

ABSTRACT
We previously demonstrated that OVE transgenic diabetic mice are susceptible to chronic complications of diabetic nephropathy (DN) including substantial oxidative damage to the renal glomerular filtration barrier (GFB). Importantly, the damage was mitigated significantly by overexpression of the powerful antioxidant, metallothionein (MT) in podocytes. To test our hypothesis that GFB damage in OVE mice is the result of endothelial oxidative insult, a new JTMT transgenic mouse was designed in which MT overexpression was targeted specifically to endothelial cells. At 60 days of age, JTMT mice were crossed with age‐matched OVE diabetic mice to produce bi‐transgenic OVE‐JTMT diabetic progeny that carried the endothelial targeted JTMT transgene. Renal tissues from the OVE‐JTMT progeny were examined by unbiased TEM stereometry for possible GFB damage and other alterations from chronic complications of DN. In 150 day‐old OVE‐JTMT mice, blood glucose and HbA1c were indistinguishable from age‐matched OVE mice. However, endothelial‐specific MT overexpression in OVE‐JTMT mice mitigated several DN complications including significantly increased non‐fenestrated glomerular endothelial area, and elimination of glomerular basement membrane thickening. Significant renoprotection was also observed outside of endothelial cells, including reduced podocyte effacement, and increased podocyte and total glomerular cell densities. Moreover, when compared to OVE diabetic animals, OVE‐JTMT mice showed significant mitigation of nephromegaly, glomerular hypertrophy, increased mesangial cell numbers and increased total glomerular cell numbers. These results confirm the importance of oxidative stress to glomerular damage in DN, and show the central role of endothelial cell injury to the pathogenesis of chronic complications of diabetes. Anat Rec, 2017. © 2017 Wiley Periodicals, Inc. Anat Rec, 300:560–576, 2017. © 2016 The Authors. The Anatomical Record published by Wiley Periodicals, Inc. on behalf of American Association of Anatomists.
Keywords: glomerular cells, glomerular filtration barrier, glomerular basement membrane, diabetic mice, electron microscopy
INTRODUCTION
Internationally, more than 25% of all persons with diabetes mellitus (DM, see Table 1 for abbreviations) will develop diabetic nephropathy (DN), the largest cause of renal decompensation and failure. Careful glycemic control markedly decreases the risk of DN in both Type I (The Diabetes Control IINA and Complications Trial Research Group, 1993) and Type II [UK Prospective Diabetes Study (UKPDS) Group, 1998] diabetes. However, many diabetics with poor blood glucose control do not develop DN, while others, with good control progress to renal failure. These exceptions demonstrate that numerous factors, in addition to glucose, are important and we are still far from understanding some of the most basic mechanisms predisposing certain persons to chronic complications of DN. As with other diabetic complications, there are many mechanistic possibilities, including oxidative stress (Giacco and Brownlee, 2010, Kashihara et al., 2010), advanced glycation end products (AGE, Vlassara and Palace, 2002), elevated sorbitol (Wallner et al., 2001), activation of PKC (Ha et al., 2001), and increased TGFβ activity (Ziyadeh, 2004). It is likely that several of these processes are involved, and though it is not known which is most dominant, it is clear that oxidative stress is a major player in the development of chronic diabetic complications (Brownlee, 2005.)
Table 1.
Abbreviations
| FVB | Friend virus B mouse genotype which often serves as background mice for the development of transgenic mice. It also is a natural non‐transgenic control animal in experiments employing transgenic mice that utilized the FVB as background. |
| GBM | Glomerular Basement Membrane is an extracellular matrix layer comprised mainly of "type IV collagen, laminin, perlecan, and nidogen. It is produced by glomerular podocytes and endothelial cells, between which it is located. It serves as a permselective macromolecular filter of plasma in the glomerular capillary circulation. |
| Nmt | Transgenic mouse genotype (Zheng et al. 2008). They specifically overexpress the antioxidant metallothionein in glomerular podocytes but is phenotypically unaffected/asymptomatic. |
| OVE | Transgenic mouse genotype (Epstein et al. 1989). They are diabetic as a result of severe calmodulin overexpression in pancreatic ß cells, the line is hyperglycemic (blood sugar > 500mg%), profoundly albuminuric (60,000µg albumin/24 hr), yet fertile, long‐lived, and does not require exogenous insulin to thrive. |
| OVENmt | Bi‐transgenic mouse genotype that are progeny of OVE and Nmt mice (Zheng et al. 2008). They are profoundly diabetic but show remarkable renoprotection against diabetic nephropathic features, which is believed to be the result of free radical scavenging by podocyte‐laden metallothionein |
| JTMT | Transgenic mouse genotype (Teiken et al.,2011). They specifically overexpress the antioxidant metallothionein in vascular endothelial cells, but is phenotypically unaffected/asymptomatic. |
