

Abstract
Background
In addition to lowering low density lipoprotein-cholesterol (LDL-C), statin therapy also raises high density lipoprotein-cholesterol (HDL-C) levels. Inter-individual variation in HDL-C response to statins may be partially explained by genetic variation.
Methods and Results
We performed a meta-analysis of genome-wide association studies (GWAS) to identify variants with an effect on statin-induced HDL-C changes. The 123 most promising signals with P<1×10−4 from the 16,769 statin-treated participants in the first analysis stage were followed up in an independent group of 10,951 statin-treated individuals, providing a total sample size of 27,720 individuals. The only associations of genome-wide significance (P<5×10−8) were between minor alleles at the CETP locus and greater HDL-C response to statin treatment.
Conclusion
Based on results from this study that included a relatively large sample size, we suggest that CETP may be the only detectable locus with common genetic variants that influence HDL-C response to statins substantially in individuals of European descent. Although CETP is known to be associated with HDL-C, we provide evidence that this pharmacogenetic effect is independent of its association with baseline HDL-C levels.
Keywords: Pharmacogenetics, HDL-Cholesterol, Statins, Genome-wide association study
Introduction
The drug class of 3-hydroxymethyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, better known as “statins”, are widely prescribed and effective for the prevention and management of cardiovascular disease (CVD).[1] While the major CVD benefit of statins is due to reduction in plasma low density lipoprotein cholesterol (LDL-C)[2], statins also produce moderate increases, ranging from 4 to 10%, in levels of high density lipoprotein cholesterol (HDL-C).[3, 4] This is of particular interest since HDL-C levels are inversely related to CVD risk in the general population and in patients treated with statins.[5, 6] However, a causal role of low HDL-C as a determinant of increased CVD risk is controversial.[7]
The increase in HDL-C after statin therapy varies among individuals.[3] This might be partly due to genetic variation. Previous studies that have investigated associations between genotype and statin-induced changes in HDL-C[8–10] have focused primarily on variants within the CETP gene that are known to affect circulating HDL-C levels[11] and risk of coronary artery disease.[12] To address whether additional loci have an effect on statin-induced changes in HDL-C levels, we conducted a large-scale meta-analysis of genome-wide association studies (GWAS) using datasets from both randomized controlled trials (RCTs) and cohort studies in the large Genomic Investigation of Statin Therapy (GIST) consortium that previously identified four loci associated with LDL-C response to statins.[13]
Methods
Study populations
The GIST consortium assembled data from seven RCTs and eleven prospective population-based studies. The initial analysis (first stage) was performed in 16,769 statin-treated individuals; 8,506 individuals from six RCTs (ASCOT UK, CARDS, CAP, PRINCE, PROSPER, and TNT) and 8,263 statin-treated individuals from ten observational studies (AGES, ARIC, ASCOT UK-observational, BioVU, CHS, FHS, Health ABC, HVH, MESA, and the Rotterdam Study). Further investigation (second stage) was performed in 10,951 statin-treated individuals from two RCTs (ASCOT Scandinavia and JUPITER) and two observational studies (ASCOT Scandinavia – observational and GoDARTS), which were used to test for replication of findings from the first stage. Details of the first and second stage studies, including their genotyping and quality control (QC) information, can be found in the Supplementary Notes 1, 2 and 3 and Supplementary tables 1 and 2.
Subjects
Response to statin treatment was principally studied in statin-treated individuals only. Those treated with placebo were excluded from the analyses of RCTs and those not treated with statins were excluded from observational studies. HDL-C measurements were obtained before and after start of statin treatment. Only subjects with non-missing phenotypes and covariates were included. Those of reported or suspected non-European ancestry were excluded.
Outcome measurements
The response to statin treatment was defined as the difference between the natural log-transformed on- and off-treatment HDL-C levels (ln (on-treatment HDL-C) – ln (off-treatment HDL-C)). The corresponding linear regression coefficients thus reflect the fraction of differential HDL-C increase (relative increase) per copy of the coded allele in the additive genetic model. For observational studies, on-treatment HDL-C levels were calculated for all different prescribed statins, at any dosage, for any indication, and for any treatment episode extending at least four weeks prior to on-treatment HDL-C measurement. Characteristics of on- and off-treatment HDL-C levels and statins used in each study are shown in Supplementary Table 2. For each individual, at least one off-treatment HDL-C measurement and at least one on-treatment measurement were required. Subjects with missing on- or off-treatment measurements were excluded, with the exception of the GoDARTS study for which missing off-treatment HDL-C levels were estimated using imputation methods, as described previously.[14] In RCTs, when multiple on- or off-treatment measurements were available, the mean of the measurements was used.
Genotyping and imputation
Genotyping, quality control, data cleaning and imputation were performed independently in each study using different genetic platforms and software as outlined in Supplementary Table 3. In all studies, genotyping was performed using either Illumina, Affymetrix, or Perlegen genotyping arrays. Genotype data from each study had been imputed to the HapMap phase 2 reference panel [15], except for JUPITER which was imputed to the 1000genomes pilot data, using either MACH, Impute, or BIMBAM software [16–18], resulting in a total of approximately 2.5 million SNPs for analysis.
GWAS analysis
Each study independently performed the GWAS on the difference between natural log-transformed on- and off-treatment HDL-C levels, according to a common, central analysis protocol. To reduce confounding by possible association with off-treatment HDL-C levels, analyses were adjusted for the natural log-transformed off-treatment HDL-C levels. Linear regression was used, with SNPs represented by an additive genetic model and with imputed SNPs represented by expected allele dosage. Analyses were additionally adjusted for age, sex, and study specific covariates (e.g ancestry principal components (PCs), site, or country). FHS made use of a linear mixed effects model considering the kinship matrix in the analysis, hereby accounting for familial correlations within FHS. Analyses in the observational studies were, if the information was available, additionally adjusted for the time interval between on- and off-treatment HDL-C measures (mean follow-up times per study are provided in Supplementary Table 2) and for the natural logarithm of the statin dose equivalent, as defined in Supplementary Table 4. This table shows the dose for different statins for the LDL-C response; dividing the statin dosage for an individual drug by its dose equivalent shown in Supplementary Table 4 gives the standardized statin dosage.
Quality control and Meta-analysis
Within each study, SNPs with minor allele frequency <1% or imputation quality <0.3 were excluded from the analysis. QQ-plots were assessed for each study to check that there were no between study differences nor evidence for systematic bias within studies (Supplementary Figure 1). The software package METAL was used to perform the meta-analysis.[19] A fixed effects, inverse variance weighted approach was used. To correct for possible inflation of the test statistic, e.g. due to small amounts of potential population sub-structure, genomic control was performed by adjusting the within-study findings and the meta-analysis results for the genomic inflation factor.
Second stage
SNPs with p-values <1×10−4 in the first stage meta-analyses were selected for further investigation in the second stage. A maximum of two SNPs per locus (with a maximum 100 kB distance between SNPs) were selected, with the choice based on statistical significance. A total of 123 SNPs in 83 loci were selected for the second stage, which was performed in the GoDARTS study, the JUPITER trial, and the RCT and observational arm of the ASCOT Scandinavia study. GWAS data and response to statin treatment were available for these studies. Analysis was performed as for the first stage. Results of the first and second stage were combined using a fixed effects, inverse variance weighted meta-analysis using METAL.
