on the grid, with a face-on view for ~75% of particles. Although this preferred orientation precluded 3D reconstruction, the dominant face-on view provided a remarkably clear image of Chd1 domains on the nucleosome that indicated two major populations (A and B) of remodeler organization. As shown by representative class averages (Figure 6A), both populations displayed similar densities directly engaging the nucleosome, yet one uniquely possessed additional density on one side. The remodeler densities for population A, which were roughly symmetrical about the central two-fold dyad of the nucleosome, were also observed for population B and indistinguishable from Chd1118–1014 bound to nucleosomes (Figure 6B). Supported by our cross-linking results, we attribute these common densities to two copies of the chromodomains and ATPase motor bound at SHL1 and SHL2 on the nucleosome, respectively. The unique density observed for population B showed the size expected for the DBD, and its placement was consistent with cross-linking at the edge of the nucleosome. Based on the crystal structure of the DBD bound to DNA, this location of the DBD suggests that DNA would need to unwrap relative to the trajectory seen for nucleosome-alone crystal structures (Figure 6C). Notably, the two populations were observed in approximately equal proportions, with DBD density packed against the chromo-ATPase in half of the face-on views. Thus, while the DBD is not locked in place, this domain arrangement represents a significantly populated state of Chd1. Intriguingly, DBD density was never present on both molecules of Chd1, which may arise from asymmetric wrapping or dynamics due to the 601 sequence. An alternative possibility is that Chd1 binding induces asymmetry in the nucleosome, which may reduce stability or increase dynamics that prevents both remodelers from binding in an identical manner.
Figure 6. The DNA-binding domain stably packs against the Chd1 chromo-ATPase on the nucleosome.
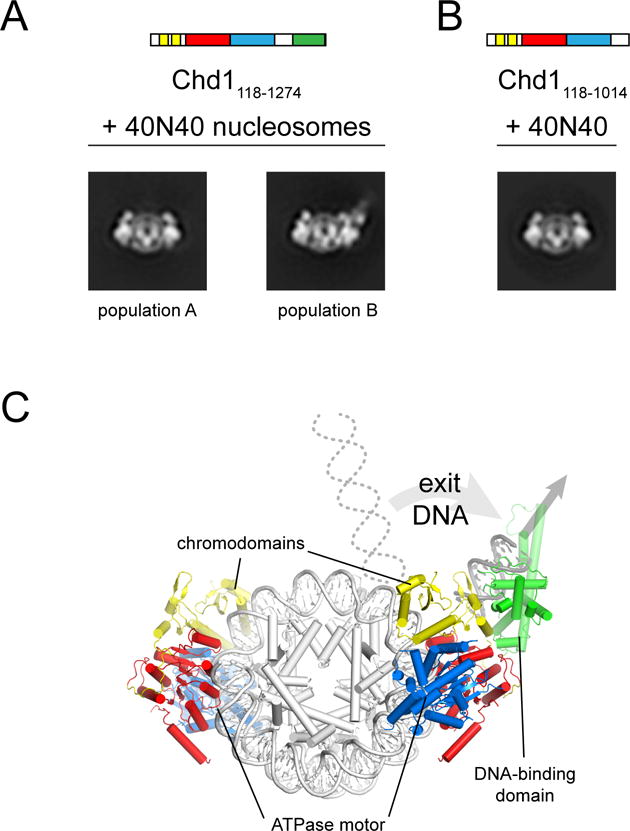
(A) Class averages from negative stain EM representing the two major populations Chd1118–1274-nucleosome complexes observed in a face-on view. Chd1118–1274 was added to 40N40 nucleosomes at a 2:1 ratio in ADP·BeF3.
(B) Class average from negative stain EM representing the only 2:1 Chd1118–1014-nucleosome population observed in face-on view. Chd1118–1014 was added to 40N40 nucleosomes at a 2:1 ratio in ADP·BeF3.
(C) A model for Chd1 domain organization on the nucleosome based on cross-linking and negative stain EM.
DNA exiting the nucleosome slows ATPase activity of Chd1
We envision that these across-the-gyre contacts allow the presence of the DBD to influence action of the ATPase motor. Since these contacts are dependent on the presence or absence of flanking DNA, we determined the levels of ATPase stimulation for different nucleosome substrates. To ensure that ATPase rates were determined before a majority of nucleosome substrates had been repositioned, we used fluorescently-labeled phosphate binding protein (PBP) and monitored ATPase activity by stopped flow (Brune et al., 1994) (Figure 7A). End-positioned 2N61 nucleosomes generated significantly higher ATPase rates than 33N26 nucleosomes, suggesting that the absence of flanking DNA on one side of the nucleosome was stimulating (Figure 7B). In support of this idea, experiments using 0N33 nucleosomes also showed higher ATPase stimulation, nearly indistinguishable from 2N61 substrates, even though the flanking DNA length on one side was comparable to 33N26. For comparison, ATPase rates were also measured with an NADH-coupled assay. Consistent with the longer time window required to determine rates, allowing a significant fraction of nucleosome substrates to be repositioned, we observed that 2N61 and 33N26 nucleosomes stimulated Chd1 ATPase activity to similar extents, whereas 0N33 was significantly weaker (Figure 7C). Thus, while nucleosomes with shorter DNA may be less stimulatory after remodeling reactions have equilibrated, the initial absence of flanking DNA on one side of the nucleosome is a highly stimulatory characteristic.
Figure 7. The presence of exit DNA, communicated via the DNA-binding domain, reduces ATPase activity of Chd1.
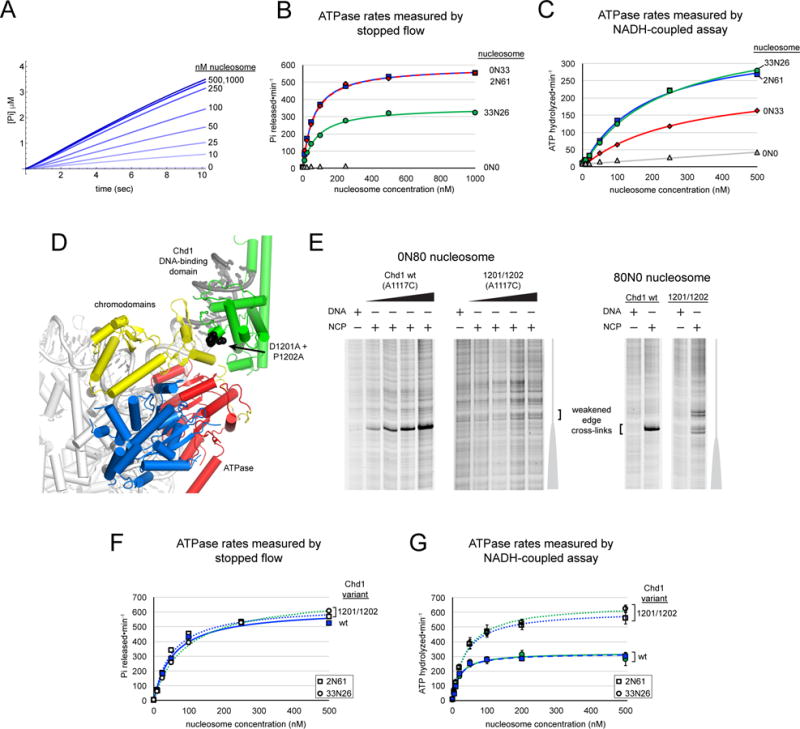
(A) Chd1 ATPase activity measured by a stopped flow phosphate binding assay. Shown is a representative titration with 2N61 nucleosomes, with 3–7 technical replicates averaged for each trace.
