

Abstract
Post-translational modifications (PTMs) affect protein function, localization, and stability, yet very little is known about the ratios of these modifications. Here, we describe a novel method to quantitate and assess the relative stoichiometry of Lys and Arg modifications (QuARKMod) in complex biological settings. We demonstrate the versatility of this platform in monitoring recombinant protein modification of peptide substrates, PTMs of individual histones, and the relative abundance of these PTMs as a function of subcellular location. Lastly, we describe a product ion scanning technique that offers the potential to discover unexpected and possibly novel Lys and Arg modifications. In summary, this approach yields accurate quantitation and discovery of protein PTMs in complex biological systems without the requirement of high mass accuracy instrumentation.
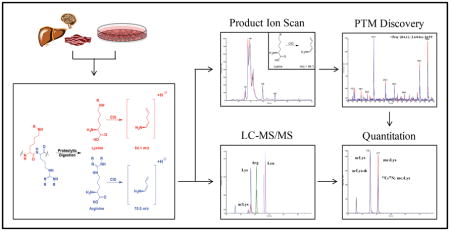
Graphical Abstract
INTRODUCTION
Post-translational modifications (PTMs) play an integral role in the folding, stability, activity, and localization of a protein.1,2 To date, nearly 90,000 PTMs have been validated with another ~230,000 predicted.2 PTMs are of particular importance in epigenetic research; histone PTMs regulate protein transcription through chromatin remodeling and/or the recruitment of transcription factors.3 The number of known PTM sites and modifications is extensive, including acetylation (Lys), methylation (Lys/Arg), phosphorylation (Ser/Thr), and ubiquitination (Lys) and is collectively referred to as the “Histone Code”. 4–7 As a result, histone PTMs are tightly regulated through the action of “writers” (e.g. histone acetyltranseferases (HATs)) and “erasers” (e.g. histone deacetylases (HDACs)). These proteins have been shown to be of critical importance in maintaining appropriate gene expression.8,9 A disruption in the homeostatic on/off rates of histone PTMs often leads to aberrant gene expression which has been implicated in nearly every disease state.10
Despite decades of research, the number of known, and critically important, PTMs is still expanding.11–14 Whereas much is known about the broad role for many PTMs, very little is known about the stoichiometry and relative abundance of these modifications. For example, it has been estimated that >20% of all mitochondrial proteins contain acetylated Lys (acLys); however, these estimations were based on immunoprecipitation procedures which do not provide any information on the relative abundance of other modifications.15–17 Current technologies are largely limited to either qualitative assessment of an individual PTM (Western blotting) or specificity of modifications (proteomics). Although quantitative measurements can be achieved, this often requires the use of either synthetic peptides, chemical labeling/derivatization, stable isotope labeling of amino acids in cell culture (SILAC), and/or immunoprecipitation procedures.18,19 Top-down MS has also been utilized to quantify relative differences between samples; however, this requires the use of purified proteins from biological samples (e.g. histones).20 Lastly, these approaches are often unable to quantitatively compare the levels of various PTMs in a given sample. It was our goal to bridge this gap in our understanding of protein PTMs by developing a label-free application for the quantitative analysis and discovery of Lys and Arg modifications (QuARKMod) from complex biological samples (Figure 1). Using exhaustive proteolytic digestion and a novel LC-MS/MS platform, we show broad applications for this methodology ranging from the determination of recombinant protein activity to the identification and characterization of unknown Lys and Arg PTMs.
Figure 1. A generalized scheme of the QuARKMod platform.

QuARKMod has been validated for use with a broad range of cell and tissue samples. Following protein digestion, the absolute concentrations of PTMs can be determined in a given sample. In addition, using data-dependent scanning, we outline a method for the discovery of novel or low-abundance Lys and Arg PTMs in the same biological samples.
EXPERIMENTAL SECTION
Materials and reagents
Information for materials and reagents and the synthesis of stable-isotope internal standards is included in Supporting Information.
Cell culture
HEK293 cells (ATCC) were cultured in low glucose Dulbecco’s Modified Eagle Medium supplemented with 10% FBS. Cells were incubated at 37 °C under 5% CO2. Cells (10 × 106) were treated for 24 h with either TSA (1 μM) or IOX1 (100 μM). Following treatments, cells were washed once and scraped into ice-cold PBS containing 5 mM sodium butyrate to prevent exogenous histone deacetylation. Cells were recovered via centrifugation at 1000 × g and frozen at −20 °C until further analysis. RAW264.7 cells (ATCC) were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% FBS. Cells (5 × 106) were treated with 100 ng/mL KLA (Avanti Polar Lipids, Alabaster, AL) for 24 h in serum-free medium. Following treatments, cells were harvested as described above.