Interaction analysis
The interaction effect of the lead CETP SNP rs247616 with the binary treatment indicator for statin versus placebo allocation was assessed in five of the participating RCTs (ASCOT Scandinavia, ASCOT UK, CARDS, JUPITER, and PROSPER). For these analyses, placebo treated individuals in the RCTs were included. The total sample size was 17,857, with 8,978 statin treated individuals and 8,879 placebo treated individuals. Regression models were applied to the combined population of statin and placebo treated subjects by adding to the model extra terms including treatment (statin (=1) or placebo (=0)) allocation and the product of treatment allocation with SNP minor allele dose.[20] Interaction coefficients of the five studies were combined in a fixed effects, inverse variance weighted meta-analysis using METAL. In addition, we also performed our main analysis for the CETP SNP rs247616 in only the placebo users of the five RCTs included in the interaction analysis.
Effect of genetic determinants of HDL-C levels on statin-induced HDL-C response
We performed a look-up in our GWAS results for all known genome-wide significant genetic variants associated with HDL-C levels, obtained from the most recent Global Lipids Genetics Consortium (GLGC) paper.[11] Of the 80 variants, 78 were available in our GWAS on statin induced HDL-C response. Subsequently, we examined whether a multi-SNP genotypic risk score constructed from these GLGC variants was associated with the level of statin induced HDL-C response, using publicly available summary level data from the GLGC (http://csg.sph.umich.edu//abecasis/public/lipids2013/). The joint effect of the 78 genetic variants on statin-induced HDL-C response was examined by means of a data-driven inverse-variance weighted approach, described previously by Dastani et al, [21] and accomplished through the gtx-package [22] (Genetics ToolboX, http://cran.r-project.org/web/packages/gtx) in the R statistical software environment.[23] Analogous to deriving a pooled estimate from the results of individual studies in conventional meta-analysis, this approach combines the causal estimates of multiple genetic variants, defined as the ratio of their association with statin response to their association with HDL-C levels.
Conditional analysis
Conditional analysis were performed in two of the participating studies, ASCOT UK (both RCT and observational – genotype data available for n=3,804) and CARDS (genotype data available for n=1194). Conditional analysis was conducted within GCTA software[24], using the –cojo method, which performs conditional and joint analysis with model selection. The genome-wide meta-analysis summary statistics from the combined analysis of both first-stage and second-stage data were used as the input data. Analysis was restricted to chromosome 16, containing the only genome-wide significant result from the meta-analysis, in order to determine whether the CETP region contains more than one independent signal of association. Within the GCTA analysis, MAF was restricted to ≥1% and a p-value cut-off of 5×10−7 was used as the selection threshold. LD was calculated between pairwise SNPs, but any SNPs further than 10 Mb apart were assumed to be in linkage equilibrium.
Variance explained
Two secondary analyses were performed to investigate the heritability of this pharmacogenetic trait. Firstly, the genome-wide heritability was calculated in GCTA[24] by estimating h2 using GREML analysis, according to all HapMap SNPs with MAF ≥ 1%, with reference to the genomic relatedness matrix generated within GCTA. Secondly, the percentage variance explained for the HDL-C response to statins adjusted for baseline HDL-C was calculated specifically for the lead CETP SNP rs247616 using R software[23] by including the dosage data for this SNP as a continuous predictor variable within the model. Firstly, the HDL-C response trait was regressed against all non-genetic covariates. The residuals from this model were used as the residual trait. In a second stage linear regression analysis the residual trait was regressed against the lead SNP and PCs. The R2 calculated from this second fitted linear regression model was used to estimate the percentage of the trait variance explained. Both analyses were performed using the ASCOT-UK dataset, as individual level raw genotype data are required. The combination of both the RCT and observational sub-cohorts of ASCOT-UK gave a total sample size of N = 2,055 statin-treated participants. The explained variance analysis in R was additionally performed in the CARDS study, including 1,194 statin-treated participants. The linear regression models used exactly the same data and covariates as from the primary GWAS analysis.
Results
First-stage meta-analysis
In the first stage of this analysis, six randomized controlled trials (n=8,506 statin recipients) and ten observational studies (n=8,263 statin recipients) were included (Supplementary Notes 1 and 2 and Supplementary Tables 1 and 2). Three SNPs at the CETP locus (chromosome 16) were identified as genome-wide significant (P<5×10−8) for their association with HDL-C response to statin treatment (Figures 1 and 2 and Table 1). The most significant association was for SNP rs247616 (MAF=0.324, β=0.011, SE=0.002, P=5.95×10−10) (Figure 3), indicating that carriers of the minor allele of this SNP respond to statins with a 1.1% greater per-allele increase in HDL-C compared with non-carriers. The average increase in HDL-C during statin treatment across all studies was 0.045 mmol/L. This additional 1.1% per-allele increase in HDL-C is equivalent to a 0.046 mmol/L increase for carriers of one copy of the CETP SNP. We found no other loci associated with HDL-C response to statin treatment at a genome-wide significant level at this first stage.
Figure 1.
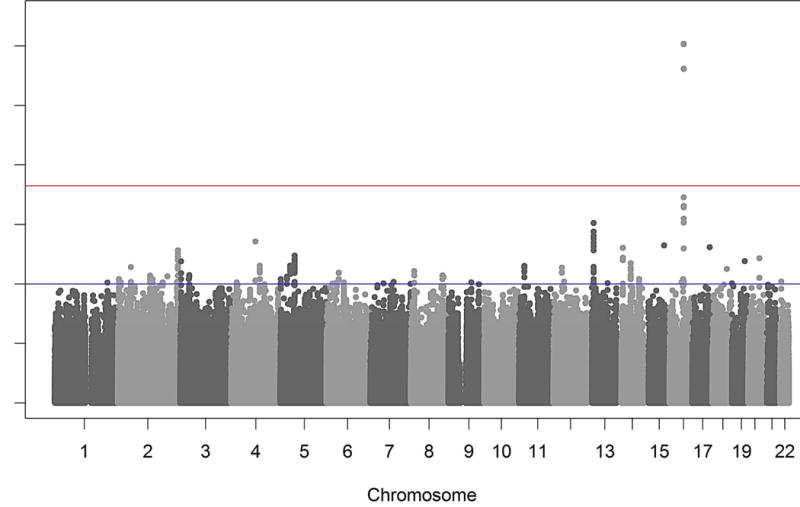
Results of the GWAS meta-analysis. Manhattan plot presenting the –log10 P-values from the combined stage 1 and 2 meta-analysis on HDL-C response to statin treatment. The top (red) line represents the genome-wide significant P-value 5×10−8, the second (blue) line represents the P-value 1×10−4, the threshold used for selecting SNPs to take forward to the second stage. Hence the results of these SNPs come from the lager combined meta-analysis, whereas all other results are taken from the stage 1 discovery meta-analysis.
Figure 2.