(B) End-positioned nucleosomes stimulate the Chd1 ATPase more than nucleosomes with flanking DNA on both sides. Stopped flow ATPase activities were calculated from a 2–3 sec window after mixing. Michaelis Menten fits yielded kcat values of 592 ± 17 min−1 (0N33 nucleosomes, red diamonds), 587 ± 8 min−1 (2N61 nucleosomes, blue squares), and 360 ± 50 min−1 (33N26 nucleosomes, green circles).
(C) ATPase activities measured by an NADH-coupled assay. Michaelis Menten fits to the data yielded kcat values of 371 ± 10 min−1 (2N61 nucleosomes, blue squares), 409 ± 13 min−1 (33N26 nucleosomes, green circles), and 260 ± 30 min−1 (0N33 nucleosomes, red diamonds). The titration series for 0N0 nucleosomes (gray triangles) is shown with a linear fit.
(D) A view of the domain-docked model of Chd1 on the nucleosome that highlights the position of residues Asp1201 and Pro1202 (black spheres), which lie at the interface with the chromo-ATPase.
(E) Cross-linking of the DBD (A1117C) to the nucleosome edge is lost (0N80) or weakened (80N0) with the D1201A/P1202A substitution. DNA and nucleosome concentrations were 150 nM, and Chd1 concentrations were 600 nM (for DNA alone), 75 nM, 150 nM, 300 nM, and 600 nM for 0N80 and 450 nM for 80N0.
(F) The D1201A/P1202A variant shows similar ATPase activation as wild type on end-positioned nucleosomes, yet unlike wild type is insensitive to the presence of exit-side DNA. Michaelis Menten fits yielded kcat values of 641 ± 9 min−1 (2N61 with D1201A/P1202A variant), 618 ± 6 min−1 (2N61 for wildtype Chd1), and 718 ± 8 min−1 (33N26 with D1201A/P1202A variant).
(G) ATP hydrolysis activity of the D1201A/P1202A variant of Chd1 is insensitive to the presence of flanking DNA. Michaelis Menten fits yielded kcat values of 610 ± 50 min−1 and 660 ± 30 min−1 for 2N61 (squares) and 33N26 (circles) nucleosomes, respectively, compared with 320 ± 20 min−1 and 320 ± 40 min−1 values for wildtype Chd1. Data reported in (B), (C), and (G) are averages of three or more independent experiments, with error bars (sometimes obscured by symbols) indicating ±S.D.; data points in (F) are averages from duplicate independent experiments, with the value ranges obscured by the symbols.
To weaken the predicted interface between the DBD and chromo-ATPase, we substituted two alanines for a highly conserved pair of residues (D1201/P1202) on the DBD (Figure 7D). This double substitution was introduced into the A1117C cross-linking background that reports on placement of the DBD at the nucleosome edge. Compared with the A1117C variant possessing an otherwise wild type DBD, cross-linking for the D1201A/P1202A variant was either not detectable (0N80) or gave rise to additional cross-links farther from the edge (80N0), consistent with weakening of interactions with the chromo-ATPase (Figure 7E).
To see if the loss of cross-linking also correlated with disrupted interdomain communication, we measured ATPase activities of the D1201A/P1202A variant in a wild-type background. Stopped flow measurements showed that the D1201A/P1202A variant was stimulated by end-positioned 2N61 equally to wild type Chd1, yet unlike wild type, the D1201A/P1202A variant showed elevated hydrolysis rates with 33N26 nucleosomes comparable to end-positioned nucleosomes (Figure 7F). These results suggested that the D1201A/P1202A substitution prevented the remodeler from differentiating between end-positioned and centered nucleosomes. This idea was confirmed by ATPase measurements using the NADH-coupled assay, which showed higher ATPase rates with both 2N61 and 33N26 nucleosomes that were remarkably similar to stopped flow measurements with 2N61 nucleosomes (Figure 7G). These experiments show a direct connection between disruption of interdomain communication and stimulation of the Chd1 ATPase, and support a model where the DBD slows ATPase activity when engaged with flanking DNA on the exit side.
Discussion
Chromatin remodelers are complex multidomain machines that specifically reorganize nucleosome substrates, and a key question is how the core ATPase motor integrates information from auxiliary domains. Our work here with Chd1 shows an unexpected communication between the chromo-ATPase at SHL2 with the DBD bound to exit DNA. This organization of chromatin remodeler domains on the nucleosome has important implications for remodeler regulation and action.
Our cross-linking revealed a unique position and orientation of the ATPase motor at SHL2 that is consistent with shifting DNA toward the nucleosome dyad using 3′→5′ directionality. Chromatin remodelers constitute one subclass of the broad superfamily 2 (SF2) of helicase-like enzymes, which have a characteristic architecture where DNA or RNA is bound over a central ATP-binding cleft defined by two RecA-like ATPase lobes (Singleton et al., 2007). Regardless of their preference for double- or single-stranded nucleic acid substrates, translocating SF2 enzymes always walk along one strand, called the “tracking strand.” Interestingly, SF2 ATPases universally engage the tracking strand with lobe 1 binding the 3′ side and lobe 2 on the 5′ side, yet this binding polarity does not dictate whether translocation occurs in a 3′→5′ or 5′→3′ direction (Saikrishnan et al., 2009). For Chd1, our cross-linking experiments have defined the tracking strand on the nucleosome, which is oriented 5′→3′ from SHL2 toward the dyad (Figure 4B). Previous ssDNA gap experiments showed that Chd1 shifts DNA from SHL2 toward the dyad (McKnight et al., 2011), which together with the orientation of the ATPase motor indicate that Chd1 is a 3′→5′ translocase. This sliding directionality is in agreement with that of the RSC remodeler (Saha et al., 2005), and suggests that intrinsic 3′→5′ translocation is likely a common characteristic shared among distinct remodeler families.
It is well established that Chd1 and ISWI remodelers slide nucleosomes away from bound transcription factors (Kang et al., 2002; Li et al., 2015; Nodelman et al., 2016; Pazin et al., 1997) and generate evenly spaced arrays (Gkikopoulos et al., 2011; Ito et al., 1997; Lusser et al., 2005; Ocampo et al., 2016; Pointner et al., 2012; Tsukiyama et al., 1999). These characteristics are reflected by a preference for sliding mononucleosomes away from DNA ends, and indicate the ability to respond to DNA outside the nucleosome core (Kagalwala et al., 2004; McKnight et al., 2011; Stockdale et al., 2006; Yang et al., 2006). Although the nucleosome has two symmetrically related halves, the DNA flanking each side of the nucleosome is in a distinct position relative to the ATPase motor bound at SHL2. Since Chd1 unidirectionally shifts DNA from SHL2 toward the dyad, the DNA segment preceding SHL2 on the same DNA gyre will be pulled further onto the nucleosome, whereas DNA on the other side of the dyad will be shifted toward the edge and off the histone core. Due to the characteristic wrapping of DNA around the histone core, this directional sliding places the segment of DNA shifting off the nucleosome (exit side DNA) spatially close to the active SHL2, whereas flanking DNA that shifts onto the nucleosome (entry side DNA) is on the far side of the nucleosome disk. Here, we show interdomain communication of Chd1 occurs between SHL2 and exit DNA.
