

Abstract
Most protein kinases phosphorylate multiple substrates, each of which induces different and sometimes opposing functions. Determining the role of phosphorylation of each substrate following a specific stimulus is a challenge but is essential to elucidate the role of that substrate in the signaling event. Here we describe a rational approach to identify inhibitors of delta protein kinase C (δPKC), each inhibiting the phosphorylation of only one of δPKC’s substrates. δPKC regulates many signaling events and we hypothesized that docking inhibitor of a given substrate to δPKC should selectively abrogate the phosphorylation of only that substrate, without affecting phosphorylation of the other δPKC substrates. We describe here the development of selective inhibitors of three δPKC substrates (in vitro kD ~3 nM); two greatly reduced ischemia-induced cardiac injury with an IC50 of ~200 nM and the third had no effect, indicating that its respective substrate phosphorylation by δPKC has no role in the response to cardiac ischemia and reperfusion (I/R). The three inhibitors are highly specific; even at 1 μM, the phosphorylation of other δPKC protein substrates was unaffected. The rationale we describe is likely applicable for the development of other substrate-specific inhibitors.
Keywords: Cardiac ischemia and reperfusion, docking site, peptides, protein kinase C (PKC), protein-protein interaction
Graphical Abstract
A rationally designed peptide inhibitors of delta protein kinase C inhibit the enzyme’s interaction with only one of its substrates and identifies the substrate’s role in cardiac protection from ischemia.
Delta protein kinase C (δPKC), a member of the novel PKC subclass, is expressed ubiquitously in all cells and tissues.[1] This isozyme is involved in various signal transduction pathways, regulating both physiological and pathological conditions including cancer,[2] neurodegenerative diseases,[3] and ischemic heart disease[4] by phosphorylating multiple protein substrates. Here we describe a rational approach to developing inhibitors of δPKC that selectively abrogate its interaction with and phosphorylation of one protein substrate at a time.[5] We designed three peptides to inhibit the docking and phosphorylation of three δPKC substrates: myristoylated alanine-rich C-kinase substrate (MARCKS), dynamin-related protein 1 (Drp1) and insulin receptor substrate 1 (IRS1). These peptide inhibitors, which bind to δPKC with high affinity in vitro, selectivity inhibit the phosphorylation of only the corresponding substrate and differentially affect δPKC function in both an in culture and ex vivo models for myocardial infarction (MI).
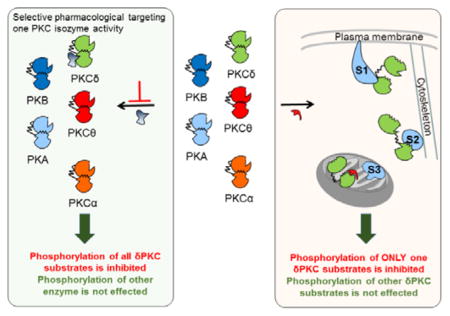
If there is a unique substrate-docking site on δPKC for an individual substrate, an inhibitor of this docking site should specifically inhibit δPKC-mediated phosphorylation of that substrate without affecting the phosphorylation of other protein substrates. To identify such inhibitors, we assumed that at least some of the substrate-docking sites on the kinase form selective intra-molecular interactions in the kinase that become available for substrate docking when δPKC is activated (Figure 1a–b). A peptide corresponding to such docking site should inhibit the interaction of δPKC with that protein substrate without affecting other protein substrates.
Figure 1.

A scheme representing the design of an inhibitor that is selective for the phosphorylation of one protein substrate of a multi-substrate kinase, δPKC.(a) In an inactive state (left), the substrate-docking site on δPKC interacts with another δPKC sequence, which mimics the kinase-binding site on the substrate, termed the pseudo-MARCKS site (ψMARCKS site, red). Upon activation, δPKC undergoes a conformational change, exposing its catalytic site, as well as disrupting the intra-molecular interaction covering the substrate-specific docking site. As a result, the substrate-specific docking site is available for binding (shown are docking sites for MARKCS and substrate XX (sub. XX) on δPKC, two of several protein substrates of this protein kinase). Specific protein-protein interactions between a substrate and its kinase increase the access of the substrate to the catalytic site, resulting in substrate phosphorylation (P). (b) A peptide corresponding to the δPKC-like sequence on MARCKS, ψMARCKS, is a competitive inhibitor for docking to and phosphorylation of MARCKS by δPKC without inhibiting docking and phosphorylation of other PKC substrates (e.g., sub. XX).