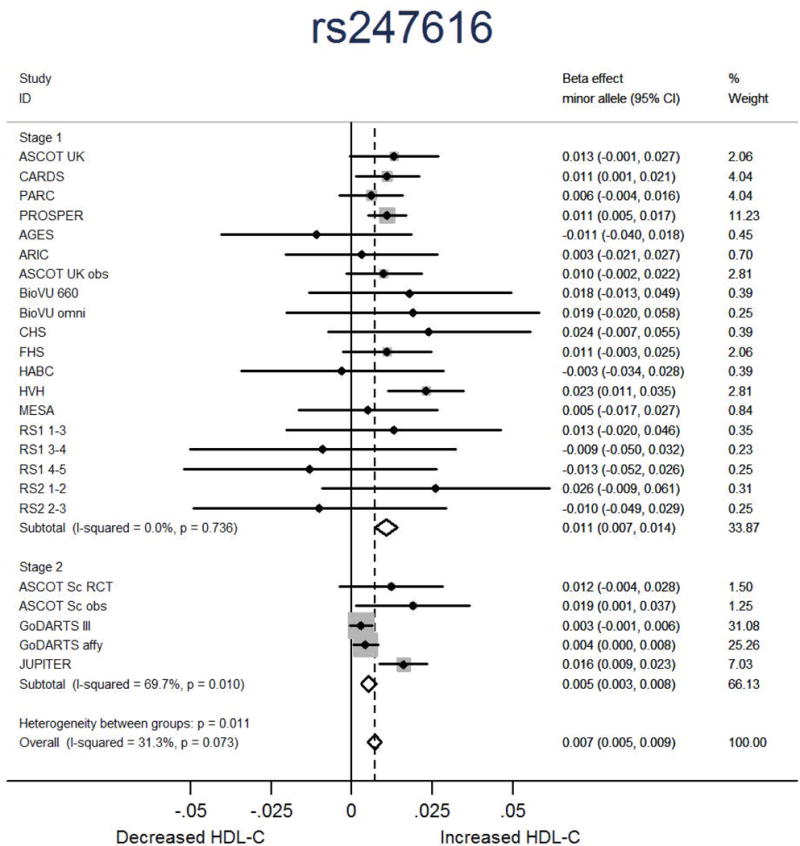
Forest plot showing the association in each study and overall association of the lead CETP SNP rs247616 with HDL-C response to statin treatment. Beta represents fractional HDL-C change for each copy of the minor allele.
Table 1.
Association of CETP SNP rs247616 (chromosome 16, bp 55547091) with HDL-C response after statin treatment in the stage 1, stage 2, and combined GWAS meta-analyses.
| Phase | N | Coding allele | Non-coding allele | Frequency coding allele | Beta* | SE | % extra increase# | P-value |
|---|---|---|---|---|---|---|---|---|
| Stage 1 | 14693 | T | C | 0.324 | 0.011 | 0.002 | 1.1 | 5.95×10−10 |
| Stage 2 | 10961 | T | C | 0.327 | 0.005 | 0.001 | 0.5 | 1.59×10−5 |
| Combined | 25654 | T | C | 0.326 | 0.007 | 0.001 | 0.7 | 8.52×10−13 |
Beta for difference between the natural log transformed on- and off-treatment HDL-C levels, adjusted for natural log transformed off-treatment HDL-C, age, sex, and study specific covariates. The beta reflects the fraction of differential HDL-C lowering in carriers vs. non-carriers of the SNP; a positive beta indicates a better statin response (larger HDL-C increase).
This percentage reflects the % extra HDL-C increase in carriers vs. non-carriers of the SNP.
Figure 3.
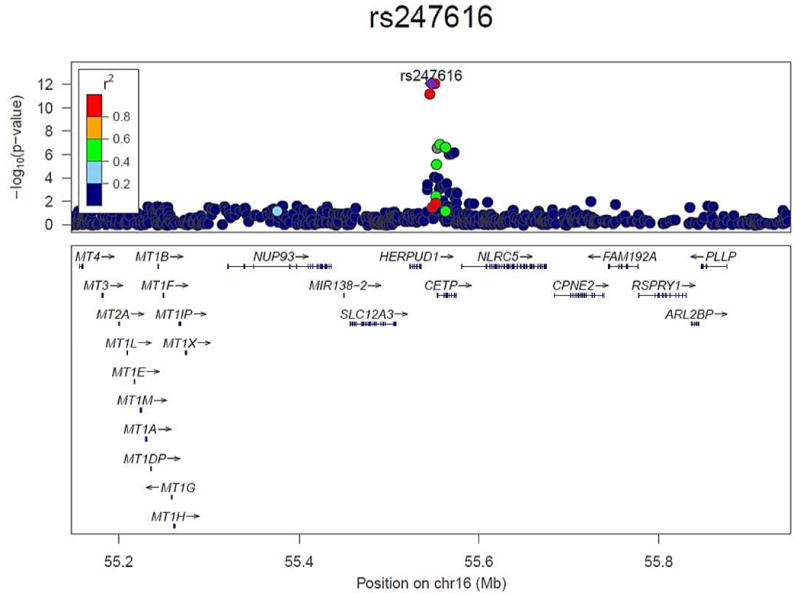
Regional association plot of the CETP region that was genome-wide significant for association with HDL-C response to statin treatment, using the results of the combined meta-analysis (generated using LocusZoom [39]). The color of each SNP is based on the LD (r2) with the lead SNP rs247616 (shown in purple). The RefSeq genes in the region are shown in the lower panel.
Second-stage meta-analysis
We selected 123 SNPs from 83 loci with P<1×10−4 in the first stage meta-analysis for further investigation in the second stage, which included 10,951 statin-treated individuals from two RCTs and two observational studies (Supplementary Note 3 and Supplementary Tables 1 and 2). The second stage meta-analysis confirmed the significant association between genetic variants within the CETP loci and HDL-C response from the first stage meta-analysis (rs247616: MAF=0.327, β=0.005, SE=0.001, P=1.59×10−5) as P<6×10−4, the Bonferroni p-value threshold for testing 123 SNPs (Table 1, Figure 2, and Supplementary Table 5). The combined effect from the first and second stage meta-analysis for the CETP rs247616 SNP was genome-wide significant (MAF=0.326, β=0.007, SE=0.001, P=8.52×10−13) (Table 1, Figure 2, and Supplementary Table 5). No other locus reached statistical significance (P<4×10−4) in the second stage meta-analysis or in the combined meta-analysis (P<5×10−8) for association with HDL-C response to statin treatment (Figure 1 and Supplementary Table 5). Indeed, Supplementary Table 5 (ordered by the combined meta-analysis p-values) shows that the three SNPs within CETP which were genome-wide significant in the first stage, were the only SNPs that reached Bonferroni significance in the second stage and genome-wide significance in the combined meta-analysis.
Interaction analysis
To exclude the possibility of confounding in the association between CETP and HDL-C response to statin treatment, two analyses were performed. First the main analysis for the CETP SNP rs247616 was repeated in the placebo users using data from five of the participating RCTs. In addition, in the same studies we tested for interaction between the CETP lead SNP rs247616 and randomized statin or placebo allocation. Supplementary Figure 2 shows the results for the association between HDL-C change during follow-up and rs247616 stratified for placebo and statin users. Table 2 shows a significant P-value for interaction in the meta-analysis combining the five studies (P-3.52×10−3, β=0.007, SE=0.002) for the CETP SNP, indicating that genetic effects of CETP on baseline HDL-C contribute at most only in part to genetic effects on HDL-C response in the statin-treated group, as the genetic effect is modified by the use of statin treatment.
Table 2.
Interaction between CETP rs247616 and statin vs. placebo allocation on HDL-C response. Meta-analysis of data from 5 RCTs.
| SNP | N | Coding allele | Non-coding allele | Frequency coding allele | Interaction Beta | Interaction SE | Interaction P-value |
|---|---|---|---|---|---|---|---|
| rs247616 | 17857 | T | C | 0.341 | 0.007 | 0.002 | 3.52×10−3 |
Interaction beta and SE refer to statistics from linear regression modelling the difference between the natural log transformed on- and of-treatment HDL-C levels adjusted for natural log transformed off-treatment HDL-C, age, sex, and study specific covariates, and including an interaction term between SNP and statin or placebo allocation. The interaction p-value refers to the significance of the SNP-by-statin or placebo allocation interaction term in the regression model.