Based on Chd1 characteristics, how might this domain organization be expected to influence the direction of sliding? Because this organization is limited to sensing exit DNA, the remodeler would be blind to the entry DNA being pulled onto the nucleosome. In the case of mononucleosomes, if exit side DNA activated Chd1, the shortening of entry side DNA could allow the end of DNA to be pulled past the nucleosome edge and onto the histone core, resulting in the histone core being “off the end” of DNA. However, Chd1 is known to slide mononucleosomes away from DNA ends and thus favors movement of the histone octamer to more central locations on DNA (Stockdale et al., 2006), a characteristic that requires the DBD (McKnight et al., 2011). In fact, replacement of the DBD with monomeric streptavidin allows Chd1 to slide biotinylated histone octamers off the ends of DNA, indicating that the Chd1 DBD normally prevents mononucleosomes from shifting off DNA ends (Patel et al., 2013). By this logic, we propose that instead of representing a stimulating state, communication with exit DNA would antagonize nucleosome sliding. We show that ATPase activity of Chd1 is stimulated by the absence of linker DNA on one side, or when communication with the DBD is disturbed (Figure 7). Coupling the availability of exit DNA to dampening ATPase activity makes sense for a nucleosome spacing factor such as Chd1, as increasing activity when exit-side DNA is occupied would bias nucleosome sliding away from bound factors or other nucleosomes. This model for exit-side inhibition would explain the faster nucleosome sliding we observed when one side of the nucleosome was bound by LacI (Nodelman et al., 2016). In that study, the presence of LacI at a LacO(−11) site, which would displace the Chd1 DBD from its position on exit DNA described here, increased the rate that Chd1 shifted nucleosomes away from the LacO site by 5- to 6-fold. The wrapping of DNA into nucleosomes is antagonistic to most transcription factors, both for reducing accessibility but also accelerating dissociation of bound factors (Luo et al., 2014). Thus, rapidly shifting the histone core away from DNA-bound factors at the nucleosome edge would greatly increase transcription factor occupancy (Nodelman et al., 2016). The sensitivity of Chd1 for exit-side availability would also be expected to promote regular nucleosome spacing by shifting closely packed or overlapping nucleosomes away from their neighbors.
The exit-side communication described here is complementary to models for directional sliding by ISWI remodelers, where the ATPase motor is stimulated by entry side DNA. For the ISWI remodeler ACF, nucleosome sliding activity has been found to be allosterically coupled to the length of entry-side DNA, which explains the preference for sliding nucleosomes toward longer segments of DNA (Hwang et al., 2014; Yang et al., 2006). Interestingly, despite these different observations showing an ability to sense entry or exit DNA, ISWI and Chd1 share a functionally similar domain architecture. Both ISWI and Chd1 remodelers have a similarly folded DBD C-terminal to the ATPase motor (Grüne et al., 2003; Ryan et al., 2011), and previous cross-linking and footprinting studies of the ISWI DBD (called HAND-SANT-SLIDE or HSS) show a position at the nucleosome edge that is similar to the location we describe here for the Chd1 DBD (Dang and Bartholomew, 2007; Yamada et al., 2011). ISWI remodelers also possess a negative regulatory domain (‘Auto-N’) N-terminal to the ISWI ATPase motor that appears to be functionally analogous to the Chd1 chromodomains (Clapier and Cairns, 2012; Hauk et al., 2010; Manning and Peterson, 2013). Our cross-linking revealed an unexpected interaction between the Chd1 chromodomains and nucleosomal DNA at SHL1, indicating that the chromodomains remain close to the ATPase motor bound at SHL2, and thus suggest the possibility of regulation on the nucleosome. Both the chromodomains and ATPase motor of Chd1 are physically close to the DBD on exit DNA, and we anticipate that the analogous domains of ISWI would enable a similar domain-domain communication between SHL2 and the DBD on exit DNA.
Another possibility for the domain organization described here is that these cross-gyre contacts could facilitate nucleosome assembly. Chd1 has been shown to assemble nucleosomes in vitro from so-called pre-nucleosomes, which lack the characteristic DNA protection and negative supercoiling of nucleosomes (Fei et al., 2015; Lusser et al., 2005; Torigoe et al., 2013). In addition to exit DNA being bound by the DBD, our modeling based on cross-linking and EM suggests that lobe 1 of the ATPase motor also contacts exit DNA. With contacts between exit DNA and SHL2, Chd1 bridges a ~90 bp loop of nucleosomal DNA. Stabilization of a left-handed superhelical DNA loop with geometry that matches the nucleosome could help direct the proper wrapping of pre-nucleosomal particles. In addition, these bridging interactions would be expected to help maintain or ensure proper histone-DNA architecture during nucleosome sliding.
By preventing binding of the ATPase motor to one SHL2 site, the streptavidin block experiments indicate that cross-gyre interactions can be accomplished with a single remodeler (Figure 5). Previous work showed that hybrid Chd1 remodelers targeted through single binding sites on entry side DNA could slide nucleosomes, indicating that nucleosome repositioning can be carried out by a monomeric remodeler (McKnight et al., 2011; Patel et al., 2013). Taken together, these data suggest that the chromo-ATPase and DBD from the same Chd1 molecule can bind to either the same or opposite DNA gyres, and thus different arrangements of a monomeric enzyme on the nucleosome likely explain the nucleosome sliding characteristics of Chd1. While nucleosomes can be repositioned by monomeric Chd1, our EM class averages show that two Chd1 molecules can simultaneously bind to the nucleosome. Importantly, the chromo-ATPase was shown to directly communicate with a DBD added in trans, indicating that interdomain interactions can occur between molecules that are not covalently linked (Figure 2). These interactions could provide a simple means for coordinating activity of two remodelers on the same nucleosome.
Methods and Resources
CONTACT FOR REAGENT AND RESOURCE SHARING
Please contact Gregory Bowman gdbowman@jhu.edu for requests for reagents or further information.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
cDNAs used for expression of Chd1 recombinant protein were of Saccharomyces cerevisiae origin.
cDNAs used for expression of recombinant histone proteins were of Xenopus laevis origin.