SDS-PAGE and immunoblotting
Samples were denatured in SDS loading buffer and heated at 95 °C for 5 min. Proteins were resolved via SDS-PAGE and transferred to nitrocellulose membranes (BioRad, Hercules, CA). Membranes were blocked with Odyssey Blotting Buffer (Li-Cor Biosciences, Superior, NE) for 45 min at room temperature and then incubated with primary antibodies overnight at 4 °C as follows: H3 (Cell Signaling Technologies, Danvers, MA, 1:5000); H2B (Cell Signaling Technologies, 1:2000); H2A (Abcam, Cambridge, MA, 1:2000); H4 (Abcam, 1:1000); pan-acetyl lysine (Abcam 1:2500); pan-trimethyl lysine (PTM biolabs, Chicoago, IL, 1:5000); pan-butyryl lysine (PTM biolabs, 1:1000). Following three washes with Tris-buffered saline + 0.1% Tween-20 (TBST), infrared secondary antibodies (Li-Cor) were added in blocking buffer (1:5000) for 45 min. Blots were developed following 3 additional washes with TBST using the Odyssey Infrared Imaging System (Li-Cor).
Chromatin extraction
Methods were adapted from Torrente et al. and Shechter et al21,22. Briefly, cell pellets were suspended in a hypotonic lysis buffer containing 10 mM HEPES/KOH (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 5 mM sodium butyrate, and 0.5% IGEPAL supplemented with protease and phosphatase inhibitor cocktails. The suspension was placed on ice for 30 min to allow hypotonic swelling, and nuclei were collected via centrifugation (1500 × g for 20 min at 4 °C)23. Supernatants were discarded, and chromatin was extracted from the pellet using a buffer consisting of 20 mM HEPES/KOH (pH 7.9), 25% glycerol, 420 mM KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 5 mM butyrate, and inhibitor cocktails. Pellets were sonicated on ice for 30 s and rotated end-over-end at 4 °C for 16 h. Chromatin was pelleted (1500 × g for 30 min at 4 °C), and resuspended in 20 mM HEPES/KOH (pH 7.9), 1.5 mM MgCl2, 0.2 mM EDTA, 5 mM butyrate, and inhibitor cocktails. Pellets were sonicated via 10 × 1 second pulses. Purity was evaluated via SDS-PAGE, Coomassie blue staining, and immunoblotting.
HPLC isolation of histones
To isolate purified histones, 200 μg of chromatin was injected onto a Phenomenex Jupiter C18 column (250 × 10.0 mm, 5 μm, 300 Å) in line with a Waters 2695 Separations module HPLC interfaced to a Waters 2487 dual detector at a flow rate of 4.0 mL/min. Buffer A was ddH2O with 0.1% trifluoroacetic acid. Buffer B was acetonitrile (ACN) with 0.1% trifluoroacetic acid. A typical gradient was as follows: 0 min (4% B) → 1 min (35% B) → 45 min (60% B) → 50 min (100% B) → 58 min (100% B) → 60 min (4% B). The column was washed with a solution containing ACN:isopropanaol:MeOH (1:1:1) for 10 min between each run and then re-equilibrated to 4% B for 10 min prior to the next injection. Absorbance was monitored at 225 and 278 nm and histones were collected at the following elution times: H2B (17.7 min), H4 (19.9 min), H2A (22.6 min), H3.2/H3.3 (29.1 min), and H3.1 (33.6 min). Purified histones were dried to completion in vaccuo and resuspended in 100 μL of 50 mM NH4HCO3.
Exhaustive proteolytic digestion of proteins
Isolated proteins (100 μg) were precipitated with ice-cold MeOH for 1 h at −20 °C. Proteins were then pelleted via centrifugation at 15,000 × g for 10 min at 4 °C. MeOH was removed, and pellets were dried to completion under N2 gas. Protein was resuspended in 153 μL of 50 mM NH4HCO3. Samples were then spiked with 10 μL of a master mix containing internal standards of interest. Due to the varying quantities of PTMs in different cellular compartments, the quantity of internal standards was sample-dependent. Proteins were digested via the addition of 10 μL of trypsin (1:100 w/w) for 16 h with end-over-end rotation at 37 °C. The use of sequencing grade trypsin is required, as N-Pep will not recognize the N-terminal acetylation present. Thus, this greatly reduces the background enzymatic digestion. Trypsin was denatured via heating at 95 °C for 10 min, and samples were cooled to room temperature. N-Pep (25 μL; 2 U) was added to each sample, yielding a final sample volume of 198 μL, and samples were again incubated for 16 h with end-over-end rotation at 37 °C. N-Pep was denatured via heating at 95 °C for 10 min, and samples again were cooled to room temperature. The ion-pairing agent, HFBA (2 μL) was added to each sample to yield a final concentration of 50 mM. Undigested proteins and any particulates were removed by centrifugation at 15,000 × g for 5 min at room temperature, and amino acids were further analyzed via LC-MS/MS.
Analysis of Lys and Arg PTMs via LC-MS/MS
The quantitation of Lys and Arg PTMs was achieved using a Shimadzu Nexera LC system in line with a Sciex 6500 QTrap MS equipped with a Phenomenex Luna C8 column (2.1 × 50 mm, 3.5 μm) (Agilent Technologies, Santa Clara, CA) or equivalent. The solvent system consisted of H2O with 10 mM HFBA (solvent A) and ACN with 10 mM HFBA (solvent B) and a flow rate of 0.325 mL/min. Analytes were chromatographically separated using a gradient elution profile which typically was as follows: 0.5 min (2.5% B) → 5.5 min (50% B) → 6.0 min (80% B) → 9.0 min (80% B) → 9.5 min (50% B). The column was held at initial conditions (2.5% B) for 3 min prior to each injection and the autosampler injection needle was washed between each injection with MeOH containing 25 mM NH4HCO3.