Effect of genetic determinants of HDL-C levels on statin-induced HDL-C response
SNPs previously shown to be associated with HDL-C levels (n=78)[11] were assessed for their association with statin-induced HDL-C response in our meta-analysis. After Bonferroni correction, rs3764261 (CETP) was the sole genetic variant associated with statin-induced HDL-C response amongst the 78 examined variants (Supplementary Table 5). Joint analysis of the HDL-C associated variants demonstrated that predisposition to high HDL-C levels is associated with increased statin-induced HDL-C response (Figure 4). This amounted to a 2.9% fractional increase (β=0.029, SE=0.003, P=1×10−19) in statin-induced HDL-C response per SD increase in genetically raised HDL-C levels. Excluding the CETP SNP (rs3764261) from the model did not materially change the results (β=0.029, SE=0.005, P=1×10−8). Testing for heterogeneity did not reveal any indication of pleiotropic effects(P=0.64).
Figure 4.
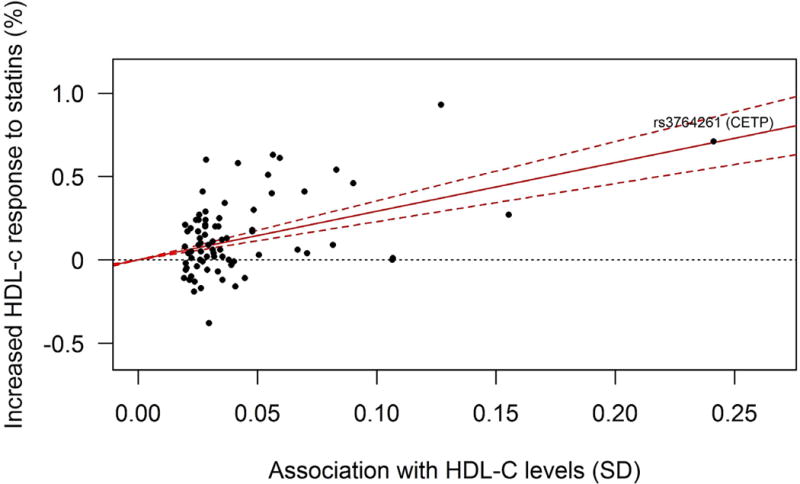
Plot of the per-allele association of genetic variants with HDL-C levels (x-axis, per allele in SD units, as reported by Willer et al. [11]) against the association with HDL-C response to statin treatment (y-axis, percentage) (generated using [22]). The regression line shows the linear relationship between these two variables, with 95% confidence boundaries.
Conditional analysis
The conditional analysis within GCTA resulted in only one remaining SNP selected in the model, namely the lead SNP rs247616 within the CETP locus, with a joint p-value of 9.96×10−10 and joint β=0.0104, equal to its unconditional effect size estimate. As can be seen from the locus zoom plot in Figure 3, the other two genome-wide significant hits are in high LD with the lead SNP, and after conditioning on the lead SNP, the GCTA conditional analysis results show that no other SNPs within chromosome 16 have significant residual association, with the minimum conditional p-value being p~3×10−5. Hence we conclude that there is only one independent signal within the CETP association.
Variance explained
From genome-wide data of the ASCOT-UK datasets, the trait heritability for HDL-C response to statins was estimated as h2 = 17.8% (SE = 0.154) although this was non-significant (p=0.125). There was insufficient power to run the GCTA analysis in the CARDS dataset, due to smaller sample size. The trait variance explained by the lead CETP SNP rs247616 alone was calculated to be 0.04% from ASCOT-UK and 0.01% from CARDS, both non-significant (p=0.38 and p=0.54, respectively).
Discussion
In this study we have performed a meta-analysis of GWAS including over 27,700 statin-treated individuals, investigating genetic variants associated with variation in HDL-C response to statin treatment. We identified three genetic variants in the CETP locus that were highly significantly associated with a larger HDL-C response to statin treatment. No other SNPs met the genome-wide criterion for association of HDL-C change with statin use.
CETP plays an important role in HDL-C metabolism by promoting the exchange of cholesteryl esters in HDL particles with triglycerides in apolipoprotein B-containing particles, leading to increased HDL catabolism and lower HDL-C levels. Increases in HDL-C levels after statin treatment are probably partly the result of a reduction in CETP mediated lipid transfer[25], as was also shown in mice expressing human CETP.[26] Statin treatment decreases CETP activity up to 30%.[27, 28] Previously it has been shown that genetic variants within CETP are associated with differences in CETP concentration.[29] The three SNPs associated with HDL-C response to statins in the present study are located 2.5–7 kb upstream of the CETP gene and are in high linkage disequilibrium (Figure 3).[30] The minor alleles of these SNPs have been shown to be associated with lower CETP mRNA expression levels in liver tissue and with higher HDL-C levels.[30, 31]
Previous studies investigating the association between SNPs in the CETP locus and the HDL-C response to statin treatment have yielded inconsistent results. Several studies showed associations with a greater HDL-C response [8, 10], whereas others showed no significant associations.[12, 32–34] These discrepancies could be explained by limited sample sizes and by the investigation of different genetic variants in these studies. An alternative explanation could be the fact that the effect of statins on HDL-C response is relatively small and depends on statin dose and type.[3, 4] Since the power to detect genetic effects on these small variations is low in single studies, the results from the present large meta-analysis, with replication, provide strong evidence that genetic variation at the CETP locus is associated with HDL-C response.
The results of six randomized clinical trials and ten observational studies were combined in the first stage of the current study. Different statins were investigated in the trials and used within the observational studies, resulting in combining several types of statins in our analysis. This and the variation in statin dosages during follow-up for an individual are a limitation of the current study, since the pharmacogenetic impact might be dependent on specific statin types and dose. To address this possible limitation, the individual study analyses were adjusted for statin equivalent dose based on effect on LDL-C levels, making the different statin types likely more comparable with respect to clinical effectiveness on HDL-C levels. Combining RCTs and observational cohort might also result in heterogeneity between the study types. To reduce the possibility of large heterogeneity we aimed to mimic the design of a RCT in the observational studies, by including only new statin-users. Comparing heterogeneity of the RCTs and observational studies included in the first stage showed no evidence of large heterogeneity (p=0.761, data not shown).
Another possible limitation of the current study is the association of the identified genetic variant with baseline HDL-C concentration. As shown in previous large GWAS studies, the CETP SNP rs3764261 is strongly associated with HDL-C levels.[11, 31] In pharmacogenetic studies investigating lipid responses to drug exposure, it is important to eliminate the effect of the association between baseline lipid levels and the investigated genetic variants.[13] To reduce the impact of these possible confounding effects, our response to treatment analyses were adjusted for baseline HDL-C levels. In addition, interaction analyses in five of the RCTs, with direct modeled comparison with a random assignment to a placebo group, suggested little or no influence of the association between the CETP SNPs and baseline HDL-C levels on the genetic effect on HDL-C response to statin treatment. It is, however, possible that mechanisms underlying the effects of CETP on HDL-C levels are also involved in mediating statin effects on HDL-C.
All genetic data in the current study was imputed to up to 2.5 million autosomal SNPs based on data from the HapMap project.[15] In addition, in our analysis we excluded genetic variants with a minor allele frequency <1%, restricting our analysis to common genetic variants. Imputation based on the more recent 1000 Genomes project could reveal more associations with rare genetic variants.[35] Future studies using exome sequencing data and investigating rare variants may identify additional associations between genetic variants and statin-induced HDL-C response. However, the non-significant estimate of heritability attributable to common variation in our analysis may indicate that the observed increase in HDL-C levels after statin-treatment may be mainly due to environmental rather than genetic effects.