Focusing on δPKC-mediated phosphorylation of specific protein substrates, we used lalign[6] to identify short sequences of homology between δPKC and each substrate (MARCKS, Drp1 and IRS1) (Figure 2a, Figure S1a, and Figure S2a). We reasoned that these homology regions may represent the sites in δPKC and in MARCKS, for example, which interact with the MARCKS-docking site on δPKC in an intra- or inter-molecular fashion, respectively. The substrate-like short sequences were found in either the C2 regulatory domain (for MARCKS) or the V5 catalytic domain of δPKC (for Drp1 and IRS1). To further evaluate the importance of these sequences for protein-protein interactions (PPIs), we determined the evolutionary conservation of these sequences; sequences that are critical for PPIs are expected to be conserved in many species.[7] We found that all sequences identified are conserved in all species that have δPKC (Figure 2b–c, Figure S1b–c, and Figure S2b–c).
Figure 2.

Rational design of inhibitors for MARCKS phosphorylation by δPKC, based on the identification of short sequence of homology between the kinase and its substrate. (a) Sequence alignment of human δPKC and MARCKS identified a short sequence of homology, RAAEEP/KAAEEP, between these two proteins. (b) Conservation of the RAAEEP sequence in δPKC and the KAAEEP in MARCKS (c) in a variety of species. (d) RAAEEP sequence is located in the C2 domain of δPKC. The RAAEEP sequence is not present in other PKC isozymes, including θPKC, the most homologous PKC isozyme to δPKC, εPKC, another member of the novel PKC class, βPKC, member of the conventional PKC class, or ξPKC, member of the atypical PKCs. (e) RAAEEP and KAAEEP sequences are found in 6 human proteins (MARCKS, δPKC and 4 other proteins). A heat map of the RAAEEP and KAAEEP conservation in orthologs of these proteins shows that RAAEEP and KAAEEP are conserved only in δPKC and MARCKS. (f) the sequence RAAEEP (red) in δPKC (PDB: 1BDY) is exposed and thus likely available for protein-protein interactions. * denotes identity and : denotes homology.
The substrate-homologous sequences in δPKC were less conserved or absent in other PKC isozymes (Figure 2d, Figure S1d, Figure S2d), suggesting their functional importance.
A BLAST search of the human genome identified several other proteins with homologous sequences. However, these sequences were not evolutionarily conserved (Figure 2e, Figure S1e, and Figure S2e). Moreover, whenever the protein structure was available, we verified that the docking site sequence was located on an exposed region of the substrate protein and therefore available for PPIs (Figure 2f, and Figure S1f). Together, these results suggest that these short sequences have potential functional importance and selectivity for the PPIs between δPKC and each of these substrates.
We synthesized peptides corresponding to the sequences in δPKC (Figure 3 and Table S1) and termed them pseudo-MARCKS-Cargo (ψMARCKS-Cargo), ψDrp1-Cargo and ψIRS1-Cargo, for the respective protein substrates.
Figure 3.

Chemical structure of the peptides used in the study.(a) Chemical structure of the ψMARCKS-Cargo, ψIRS1-Cargo, ψDrp1-Cargo and its derivative peptides. (b) Chemical structure of the ψMARCKS, ψIRS1, ψDrp1 and its derivative peptides. The peptides for the cell based and animal studies were conjugated to TAT47-57 carrier peptide, a short positively charged peptide that is used as a carrier for the delivery of the peptides into the cell, using a Gly-Gly spacer.
We evaluated the binding of these peptides to δPKC as well as to εPKC, another PKC isozyme with high homology to δPKC. Each peptide bound to δPKC with high affinity (ψMARCKS-Cargo Kd = 8.2 ± 2.4 nM; ψDrp1-Cargo Kd = 1.4 ± 0.3 nM; ψIRS1-Cargo Kd = 10.2 ± 3.0 nM), but not to the another PKC isozyme, εPKC (Figure 4; Kd values are average of three independent experiments). We also measured the dissociation rate of ψMARCKS-Cargo, ψDrp1-Cargo and ψIRS1-Cargo (Figure S3).