The 6500 QTrap was configured for IonSpray operation and the analytes were detected using multiple reaction monitoring (MRM) in the positive ion mode. Table 1 gives the transitions of all analytes reported here as well as their collision energies (CE).
Table 1.
MRM transitions and CE utilized for all analytes in these studies.
| Species | Q1 (m/z) | Q3 (m/z) | CE (v) |
|---|---|---|---|
| Lys | 147.1 | 84.1 | 22 |
| 13C615N2 Lys | 155.1 | 90.1 | 22 |
| Arg | 175.1 | 70.1 | 26 |
| 13C615N4 Arg | 185.1 | 75.1 | 26 |
| acLys/me3Lys | 189.2 | 84.1 | 29 |
| acLys-d8 | 197.2 | 91.1 | 29 |
| 13C615N2 me3Lys | 197.2 | 90.1 | 29 |
| Leu | 132.1 | 86.1 | 13 |
| 13C615N Leu | 139.1 | 93.1 | 13 |
| meLys | 161.1 | 84.1 | 26 |
| me2Lys | 175.1 | 84.1 | 26 |
| S/ADMA | 203.1 | 70.1 | 29 |
| ADMA-d7 | 210.1 | 77.1 | 29 |
Values of additional QTrap parameters are as follows: curtain gas (CUR) = 25; collision gas (CAD) = High; IonSpray voltage (IS) = 5500; temperature (TEM) = 350; ion source gas 1 (GS1) = 35; ion source gas 2 (GS2) = 40, declustering potential (DP) = 45; entrance potential (EP) = 7; collision cell exit potential (CXP) = 9. These values were determined empirically by infusing selected standards into the 6500 QTrap and adjusting the values to obtain the maximal response. The resolution of Q1 and Q3 was ‘unit’ and there was a 5 ms delay between mass ranges. The dwell time for each analyte was between 10 and 50 ms, and was adjusted based on the anticipated abundance of the analyte. The total cycle time was kept to ~0.3 s, which typically gave at least 14 points across all chromatographic peaks.
Analytes were quantified using their respective stable isotope standards. All methylated lysine species (meLys, me2Lys, me3Lys) are quantified against 13C615N2 me3Lys. Lys and Arg levels are measured to confirm consistent ratios between samples, indicating minimal changes to amino acid composition. The linearity, dynamic range, and limits of detection for this LC-MS/MS system were assessed by injecting a series of standard mixtures (13C6, 15N2 Lys, meLys, me2Lys, me3Lys, Arg and ADMA) and plotting the amount of analyte versus the chromatographic peak area of that analyte. The points were fit to a linear curve and an r2 > 0.99 was observed for all analytes. This indicates the instrument response to be linear over a 1250-fold range of concentrations. The upper limit of the instrument’s capacity was judged to be ~50,000 fmol on-column, while the lower limit was ~40 fmol on-column. This lower limit yielded a signal:noise ratio of ~50:1, which suggests a limit of detection of ~5–10 fmol on-column. However, these values can vary significantly between instruments and can also be affected by adjustments to the acquisition method.
For IDA of Lys PTMs, the QTrap survey scan was a Precursor Ion scan with a precursor of 84 (Da). Several masses were excluded from Enhanced Product Ion (EPI) analysis as they were determined to be background masses specific to the system. These were 278, 296, 314, 337, 338, 319 and 320 m/z.
Analysis of recombinant demethylase activity
Recombinant JMJD2A protein (Cayman Chemical) reactions were carried out in enzyme assay buffer consisting of 50 mM HEPES (pH 7.5), 50 mM NaCl, 1 mM ascorbic acid, 50 mM ferrous ammonium sulfate, and 1 mM α-ketoglutarate with 4.72 μM JMJD2A and 100 μM H3K9me3 peptide (with sequence ARTKQTARKme3STGGKA, Cayman Chemical). Recombinant KDM3A, KDM5B, and KDM4B protein (Active Motif, Carlsbad, CA) reactions were carried out in enzyme assay buffer consisting of 50 mM HEPES (pH 7.5), 0.02% (v/v) Triton X-100, 100 μM ascorbic acid, 50 μM ferrous ammonium sulfate, 100 μM α-ketoglutarate, and 1 mM TCEP. Recombinant KDM3A (50 nM) was incubated with 5 μM H3K9me peptide (with sequence ARTKQTARKmeSTGGKAPRKQLA, AnaSpec). Recombinant KDM5B (100 nM) was incubated with 5 μM H3K4me3 peptide (ARTKme3QTARKSTGGKAPRKQLA, AnaSpec). Recombinant KDM4B (25 nM) was incubated with 5 μM H3K9me3 peptide. Reactions with JMJD2A were conducted at 37 °C for 4 h, whereas reactions with KDM3A, KDM5B, and KDM4B were conducted at room temperature for 2 h. All reactions were stopped by heat-denaturation of the enzymes at 95 °C for 10 min. Isotopically labeled standards were added to each sample prior to digestion, which was performed as described above, and samples were analyzed for Leu, Lys, Arg, and respective PTMs.