The implications of the present findings regarding genetic effects on the efficacy of statins for reductions in risk of CVD are uncertain. Based on the strong inverse relationship of HDL-C with CVD, the greater statin-induced increase in HDL-C among carriers of the minor vs. major alleles of the three CETP SNPs reported here may confer a greater protective effect of statins on CVD in patients carrying the minor allele. However, a recent study employing Mendelian randomization found that genotypes associated with plasma HDL-C levels were not associated with the impact on CVD risk that would be predicted by the magnitude of the genotypic effects on HDL-C.[7] Moreover, two large clinical trials have failed to show reduction of CVD events by nicotinic acid-induced increases in HDL-C in patients with well-controlled LDL-C levels.[36, 37] Hence, whether greater genetically-mediated HDL-C increases with statin treatment confer increased protection from CVD remains unknown.
In conclusion, this study is the largest meta-analysis of GWAS for HDL-C response to statin treatment conducted to date. The findings suggest that CETP may be the only locus in which common genetic variants are significantly associated with a substantial HDL-C response to statin treatment in individuals of European descent.
Supplementary Material
Acknowledgments
PROSPER/PHASE
The Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) trial was supported by an investigator initiated grant from Bristol-Myers Squibb, USA. The study was conducted, analysed, and reported independently of the company. The GWAS project PHASE has received funding from the European Union’s Seventh Framework Programme (FP7/2007–2013) under grant agreement HEALTH-F2-2009-223004. A part of the genotyping was funded by The Netherlands Consortium for Healthy Ageing (NGI: 05060810). Prof. Dr. J. W. Jukema is an established clinical investigator of The Netherlands Heart Foundation (2001 D 032).
ASCOT
The Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) trial was funded by an investigator initiated grant from Pfizer USA. The study was investigator-led and was conducted, analyzed, and reported independently of the company. The Genomewide Association Scan was funded by the National Institutes for Health Research (NIHR) as part of the portfolio of translational research of the NIHR Biomedical Research Unit at Barts and the NIHR Biomedical Research Centre at Imperial College, the International Centre for Circulatory Health Charity and the Medical Research Council through G952010.
CARDS
The authors thank the other investigators, the staff, and the participants of the CARDS study. A full list of CARDS investigators can be found in original CARDS paper [38]. CARDS was funded by grants to the Universities of London and Manchester by Pfizer, Diabetes UK and the Department of Health.
PARC
This research was supported by the National Institutes of Health: grant U19 HL069757 from the National Heart, Lung, and Blood Institute; and grant UL1TR000124 from the National Center for Advancing Translational Sciences.
TNT
The TNT study was funded by Pfizer, who also provided support for genotyping.
AGES
This study has been funded by NIH contract N01-AG-1-2100, the NIA Intramural Research Program, Hjartavernd (the Icelandic Heart Association), and the Althingi (the Icelandic Parliament). The study is approved by the Icelandic National Bioethics Committee, VSN: 00-063. The researchers are indebted to the participants for their willingness to participate in the study.
ARIC
The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. The authors thank the staff and participants of the ARIC study for their important contributions. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
BioVU
BioVU receives support through the National Center for Research Resources UL1 RR024975, which is now the National Center for Advancing Translational Sciences, 2 UL1 TR000445. Genotyping was supported via grant U01-HG04603 from the National Human Genome Research Institute and RC2-GM092318 from the National Insitute of General Medical Sciences.
CHS
This CHS research was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086; and NHLBI grants U01HL080295, R01HL087652, R01HL105756, R01HL103612, and R01HL120393 with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided through R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR000124, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Framingham HS
The Framingham Heart Study work was supported by the National Heart Lung and Blood Institute of the National Institutes of Health and Boston University School of Medicine (Contract No. N01-HC-25195), its contract with Affymetrix, Inc. for genotyping services (Contract No. N02-HL-6-4278), and based upon analyses by Framingham Heart Study investigators participating in the SNP Health Association Resource (SHARe) project. A portion of this research was conducted using the Linux Cluster for Genetic Analysis (LinGA-II) funded by the Robert Dawson Evans Endowment of the Department of Medicine at Boston University School of Medicine and Boston Medical Center. Also supported by R01HL103612 (PI Psaty, subcontract PI, Vasan)
Health ABC
The Health ABC study was supported by NIA contracts N01AG62101, N01AG62103, and N01AG62106. The genome-wide association study was funded by NIA grant 1R01AG032098-01A1 to Wake Forest University Health Sciences and genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C. This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging.
HVH
This Heart and Vascular Health Study research was supported by NHLBI grants HL085251, HL073410, HL085251, and HL068986.
MESA
The Multi-Ethnic Study of Atherosclerosis (MESA) and MESA SNP Health Association Resource (SHARe) are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support is provided by grants and contracts N01 HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169 and RR-024156. Additional funding was supported in part by the Clinical Translational Science Institute grant UL1RR033176 and is now at the National Center for Advancing Translational Sciences, CTSI grant UL1TR000124. The authors thank the other investigators in the Pharmarcogenetics Working Group, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org.
Rotterdam Study
The Rotterdam Study is supported by the Erasmus Medical Center and Erasmus University Rotterdam; the Netherlands Organization for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly; the Ministry of Education, Culture and Science; the Ministry of Health Welfare and Sports; the European Commission and Municipality of Rotterdam. This work was supported by the Netherlands Genomics Initiative (NGI) Netherlands Organization for Scietific Research (NOW; 050-060-810).
JUPITER
Genetic analysis in JUPITER was supported by a research grant from AstraZeneca to DC and PM.
GoDARTS
We are grateful to all the participants who took part in this study, to the general practitioners, to the Scottish School of Primary Care for their help in recruiting the participants, and to the whole team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses. The Wellcome Trust provides support for Wellcome Trust United Kingdom Type 2 Diabetes Case Control Collection (GoDARTS) and informatics support is provided by the Chief Scientist Office. The Wellcome Trust funds the Scottish Health Informatics Programme, provides core support for the Wellcome Trust Centre for Human Genetics in Oxford and funds the Wellcome Trust Case Control Consortium 2. This research was specifically funded by Diabetes UK (07/0003525) and the Wellcome Trust (084727/Z/08/Z, 085475/Z/08/Z, 085475/B/08/Z). Genotyping of the GoDARTS samples (sample 2) was also funded as part of the EU IMI-SUMMIT programme.
Footnotes
Author contributions
Writing and analysis group: IP, HRW, ST, BJA, DIC, HAD, XL, RAJS, DIC, GKH, PBM, MJC, HMC, LAC, GH, CNAP, BMP, CMS, JWJ, JIR, RMK.
IP and ST performed quality control on the individual study summary results.
IP and ST performed meta-analysis.
IP, HRW, and RAJS performed additional analyses.
All analysis and writing group authors extensively discussed the analysis, results, interpretation and presentation of results.
All authors contributed to the research and reviewed the manuscript.
Study concept and design of contributing studies: (PROSPER) JWJ, DJS, BMB, IF, NS, RGJW; (ASCOT) MJC, PS, NP, AS, DCS, EO; (CARDS) HAD, HMC, PMM, JB, PND, AD, GH; (PARC) XL, YDIC, JIR, RMK; (TNT) JJPK; (AGES) LJL, TBH, VG; (ARIC) CLA, EAW, TS, EB, CMB; (BioVU) QF, WW, CMS, RAW, JCD; (CHS, HVH) NS, KR, TL, BMP; (FHS) LAC, VR; (HABC) SRC, YL; (MESA) XG, SRH, WP, JIR, SSR; (Rotterdam Study) CEK, BHS, AGU, AH, FR; (JUPITER) DIC, BJB, FN, PMR; (GoDARTS) CNAP, HMC.