Figure 4.

Binding activity and selectivity of the peptides in vitro. (a, c, e) Binding curves of δPKC and εPKC, at ~ 75 μg/mL (~1 μM), to the ψMARCKS-Cargo (a), ψDrp1-Cargo (c), and ψIRS1-Cargo (e) peptides. Each peptide selectivity binds to δPKC and not to another novel PKC, εPKC. (b, d, f) Binding assay of increasing amounts of δPKC to ψMARCKS-Cargo (b), ψDrp1-Cargo (d), and ψIRS1-Cargo (f) peptides. Each peptide selectivity binds to δPKC (ψMARCKS-Cargo Kd 8.2 ± 2.3 nM; ψDRP1-Cargo Kd 1.4 ± 0.3 nM; ψIRS1-Cargo Kd 10.2 ± 3.0 nM). The results are from three independent experiments (Table S2). (g–i) Binding assay of increasing amounts of δPKC to ψMARCKS (g), ψDrp1 (h), and ψIRS1 (i), peptides. Each peptide selectivity binds to δPKC (ψMARCKS Kd 3.3 ± 0.9 nM; ψDRP1 Kd 2.9 ± 1.1 nM; ψIRS1 Kd 2.9 ± 1.1 nM).
To confirm that ψDrp1-Cargo, for example, is an inhibitor of the protein-protein interaction between δPKC and Drp1, we determined its effect on co-immunoprecipitation of recombinant δPKC with Drp1. ψDrp1-Cargo peptide blocked δPKC binding to Drp1 (Figure S4). Since the docking site sequence for IRS1 on δPKC is identical to a sequence in another PKC isozyme, θPKC, we also measured the binding of θPKC to ψIRS1-Cargo peptide. There was a 20 fold higher Kd measured for ψIRS1-Cargo peptide to θPKC, (200 ± 18) nM, as compare to δPKC (10.2 nM) (data not shown). Importantly and relevant to our study, whereas δPKC is expressed in most tissues and cell types, θPKC is mainly expressed in T lymphocytes.
To test the biological activity of the peptides, the peptides were conjugated to the TAT-derived cell permeating peptide, TAT47-57,[8] which enables safe and effective delivery of peptides into cells in culture, in vivo,[9] and even in humans.[1b, 10] We first confirmed that the affinity of these peptides for δPKC when conjugated to a TAT carrier is not inhibited and found that with or without TAT carrier, the peptides have similar affinities to δPKC (ψMARCKS Kd = 3.3 ± 0.9 nM; ψDrp1 Kd = 2.9 ± 1.1 nM; ψIRS1 Kd = 2.9 ± 1.1 nM).
We then determined whether the peptides could rescue mitochondrial function following ischemic injury in an H9c2 heart myoblast cell culture used as a model of myocardial infarction (MI). Ischemic damage increased the production of excessive mitochondrial reactive oxygen species (ROS), reduced overall mitochondrial function, and decreased cell viability. Treatment with ψDrp1 and ψIRS (2 μM) throughout reoxygenation decreased excessive mitochondrial ROS production (Figure 5). In addition, treatment with these two peptides rescued mitochondrial function and cell viability (Figures S5 and S6). Therefore, treatment with ψDrp1 and ψIRS1 reduces mitochondrial dysfunction and cell death following ischemia and reoxygenation damage in culture.
Figure 5.

ψDrp1 and ψIRS reduce the production of excessive mitochondrial reactive oxygen species. H9c2 cells were subjected to 24 hours of ischemia and 2 hours of reoxygenation. Peptide were added in during both ischemia and reoxygenation (2 μM). Mitochondrial production of ROS was measured (arbtirary fluoresence units) by MitoSox dye, shown in green, relative to cell count, measured by hoechst 3342 stain, shown in blue. As shown in the images and the bar graph, ischemia and reoxygenationIR damage results in an increased mitochondrial ROS production per cell. Treatment with ψDrp1 and ψIRS1, but not with ψMARCKS reduced the production of mitochondrial ROS by 25% and 20%, respectveily. *p<0.05 and **p<0.01 compared to TAT control.