Statistical analysis
Statistical analysis and generation of graphs were performed using GraphPad Prism 4.02 (GraphPad Software, San Diego, CA). Differences between control and treated samples (unpaired Student’s t-test) or three or more groups (one-way ANOVA) were considered significant if P < 0.05 (*), P < 0.01 (**), P < 0.001 (***).
RESULTS AND DISCUSSION
Lys and Arg display consistent CID fragmentations regardless of their PTM status
In the development of this method, we sought to devise a strategy to not only quantitate, but also discover potentially novel, low abundance Lys and Arg PTMs from complex biological samples. To accomplish this goal, we first assessed the fragmentation of unmodified Lys (146.2 Da) via collisionally induced dissociation (CID) MS. The parent ion with an m/z of 147.1 produced major fragments at 130.1 and 84.0 m/z (Figure S1a in the Supporting Information). The CID spectrum of acLys (189.1 m/z) similarly displays fragments at 130.1 and 84.0 m/z (Figure S1a in the Supporting Information). This suggests that Lys and modified Lys residues lose the α-carboxylate moiety as well as the ε-amino group, generating the proposed fragment in Figure S1b which, when complexed with a proton, has an m/z of 84.0. Of the twelve ε-modified Lys authentic standards examined in this manner, all have displayed a major fragment at 84.0 m/z. This includes mono-, di- and trimethylLys (meLys, me2Lys, and me3Lys, respectively) and carboxymethylLys (CML) (CID spectra of all discussed species are shown in Figure S1a in the Supporting Information). Much akin to Lys, the CID spectrum of protonated Arg (175.1 m/z) displayed a fragment of 70.0 m/z. This fragment is also seen in the CID spectra of asymmetric and symmetric dimethyl arginine (ADMA and SDMA, respectively) (Figure S2a in the Supporting Information). This potentially corresponds to the loss of the α-carboxylate and the guanidinium moiety (Figure S2b in the Supporting Information).
This mass spectral data provides fragmentation information that may be employed in the selected reaction monitoring of Lys, Arg, and their PTMs. The transitions used for QuARKMod analysis are given in the Experimental Section. Additionally, these data suggest that information-dependent acquisition (IDA) methods may be developed to interrogate samples for unknown or unexpected Lys and Arg PTMs. Specifically, the survey scan of an IDA method may be a product ion scan where the product is 84.0 m/z for Lys PTMs or 70.0 m/z for Arg PTMs. Thus, this MS technique allows for the identification and quantitation of Lys and Arg PTMs by monitoring only one product ion in each case, independent of the parent mass (Figure 1).
Chromatographic separation of amino acids
Many Lys and Arg PTMs have nearly identical masses and, therefore, cannot be distinguished by MS without high-resolution instrumentation. This is especially evident with acLys and me3Lys, which under the MS conditions utilized both display a parent ion of 189.1 m/z, yet have distinctly different physiological roles.3,10,24 We therefore sought to develop an LC system for QuARKMod, which would allow these analytes to be chromatographically separated. Individual amino acid residues are quite hydrophilic and not normally amenable to reverse-phase chromatography; however, the inclusion of the ion-pairing agent heptafluorobutyric acid (HFBA) allows these analytes to be retained and separated on a C8 column. Figure 1 shows a representative chromatogram of a digested protein sample. Here, Lys, acLys, Arg, Leu, and Ile are well-separated. In addition, Figure S3 in the Supporting Information illustrates the chromatographic behavior of isobaric pairs of analytes: (S3a) acLys and me3Lys and (S3b) Arg and me2Lys. In both cases, chromatographic separation is achieved, which is crucial in the case of acLys and me3Lys, as they are detected via the same SRM transition. Analyte quantitation can then be achieved via stable isotope dilution MS, whereby stably labeled compound analogs serve as internal standards and are incorporated into each sample prior to the initiation of the digestion procedure. These procedures provide the foundation for the analysis and quantitation of Arg and Lys modifications.