| OVE‐JTMT | Bi‐transgenic mouse genotype that are progeny of OVE and Nmt mice (Teiken, 2011). They are profoundly diabetic but show remarkable renoprotection against diabetic nephropathic features, which is believed to be the result of free radical scavenging by vascular endothelial cell‐laden metallothionein |
| PGBM | Peripheral Glomerular Basement Membrane is a subset of all glomerular basement membrane. It is the portion of the membrane covered by podocyte foot processes on its external surface and by endothelial cells on its internal surface. It specifically excludes “stalk” and “mesangial regions” of the glomerular basement membrane. |
| ROS | Reactive Oxygen Species are “free radicals” and include atoms or molecules that have an odd number of electrons. They can cause damage to cells and DNA, and are associated with creating an environment within the body that may contribute to chronic diseases such as cancer and heart disease. |
| UAE | Urinary Albumin Excretion is the quantity (in micrograms) of albumin excreted in the urine by an organism in a 24 hr period. |
Overall, antioxidant status is reduced in diabetes (Vijayalingam et al., 1996; Wolff et al., 1991) and reactive oxygen species (ROS) are important players in many diabetic complications (Baynes, 1991; Kawaguchi, et al., 1992). Several lines of evidence implicate oxidative stress as a cause of chronic complications of diabetes (Giacco and Brownlee, 2010; Kashihara et al., 2010) These include glomerular basement membrane (GBM) thickening, increased glomerular ROS production, and expression of several biomarkers characteristic of oxidative damage. Importantly, all glomerular cell types are capable of producing free radicals (Baynes, 1991) and glomerular morphology is particularly susceptible to oxidative damage. Moreover, in animal models of several kidney diseases including DN (reviewed by Shah, 1995), oxidative stress produces glomerular injury.
In this regard, we have published several descriptions (Carlson, et al., 1997; Carlson, et al., 2003; Zheng, et al., 2004; Teiken et al., 2008; Zheng, et al., 2008; Teiken et al., 2011) of a transgenic diabetic mouse (called OVE26, herein referred to as OVE) that provides the most accurate morphological and functional model available for human DN (Zheng et al., 2004; Xu, et al., 2010). In an effort to intervene in the overproduction of ROS in this model, Zheng and co‐workers (Zheng et al., 2008, 2004) demonstrated that breeding OVE to transgenic mice (Nmt) that overexpress the antioxidant, metallothionein (MT) in glomerular podocytes produces a double transgenic diabetic mouse (OVENmt) in which the progression to advanced DN is markedly reduced. Their elegant manuscript (Zheng et al., 2008) clearly demonstrated that in OVENmt mice, MT reduced numerous cytopathological features of DN, including glomerular and mesangial volume, glomerular cell death, and upwards of a 70% reduction in urine albumin excretion (UAE) at four months of age—though this did not appear to be permanent.
These experiments formed a basis for those carried out by us in a subsequent unbiased TEM stereometric investigation (Carlson et al., 2013) in which we sought to clarify the quantitative aspects of the OVENmt analysis. Our TEM investigation affirmed the results of the study by Zheng et al. (2008) and provided rigorous numerical evidence that oxidative damage to podocytes induces a number of primary DN features, complications of which were significantly mitigated by targeted MT overexpression in podocytes.
Recently we developed a new transgenic mouse (JTMT) that overexpresses MT specifically and ubiquitously in vascular endothelial cells (EC, Teiken et al., 2011, Abstract). Furthermore, we now have sufficient numbers of 150 day‐old OVE‐JTMT mice (progeny of male OVE and female JTMT adults), to realistically examine and catalog their renoprotective potential against hyperglycemia‐driven oxidative damage. Accordingly, in the current TEM morphometric study, we compare several important DN parameters in OVE and OVE‐JTMT mice. Our data provide significant evidence for a direct role of oxidative damage to the glomerular endothelium in diabetic mice. This strongly supports our working hypothesis that oxidative insult to glomerular cells is a primary cause of DN chronic complications. Furthermore, data in the current study demonstrate that OVE‐JTMT mitigation of DN features is remarkably similar to the renoprotection provided by the Nmt transgene in podocytes of OVENmt mice (Carlson et al., 2013). It seems possible, therefore, that the development of a future tri‐transgenic (OVE‐Nmt‐JTMT) mouse could be an even more powerful new tool in the renoprotective armamentarium of future diabetes researchers.
MATERIALS AND METHODS
Experimental Animals
Four genotypes, including at least five age‐matched 150 day‐old FVB, JTMT, OVE, and OVE‐JTMT, were used in the current study. These mice were physically indistinguishable with the exception that OVE and OVE‐JTMT had “small eyes” owing to the coinjection and cointegration of a cataract‐inducing GR19 gene inserted when the OVE mice were developed (Epstein et al., 1989). Accordingly, by two weeks of age, diabetic (OVE and OVE‐JTMT) mice were easily identified within the colony. All mice were highly active, and exhibited sleek shiny coats.