We next determined the effect of each peptide on cardiac protection following ischemia and reperfusion (I/R) injury using the Langendorff animal model of myocardial infarction[11] (Figure 6a). Either ψDrp1 or ψIRS1 peptides (1 μM) reduced cardiac damage by ~65%, which was demonstrated by decreased infarct size (Figure 6b–c) and reduced levels of creatine kinase release (CK, an enzyme that is released from dead cardiac cells and used as an indicator for myocardial damage in humans with MI; Figure 6d). On the other hand, ψMARKCS peptide had no significant effect on infarct size or CK release in the same heart attack model (Figure 6b–d). δV1-1 (1 μM), a general inhibitor of δPKC, decreased infarct size by ~70%, as compared to treatment with hearts treated with TAT peptide. We also tested the effect of peptide cargos without TAT, to further validate the importance of TAT for the intracellular delivery of the peptides. None of the peptides lacking TAT affected cardiac injury from I/R (Figure S7).
Figure 6.

Cardioprotective activity of the peptides as measured in whole hearts subjected to simulated myocardial infarction, ex vivo.
(a) Protocol of the myocardial infarction model using isolated hearts subjected to I/R (simulated myocardial infarction; MI) or normoxia (Nor). Horizontal bars indicate the length (in minutes) of each treatment (eq = equilibration). Rat hearts were subjected to 30 minutes of ischemia followed by 60 minutes of reperfusion with or without peptide treatment for the first 20 minutes of reperfusion. (b–c) triphenyltetrazolium chloride solution (TTC) staining (red indicates live tissue and white indicated dead tissue; n=6/hearts per treatment), and (d) release of cardiac creatine kinase (CK) (n=6). ****p<0.001 compared to TAT control.
Using the same model, we confirmed that each peptide inhibited only the phosphorylation of the corresponding substrate (Figure 7). Importantly, δPKC-mediated phosphorylation of other δPKC substrates was unaffected by treatment with each of the novel peptides. Meanwhile, all these phosphorylation events were inhibited by treatment with δV1-1, the pan inhibitor of all δPKC-mediated phosphorylations[4a] (Figure 7 and S8). Thus, our novel peptides are highly selective inhibitors of δPKC-mediated phosphorylation of the corresponding substrate.
Figure 7.

Selectivity of each peptide as a phosphorylation inhibitor; phosphorylation of four δPKC substrates following simulated myocardial infarction. Phosphorylation of MARCKS (a), Drp1 (b), IRS1 (c), or STAT (d) after cardiac I/R in the presence of control or the indicated peptides (1 μM). Only the phosphorylation of the substrate corresponding to the peptide was inhibited by the treatment. Phosphorylation is expressed as percent change from control (TAT)-treated hearts subjected to I/R, as in Figure 6.
The IC50 for ψDrp1 peptide in reducing cardiac injury ex vivo was ~100 nM and the IC50 for ψIRS1 was ~350 nM, as measured by infarct size and cardiac CK release, a clinical biomarker for heart attack (Figure 8).
Figure 8.
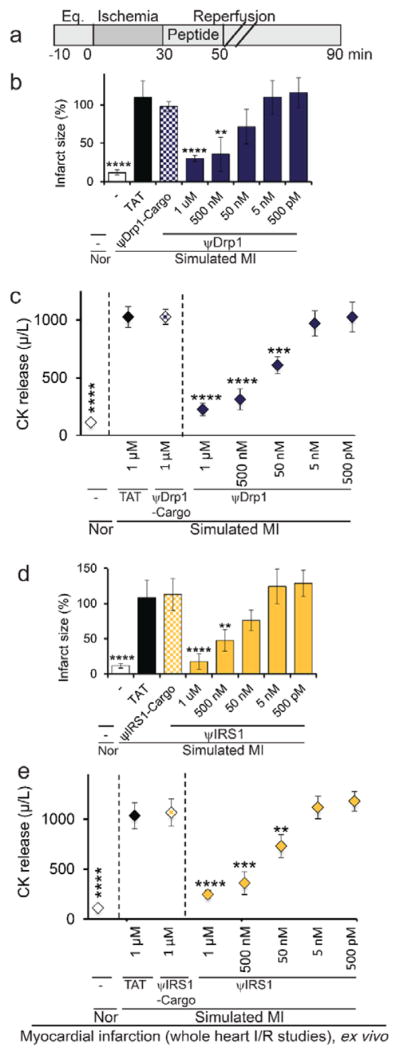
Dose-dependent cardio-protective effect of ψDrp1 and ψIRS1, as measured in whole heart subjected to simulated myocardial infarction, ex vivo. (a) Protocol of myocardial infarction model using isolated hearts subjected to I/R (simulated myocardial infarction; MI) or normoxia (Nor). Horizontal bars indicate the length (in minutes) of each treatment (eq = equilibration). Rat hearts were subjected to 30 minutes of ischemia followed by 60 minutes of reperfusion with or without the indicated peptide at the indicated concentration added for the first 20 minutes of reperfusion. (b, d) infarct size and (c, e) release of cardiac creatine kinase (CK) (n=4/group). **p<0.01, ***p<0.005, ****p<0.001 compared to TAT control.