Phenotype data acquisition of contributing studies: (PROSPER) JWJ, DJS, BMB, IF, AJMC, NS, RGJW; (ASCOT) MJC, PBM, PS, NP, AS, DCS, EO, SSH; (CARDS) HAD, HMC, PMM, JB, PND, AD, GH; (PARC) XL, YDIC, JIR, RMK; (TNT) JJPK; (AGES) GE; (ARIC) CMB; (BioVU) WW, CMS; (CHS, HVH) KLW, JCB, AMA, NLS, BMP; (FHS) LAC, CJO, VR; (GoDARTS) CNAP, HMC; (HABC) SBK; (MESA) SRH, JIR; (Rotterdam Study) CEK, BHS, AH, OHF; (JUPITER) DIC, PMR.
Genotype data acquisition of contributing studies: (PROSPER) ST, JWJ, AJMC, PES; (ASCOT) MJC, HRW, PBM, PS, AS, SSH; (CARDS) HAD, HMC, PMM, PND, AD, GH; (PARC) YDIC, JIR, DAN, JDS; (TNT) BJA, MPD, SMB, GKH, JCT; (AGES) AVS; (ARIC) EB; (BioVU) QF, JCD; (CHS, HVH) JCB; (FHS) CJO; (GoDARTS) CNAP, HMC; (HABC) YL; (MESA) KDT, JIR; (Rotterdam Study) AGU, FR; (JUPITER) DIC, BJB, FN, PMR.
Primary analysis from contributing studies: (PROSPER) IP, ST, AJMC, RAJS, PES; (ASCOT) HRW; (CARDS) HAD, HMC, PMM; (PARC) XL, YDIC, JIR; (TNT) BJA, MPD, SMB, GKH, JCT; (AGES) AVS; (ARIC) CLA, EAW, TS; (BioVU) QF, WW; (CHS, HVH) KLW, KR, TL; (FHS) LAC, FS; (GoDARTS) CNAP, HMC; (HABC) DSE, JMS; (MESA) KDT, XG, JIR; (Rotterdam Study) CEK, BHS; (JUPITER) DIC.
Competing financial interests: BMP serves on the Data and Safety Monitoring Board of a clinical trial funded by the device manufacturer (Zoll LifeCor) and serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. NP and AS received funding from Pfizer for the extended follow-up of the ASCOT UK participants. DIC and PMR received research support for independent genetic analysis in JUPITER from AstraZeneca. FN and BJB have employment, stock and stock options in AstraZeneca, a for-profit company engaged in the discovery, development, manufacture and marketing of proprietary therapeutics such as rosuvastatin, but do not consider that this creates any conflict of interest with the subject-matter of this publication. RMK serves on the Merck Global Atherosclerosis Advisory Board. The remaining authors declare no competing financial interests.
References
- 1.Davidson MH, Toth PP. Comparative effects of lipid-lowering therapies. Prog Cardiovasc Dis. 2004;47(2):73–104. doi: 10.1016/j.pcad.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670–81. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McTaggart F, Jones P. Effects of statins on high-density lipoproteins: a potential contribution to cardiovascular benefit. Cardiovasc Drugs Ther. 2008;22(4):321–38. doi: 10.1007/s10557-008-6113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicholls SJ, Tuzcu EM, Sipahi I, Grasso AW, Schoenhagen P, Hu T, Wolski K, Crowe T, Desai MY, Hazen SL, Kapadia SR, Nissen SE. Statins, high-density lipoprotein cholesterol, and regression of coronary atherosclerosis. JAMA. 2007;297(5):499–508. doi: 10.1001/jama.297.5.499. [DOI] [PubMed] [Google Scholar]
- 5.Di AE, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boekholdt SM, Arsenault BJ, Hovingh GK, Mora S, Pedersen TR, Larosa JC, Welch KM, Amarenco P, Demicco DA, Tonkin AM, Sullivan DR, Kirby A, Colhoun HM, Hitman GA, Betteridge DJ, Durrington PN, Clearfield MB, Downs JR, Gotto AM, Jr, Ridker PM, Kastelein JJ. Levels and changes of HDL cholesterol and apolipoprotein A-I in relation to risk of cardiovascular events among statin-treated patients: a meta-analysis. Circulation. 2013;128(14):1504–12. doi: 10.1161/CIRCULATIONAHA.113.002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de FU, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, M-vdZ AH, Peters BJ, de BA, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, Konig IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schafer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O’Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380(9841):572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anagnostopoulou K, Kolovou G, Kostakou P, Mihas C, Mikhailidis D, Cokkinos DV. Pharmacogenetic study of cholesteryl ester transfer protein gene and simvastatin treatment in hypercholesterolaemic subjects. Expert Opin Pharmacother. 2007;8(15):2459–63. doi: 10.1517/14656566.8.15.2459. [DOI] [PubMed] [Google Scholar]
- 9.Bercovich D, Friedlander Y, Korem S, Houminer A, Hoffman A, Kleinberg L, Shochat C, Leitersdorf E, Meiner V. The association of common SNPs and haplotypes in the CETP and MDR1 genes with lipids response to fluvastatin in familial hypercholesterolemia. Atherosclerosis. 2006;185(1):97–107. doi: 10.1016/j.atherosclerosis.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 10.Leusink M, Onland-Moret NC, Asselbergs FW, Ding B, Kotti S, van Zuydam NR, Papp AC, Danchin N, Donnelly L, Morris AD, Chasman DI, Doevendans PA, Klungel OH, Ridker PM, van Gilst WH, Simon T, Nyberg F, Palmer CN, Sadee W, van der Harst P, de Bakker PI, de BA, Verstuyft C, M-vdZ AH. Cholesteryl ester transfer protein polymorphisms, statin use, and their impact on cholesterol levels and cardiovascular events. Clin Pharmacol Ther. 2014;95(3):314–20. doi: 10.1038/clpt.2013.194. [DOI] [PubMed] [Google Scholar]
- 11.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Do R, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkila K, Hypponen E, Isaacs A, Jackson AU, Johansson A, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikainen LP, Magnusson PK, Mangino M, Mihailov E, Montasser ME, Muller-Nurasyid M, Nolte IM, O’Connell JR, Palmer CD, Perola M, Petersen AK, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney AS, Doring A, Elliott P, Epstein SE, Eyjolfsson GI, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJ, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimaki T, Lin SY, Lindstrom J, Loos RJ, Mach F, McArdle WL, Meisinger C, Mitchell BD, Muller G, Nagaraja R, Narisu N, Nieminen TV, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stancakova A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YD, Collins FS, Cooper RS, Danesh J, Dedoussis G, de FU, Feranil AB, Ferrieres J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Jarvelin MR, Jula A, Kahonen M, Kaprio J, Kesaniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, Marz W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njolstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PE, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BH, Ordovas JM, Boerwinkle E, Palmer CN, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–83. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boekholdt SM, Sacks FM, Jukema JW, Shepherd J, Freeman DJ, McMahon AD, Cambien F, Nicaud V, de Grooth GJ, Talmud PJ, Humphries SE, Miller GJ, Eiriksdottir G, Gudnason V, Kauma H, Kakko S, Savolainen MJ, Arca M, Montali A, Liu S, Lanz HJ, Zwinderman AH, Kuivenhoven JA, Kastelein JJ. Cholesteryl ester transfer protein TaqIB variant, high-density lipoprotein cholesterol levels, cardiovascular risk, and efficacy of pravastatin treatment: individual patient meta-analysis of 13,677 subjects. Circulation. 2005;111(3):278–87. doi: 10.1161/01.CIR.0000153341.46271.40. [DOI] [PubMed] [Google Scholar]