Animal Genotype Production and Selection
Transgenic mice designed to overexpress MT specifically and ubiquitously in endothelial cells were produced with an EC targeted transgene (MT) containing the human MTII gene, and regulated by the murine TIE‐2 promoter (Teiken et al., 2011; Abstract). Early attempts to produce transgenic founder mice resulted in the generation of five viable MT‐positive genotypes (called JTMT‐1, ‐2, ‐3, ‐4, and ‐5; Fig. 1A). While genotyping confirmed that JTMT transgenic progeny contained the JTMT transgene, the glomerular MT expression was determined by Western Blot (Fig. 1B) as described by Wang and associates (Wang et al., 2006). Quantitative assessment of glomerular MT expression was carried out as previously reported (Takemoto et al., 2002).
Figure 1.
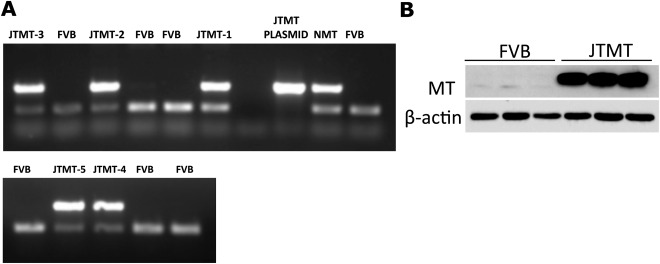
A. PCR genotyping of DNA samples from JTMT founder mice. Primers corresponding to the human MTII gene resulted in a 536 bp DNA band, and control primers designed for a region of the FVB background DNA resulted in a 200 bp band. DNA containing the transgene displayed two DNA bands, and the corresponding animals were given the nomenclature JTMT‐1–5. B. Glomerular MT protein expression. Glomeruli were isolated from both wild type FVB and JTMT transgenic mice. The MT protein expression was detected by Western blot as previously reported (Wang et al., 2006).
Of five original genotypes, three (JTMT‐2, ‐3, ‐5) were discontinued due to lack of insertion of the transgene in mating progeny. To determine which of the remaining viable genotypes would be used in our experimental work, male diabetic OVE mice were cross‐bred with (JTMT‐1 and JTMT‐4) to produce double transgenic progeny (OVE‐JTMT‐1 and OVE‐JTMT‐4 mice), and the possible protective effects of MT overexpression on DN were assessed in each type. Preliminary experiments clearly favored the JTMT‐1 mouse as a more effective experimental tool for our proposed experiments against hyperglycemia‐driven damage in the 150 day murine glomerulus. Accordingly, four genotypes, including FVB, JTMT‐1, now referred to simply as “JTMT”, OVE, and OVE‐JTMT‐1 (now “OVE‐JTMT”), were selected for the current study. All mice were maintained in the laboratories of Dr. Edward Carlson, Department of Biomedical Sciences, University of North Dakota, Grand Forks, North Dakota and of Dr. Paul Epstein, Department of Pediatrics, University of Louisville, Louisville, Kentucky. Dr. Carlson's laboratory has maintained diabetic OVE mice on the FVB background for 13 years and Dr. Epstein's laboratory has 27 years of experience with this strain (Epstein et al., 1989). JTMT mice were developed on the FVB background in the Carlson laboratory (Teiken et al., 2011, Abstract) and have also been maintained by Dr. Carlson for four years. No insulin therapy was given to diabetic animals, and all mice had free access to food and tap water. All animal procedures adhered to the guidelines of the NIH Guide for the Care and Use of Laboratory Animals and were approved by the IACUC committees of the University of North Dakota and the University of Louisville.
Determination of MT Overexpression and Co‐Localization in EC
Immunohistochemistry
To assess the co‐localization of MT with EC in renal tissues, five, 60 day‐old FVB and six, 60 day‐old JTMT mice (Table 2) were euthanized and 5‐20 tissue samples were prepared for light microscopic (LM) immunohistochemical observation. A monoclonal mouse anti‐horse MT‐I/II antibody (Dako, 13 mg/L) was used to visualize MT by an HRP‐conjugated anti‐mouse IgG secondary antibody on sections of formalin‐fixed paraffin‐embedded renal tissue. Some slides were incubated with polyclonal goat anti‐mouse PECAM‐1 antibody to label vascular endothelium. Negative control slides did not receive primary antibody, but were incubated with HRP conjugated secondary antibody.
Table 2.