Hydrolysis of proteins to single amino acids
To apply this LC-MS/MS platform to biological samples requires efficient protein hydrolysis while conserving intact PTMs. We collected Lys- and Arg-rich chromatin fractions isolated from HEK293 cells and hydrolyzed the proteins to single amino acids through exhaustive proteolytic digestion using trypsin and aminopeptidase M (N-Pep) (Figure S4a in the Supporting Information). Whereas trypsin is a relatively high fidelity protease (C-terminal of unmodified Lys and Arg), N-Pep non-specifically hydrolyzes amino acids from peptides lacking N-terminal modifications. Due to the presence of post-translational N-terminal acetylations on many proteins, most notably histones, it is necessary to treat proteins first with trypsin. These methods resulted in near complete digestion of isolated chromatin as visualized by Coomassie blue staining and western blotting, while intact proteolytic enzymes can still be observed (Figure S4a in the Supporting Information). In addition, we also quantified Lys, Arg, Leu, acLys, and me3Lys using stable isotope dilution mass spectrometry to assess the digestion conditions (Figure S4b in the Supporting Information). Collectively, the data demonstrated efficient digestion of proteins while minimizing the background resulting from the proteolytic enzymes used. Although complete digestion of intact proteins is observed, it is unlikely that all peptides are hydrolyzed to single amino acids. We have digested synthetic peptides using this method to quantify the digestion efficiency. In addition, we have also tested the digestion efficiency against purified histone H2B, observing 64, 66, and 62% of the expected Lys, Arg, and Leu, respectively. These data may indicate a relative sequence specificity of N-Pep; however, the use of this digestion procedure results in a quantitative measurement that is representative of the population of PTMs in any given sample, which is especially useful when dealing with complex biological samples (e.g. chromatin). Furthermore, to generate consistent quantitation and stoichiometric determination of Lys and Arg PTMs, each digestion is normalized to Leu, as this amino acid lacks any notable, or abundant, PTM, thus providing an internal control for digestion efficiency. Previous studies have normalized Lys PTMs to the total unmodified Lys content.25 The latter normalization risks falsely skewing the results due to the fact that the unmodified Lys levels will change reciprocally to the total quantity of PTMs.
The application of QuARKMod for the study of PTM-modifying enzyme activity
A potential application of QuARKMod is to screen for the activity of PTM writers and erasers. To test this application, we evaluated the activity of the recombinant histone demethylase, JMJD2A, using 2 nmol of a synthetic peptide containing a single me3Lys residue. As shown in Figure 2, we observed quantitative yield of the analyte, detecting 2 nmol of me3Lys. Following incubation with, JMJD2A, we were able to measure substantial reductions in me3Lys with compensatory increases in me2- and meLys, consistent with the proposed function of the enzyme.26,27 This application has been tested with numerous peptide substrates and enzymes, validating QuARKMod for this application (Figure S5 in the Supporting Information). Although complete digestion was not observed with all peptide substrates tested, implying some degree of sequence specificity for N-Pep, relative efficiency is consistent for any given peptide. Hence, quantitative enzymatic activity can still be measured (Figure S5 in the Supporting Information). This method allows for the quantitative measurement of both enzymatic activity as well as the product profile, representing a major improvement over previous epigenetic screens, which rely on a coupled fluorophore to measure activity.28
Figure 2. The application of QuARKMod for the study of in vitro enzyme activity.

QuARKMod was used to investigate the overall methylation status of a synthetic peptide (2 nmol) containing a single me3Lys following incubation with a recombinant Lys demethylase, JMJD2A. A quantitative yield was observed in total methylated Lys following incubation with JMJD2A with me2Lys being the predominant PTM. The peptide sequence was as follows: ARTKQTARKme3STGGKA.
The application of QuARKMod for the study of histone-specific modifications in cells
The analysis of PTMs in histones is typically accomplished by western blotting using antibodies directed against the modifications of interest. This approach is, at best, only semi-quantitative, and it has been plagued by issues of poor antibody specificity. Although quantitative methods have been established, these aim to quantify site-specific modifications and therefore require the use of synthetic labeled peptides bearing the PTM of interest and high resolution MS.29,30 The application of QuARKMod to these analyses provides a bridge for this glaring gap in our understanding of PTM abundance and regulation. To evaluate QuARKMod in this application, we analyzed PTMs in histone-rich chromatin fractions isolated from HEK293 cells treated with a histone deacetylase inhibitor, trichostatin A (TSA), a demethylase inhibitor (IOX1), or the vehicle (DMSO). We isolated the chromatin using a method that avoids the acidic conditions (e.g., 0.2 N H2SO4) that are typically employed for this purpose in order to preserve the structure of labile modifications.21,22 Western blotting of the chromatin confirmed the expected increases in acLys and me3Lys in cells treated with TSA (Figure S6a in the Supporting Information) and IOX1 (Figure S6b in the Supporting Information), respectively. These changes were confirmed by QuARKMod, which also revealed a marked increase in me2Lys in IOX1-treated samples (Figure S6c in the Supporting Information). In fact, me2Lys was the most abundant of the methylated Lys species. In addition, the levels of ADMA were significantly increased in both TSA- and IOX1-treated samples. Clearly, QuARKMod represents a vast improvement over traditional techniques to measure Lys and Arg PTMs. Multiple species can be measured in a single MS run, structurally different PTMs can be positively identified, and critical information on the absolute quantity and stoichiometry of the modifications is obtained.