Since ψDrp1 was cardio-protective (as compared with ψMARCKS) and has better in vitro and ex vivo bioactivity (as compared to ψIRS1), we did a small structure activity relationship study by substituting specific amino acids of the cargo of ψDrp1 (YTDFDE) with alanine (Figure 3). Substitutions of Asp3 to Ala only slightly reduced its protection from cardiac damage relative to the original peptide ψDrp1, from 75% for ψDrp1 (Figure 8c) to 65% for ψDrp1 Asp3 Ala (YTAFDE-TAT, Figure 9b–d). However, YTDFAA-TAT peptide (1μM) did not reduce any cardiac damage (Figure 9b–d), and it had also an approximately 500-fold lower affinity for δPKC (Kd =1.44 μM as compared to active ψDrp1 with Kd of 2.9 nM). We also confirmed the activity of the peptides on the designated target. Indeed treatment with YTAFDE-TAT peptide inhibited Drp1 phosphorylation. However, treatment with YTDFAA-TAT peptide did not reduce the phosphorylation of Drp1 (Figure S9).
Figure 9.
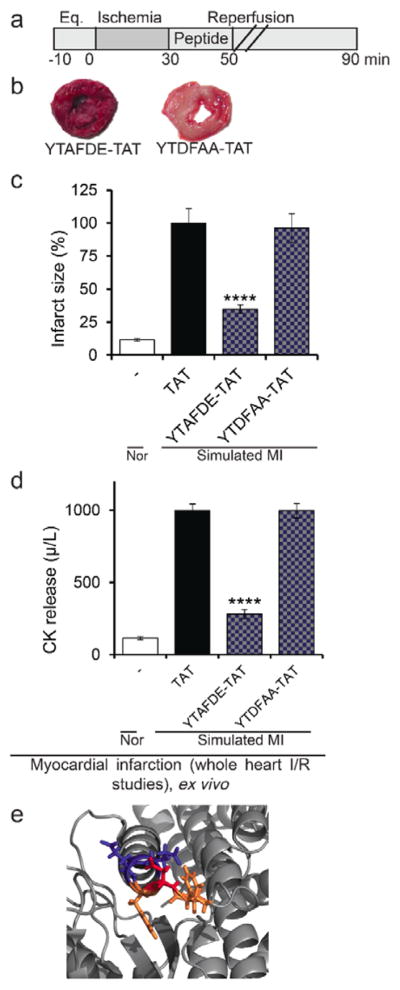
ψDrp1 peptide structure activity studies.(a) Protocol of the myocardial infarction model using isolated hearts subjected to I/R (simulated myocardial infarction; MI) or normoxia (Nor). Horizontal bars indicate the length (in minutes) of each treatment (eq = equilibration). Rat hearts were subjected to 30 minutes of ischemia followed by 60 minutes of reperfusion with or without peptide treatment for the first 20 minutes of the reperfusion only. (b–c) triphenyltetrazolium chloride solution (TTC) staining (red indicates live tissue and white indicates dead tissue), (d) release of cardiac creatine kinase (CK; n=4/hearts per treatment). ****p<0.001 compared to TAT control. (e) The δPKC-binding site in Drp1, YTDFDE, (PDB: 3ZVR) presented with the amino acids of the ψDrp1 in stick presentation. The two amino acids at the C-terminus that whose substitution to Ala abolish the biological activity are in blue (D and E). The D to A substitution that had prevailing biological activity is colored in red, and the rest amino acids of the peptide are colored in orange.