METHOD DETAILS
Protein constructs and purification
For cross-linking, a cysteine-free variant of Chd1118–1274 was generated (C207A, C246A, C453A, C455A, C1063A), into which single cysteines were individually substituted at the following locations (* indicates weak or no cross-linking): R237*, G238, N459, T491*, E493, S523*, S524*, G643*, F646*, N650, V721, G743, S770*, A1117, L1119*, N1129*, A1250, and H1252. All amino acid changes were generated using Phusion DNA polymerase (NEB). His-tagged, truncated forms of Saccharomyces cerevisiae Chd1 were expressed in BL21(DE3) CodonPlus-RIL cells with Trigger chaperone and purified as previously described (Nodelman and Bowman, 2013; Patel et al., 2011; Sharma et al., 2011). Each Chd1 construct was transformed into E.coli cells harboring the BL21(DE3)codon-plus-RIL plasmid and an expression plasmid carrying Trigger chaperone, grown in 8L of Terrific Broth media, induced with 0.35mM IPTG at an A600nm=0.6–0.8 and incubated overnight at 18°C. The next morning cells were pelleted and resuspended in 180ml of HisBind A Buffer (50mM Tris-HCl, pH 7.6, 500mM NaCl, 10mM imidazole, pH 8.0, 10% glycerol), and flash frozen. The resuspended cell pellets were thawed and incubated on ice for 30 minutes after addition of 0.2mM PMSF, 2mM MgCl2, 10μg/ml DNaseI, 5mM ß-mercaptoethanol, and 1mg/ml lysozyme. Cells were sonicated on ice for 3–4 rounds, 50 pulses, output=4, duty cycle=50% then clarified by spinning for 40 minutes, 4°C, 45,000×g. Supernatant was passed through a glass fiber filter and then applied to a nickel affinity column (GE HisPrep FF 16/10 column). Bound fraction was eluted in a 35% HisBind Buffer B (HisBind Buffer A + 500mM imidazole) bump. Pooled peak fractions were slowly diluted 5-fold with TG buffer (30mM Tris-HCl, pH 7.6, 10% glycerol) to reduce NaCl concentration to 100mM and then loaded onto a 5ml SP FF column (with the exception of Chd1118–1014 which was applied to a 5ml Q FF) pre-equilibrated in TG buffer + 100mM NaCl. Protein was eluted with a linear gradient from 0–50%B (Buffer B= TG buffer +1M NaCl) over 23 column volumes. Pooled fractions were incubated on ice overnight with Precission Protease, and the next morning, imidazole, pH 8.0 was added to a final concentration of 7–10mM then passed over a 5ml HisTrap HP (nickel affinity) pre-equilibrated in HisBindA buffer. Flow through was concentrated and loaded onto a prep grade HiLoad 16/600 Superdex 200 pre-equilibrated in TG buffer + 150mM NaCl. Peak fractions were pooled, concentrated, and flash frozen.
Xenopus laevis histones were expressed and purified almost entirely as previously described (Luger et al., 1999). Each histone construct was grown pLysS cells in 4L of 2×TY at 37°C, induced at an A600nm=0.6 with 0.3mM IPTG for 3–4 hours 37°C. Cells were spun down at 18°C for 15 mins at 3000rpm in an RC3C+ centrifuge. Pellets were resuspended in 40ml of wash buffer (50mM Tris-HCl, pH 7.5, 100mM NaCl, 1mM EDTA, pH 8.0) and frozen in −80°C for further processing. Pellets were thawed and 1mM benzamidine was added. Cells were sonicated on ice for 3–4 rounds and spun at 23,000×g, 4°C, for 20 minutes. Supernatant was decanted and inclusion body pellet was resuspended in 90ml wash buffer with 1% Triton X-100, and spun at 15,000 rpm (SA-300 rotor, Evolution centrifuge), 10 minutes, 4°C. This step was repeated once again with wash buffer plus detergent, and then repeated two times with wash buffer alone. The final pellet was smeared with a spatula over the inside of a 50ml conical tube and stored at −20°C. For purification, the histone smear was incubated for 30 minutes at room temperature with 1ml of DMSO followed by an incubation with 40ml of unfolding buffer (7M guanidine-HCl, 20mM Tris-HCl, pH 7.5, 10mM DTT) and agitation to solubilize unfolded histone. The histone protein was then passed over a HiPrep 26/10 desalting column (GE) in 2–3 injections to exchange into 7M urea, 10mM Tris-HCl, pH 7.5, 100mM NaCl, 1mM EDTA, pH 8.0. Peaks were pooled and loaded onto a tandem Q-S column (GE HiPrep 16/10 SPFF; GE HiPrep 16/10 Q FF; as described in online protocols from Tsukiyama lab, http://research.fhcrc.org/content/dam/stripe/tsukiyama/files/Protocols/expression.pdf) preequilibrated in same buffer as desalting column. Columns were detached after baseline was acquired and histone protein was eluted with a linear salt gradient from the S column, pooled and dialyzed into water + 5mM β-mercaptoethanol. Histones were lyophilized in 2mg aliquots.
The A197C variant of E. coli phosphate binding protein (PBP) (Brune et al., 1994) was expressed in BL21(DE3)star cells. Per 0.5 liter of growth, phosphate binding protein was purified on a HiPrep 16/10 Q-FF anion exchange column using a shallow salt gradient (Buffer A: 10 mM Tris-HCl, pH 7.5, 1 mM DTT; Buffer B: Buffer A plus 100 mM NaCl), and then dialyzed against 10mM Tris-HCl, pH 7.5. Multiple batchs (equivalent to 3L of growth) of purified phosphate binding protein were combined (220mg), concentrated to 50 ml and incubated with 0.2U/ml purine nucleoside phosphorylase and 0.2mM 7-methylguanosine for 1 hour at room temperature. To 160μM PBP, N- [2-(l-maleimidyl)ethyl]-7-(diethylamino)coumarin-3- carboxamide (MDCC; a 5mg bottle was resuspended in DMF to Cf=25mM) was added at a molar ratio of 1.4 MDCC:1 PBP, and incubated in the dark for 3 hours at room temperature. Labeled PBP was exchanged 3× into 10mM Tris-HCl, pH 8.0 using a 3kDa MWCO concentrator and volume was increased to 50ml (concentration of label was brought below 1 μM) to load onto a HiPrep 16/10 Q FF pre-equilibrated in Buffer A (10mM Tris-HCl, pH 8.0). Bound MDCC-PBP was eluted with a 0–30%B (Buffer A + 100mM NaCl) linear gradient, then fractions where the labeling efficiency was at least 80% as determined by dividing the molar ratio of MDCC label to PBP (using MDCC ε=46800 and PBP ε=61880 A430nm/A280nm) were pooled, concentrated to ~167μM, and flash frozen in liquid nitrogen.
Nucleosome constructs and preparation
Nucleosomal DNA was generated by large-scale PCR, using the Widom 601 sequence (Lowary and Widom, 1998), with some constructs possessing a LacO sequence on one side of the 601 sequence as described (Nodelman et al., 2016). Primers and templates for PCR are given in Tables S2 and S3. Fluorescent DNA primers were synthesized and 5′ end-labeled by IDT. The biotin moiety was introduced using a primer containing an internal biotin-dT modification. DNA was incorporated into nucleosomes by salt gradient dialysis, and both naked DNA and nucleosomes were purified by Mini Prep Cell or Prep Cell as previously described (Luger et al., 1999). Sequence information of 601-array plasmids used to generate DNAs for reconstitution of nucleosomes for ATP hydrolysis or nucleosome binding experiments is given in Table S4.