Animal genotype, number of animals, number of paraffin and epoxy blocks sectioned, and number of LM and TEM glomerular profiles photographed for mean glomerular profile area, glomerular volume, and glomerular cell number calculation
| For Immuno‐ Histochem. Fluorem. | For Glomerular Morphometry | For Calculation of Glomerular Volume | For Calculation of Glomerular Cell Number | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Age in Days | Total Number of Animals | Total Number of Blocks Sectioned | Age in Days | Total Number of Animals | Total Number of Blocks Sectioned | Age in Days | Total LM Glomerular Profiles per Animal | Total LM Glomerular Profiles | Age in Days | Total TEM Glomerular Profiles per Animal | Total 1EM Glomerular Profiles |
| EVE | 60 | 6 | 24 | 150 | 8 | 8 | 150 | 8–39 | 227 | 150 | 1–5 | 32 |
| J1MT | 60 | 5 | 20 | 150 | 5 | 10 | 150 | 4–12 | 32 | 150 | 4–12 | 32 |
| OVE | – | – | – | 150 | 8 | 18 | 150 | 15–18 | 138 | 150 | 1–7 | 36 |
| JTMT | – | – | – | 150 | 5 | 10 | 150 | 4–6 | 24 | 150 | 2–5 | 24 |
| Total | 11 | 44 | 26 | 46 | 421 | 124 | ||||||
Immunofluorescence
To further assess the co‐localization of MT to the EC in renal tissues by immunofluorescence, six, 60 day‐old FVB and five, 60 day‐old JTMT mice (Table 2) were euthanized and their kidneys were conventionally prepared for optimal visualization by confocal microscopy. Frozen sections were transferred to small glass vials containing either Texas Red conjugated Lycopersicon esculentum (Tomato) lectin (1:1000, Vector Laboratories) for the labeling of the vascular endothelium or phosphate buffered saline containing 0.3% Triton X‐100 (PBS‐T). These sections were then incubated for 36 hrs in monoclonal mouse anti‐horse MTI/II primary antibody (Dako, 13 mg/L), diluted 1:20 in PBS, followed by incubation for ∼36 hr at room temperature with slow rotation in FITC conjugated anti‐mouse IgG diluted 1:100 in PBS‐T. Negative control tissue sections did not receive MTI/II primary antibody, but received the secondary antibody. Unstained tissues incubated solely in PBS‐T for the entire staining protocol were observed for the presence of autofluorescence.
Sections were imaged with a 40× objective on a Zeiss LSM‐510 Meta confocal microscope using lasers set to the appropriate wavelengths necessary for visualization of the fluorochromes.
Tissue Preparation and Electron Microscopic Technique
At least five 150 day‐old animals of each of the four mouse genotypes central to the current study were used (Table 2). Prior to sacrifice, all mice were weighed and non‐fasted blood glucose levels were determined by Lifescan glucometer. Percentages of HbA1c were quantified using an A1CNow+ kit (Bayer HealthCare).
All animals destined for TEM morphometric analysis were sacrificed by vascular perfusion with PIPES fixative (Baur and Stacey, 1977). These procedures were carried out exclusively in the Carlson laboratory. By this method, mice were perfused with a 0.9% saline washout solution until the effluent was clear. This was followed by infusion of 20 mL of warm, then 40 mL of cold PIPES fixative (1% paraformaldehyde with 1% gluteraldehyde and 5% dextran in 0.1 M piperazine‐N‐N, bis 2‐ethane sulphonic acid) buffered with NaOH (pH 7.6) as previously described (Carlson et al., 2013). Animals destined for qualitative TEM only (Fig. 5) were sacrificed by vascular perfusion with cacodylate buffers as previously described (Carlson et al., 2003). In an effort to retain only scrupulously unbiased samples at each experimental step, rigid sampling procedures as established in our prequel studies (Carlson et al., 2013) were rigorously enforced.
Figure 5.
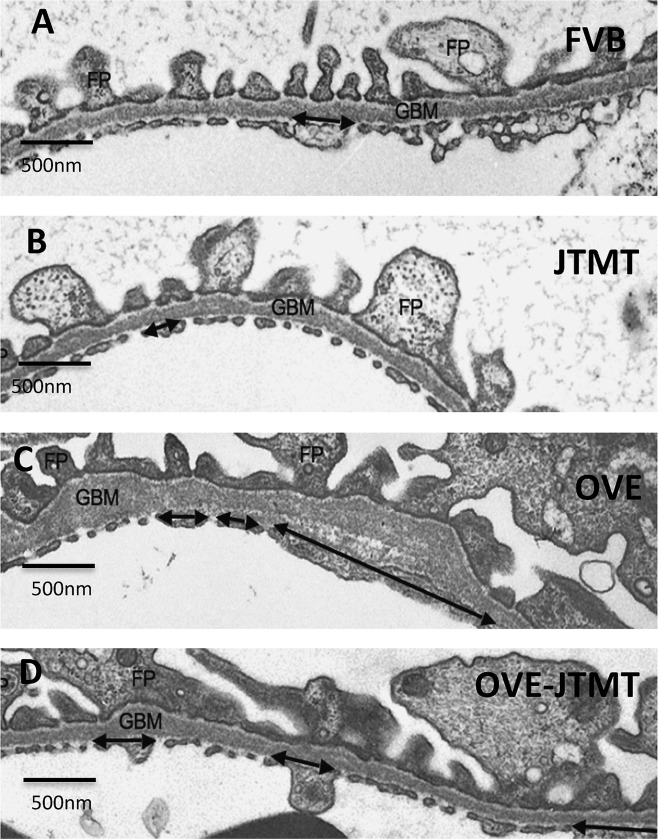
Representative transmission electron micrographs of the GFB in 150 day‐old FVB, JTMT, OVE and OVE‐JTMT mice. FP, podocyte foot process: GBM, glomerular basement membrane; Double arrow, non‐fenestrated capillary endothelium.