Examination of the structure of the C2 domain shows that the δPKC-binding site on Drp1 is located on an alpha-helix (the C-terminus, D and E) and has no defined structure for the rest of the amino acids (YTDF). That fits with our observation that the substitution of the two amino acids on the C-terminus to Ala significantly reduced the bioactivity of the peptides (D and E). These two amino acids are part of an alpha-helix and are facing the surface of the protein (Figure 9E, amino acids colored in red). In addition, the Ala substitution of the Asp amino acid, which is in the middle of the peptide and is less exposed to the surface had a mild effect on the bioactivity.
We next determined that the pseudo-peptides do not bind to the same site on δPKC and found that δPKC binding of ψDrp1-Cargo, for example, was unaffected by the presence of another pseudo-substrate peptide, ψIRS1-Cargo (10 μM; Figure 10a). We also tested the effect of treating the hearts with both ψDrp1 and ψIRS1 together and found an additional effect on cardiac protection as compared with each peptide alone. A dose–response study demonstrated that the combination of ψDrp1 and ψIRS1 has superior activity compared to each peptide alone, with an IC50 of ~50 nM (Figure 10 and Figure S10).
Figure 10.

Binding to δPKC in vitro and ex vivo bioactivity of peptides combination. (a) Binding curves of δPKC (~ 75 μg/mL; ~1 μM) to ψDrp1-Cargo is unaffected by the presence of ψIRS1-Cargo (10 μM). (b) Protocol of myocardial infarction model using isolated hearts subjected to I/R (simulated myocardial infarction; MI) or normoxia (Nor). Horizontal bars indicate the length (in minutes) of each treatment (eq = equilibration). Rat hearts were subjected to 30 minutes of ischemia followed by 60 minutes of reperfusion without or with ψDrp1 together with ψIRS1, added during the first 20 minutes of the reperfusion only. (c) infarct size and (d) release of cardiac creatine kinase (CK) (n=4/group). **p<0.01, ****p<0.001 as compared to TAT control.
It is not surprising that selective inhibition of δPKC-mediated MARCKS phosphorylation did not contribute to reducing cardiac injury. Upon cardiac injury, δPKC phosphorylates MARCKS, decreasing MARCKS association to the membrane, and leading to its translocation into the cytosol.[12] MARCKS serves as a major reservoir for calmodulin.[13] Although MARCKS phosphorylation may affect the levels of calmodulin in the cell and eventually can lead to the elevation of intracellular Ca2+ [14], this event is unlikely to contribute to the acute response of the heart to I/R injury at this time window. Our findings indicating the critical contribution of δPKC-mediated phosphorylation of IRS1 or Drp1 are also consistent with the literature. Mitochondria are an essential orchestrators of I/R injury, not only because of their role in ATP production, but by mediating cell death through reactive oxygen species (ROS) production and activation of apoptosis.[15] In cardiomyocytes, δPKC binds to and phosphorylates Drp1, a large GTPase that regulates mitochondrial fission.[3b, 16] δPKC activation of Drp1 through phosphorylation during I/R injury contributes substantially to ROS generation and results in severe diastolic dysfunction in hearts.[16–17] A decrease in δPKC-mediated Drp1 phosphorylation reduces cardiomyocyte death. Drp1 inhibition has cardio-protective effects in cells and in animal models of myocardial infarction.[16–17] δPKC also phosphorylates IRS1[18] following exposure to oxidative stress.[19] IRS1 modulates metabolic response and mitochondrial function.[20] Therefore, phosphorylation of Drp1 or IRS1 by δPKC each can mediate mitochondrial injury following I/R. These data suggest that reducing one of the damaging processes following ischemia is sufficient to reduce acute injury, presumably through the preservation of sufficiently healthy mitochondria.