Chromatin fractions are very rich in the core histones H4, H3, H2B, and H2A. Previous work has demonstrated that H3 and H4 are the most heavily modified, with the vast majority of these modifications occurring on Lys residues.3,4 This is evident in Figure S6a and S6b in the Supporting Information, where western blotting of chromatin fractions largely reveals bands containing H3 and H4, providing further evidence that on a stoichiometric basis, these histones contain the most PTMs. It should be noted, however, that H2A and H2B are also heavily modified proteins, a fact that is typically missed using standard immunoblotting techniques. To investigate the levels of histone-specific PTMs, we subjected the chromatin fractions isolated from HEK293 cells to chromatography on a C18 column, and histones were collected off-line as described in Experimental Section. Commassie staining and western blot analysis demonstrated the high quality of the resulting preparations (Figure 3), although not all fractions were completely pure. The complete separation of H4 and H2A has been reported to be a significant challenge.22,31 Our HPLC conditions are a marked improvement over previously published methods using C8 chromatography; however, trace amounts of H2A may be seen in the H4 fraction.22 In addition, due to the high degree of sequence homology between histones H3.2 and H3.3, complete chromatographic separation cannot be achieved. Using these isolated histones, we applied the QuARKMod method to measure both Lys and Arg PTMs in all five fractions (Figure 3). Based on previous reports using western blotting (in addition to Figure S6a and S6b in the Supporting Information), it was not surprising to observe that both H3 fractions (gray and black bars) contained the highest quantities of Lys and Arg PTMs. However, the relatively high quantities of methylated species, particularly me2Lys, compared to those of acLys, were interesting. As highlighted, acLys remains one of the more heavily studied histone PTMs, while the role of me2Lys has not been investigated to the same degree. These data suggest that me2Lys might have a much larger role in the regulation of chromatin than previously thought. We also measured succinylLys modification, a newly described histone PTM.32 Our data demonstrate that succinylLys is present at a relatively low abundance, with the highest levels being reported on H3.1. Lastly, the levels of ADMA were again highest on the H3 isoforms, although detectable levels were found in all five fractions. SDMA was also measured, though no detectable levels were observed, which may be reflective of its reportedly modest frequency.33
Figure 3. Quantitation of Lys and Arg PTMs on isolated histones.

(a) To measure histone-specific changes, histones were purified using an offline HPLC method, resulting in significant separation of each core histone. (b) Purified histones collected from HEK293 cells demonstrate varying degrees of Lys and Arg PTMs.
QuARKMod defines the stoichiometry of Lys and Arg modifications in subcellular compartments
As noted above, much research has investigated the role of Lys and Arg PTMs in physiological settings; however, very little is known regarding the relative abundance of these modifications, especially in subcellular compartments. As stated, it has been estimated that >20% of all mitochondrial proteins contain acLys.15–17 We aimed to define the relative abundance and ratios of Lys and Arg PTMs in mitochondrial, microsomal, nuclear, and cytosolic enriched subcellular compartments isolated from murine liver sections. As shown in Figure 4, we were able to detect varying levels of Lys and Arg PTMs in each compartment. Given the high degree of reported PTMs on nuclear proteins, it was not surprising to observe the highest levels of all analytes measured in nuclear fractions. This is perhaps most notable with ADMA, where >10-fold higher levels were detected in the nucleus as compared to any other compartment. Perhaps the most striking result from these data is the high level of me3Lys detected in all four compartments, particularly in the mitochondrial fractions. Although me3Lys has been studied in depth, particularly on histones H3 and H4, the relative levels of this PTM have not been reported. These data suggest that me3Lys may play a more predominant role in mitochondrial processes compared to acLys.
Figure 4. QuARKMod reveals distinct patterns of PTMs in subcellular fractions.

Measurement of Lys and Arg PTMs in subcellular isolations from murine liver reveals that nuclear preparations contain the highest levels of modifications. Mitochondrial fractions contain high levels of acLys, me3Lys, and ADMA.
Product-ion scanning for the discovery of Lys and Arg PTMs
As PTM research continues to expand, novel histone and non-histone PTMs are being explored.11,13,14,34 The common fragment ions of Lys (84.0 and 130.1 m/z) and Arg species (70.0 and 116 m/z) suggest that the IDA mode of the mass spectrometer may be utilized to discover novel or unexpected PTMs, circumventing problematic assignments associated with peptide mass shifts.13,14 This can be accomplished by setting the survey scan to the common fragment ion (e.g. 84.0 m/z for Lys and 70.0 m/z for Arg) and scanning for ions which gave rise to those fragments (i.e. the parent ion or Q1 mass). This represents an unbiased approach to identify Lys or Arg PTMs by simply scanning for the Q3 ion mass.
To test this application, we treated RAW264.7 macrophages with or without the lipopolysaccharide mimetic, Kdo2-lipid A (KLA).12 KLA treatment of cells is a well-characterized model for initiation of inflammatory signaling events which result in decreased histone acetylation.35 Employing this cellular model, we have previously identified histones as selective targets for adduction by the lipid peroxidation end product, 4-oxo-2 nonenal.12 Therefore, this model allows for the discovery of novel PTMs (e.g. 4-oxo-2-nonenal) as well as an internal validation of previously reported alterations in histone PTMs (e.g. acLys). Using chromatin fractions collected from KLA-treated or vehicle control RAW264.7 cells, we sought to test and validate this discovery mode. Western blotting shown in Figure S7a-c in the Supporting Information revealed a significant decrease in acLys with a concomitant increase in me3Lys in KLA-treated cells as compared to controls. We observe similar trends in chromatin PTMs using QuARKMod (Figure S7d in the Supporting Information), validating this model for investigation of Lys and Arg PTMs. Although a significant decrease in acLys was not observed via QuARKMod, this is likely due to the challenge of Western blot densitometry measurements across an entire lane. By applying QuARKMod for these measurements, any bias can be removed, thus providing a more representative quantitation of the total PTM status of a given sample.