Site-specific cross-linking
In the dark, azidophencyl bromide (APB) was dissolved in 100% DMF at a concentration of 80 mM and then diluted to 8 mM in 30 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10% glycerol, and 20% DMF. APB was then added to a 7.5 μM sample of the Chd1 variant containing a single cysteine (in 30 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10% glycerol), to achieve a final concentration of 400 μM APB and 1% DMF. Labeling was allowed to proceeded for 2–3 hours at room temperature, after which Chd1 (150–450 nM) was incubated with either nucleosomes (150 nM) or naked DNA (150 nM) for 40–60 min in 1×SlideBuffer (20 mM Tris HCl, pH 7.5–7.6; 50 mM KCl; 0.1 mg/ml BSA; 1 mM DTT; 5% sucrose; 5 mM MgCl2) in 50 μl volumes. Nucleotide conditions contained either 2 mM ADP, 2mM AMP-PNP, or 2mM ADP, 15 mM NaF, 3 mM BeCl2, and 6 mM MgCl2 (ADP·BeF3). Where indicated, a 32-fold molar excess of tetrameric streptavidin (4.8 μM, Pierce, cat# 21125) was preincubated with nucleosomes for 15 minutes at room temperature to maximize the number of singly bound nucleosomes. For cross-linking, samples were then transferred to a silanized coverslip and UV-irradiated for 15 seconds, and subsequently quenched with 100μl of 20 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mg/ml BSA, 5 mM DTT, and 5 mM EDTA, and processed as described for histone mapping (Kassabov & Bartholomew, 2004). Post-irradiation buffer (150μl; 20mM Tris-HCl, pH 8.0, 0.2% SDS, 50mM NaCl) was next added to samples, briefly vortexed, and placed at 70°C for 20 minutes. Crosslinked samples were combined with 300μl 5:1 Phenol:Chloroform, vortexed (5 sec) and spun in the microfuge for 2 minutes, max speed, at room temperature. Since the desired crosslinked molecules are at the interface between the aqueous and phenol layers, the bulk of the supernatant is carefully removed without disturbing the interface leaving 30–50μl of the upper aqueous layer which will be enriched in the crosslinked material. Four subsequent washes were performed by adding 280μl 1.0M Tris-HCl, pH 8.0, 5% SDS, vortexing, centrifuging for 2 minutes, and again most of the upper aqueous layer was removed while leaving the upper 30–50μl layer closest to the interface. Next, 1μl of 10mg/ml salmon sperm DNA, 30μl 3.0M NaAcetate, 750μl 100% EtOH was added, the samples were vortexed and left o/n on ice. The next morning, samples were spun in a microfuge at 4°C, max speed for 30 minutes. The supernatant was discarded, and pellets were washed with 70% EtOH two times. After ethanol was removed pellets were air dried by inverting on bench for 1 hour, then resuspended in 100μl of 20mM Ammonium Acetate, 2% SDS, 0.1mM EDTA, pH 8.0 by vortexing. Samples were then spun in a microfuge at room temperature at max speed for 10 minutes, and supernatants were transferred to fresh tubes and placed in heat block at 90°C for 2 minutes. Condensation was spun down and 5μl 2M NaOH was added, vortexed briefly and incubated at 90°C for 40–50 minutes. Following incubation, samples were pulse spun, and 105μl 20mM Tris-HCl, pH 8.0, 6μl 2M HCl, 1μl 2M MgCl2, and 480μl 100% EtOH were added, vortexed and placed at −20°C o/n. The next morning samples were pelleted in a microfuge, at 4°C for 30 mins, washed two times with 70% EtOH, and allowed to air dry on bench for at least 1 hour. Four microliters of deionized formamide gel loading buffer with Orange G dye was added to resuspend DNA samples for gel loading. Samples were separated on 8 M urea sequencing gels containing 5–6% (29:1) or 8% (19:1) polyacrylamide, run at 65 W. Gels were scanned within optically clear glass plates with 3mm thickness (Gel Company) and visualized on a GE Typhoon 9410 variable mode imager. Analyses of cross-linking reactions were performed using ImageJ (https://imagej.nih.gov/ij/). Sequencing ladders were generated using Thermo Sequenase Dye Primer Manual Cycle Sequencing Kit (Affymetrix).
Nucleosome Binding
Nucleosome binding experiments used Xenopus histones H3-C110A, H4-A15C, and wild type H2A and H2B and was carried out as previously described (Leonard and Narlikar, 2015; Nodelman et al., 2016). In addition to binding buffer (20 mM HEPES, pH 7.5; 5 mM MgCl2; 0.1 mM EDTA; 5% sucrose; 1 mM DTT; 0.02% Nonidet P40 Substitute (Roche); 0.1 mg/mL BSA; and 100 mM or 150 mM KCl), titrations additionally had either 1 mM adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP) or 1 mM ADP, 1.2 mM BeCl2, and 6 mM NaF (ADP·BeFX). Binding isotherms from AMP-PNP conditions were fit with two apparent dissociation constants (Nodelman et al, 2016), whereas those from ADP·BeFX conditions were fit with one apparent dissociation constant using the quadratic equation,
where Y is the signal intensity, X is the concentration of Chd1, A is the signal amplitude, Kapp is the apparent dissociation constant, N is the nucleosome concentration, and C is the signal from nucleosome alone. Fits were performed using Mathematica. Titrations for each experimental condition were performed three or more times, with the average K1/2 values and standard deviations reported in Figure 3 and Table S1.
Electron microscopy
Complexes of either 100nM Chd1118–1274 or Chd1118–1014 bound to 50nM 40-601-40 nucleosomes (2:1 ratio) were prepared in 20mM Tris-HCl, pH 7.6, 60mM KCl, 0.5mM DTT, 3% glycerol, and ADP-BeF3 (2 mM ADP, 2 mM BeCl2, 10 mM NaF, and 2 mM MgCl2) and incubated at room temperature for 15–45 minutes. Samples of the Chd1-nucleosome complex (4μl) were applied to continuous carbon film EM grids, and stained with 2% uranyl formate. EM grids were imaged in a Tecnai T12 TWIN transmission electron microscope equipped with a LaB6 filament (operated at 100 kV) and a 2K × 2K FEI Eagle CCD camera. Approximately 200 micrographs were automatically acquired using the EPU software package at a nominal magnification of 52,000, resulting in a pixel size of 4.16 Å/pixel at the specimen level. For image processing, contrast transfer function parameters were estimated with CTFFIND4 (Rohou and Grigorieff, 2015) and subsequently corrected by phase flipping using SPIDER (Frank et al., 1996). Particles were automatically picked in a reference-free manner using DOGPICKER (Voss et al., 2009), extracted with a box size of 80 × 80 pixels, and normalized with XMIPP (Scheres et al., 2008; Sorzano et al., 2004). 2D classification was performed with RELION (Scheres, 2012, 2016). The final image stacks contained 70,000 and 65,000 particles for the nucleosome-Chd1118–1274 and nucleosome-Chd1118–1014 datasets, respectively. For class averages obtained with Chd1118–1274, nucleosomes were predominantly bound by two Chd1 molecules, while for the Chd1118–1014 dataset, nucleosomes were bound by either one (~21,500 particles) or two (~17,000 particles) Chd1118–1014 molecules. For the Chd1118–1274 dataset, approximately 53,500 particles were oriented in a face-on nucleosome view, with ~27,500 particles exhibiting clear density that could be attributed to the DBD, whereas ~26,000 particles lacked DBD density and resembled all 2:1 face-on views from the Chd1118–1014 dataset. Representative class averages for the face-on view are shown in Figure 6.