The peptides in this study are derived from the C2 or V5 domains of δPKC. The C2 domain of δPKC is critical for subcellular translocation of the activated kinase to its substrates by anchoring to a scaffold protein: receptor for activated C kinase (RACK).[4a, 21] The V5 domain is part of the catalytic domain of δPKC is a highly variable region, even within the PKC family. Notably, the C2 and the V5 domains interact in the inactive conformation of PKC, but do not interact upon PKC activation, which releases both domains to participate in intermolecular PPIs. [1b, 22] Our findings that the pseudosubstrate-docking sites are derived from these two domains are consistent with the above findings.
As previously discussed, several interactions contribute to the specificity of a kinase for its substrate including kinase-substrate recognition of target phosphoacceptor residue on the substrate by kinase catalytic cleft. However, because of the high sequence and structure similarity of the catalytic site among many protein kinases, additional interaction sites, distinct from the phosphoacceptor site, increase the kinase-substrate selectivity. These docking sites, first identified for c-Jun’s interaction with its kinase JNK,[23] were shown to improve the specificity of a kinase for its individual substrates by several fold. [23] Here, we show that δPKC has at least three different docking sites, one for each substrate, and that biological activities of the peptides that interfere with docking of each substrate demonstrate that substrate docking sites are useful targets to regulate the functional consequences of δPKC activation.
In summary, we describe the development of highly potent peptide inhibitors for PPIs between δPKC and individual substrate proteins. The peptides were designed through the identification of specific docking sites for δPKC on each of three of its substrates, MARCKS, Drp1 and IRS1. Each of the peptide inhibitors demonstrated a high specificity in inhibiting the phosphorylation of its corresponding substrate in vitro, in culture, and ex vivo. The rational design described here represents a generalizable strategy that can be used to develop new selective phosphorylation inhibitors for one of many substrates of multi-substrate protein kinases.
Supplementary Material
Acknowledgments
The work was supported by National Institutes of Health grant HL52141 to D.M.-R. We thank Yoni Samuel Rubin for reviewing the manuscript. Nanomedical Diagnostics provided assistance in use of the AGILE Dev Kit binding assay and analysis of the results.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Kikkawa U, Matsuzaki H, Yamamoto T. J Biochem. 2002;132:831–839. doi: 10.1093/oxfordjournals.jbchem.a003294. [DOI] [PubMed] [Google Scholar]; b) Mochly-Rosen D, Das K, Grimes KV. Nat Rev Drug Discov. 2012;11:937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim J, Koyanagi T, Mochly-Rosen D. The Prostate. 2011;71:946–954. doi: 10.1002/pros.21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Qi X, Inagaki K, Sobel RA, Mochly-Rosen D. J Clin Invest. 2008;118:173–182. doi: 10.1172/JCI32636. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D. Mol Biol Cell. 2011;22:256–265. doi: 10.1091/mbc.E10-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, Dorn GW, 2nd, Mochly-Rosen D. Proc Natl Acad Sci U S A. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly-Rosen D. Circulation. 2003;108:2304–2307. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]; c) Steinberg SF. Physiology (Bethesda) 2012;27:130–139. doi: 10.1152/physiol.00009.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kornfeld OS, Hwang S, Disatnik MH, Chen CH, Qvit N, Mochly-Rosen D. Circ Res. 2015;116:1783–1799. doi: 10.1161/CIRCRESAHA.116.305432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qvit N, Disatnik MH, Sho J, Mochly-Rosen D. J Am Chem Soc. 2016;138:7626–7635. doi: 10.1021/jacs.6b02724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang XQ, Miller W. Advances in Applied Mathematics. 1991;12:337–357. [Google Scholar]