To explore novel Lys PTMs, chromatin digests used in the experiment shown in Figure S7 in the Supporting Information were analyzed via the IDA methodology described (Figure 5a). The IDA data revealed several known and expected PTMs. For example, the extracted ion chromatogram of 189 m/z from a vehicle-treated sample displays two peaks at 1.55 and 2.04 min (Figure S8a in the Supporting Information), which correspond to retention times of acLys and me3Lys, respectively. The CID spectrum of the 189 m/z peak from both 1.55 and 2.04 min showed the common fragment seen in Lys-derived species (84.0 and 130.1 m/z). Further interrogation of the Lys IDA data showed an apparent chromatographic peak at 2.57 min in the extracted ion chromatograph of 217.3 m/z which appeared to decrease in KLA-treated samples (Figure 5b). The CID spectrum of this peak is (Figure S8b in the Supporting Information) and the m/z of 84.0 and 130.1 suggests this to be a Lys PTM. The m/z of 217.3 represents a mass shift of 70.0 m/z from unmodified Lys, which is consistent with the reported mass shift of butyrylLys, a conserved histone mark.36 To validate this tentative identification, western blotting was performed on chromatin samples with an antibody targeting pan-butyrylation. As shown in Figure 5c, a significant decrease in butyrylated histones was observed in KLA-treated samples compared to vehicle controls, further corroborating the data presented in Figure 5b. The role of histone butyrylation has yet to be explored in inflammatory signaling and presents a novel avenue for research into the regulation of these pathways. This application can also be used for the discovery of Arg modifications by scanning for the common product ion of 70.0 m/z.
Figure 5. Product ion scanning can be utilized for the discovery of novel Lys and Arg PTMs.

(a) Profile of analytes with a product ion of 84.0 m/z (Lys) collected from chromatin isolated from KLA (red) or vehicle control (blue)-treated RAW264.7 macrophages. (b) XIC of 217.3 m/z displays a decrease in signal in KLA-treated samples compared to that in vehicle controls. (c) Validation of the observed decrease in butyrylLys in KLA-treated samples via western blotting of these chromatin fractions.
CONCLUSIONS
The need for accurate quantitation of histone-specific modifications is critical for the advancement of epigenetics research. As noted above, typical studies evaluating histone PTMs have relied on antibody-based assays to observe differences between treatments; however, poor antibody specificity has plagued this field of research. Recently, Egelhofer et al. highlighted this dilemma, reporting over 25% of the antibodies evaluated failed specificity tests.37 Aside from the reported complications regarding antibody specificity, standard immunoblotting does not provide information on the total abundance or ratios of modifications, but rather relative abundance between samples. This further necessitates a need for unbiased and accurate quantitation of these modifications from complex samples. Here, we describe a novel methodology for the quantitation of Lys and Arg PTMs. We show multiple applications for this technology, ranging from in vitro enzyme assays to quantitation in tissue extracts.
Traditional proteomic approaches require knowledge of the peptide sequence for PTM assignment.13,23,38 While these methods are powerful in their ability to study site-specific PTMs, label-free quantitation remains a significant challenge. In the development of this technique, we sought to define both the quantity and stoichiometry of all Lys and Arg PTMs in a given sample, without the need for high mass accuracy instrumentation. Using stable isotope dilution mass spectrometry, it is possible to calculate absolute concentrations in a given sample. In addition, we demonstrate the application of this method for the quantitation of enzymatic activity assays using synthetic peptides. This method is particularly advantageous when assessing the enzymatic activity of histone methyltransferases and demethylases. Typical screens for these enzymes rely on coupled fluorometric assays, which do not assess which type of methylation is occurring on a given Lys or Arg. The methods described here are capable of accurately quantifying decreases in Lys methylation using synthetic peptides, revealing precisely how these enzymes function. Thus, this technique is a highly accurate method for the evaluation of Lys and Arg PTMs and may be utilized for the screening of novel epigenetic therapeutics.
Supplementary Material
Acknowledgments
We would like to thank Carol A. Rouzer for help in scientific discussion and preparation of the manuscript. This work was supported by the following grants from the US National Institutes of Health: T32 ES007028 to J.G.; R37 CA087819 and S10OD017997 to L.M.; AA022146 to K.F.
Footnotes
Author Contributions
J.G. and P.K. conceived the method; J.G., M.M., J.C., P.H., K.F. and J.W. performed experiments; O.W. synthesized standards; J.G., M.M., J.W., P.K., and L.M. analyzed the data and wrote the paper.