ATPase assays
Steady state ATPase activities were measured using an NADH-coupled assay as previously described (Kiianitsa et al., 2003; Nodelman and Bowman, 2013). Linear titrations of freshly prepared NADH stocks were performed each day to obtain standard curve for units of absorbance/mM NADH. Reaction components were added in a 96-well microtiter plate to a final volume of 100μl in 1xbuffer (20mM Hepes-KOH, pH 7.6, 50mM KCl, 5mM MgCl2, 0.1mg/ml BSA, 1mM DTT, and 5% sucrose) in the following order: buffer, 4μl 10mg/ml NADH, 5μl 50mM PEP, 6μl PK:LDH mix (200-fold dilutions of PK (Roche 10128155001) and LDH (Roche 10127230001) from ammonium sulfate stocks), 0.5–50μM from 1μM nucleosome, 5μl 1μM Chd1, and 5μl 50mM ATP. Reactions were quickly mixed and read at A340nm every 10 seconds on a SpectraMax190 at 24°C.
Stopped flow ATPase experiments were carried out as described (Brune et al., 1994), using an SX20 stopped flow spectrometer (Applied Photophysics) with excitation at 425 nm and a 450 nm longpass filter for emission. Phosphorous standard solution (Sigma) was used to calibrate the phosphate-dependent increase in fluorescence of PBP labeled with N- [2-(l-maleimidyl)ethyl]-7-(diethylamino)coumarin-3- carboxamide (MDCC). Prior to rapid mixing, free phosphate was depleted from all 1 ml solutions using a Pi mop consisting of 0.1U/ml purine nucleotide phosphorylase (PNPase) and 0.2mM 7-methylguanosine (7-MEG). Final concentrations of components after mixing were 50nM Chd1 with varying concentrations of nucleosomes (0, 10, 25, 50, 100, 250, 500nM and 1μM), 8.5μM PBP-MDCC, buffer (20 mM HEPES, pH 7.6; 100 mM KCl, 5 mM MgCl2, 0.1 mg/ml BSA, and 2 mM DTT) and 1mM ATP. ATPase rates were calculated from data collected between 4–14 min (NADH assay) or 2–3 sec (stopped flow) and fit to the standard Michaelis Menten equation, velocity = kcat*[Chd1]*[NCP]/(KM + [NCP]) using Mathematica.
Molecular modeling
All structural alignments and visualizations were performed using PyMOL.
QUANTIFICATION AND STATISTICAL ANALYSIS
For nucleosome binding affinities, values given are the mean from 3 or more independent experiments ± SD. With the exception of data shown in Figure 7F, which were performed in duplicate, all other ATP hydrolysis rates given are the mean ± SD of 3 or more independent experiments.
Supplementary Material
Acknowledgments
We thank Marc Greenberg for advice and suggestions with cross-linking experiments, Manju Hingorani, Miho Sakato and Sarah Woodson for advice with stopped flow experiments, Li Ma and Guy Montelione for a Trigger Factor co-expression plasmid, Barry Pang for assistance with single-cysteine mutagenesis, and Sarah Woodson for generously sharing equipment. This work was supported by the National Institutes of Health (R01GM-084192, R01GM-113240 to G.D.B.; R01-CA077373 to J.M.B.).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-Azidophenacyl bromide | Sigma Aldrich | A6057 |
| 7-Diethylamino-3-[N-(2-maleimidoethyl)carbamoyl]coumarin (MDCC) | Sigma Aldrich | 05019 |
| Purine Nucleoside Phosphorylase | Sigma Aldrich | N8264 |
| 7-Methylguanosine | Sigma Aldrich | M0627 |
| 0.65mM Phosphorus Standard Solution | Sigma Aldrich | P3869 |
| Adenosine 5′-(β,γ-imido)triphosphate lithium salt hydrate (AMPPNP) | Sigma Aldrich | A2647 |
| Adenosine 5′-diphosphate sodium salt (ADP) | Sigma Aldrich | A2754 |
| Phenol:Chloroform 5:1 | Sigma Aldrich | P1944 |
| Sodium Fluoride | Fluka | 71519 |
| Beryllium Chloride | Sigma Aldrich (discontinued) | 201197 |
| Streptavidin | Pierce | 21125 |
| 3.05 mm copper grids, 400 lines/inch square mesh | Electron Microscopy Sciences | G400-Cu |
| Uranyl formate | SPI supplies | 16984-59-1 |
| Phusion DNA Polymerase | NEB | M0530 |
| Pyruvate Kinase from rabbit muscle | Roche | 10 128 155 001 |
| L-Lactate Dehydrogenase from rabbit muscle | Roche | 10 127 230 001 |
| Phospho(enol)pyruvic acid trisodium salt hydrate | Sigma Aldrich | P7002 |
| β-Nicotinamide adenine dinucleotide, reduced disodium salt hydrate | Sigma Aldrich | N8129 |
| Critical Commercial Assays | ||
| Thermo Sequenase Dye Primer Manual Cycle Sequencing Kit | Affymetrix | 79260 |
| Recombinant DNA | ||
| Cysteine-free ScChd1118–1274 | This study | N/A |
| ScChd1118–1274-A1117C | This study | N/A |
| ScChd1118–1274-H1252C | This study | N/A |
| ScChd1118–1274-A1250C | This study | N/A |
| ScChd1118–1274-E493C | This study | N/A |
| ScChd1118–1274-N459C | This study | N/A |
| ScChd1118–1274-N650C | This study | N/A |
| ScChd1118–1274-V721C | This study | N/A |
| ScChd1118–1274-G743C | This study | N/A |
| ScChd1118–1274-G238C | This study | N/A |
| ScChd1118–1274-R237C | This study | N/A |
| ScChd1118–1274-L1119C | This study | N/A |
| ScChd1118–1274-N1129C | This study | N/A |
| ScChd1118–1274-T491C | This study | N/A |
| ScChd1118–1274-S523C | This study | N/A |
| ScChd1118–1274-S524C | This study | N/A |
| ScChd1118–1274-G643C | This study | N/A |
| ScChd1118–1274-F646C | This study | N/A |
| ScChd1118–1274-S770C | This study | N/A |
| ScChd1118–1274 | McKnight et al., 2011 | N/A |
| ScChd1118–1014 | McKnight et al., 2011 | N/A |
| ScChd11006–1274 | Sharma et al., 2011 | N/A |
| XlHistone H2A | Karolin Luger Lab | N/A |
| XlHistone H2B | Karolin Luger Lab | N/A |
| XlHistone H3 | Karolin Luger Lab | N/A |
| XlHIistone H4 | Karolin Luger Lab | N/A |
| XlHistone H4-A15C | Geeta Narlikar Lab | N/A |
| XlHistone H3-C110A | Blaine Bartholomew Lab | N/A |
| E.coli Phosphate Binding Protein-A197C | Martin Webb Lab | N/A |
| pGEM-601 (see Table S3) | Jonathan Widom Lab | |
| pGEM-601(LacO-11L) (see Table S3) | Nodelman et al., 2016 | N/A |
| pGEM-601(LacO-11R) (see Table S3) | Nodelman et al., 2016 | N/A |
| pGEM-601(LacO-6R) (see Table S3) | Nodelman et al., 2016 | N/A |
| pJ201-601array-208bpx34 (33N26, 2N61) (see Table S4) | Patel et al., 2011 | N/A |
| pJ201-601array-169bpx16 (0N0) (see Table S4) | This study | N/A |
| pJ201-601array-178bpx12 (0N33) (see Table S4) | This study | N/A |
| Sequence-Based Reagents | ||
| Primers for nucleosomal DNA constructs are given in Table S2; Templates for nucleosomal DNA constructs are given in Table S3. | ||
| 80N0 DNA (see Tables S2, S3) | This study | N/A |
| 0N80 DNA (see Tables S2, S3) | This study | N/A |
| 0N63 DNA (see Tables S2, S3) | This study | N/A |
| 11N70 DNA (see Tables S2, S3) | This study | N/A |
| 29N19 DNA (see Tables S2, S3) | This study | N/A |
| 40N40 DNA (see Tables S2, S3) | This study | N/A |
| Software and Algorithms | ||
| ImageJ 1.47v | NIH | https://imagej.nih.gov/ij/ |
| PyMol, (version 1.7) | The PyMol Molecular Graphics System, Schrodinger, LLC | www.pymol.org |
| Mathematica (version 10) | Wolfram Research | https://www.wolfram.com/mathematica/ |