Following vascular perfusion fixation, kidneys were weighed and renal cortical tissue samples were conventionally dehydrated and embedded in Epon/Araldite as described previously (Teiken et al., 2008). Thick (∼250 nm) sections were stained with 1% toluidine blue (in 1% sodium borate) and examined by bright field microscopy for presence and location of renal glomeruli. Thin sections (∼70 nm) were mounted on 200 µm mesh naked copper grids (1/8 “diameter metal screen‐like sieves) that support thin sections being viewed in the TEM). Alternatively, they are supported by transluscent plastic (Formvar) coated slot grids (1/8” diameter copper circles with an etched “slot‐like opening” covered by Formvar), These may support entire TEM sections, and therefore entire glomerular profiles were viewed in the TEM. These were stained with 2% uranyl acetate and lead citrate (Venable and Coggeshall, 1965). Thin sections were examined and photographed in a Hitachi model H‐7500 TEM at 6,000 or 15,000 diameters at an accelerating voltage of 80 kV. TEM calibration was established by photographing a standard carbon grating (2,160 lines/mm) under the same conditions and initial magnifications used for the tissues.
Glomerular Filtration Barrier
Non‐fenestrated endothelial area (NFEA)
Mean capillary NFEA was determined in TEM sections by a modification of the method of Toyoda et al. (2007) as previously described (Carlson et al., 2013). Non‐fenestrated endothelium was quantified along visibly eligible segments of peripheral GBM (PGBM) on ∼100 TEM micrographs from each mouse. Glomerular endothelium was classified as non‐fenestrated when it was more than twice the thickness of the adjacent cytoplasm and exhibited no perforations. Non‐fenestrated areas on each micrograph were demarcated with a permanent marker and these were measured by ImageJ and summed. Finally, the sum of all PGBM segments for each animal was divided by the sum of its non‐fenestrated lengths to generate a group mean percentage ± S.D.
In summary, for each genotype (not less than 5 animals), the lengths of all visually eligible, non‐fenestrated PGBM segments in each TEM micrograph was summed. The sum of all non‐fenestrated PGBM segments for micrographs of each mouse was divided by the total of its PGBM lengths to generate a mean percentage ± S.D.
Glomerular basement membrane (GBM) thickness
GBM thickness was determined by a modification (Dische, 1992) of the orthogonal intercept method (Jensen et al., 1979). In summary, measurements were carried out on TEM micrographs (>1,000 total TEM micrographs; >10,000 total measurements). All tissue samples were randomized so that all glomerular planes of section were equally likely to occur. Randomized measurements were made with a measuring ruler with a quasi‐logarithmic scale utilizing a digitizing tablet and appropriate software (Bioquant, R&M Biometrics) as previously described (Carlson et al., 2013). “True” GBM thickness for each animal was generated from the measurements according to the orthogonal intercept technique. Finally, for each genotype (not less than 5 animals), the sum of all true GBM thicknesses for each animal was divided by the sum of its measurement points to generate a mean percentage ± S.D.
Foot process width (FPw)
Mean podocyte FPW was calculated by a modification of the method of Mifsud and coworkers (Mifsud et al., 2001) as previously described (Carlson et al., 2013). Foot processes per length of GBM were determined by counting the number of foot process profiles along visibly eligible segments of PGBM. The same micrographs (final magnification, 16,800× diameter) used to calculate NFEA (described above) were utilized for these studies. Foot processes were included in the study only if they were connected by slit diaphragms, and it could be demonstrated that all podocytes were interpreted as covering the GBM. For each animal, mean FPW ± S.D. was determined from the reciprocal of the number of foot processes per length of GBM.
Glomerular Morphometry
Mean glomerular volume (VG)
In order to calculate V G, at least 15 LM glomerular profiles (Table 2) from each animal genotype were chosen randomly by unbiased observers. V G was calculated using the mean area of glomerular profiles (determined by ImageJ software) by the method of Weibel and Gomez (Weibel and Gomez, 1962; Weibel, 1979) from the formula: V G = Area1.5 × ß/K, or V G = Area1.5 × 1.38/1.01, (Eq. 1) where ß, (shape coefficient for a sphere) is 1.38, and K (size distribution coefficient assuming a 10% coefficient of variation) is 1.01.