Competing Financial Interest
The authors declare no competing financial interests.
Supporting Information Available: The supporting information contains additional Materials and Reagents, the synthesis of 13C6,15N2 N-ε-trimethyllysine and 13C6,15N2 N-ε-succinyllysine, Figures S1–S10, and supporting information references. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lothrop AP, Torres MP, Fuchs SM. FEBS Lett. 2013;587:1247–1257. doi: 10.1016/j.febslet.2013.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khoury GA, Baliban RC, Floudas CA. Sci Rep. 2011;1:90. doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kouzarides T. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Latham JA, Dent SY. Nat Struct Mol Biol. 2007;14:1017–1024. doi: 10.1038/nsmb1307. [DOI] [PubMed] [Google Scholar]
- 5.Grunstein M. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 6.Strahl BD, Allis CD. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 7.Jenuwein T, Allis CD. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 8.Mariadason JM. Epigenetics. 2008;3:28–37. doi: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- 9.Dekker FJ, Haisma HJ. Drug Discov Today. 2009;14:942–948. doi: 10.1016/j.drudis.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Li XD. Curr Opin Chem Biol. 2014;24C:80–90. doi: 10.1016/j.cbpa.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Galligan JJ, Rose KL, Beavers WN, Hill S, Tallman KA, Tansey WP, Marnett LJ. J Am Chem Soc. 2014;136:11864–11866. doi: 10.1021/ja503604t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai L, Peng C, Montellier E, Lu Z, Chen Y, Ishii H, Debernardi A, Buchou T, Rousseaux S, Jin F, Sabari BR, Deng Z, Allis CD, Ren B, Khochbin S, Zhao Y. Nat Chem Biol. 2014;10:365–370. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]
- 15.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 16.Anderson KA, Hirschey MD. Essays Biochem. 2012;52:23–35. doi: 10.1042/bse0520023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Huang Y, Zhang B, Pan X, Zhu X, Li G. Anal Chem. 2014;86:12138–12142. doi: 10.1021/ac503077f. [DOI] [PubMed] [Google Scholar]
- 18.Weinert BT, Iesmantavicius V, Moustafa T, Scholz C, Wagner SA, Magnes C, Zechner R, Choudhary C. Mol Syst Biol. 2014;10:716. doi: 10.1002/msb.134766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou T, Chung YH, Chen J, Chen Y. J Proteome Res. 2016;15:1103–1113. doi: 10.1021/acs.jproteome.5b01097. [DOI] [PubMed] [Google Scholar]
- 20.Molden RC, Bhanu NV, LeRoy G, Arnaudo AM, Garcia BA. Epigenetics Chromatin. 2015;8:15. doi: 10.1186/s13072-015-0006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torrente MP, Zee BM, Young NL, Baliban RC, LeRoy G, Floudas CA, Hake SB, Garcia BA. PLoS One. 2011;6:e24747. doi: 10.1371/journal.pone.0024747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shechter D, Dormann HL, Allis CD, Hake SB. Nat Protoc. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 23.Garcia BA, Mollah S, Ueberheide BM, Busby SA, Muratore TL, Shabanowitz J, Hunt DF. Nat Protoc. 2007;2:933–938. doi: 10.1038/nprot.2007.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Proc Natl Acad Sci U S A. 2010;107:2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edrissi B, Taghizadeh K, Dedon PC. PLoS Genet. 2013;9:e1003328. doi: 10.1371/journal.pgen.1003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 27.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 28.Wegener D, Wirsching F, Riester D, Schwienhorst A. Chem Biol. 2003;10:61–68. doi: 10.1016/s1074-5521(02)00305-8. [DOI] [PubMed] [Google Scholar]
- 29.Wang C, Weerapana E, Blewett MM, Cravatt BF. Nat Methods. 2014;11:79–85. doi: 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Tallman KA, Porter NA, Liebler DC. Anal Chem. 2015;87:2535–2541. doi: 10.1021/ac504685y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Nat Chem Biol. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gayatri S, Bedford MT. Biochim Biophys Acta. 2014;1839:702–710. doi: 10.1016/j.bbagrm.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirschey MD, Zhao Y. Mol Cell Proteomics. 2015;14:2308–2315. doi: 10.1074/mcp.R114.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN. Blood. 2005;106:3114–3122. doi: 10.1182/blood-2005-02-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung MS, Day DS, Gadel S, Gorchakov AA, Gu T, Kharchenko PV, Kuan S, Latorre I, Linder-Basso D, Luu Y, Ngo Q, Perry M, Rechtsteiner A, Riddle NC, Schwartz YB, Shanower GA, Vielle A, Ahringer J, Elgin SC, Kuroda MI, Pirrotta V, Ren B, Strome S, Park PJ, Karpen GH, Hawkins RD, Lieb JD. Nat Struct Mol Biol. 2011;18:91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bremang M, Cuomo A, Agresta AM, Stugiewicz M, Spadotto V, Bonaldi T. Mol Biosyst. 2013;9:2231–2247. doi: 10.1039/c3mb00009e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.