- 13.Postmus I, Trompet S, Deshmukh HA, Barnes MR, Li X, Warren HR, Chasman DI, Zhou K, Arsenault BJ, Donnelly LA, Wiggins KL, Avery CL, Griffin P, Feng Q, Taylor KD, Li G, Evans DS, Smith AV, de Keyser CE, Johnson AD, de Craen AJ, Stott DJ, Buckley BM, Ford I, Westendorp RG, Slagboom PE, Sattar N, Munroe PB, Sever P, Poulter N, Stanton A, Shields DC, O’Brien E, Shaw-Hawkins S, Chen YD, Nickerson DA, Smith JD, Dube MP, Boekholdt SM, Hovingh GK, Kastelein JJ, McKeigue PM, Betteridge J, Neil A, Durrington PN, Doney A, Carr F, Morris A, McCarthy MI, Groop L, Ahlqvist E, Bis JC, Rice K, Smith NL, Lumley T, Whitsel EA, Sturmer T, Boerwinkle E, Ngwa JS, O’Donnell CJ, Vasan RS, Wei WQ, Wilke RA, Liu CT, Sun F, Guo X, Heckbert SR, Post W, Sotoodehnia N, Arnold AM, Stafford JM, Ding J, Herrington DM, Kritchevsky SB, Eiriksdottir G, Launer LJ, Harris TB, Chu AY, Giulianini F, MacFadyen JG, Barratt BJ, Nyberg F, Stricker BH, Uitterlinden AG, Hofman A, Rivadeneira F, Emilsson V, Franco OH, Ridker PM, Gudnason V, Liu Y, Denny JC, Ballantyne CM, Rotter JI, Adrienne CL, Psaty BM, Palmer CN, Tardif JC, Colhoun HM, Hitman G, Krauss RM, Wouter JJ, Caulfield MJ. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat Commun. 2014;5:5068. doi: 10.1038/ncomms6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donnelly LA, van Zuydam NR, Zhou K, Tavendale R, Carr F, M-vdZ AH, Leusink M, de BA, Doevendans PA, Asselbergs FW, Morris AD, Pearson ER, Klungel OH, Doney AS, Palmer CN. Robust association of the LPA locus with low-density lipoprotein cholesterol lowering response to statin treatment in a meta-analysis of 30 467 individuals from both randomized control trials and observational studies and association with coronary artery disease outcome during statin treatment. Pharmacogenet Genomics. 2013;23(10):518–25. doi: 10.1097/FPC.0b013e3283642fd6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The International HapMap Project. Nature. 2003;426(6968):789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39(7):906–13. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 18.Servin B, Stephens M. Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS Genet. 2007;3(7):e114. doi: 10.1371/journal.pgen.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–91. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chasman DI, Giulianini F, Macfadyen J, Barratt BJ, Nyberg F, Ridker PM. Genetic Determinants of Statin Induced LDL-C Reduction: The JUPITER Trial. Circ Cardiovasc Genet. 2012;5:257–64. doi: 10.1161/CIRCGENETICS.111.961144. [DOI] [PubMed] [Google Scholar]
- 21.Dastani Z, Hivert MF, Timpson N, Perry JR, Yuan X, Scott RA, Henneman P, Heid IM, Kizer JR, Lyytikainen LP, Fuchsberger C, Tanaka T, Morris AP, Small K, Isaacs A, Beekman M, Coassin S, Lohman K, Qi L, Kanoni S, Pankow JS, Uh HW, Wu Y, Bidulescu A, Rasmussen-Torvik LJ, Greenwood CM, Ladouceur M, Grimsby J, Manning AK, Liu CT, Kooner J, Mooser VE, Vollenweider P, Kapur KA, Chambers J, Wareham NJ, Langenberg C, Frants R, Willems-Vandijk K, Oostra BA, Willems SM, Lamina C, Winkler TW, Psaty BM, Tracy RP, Brody J, Chen I, Viikari J, Kahonen M, Pramstaller PP, Evans DM, St PB, Sattar N, Wood AR, Bandinelli S, Carlson OD, Egan JM, Bohringer S, van HD, Kedenko L, Kristiansson K, Nuotio ML, Loo BM, Harris T, Garcia M, Kanaya A, Haun M, Klopp N, Wichmann HE, Deloukas P, Katsareli E, Couper DJ, Duncan BB, Kloppenburg M, Adair LS, Borja JB, Wilson JG, Musani S, Guo X, Johnson T, Semple R, Teslovich TM, Allison MA, Redline S, Buxbaum SG, Mohlke KL, Meulenbelt I, Ballantyne CM, Dedoussis GV, Hu FB, Liu Y, Paulweber B, Spector TD, Slagboom PE, Ferrucci L, Jula A, Perola M, Raitakari O, Florez JC, Salomaa V, Eriksson JG, Frayling TM, Hicks AA, Lehtimaki T, Smith GD, Siscovick DS, Kronenberg F, van DC, Loos RJ, Waterworth DM, Meigs JB, Dupuis J, Richards JB, Voight BF, Scott LJ, Steinthorsdottir V, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, Amin N, Wu G, Willer CJ, Raychaudhuri S, McCarroll SA, Hofmann OM, Segre AV, van HM, Navarro P, Ardlie K, Balkau B, Benediktsson R, Bennett AJ, Blagieva R, Boerwinkle E, Bonnycastle LL, Bostrom KB, Bravenboer B, Bumpstead S, Burtt NP, Charpentier G, Chines PS, Cornelis M, Crawford G, Doney AS, Elliott KS, Elliott AL, Erdos MR, Fox CS, Franklin CS, Ganser M, Gieger C, Grarup N, Green T, Griffin S, Groves CJ, Guiducci C, Hadjadj S, Hassanali N, Herder C, Isomaa B, Jackson AU, Johnson PR, Jorgensen T, Kao WH, Kong A, Kraft P, Kuusisto J, Lauritzen T, Li M, Lieverse A, Lindgren CM, Lyssenko V, Marre M, Meitinger T, Midthjell K, Morken MA, Narisu N, Nilsson P, Owen KR, Payne F, Petersen AK, Platou C, Proenca C, Prokopenko I, Rathmann W, Rayner NW, Robertson NR, Rocheleau G, Roden M, Sampson MJ, Saxena R, Shields BM, Shrader P, Sigurdsson G, Sparso T, Strassburger K, Stringham HM, Sun Q, Swift AJ, Thorand B, Tichet J, Tuomi T, van Dam RM, van Haeften TW, van HT, van Vliet-Ostaptchouk JV, Walters GB, Weedon MN, Wijmenga C, Witteman J, Bergman RN, Cauchi S, Collins FS, Gloyn AL, Gyllensten U, Hansen T, Hide WA, Hitman GA, Hofman A, Hunter DJ, Hveem K, Laakso M, Morris AD, Palmer CN, Rudan I, Sijbrands E, Stein LD, Tuomilehto J, Uitterlinden A, Walker M, Watanabe RM, Abecasis GR, Boehm BO, Campbell H, Daly MJ, Hattersley AT, Pedersen O, Barroso I, Groop L, Sladek R, Thorsteinsdottir U, Wilson JF, Illig T, Froguel P, van Duijn CM, Stefansson K, Altshuler D, Boehnke M, McCarthy MI. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012;8(3):e1002607. doi: 10.1371/journal.pgen.1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson T. (Technical report, The Comprehensive R Archive Network).Efficient calculation for multi-SNP genetic risk scores. 2013 http://cran.r-project.org/web/packages/gtx/vignettes/ashg2012.pdf. Last accessed 28 September 2015., 2015.