Mean endothelial cell (eNV), mesangial cell (mNV), podocyte (pNV) and total glomerular (gNV) nuclear (cellular) density
All data were derived from TEM images of glomerular profiles. Assuming one nucleus per cell, nuclear density (i.e., the number of nuclei per unit of glomerular tissue) and cellular density are identical. Therefore, determining cellular nuclear density in TEM micrographs provides the desired numerical data and also conveniently obviates the difficulty of quantifying variable cell shapes. Also, ultrastructural observation enables unequivocal identification of EC, mesangial cell and podocyte nuclei.
In an effort to avoid bias produced by variable nuclear shape and size distribution the Weibel and Gomez (Weibel and Gomez, 1962; Weibel, 1979) method as described by White and Bilous (White and Bilous, 2004) was employed. This technique utilized point‐counting measurements carried out on at least one TEM micrograph of a glomerular profile per animal. Measurements were performed on 13–21 TEM sectional profiles from animals of each animal genotype (124 total TEM images; Table 2).
To determine N V for endothelial cell (eNV) mesangial cell (mNV), podocyte (pNV) and the total cell density (gNV), we employed the formula: N V = K/ß√ ( /V V) (Eq. 2). A transparent plastic grid was superimposed on each TEM glomerular profile, and the number of EC, mesangial cell, podocyte, or total nuclear profiles and fine points hitting the glomerular tuft (P tuft) were counted (Table 3) in order to estimate nuclear profile area (N A): N A = Σn/[Σ P tuft × area per point] (Eq. 3). The number of fine points hitting EC (E nucleus), mesangial cell (M nucleus) or podocyte (P nucleus) nuclear profiles were then counted to calculate the volume fraction of nuclei of each cell type in the glomerular tuft (V V): V V = ΣP nucleus/ΣP tuft) (Eq. 4). Equation 2, with a size distribution (K) of 1.01 and a shape constant (ß) of 1.55, was used to calculate eN V, mN V , and pN V.
Table 3.
Animal genotype, number, and number of fine points on TEM glomerular profiles for calculating cell number per glomerulus
| Genotype | (n) | P tuft | P n | P nucleus | M n | M nucleus | E n | E nucleus | T n | T nucleus |
|---|---|---|---|---|---|---|---|---|---|---|
| FVB | 32 | 145.0 | 8.1 | 6.4 | 13.8 | 6.9 | 11.3 | 7.1 | 32.9 | 20.5 |
| JTMT | 32 | 124.0 | 8.7 | 6.5 | 13.8 | 5.8 | 15.2 | 9.8 | 37.7 | 22.0 |
| OVE | 36 | 257.0 | 7.3 | 6.2 | 18.6 | 10.3 | 13.0 | 9.4 | 39.0 | 25.9 |
| OVE‐JTMT | 24 | 219.1 | 8.1 | 6.4 | 14.2 | 7.7 | 15.9 | 9.0 | 38.2 | 23.1 |
(n) = number of TEM glomerular profiles.
P tuft = mean number of fine points on glomerular profiles.
P n = mean number of podocyte nuclei.
P nucleus = mean number of fine points on podocyte nuclei.
M n = mean number of mesangial nuclei.
M nucleus = mean number of fine points on mesangial nuclei.
E n = mean number of endothelial cell nuclei.
Mean endothelial cell (eN), mesangial cell (mN), podocyte (pN) and total (gN) cell number per glomerulus: (N)
For determination of N, we employed the general formula for numerical density: N = V G × N V, using V G from Equation 1, and eN V, mN V, pN V , and gN V from Equation 2.
Statistics
In those cases where data did not meet the specific assumptions of normal distribution and equal variance required for parametric analysis, the Wilcoxon‐ Mann‐Whitney Rank‐Sum test was employed to compare experimental and age‐matched control mice. Likewise, the one‐way analysis of variance (ANOVA) and the non‐parametric Kruskal‐Wallis analysis of variance on ranks were used to compare data for each mouse genotype. Statistical analyses were carried out using Sigmastat 3.0 (SPSS, Inc.) and Kaleidagraph. Level of significance for all tests was set at P <0.05 and power was set at 0.8.
RESULTS
Determination of MT Overexpression and Co‐Localization in EC
Immunohistochemistry and immunofluorescence
Transgenic (JTMT) and FVB progeny from the JTMT founder mouse lines were used to determine the location of MT in renal tissues (Figs. 2 and 3). In immunohistochemical preparations, MT staining was not observed in the vasculature of control FVB mice, but was exhibited occasionally in proximal tubules throughout the cortex (Fig. 2B). In contrast, JTMT mice displayed strong MT staining in glomerular capillary tufts and peritubular capillaries (Fig. 2F) in a pattern similar to that seen in PECAM‐1 stained tissues (Fig. 2D). Negative control sections showed little to no staining (Fig. 2A,C,E). Immunofluorescent labeling of renal tissues with Tomato lectin and the MT antibody showed similar distribution patterns (Fig. 3).
Figure 2.