| EPU software package | FEI | |
| CTFFIND4 | Rohou and Grigorieff, 2015 | http://grigoriefflab.janelia.org/ctffind4 |
| DOGPICKER | Voss et al., 2009 | http://emg.nysbc.org/redmine/projects/software/wiki/DoGpicker |
| SPIDER | Frank et al., 1996 | http://spider.wadsworth.org/spider_doc/spider/docs/spider.html |
| XMIPP 2.4 | Scheres et al, 2008 | http://xmipp.cnb.csic.es/twiki/bin/view/Xmipp/WebHome |
| RELION 1.4 | Scheres, 2012; Scheres, 2016 | http://www2.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| Other | ||
| HisPrep FF 16/10 (Nickel affinity) | GE | 28-9365-51 |
| HisTrap HP, 5ml (Nickel affinity) | GE | 17-5248-01 |
| HiTrap SP FF, 5ml | GE | 17-5157-01 |
| HiTrap Q FF, 5ml | GE | 17-5156-01 |
| HiLoad 16/600 Superdex 200, prep grade | GE | 28-9893-35 |
| HiPrep 26/10 Desalting | GE | 17-5087-01 |
| HiPrep 16/10 Q FF | GE | 17-5190-01 |
| HiPrep 16/10 SP FF | GE | 17-5192-01 |
Footnotes
Author Contributions
Conceptualization, I.M.N. and G.D.B; Methodology, I.M.N. and G.D.B.; Investigation, I.M.N., F.B., A.P., R.R., and K.C.H.; Resources, I.M.N., A.P., R.R., and K.C.H.; Writing – Original Draft, I.M.N. and G.D.B.; Writing – Review and Editing, I.M.N., F.B., A.P., R.R., K.C.H., J.M.B., and G.D.B.; Funding Acquisition, J.M.B. and G.D.B.; Supervision, J.M.B. and G.D.B.
References
- Brune M, Hunter JL, Corrie JE, Webb MR. Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry. 1994;27:8262–8271. doi: 10.1021/bi00193a013. [DOI] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. Regulation of ISWI involves inhibitory modules antagonized by nucleosomal epitopes. Nature. 2012;7428:280–284. doi: 10.1038/nature11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang W, Bartholomew B. Domain architecture of the catalytic subunit in the ISW2-nucleosome complex. Mol Cell Biol. 2007;23:8306–8317. doi: 10.1128/MCB.01351-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J Mol Biol. 2002;5:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- Dechassa ML, Hota SK, Sen P, Chatterjee N, Prasad P, Bartholomew B. Disparity in the DNA translocase domains of SWI/SNF and ISW2. Nucleic Acids Res. 2012;10:4412–4421. doi: 10.1093/nar/gks007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas V, Stokes DG, Perry RP. A mammalian DNA-binding protein that contains a chromodomain and an SNF2/SWI2-like helicase domain. Proc Natl Acad Sci U S A. 1993;6:2414–2418. doi: 10.1073/pnas.90.6.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürr H, Korner C, Muller M, Hickmann V, Hopfner KP. X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell. 2005;3:363–373. doi: 10.1016/j.cell.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Fei J, Torigoe SE, Brown CR, Khuong MT, Kassavetis GA, Boeger H, Kadonaga JT. The prenucleosome, a stable conformational isomer of the nucleosome. Genes Dev. 2015;24:2563–2575. doi: 10.1101/gad.272633.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaus A, Martin DM, Barton GJ, Owen-Hughes T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006;10:2887–2905. doi: 10.1093/nar/gkl295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. Journal of Structural Biology. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Gaspar-Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, Ramalho-Santos J, McManus MT, Plath K, Meshorer E, Ramalho-Santos M. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;7257:863–868. doi: 10.1038/nature08212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, Workman JL, Barton GJ, Owen-Hughes T. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science. 2011;6050:1758–1760. doi: 10.1126/science.1206097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grüne T, Brzeski J, Eberharter A, Clapier CR, Corona DF, Becker PB, Müller CW. Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol Cell. 2003;2:449–460. doi: 10.1016/s1097-2765(03)00273-9. [DOI] [PubMed] [Google Scholar]
- Hauk G, Bowman GD. Structural insights into regulation and action of SWI2/SNF2 ATPases. Curr Opin Struct Biol. 2011;6:719–727. doi: 10.1016/j.sbi.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauk G, McKnight JN, Nodelman IM, Bowman GD. The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol Cell. 2010;5:711–723. doi: 10.1016/j.molcel.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;7280:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WL, Deindl S, Harada BT, Zhuang X. Histone H4 tail mediates allosteric regulation of nucleosome remodelling by linker DNA. Nature. 2014;7513:213–217. doi: 10.1038/nature13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Bulger M, Pazin MJ, Kobayashi R, Kadonaga JT. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell. 1997;1:145–155. doi: 10.1016/s0092-8674(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Kagalwala MN, Glaus BJ, Dang W, Zofall M, Bartholomew B. Topography of the ISW2-nucleosome complex: insights into nucleosome spacing and chromatin remodeling. EMBO J. 2004;10:2092–2104. doi: 10.1038/sj.emboj.7600220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JG, Hamiche A, Wu C. GAL4 directs nucleosome sliding induced by NURF. EMBO J. 2002;6:1406–1413. doi: 10.1093/emboj/21.6.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassabov S, Bartholomew B. Site-directed histone-DNA contact mapping for analysis of nucleosome dynamics. Methods Enzymol. 2004;375 doi: 10.1016/s0076-6879(03)75013-7. [DOI] [PubMed] [Google Scholar]
- Kiianitsa K, Solinger JA, Heyer WD. NADH-coupled microplate photometric assay for kinetic studies of ATP-hydrolyzing enzymes with low and high specific activities. Anal Biochem. 2003;2:266–271. doi: 10.1016/s0003-2697(03)00461-5. [DOI] [PubMed] [Google Scholar]
- Koh FM, Lizama CO, Wong P, Hawkins JS, Zovein AC, Ramalho-Santos M. Emergence of hematopoietic stem and progenitor cells involves a Chd1-dependent increase in total nascent transcription. Proc Natl Acad Sci U S A. 2015;14:E1734–43. doi: 10.1073/pnas.1424850112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;3:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- Leonard JD, Narlikar GJ. A nucleotide-driven switch regulates flanking DNA length sensing by a dimeric chromatin remodeler. Mol Cell. 2015;5:850–859. doi: 10.1016/j.molcel.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hada A, Sen P, Olufemi L, Hall MA, Smith BY, Forth S, McKnight JN, Patel A, Bowman GD, Bartholomew B, Wang MD. Dynamic regulation of transcription factors by nucleosome remodeling. Elife. 2015 doi: 10.7554/eLife.06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JJ, Lehmann LW, Bonora G, Sridharan R, Vashisht AA, Tran N, Plath K, Wohlschlegel JA, Carey M. Mediator coordinates PIC assembly with recruitment of CHD1. Genes Dev. 2011;20:2198–2209. doi: 10.1101/gad.17554711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;1:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Richmond TJ. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- Luo Y, North JA, Rose SD, Poirier MG. Nucleosomes accelerate transcription factor dissociation. Nucleic Acids Res. 2014;5:3017–3027. doi: 10.1093/nar/gkt1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusser A, Urwin DL, Kadonaga JT. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat Struct Mol Biol. 2005;2:160–166. doi: 10.1038/nsmb884. [DOI] [PubMed] [Google Scholar]