- 7.a) Lichtarge O, Bourne HR, Cohen FE. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]; b) Caffrey DR, Somaroo S, Hughes JD, Mintseris J, Huang ES. Protein Sci. 2004;13:190–202. doi: 10.1110/ps.03323604. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Guharoy M, Chakrabarti P. BMC Bioinformatics. 2010;11:286. doi: 10.1186/1471-2105-11-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gump JM, Dowdy SF. Trends Mol Med. 2007;13:443–448. doi: 10.1016/j.molmed.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 9.a) Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]; b) Begley R, Liron T, Baryza J, Mochly-Rosen D. Biochem Biophys Res Commun. 2004;318:949–954. doi: 10.1016/j.bbrc.2004.04.121. [DOI] [PubMed] [Google Scholar]
- 10.a) Bates E, Bode C, Costa M, Gibson CM, Granger C, Green C, Grimes K, Harrington R, Huber K, Kleiman N, Mochly-Rosen D, Roe M, Sadowski Z, Solomon S, Widimsky P. Circulation. 2008;117:886–896. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]; b) Johnson RM, Harrison SD, Maclean D. Methods Mol Biol. 2011;683:535–551. doi: 10.1007/978-1-60761-919-2_38. [DOI] [PubMed] [Google Scholar]; c) Rizzuti M, Nizzardo M, Zanetta C, Ramirez A, Corti S. Drug Discov Today. 2015;20:76–85. doi: 10.1016/j.drudis.2014.09.017. [DOI] [PubMed] [Google Scholar]; d) Lonn P, Dowdy SF. Expert Opin Drug Deliv. 2015;12:1627–1636. doi: 10.1517/17425247.2015.1046431. [DOI] [PubMed] [Google Scholar]
- 11.Langendorff O. Pflügers Archiv European Journal of Physiology. 1895;61:291–332. [Google Scholar]
- 12.Xu XH, Deng CY, Liu Y, He M, Peng J, Wang T, Yuan L, Zheng ZS, Blackshear PJ, Luo ZG. Cell Res. 2014;24:576–594. doi: 10.1038/cr.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallant C, You JY, Sasaki Y, Grabarek Z, Morgan KG. J Cell Sci. 2005;118:3595. doi: 10.1242/jcs.02493. [DOI] [PubMed] [Google Scholar]
- 14.Tang LH, Xia ZY, Zhao B, Wei XD, Luo T, Meng QT. J Biomed Biotechnol. 2011;2011:4. doi: 10.1155/2011/107091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forini F, Nicolini G, Iervasi G. Int J Mol Sci. 2015;16:6312–6336. doi: 10.3390/ijms16036312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaja I, Bai X, Liu Y, Kikuchi C, Dosenovic S, Yan Y, Canfield SG, Bosnjak ZJ. Biochem Biophys Res Commun. 2014;453:710–721. doi: 10.1016/j.bbrc.2014.09.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]; b) Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL. FASEB J. 2014;28:316–326. doi: 10.1096/fj.12-226225. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zepeda R, Kuzmicic J, Parra V, Troncoso R, Pennanen C, Riquelme JA, Pedrozo Z, Chiong M, Sanchez G, Lavandero S. J Cardiovasc Pharmacol. 2014;63:477–487. doi: 10.1097/FJC.0000000000000071. [DOI] [PubMed] [Google Scholar]
- 18.Greene MW, Ruhoff MS, Roth RA, Kim JA, Quon MJ, Krause JA. Biochem Biophys Res Commun. 2006;349:976–986. doi: 10.1016/j.bbrc.2006.08.158. [DOI] [PubMed] [Google Scholar]
- 19.Archuleta TL, Lemieux AM, Saengsirisuwan V, Teachey MK, Lindborg KA, Kim JS, Henriksen EJ. Free Radic Biol Med. 2009;47:1486–1493. doi: 10.1016/j.freeradbiomed.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, Bevins J, Valdez S, Noh J, Kim BJ, Moreira AB, Weatherford ET, Manivel R, Rawlings TA, Rech M, White MF, Abel ED. Mol Cell Biol. 2014;34:3450–3460. doi: 10.1128/MCB.00426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. J Biol Chem. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- 22.a) Kheifets V, Mochly-Rosen D. Pharmacol Res. 2007;55:467–476. doi: 10.1016/j.phrs.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qvit N, Mochly-Rosen D. Drug Discov Today Dis Mech. 2010;7:e87–e93. doi: 10.1016/j.ddmec.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Stebbins EG, Mochly-Rosen D. J Biol Chem. 2001;276:29644–29650. doi: 10.1074/jbc.M101044200. [DOI] [PubMed] [Google Scholar]; d) Newton AC. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Banci L, Cavallaro G, Kheifets V, Mochly-Rosen D. J Biol Chem. 2002;277:12988–12997. doi: 10.1074/jbc.M106875200. [DOI] [PubMed] [Google Scholar]
- 23.Adler V, Unlap T, Kraft AS. J Biol Chem. 1994;269:11186–11191. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.