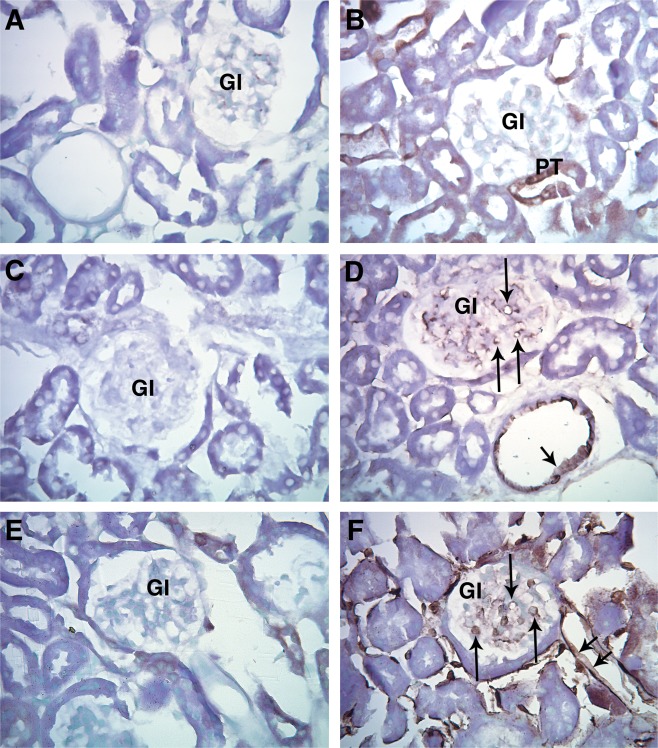
Representative light micrographs of paraffin embedded renal cortex immunohistochemically stained for MT (A, B, E, F) and PECAM‐1 (C, D.). A. FVB: Negative control, primary antibody omitted. B. FVB: Endogenous MT staining was not observed in the vasculature, but was occasionally observed in proximal tubules (PT). C. FVB: Negative control, primary antibody omitted. D. FVB Staining was observed in the cytoplasm of EC in the capillaries of the glomerulus (arrow) and in renal cortical arterioles (arrowhead). E. JTMT: Negative control, primary antibody omitted. F. JTMT: Cytoplasm of glomerular capillaries (arrow), peritubular capillaries (double arrowheads), and and other cortical microvascular structures displayed intense MT stain. Gl; glomerulus.
Figure 3.
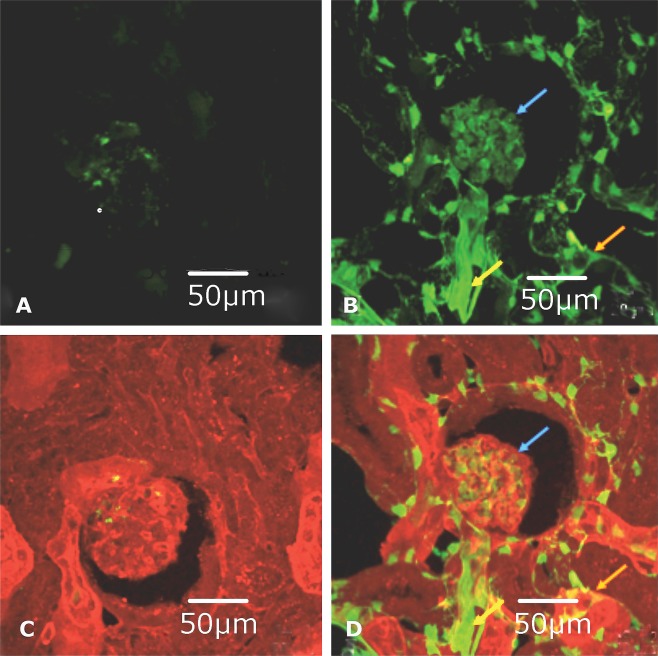
Representative confocal micrographs of FVB and JTMT renal cortex frozen sections immunofluorescently stained for MT and EC. A. FVB: MT expression visualized with FITC (green) immunofluorescence. There was little to no MT expression in tissue sections from the control animal. B. JTMT: MT expression visualized with FITC (green) immunofluorescence. In the transgenic mouse, strong MT staining was observed in the glomerulus (blue arrow), peritubular capillaries (orange arrow) and arterioles (yellow arrow). C. FVB: Merged tomato lectin (red) and MT (green) immunofluorescent staining. The control mouse displayed limited MT expression and it was not associated with the vasculature. D. JTMT: Merged tomato lectin (red) and MT (green) immunofluorescent staining. Due to the somewhat “leakiness” of the TIE‐2 promoter (tends to promote staining of hematopoietic cells) and its ability to bind non‐vascular structures in the kidney, there was limited co‐distribution (yellow staining) of MT with the tomato lectin. However, the MT overexpression was localized to EC in the glomerulus (blue arrow), peritubular capillaries (orange arrow), and arterioles (yellow arrow).