- Manning BJ, Peterson CL. Releasing the brakes on a chromatin-remodeling enzyme. Nat Struct Mol Biol. 2013;1:5–7. doi: 10.1038/nsmb.2482. [DOI] [PubMed] [Google Scholar]
- McKnight JN, Jenkins KR, Nodelman IM, Escobar T, Bowman GD. Extranucleosomal DNA Binding Directs Nucleosome Sliding By Chd1. Mol Cell Biol. 2011;23:4746–4759. doi: 10.1128/MCB.05735-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodelman IM, Bowman GD. Nucleosome sliding by Chd1 does not require rigid coupling between DNA-binding and ATPase domains. EMBO Rep. 2013;12:1098–1103. doi: 10.1038/embor.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodelman IM, Horvath KC, Levendosky RF, Winger J, Ren R, Patel A, Li M, Wang MD, Roberts E, Bowman GD. The Chd1 chromatin remodeler can sense both entry and exit sides of the nucleosome. Nucleic Acids Res. 2016;16:7580–7591. doi: 10.1093/nar/gkw406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo J, Chereji RV, Eriksson PR, Clark DJ. The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacing in vivo. Nucleic Acids Res. 2016;44:4625–35. doi: 10.1093/nar/gkw068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Chakravarthy S, Morrone S, Nodelman IM, McKnight JN, Bowman GD. Decoupling nucleosome recognition from DNA binding dramatically alters the properties of the Chd1 chromatin remodeler. Nucleic Acids Res. 2013;3:1637–1648. doi: 10.1093/nar/gks1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazin MJ, Bhargava P, Geiduschek EP, Kadonaga JT. Nucleosome mobility and the maintenance of nucleosome positioning. Science. 1997;5313:809–812. doi: 10.1126/science.276.5313.809. [DOI] [PubMed] [Google Scholar]
- Pendergrast PS, Chen Y, Ebright YW, Ebright RH. Determination of the orientation of a DNA binding motif in a protein-DNA complex by photocrosslinking. Proc Natl Acad Sci U S A. 1992;21:10287–10291. doi: 10.1073/pnas.89.21.10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pointner J, Persson J, Prasad P, Norman-Axelsson U, Stralfors A, Khorosjutina O, Krietenstein N, Peter Svensson J, Ekwall K, Korber P. CHD1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in S. pombe. EMBO J. 2012;23:4388–4403. doi: 10.1038/emboj.2012.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan A, Wang F, Mizuguchi G, Wei D, Huang Y, Wu C. H2A histone-fold and DNA elements in nucleosome activate SWR1-mediated H2A.Z replacement in budding yeast. Elife. 2015:e06845. doi: 10.7554/eLife.06845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. Journal of Structural Biology. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DP, Sundaramoorthy R, Martin D, Singh V, Owen-Hughes T. The DNA-binding domain of the Chd1 chromatin-remodelling enzyme contains SANT and SLIDE domains. EMBO J. 2011;13:2596–2609. doi: 10.1038/emboj.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Wittmeyer J, Cairns BR. Chromatin remodeling through directional DNA translocation from an internal nucleosomal site. Nat Struct Mol Biol. 2005;9:747–755. doi: 10.1038/nsmb973. [DOI] [PubMed] [Google Scholar]
- Saikrishnan K, Powell B, Cook NJ, Webb MR, Wigley DB. Mechanistic basis of 5′-3′ translocation in SF1B helicases. Cell. 2009;5:849–859. doi: 10.1016/j.cell.2009.03.036. [DOI] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. Processing of Structurally Heterogeneous Cryo-EM Data in RELION. Methods in Enzymology. 2016;579:125–157. doi: 10.1016/bs.mie.2016.04.012. [DOI] [PubMed] [Google Scholar]
- Scheres SH, Nunez-Ramirez R, Sorzano CO, Carazo JM, Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nature Protocols. 2008;3:977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanbeck R, Xiao H, Wu C. Spatial contacts and nucleosome step movements induced by the NURF chromatin remodeling complex. J Biol Chem. 2004;38:39933–39941. doi: 10.1074/jbc.M406060200. [DOI] [PubMed] [Google Scholar]
- Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;2:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- Sharma A, Jenkins KR, Héroux A, Bowman GD. Crystal structure of the chromodomain helicase DNA-binding protein 1 (Chd1) DNA-binding domain in complex with DNA. J Biol Chem. 2011;49:42099–42104. doi: 10.1074/jbc.C111.294462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, Washburn MP, Workman JL. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol. 2012;9:884–892. doi: 10.1038/nsmb.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorzano CO, Marabini R, Velazquez-Muriel J, Bilbao-Castro JR, Scheres SH, Carazo JM, Pascual-Montano A. XMIPP: a new generation of an open-source image processing package for electron microscopy. Journal of Structural Biology. 2004;148:194–204. doi: 10.1016/j.jsb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of Nucleosome Repositioning by Yeast ISWI and Chd1 Chromatin Remodeling Complexes. J Biol Chem. 2006;24:16279–16288. doi: 10.1074/jbc.M600682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torigoe SE, Patel A, Khuong MT, Bowman GD, Kadonaga JT. ATP-dependent chromatin assembly is functionally distinct from chromatin remodeling. Elife. 2013:e00863. doi: 10.7554/eLife.00863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama T, Palmer J, Landel CC, Shiloach J, Wu C. Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev. 1999;6:686–697. doi: 10.1101/gad.13.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. Journal of Structural Biology. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Frouws TD, Angst B, Fitzgerald DJ, DeLuca C, Schimmele K, Sargent DF, Richmond TJ. Structure and mechanism of the chromatin remodelling factor ISW1a. Nature. 2011;7344:448–453. doi: 10.1038/nature09947. [DOI] [PubMed] [Google Scholar]
- Yang JG, Madrid TS, Sevastopoulos E, Narlikar GJ. The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol. 2006;12:1078–1083. doi: 10.1038/nsmb1170. [DOI] [PubMed] [Google Scholar]
- Zofall M, Persinger J, Kassabov SR, Bartholomew B. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat Struct Mol Biol. 2006;4:339–346. doi: 10.1038/nsmb1071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.