- 23.R Core Team: A language and environment for statistical computing. R Foundationfor Statistical Computing. Vienna, Austria: 2015. https://www.R-project.org., 2015. [Google Scholar]
- 24.Yang J, Ferreira T, Morris AP, Medland SE, Madden PA, Heath AC, Martin NG, Montgomery GW, Weedon MN, Loos RJ, Frayling TM, McCarthy MI, Hirschhorn JN, Goddard ME, Visscher PM. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44(4):369–3. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rensen PC, Havekes LM. Cholesteryl ester transfer protein inhibition: effect on reverse cholesterol transport? Arterioscler Thromb Vasc Biol. 2006;26(4):681–84. doi: 10.1161/01.ATV.0000214979.24518.95. [DOI] [PubMed] [Google Scholar]
- 26.de Haan W, van der Hoogt CC, Westerterp M, Hoekstra M, Dallinga-Thie GM, Princen HM, Romijn JA, Jukema JW, Havekes LM, Rensen PC. Atorvastatin increases HDL cholesterol by reducing CETP expression in cholesterol-fed APOE*3-Leiden. CETP mice. Atherosclerosis. 2008;197(1):57–63. doi: 10.1016/j.atherosclerosis.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Ahnadi CE, Berthezene F, Ponsin G. Simvastatin-induced decrease in the transfer of cholesterol esters from high density lipoproteins to very low and low density lipoproteins in normolipidemic subjects. Atherosclerosis. 1993;99(2):219–28. doi: 10.1016/0021-9150(93)90024-o. [DOI] [PubMed] [Google Scholar]
- 28.Guerin M, Egger P, Soudant C, Le GW, van TA, Dupuis R, Chapman MJ. Dose-dependent action of atorvastatin in type IIB hyperlipidemia: preferential and progressive reduction of atherogenic apoB-containing lipoprotein subclasses (VLDL-2, IDL, small dense LDL) and stimulation of cellular cholesterol efflux. Atherosclerosis. 2002;163(2):287–96. doi: 10.1016/s0021-9150(02)00037-0. [DOI] [PubMed] [Google Scholar]
- 29.Kuivenhoven JA, Jukema JW, Zwinderman AH, de KP, McPherson R, Bruschke AV, Lie KI, Kastelein JJ. The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. The Regression Growth Evaluation Statin Study Group. N Engl J Med. 1998;338(2):86–93. doi: 10.1056/NEJM199801083380203. [DOI] [PubMed] [Google Scholar]
- 30.Papp AC, Pinsonneault JK, Wang D, Newman LC, Gong Y, Johnson JA, Pepine CJ, Kumari M, Hingorani AD, Talmud PJ, Shah S, Humphries SE, Sadee W. Cholesteryl Ester Transfer Protein (CETP) polymorphisms affect mRNA splicing, HDL levels, and sex-dependent cardiovascular risk. PLoS One. 2012;7(3):e31930. doi: 10.1371/journal.pone.0031930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin CY, Jin GM, Jin KY, Lee JY, Park T, Kim K, Sim X, Twee-Hee OR, Croteau-Chonka DC, Lange LA, Smith JD, Song K, Hua ZJ, Yuan X, Luan J, Lamina C, Ziegler A, Zhang W, Zee RY, Wright AF, Witteman JC, Wilson JF, Willemsen G, Wichmann HE, Whitfield JB, Waterworth DM, Wareham NJ, Waeber G, Vollenweider P, Voight BF, Vitart V, Uitterlinden AG, Uda M, Tuomilehto J, Thompson JR, Tanaka T, Surakka I, Stringham HM, Spector TD, Soranzo N, Smit JH, Sinisalo J, Silander K, Sijbrands EJ, Scuteri A, Scott J, Schlessinger D, Sanna S, Salomaa V, Saharinen J, Sabatti C, Ruokonen A, Rudan I, Rose LM, Roberts R, Rieder M, Psaty BM, Pramstaller PP, Pichler I, Perola M, Penninx BW, Pedersen NL, Pattaro C, Parker AN, Pare G, Oostra BA, O’Donnell CJ, Nieminen MS, Nickerson DA, Montgomery GW, Meitinger T, McPherson R, McCarthy MI, McArdle W, Masson D, Martin NG, Marroni F, Mangino M, Magnusson PK, Lucas G, Luben R, Loos RJ, Lokki ML, Lettre G, Langenberg C, Launer LJ, Lakatta EG, Laaksonen R, Kyvik KO, Kronenberg F, Konig IR, Khaw KT, Kaprio J, Kaplan LM, Johansson A, Jarvelin MR, Janssens AC, Ingelsson E, Igl W, Kees HG, Hottenga JJ, Hofman A, Hicks AA, Hengstenberg C, Heid IM, Hayward C, Havulinna AS, Hastie ND, Harris TB, Haritunians T, Hall AS, Gyllensten U, Guiducci C, Groop LC, Gonzalez E, Gieger C, Freimer NB, Ferrucci L, Erdmann J, Elliott P, Ejebe KG, Doring A, Dominiczak AF, Demissie S, Deloukas P, de Geus EJ, de FU, Crawford G, Collins FS, Chen YD, Caulfield MJ, Campbell H, Burtt NP, Bonnycastle LL, Boomsma DI, Boekholdt SM, Bergman RN, Barroso I, Bandinelli S, Ballantyne CM, Assimes TL, Quertermous T, Altshuler D, Seielstad M, Wong TY, Tai ES, Feranil AB, Kuzawa CW, Adair LS, Taylor HA, Jr, Borecki IB, Gabriel SB, Wilson JG, Holm H, Thorsteinsdottir U, Gudnason V, Krauss RM, Mohlke KL, Ordovas JM, Munroe PB, Kooner JS, Tall AR, Hegele RA, Kastelein JJ, Schadt EE, Rotter JI, Boerwinkle E, Strachan DP, Mooser V, Stefansson K, Reilly MP, Samani NJ, Schunkert H, Cupples LA, Sandhu MS, Ridker PM, Rader DJ, van Duijn CM, Peltonen L, Abecasis GR, Boehnke M, Kathiresan S. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barber MJ, Mangravite LM, Hyde CL, Chasman DI, Smith JD, McCarty CA, Li X, Wilke RA, Rieder MJ, Williams PT, Ridker PM, Chatterjee A, Rotter JI, Nickerson DA, Stephens M, Krauss RM. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS One. 2010;5(3):e9763. doi: 10.1371/journal.pone.0009763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Zhang L, Xie NZ, Deng B, Lv LX, Zheng LQ. Relationship between the cholesterol ester transfer protein TaqIB polymorphism and the lipid-lowering effect of atorvastatin in patients with coronary atherosclerotic heart disease. Genet Mol Res. 2014;13(1):2140–48. doi: 10.4238/2014.March.24.21. [DOI] [PubMed] [Google Scholar]
- 34.Singer JB, Holdaas H, Jardine AG, Fellstrom B, Os I, Bermann G, Meyer JM. Genetic analysis of fluvastatin response and dyslipidemia in renal transplant recipients. J Lipid Res. 2007;48(9):2072–78. doi: 10.1194/jlr.M700076-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–67. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 37.Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, Armitage J. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203–12. doi: 10.1056/NEJMoa1300955. [DOI] [PubMed] [Google Scholar]
- 38.Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, Thomason MJ, Mackness MI, Charlton-Menys V, Fuller JH. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364(9435):685–96. doi: 10.1016/S0140-6736(04)16895-5. [DOI] [PubMed] [Google Scholar]
- 39.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–37. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.