Body Features
Body and kidney weights
JTMT mice weighed slightly less than the other genotypes (Fig. 4A,B), which were not significantly different from each other. Left and right kidney weights were somewhat variable, but their combined weights in OVE (diabetic) mice were significantly greater (∼80%) than FVB controls. Interestingly, diabetic nephromegaly was reduced (protected) in OVE‐JTMT mice and their combined kidney weights were significantly less (∼29%) than age‐matched OVE.
Figure 4.
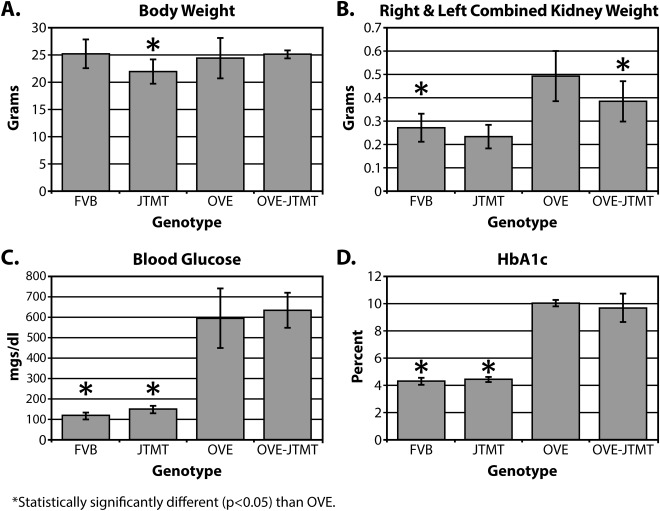
Body Features of 150 day‐old Mice. A. Body Weight: Although JTMT mice weighed slightly less than other genotypes, of the four genotypes utilized in the current study, no group varied more than 15% from any other. B: Combined weight of left and right kidneys. Total renal weight was significantly greater (∼86%) in OVE animals than in non‐diabetic controls, but diabetic nephromegaly was mitigated significantly by breeding with JTMT mice, reducing the average weight in OVE‐JTMT mice by ∼29%. C. Blood Sugar. Serum glucose frequently reached 600mg/dl in OVE mice, and these levels were not mitigated by overexpression of MT. D: Likewise, HbA1c rose to more than 10% in severely diabetic OVE mice, and was maintained at highest levels with or without EC targeted MT.
Blood glucose and HbA1c
The transgene did not reduce blood glucose in fed adult OVE mice. As expected, non‐fasting blood glucose levels in OVE and OVE‐JTMT animals were significantly greater than non‐diabetic controls and often exceeded them by ∼400 mg/dl (Fig. 4C). To confirm that chronic glycemia was unaffected, we also measured glycosylated hemoglobin A1c. This was elevated to the same extent as blood glucose in OVE (10.43%) and OVE‐JTMT (9.65%), which was more than two‐fold higher than non‐diabetic controls (Fig. 4D). Therefore, MT‐induced changes in diabetic nephropathic features could not be due to less severe diabetes.
Glomerular Filtration Barrier
In an effort to determine which specific renal morphological features might be most susceptible to diabetes‐induced alterations, a preliminary TEM examination of renal glomeruli from the four murine genotypes in the current study was carried out (Fig. 5). Early observations from 150 day‐old severely diabetic OVE mice suggested that the area of the capillary luminal wall occupied by endothelial fenestrations seemed markedly reduced. Also, GBMs were unmistakably wider than those typically observed at 150 days. In addition, podocyte foot processes appeared somewhat wider than those in most normoglycemic mice (Fig. 5C).
To confirm these preliminary visual impressions, we carried out an unbiased, TEM morphometric analyses on the major components of the glomerular filtration barrier (GFB), including mean non‐fenestrated endothelial area (NEFA), GBM thickness, and podocyte foot process width (FPw), in 150 day old FVB, JTMT, OVE, and OVE‐JTMT mice (Fig. 6).
Figure 6.
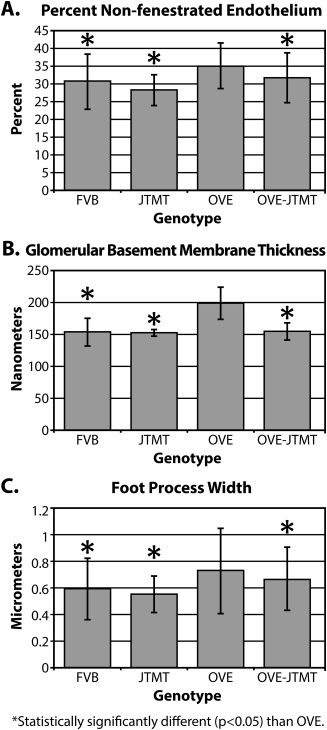
Morphometric differences in components of the GFB in 150 day old FVB, JTMT, OVE and OVE‐JTMT mice. A. Mean percent non‐fenestrated endothelium ± S.D: FVB vs. OVE, JTMT vs. OVE, and OVE‐JTMT vs. OVE all show statistically significant differences. OVE mice show greatest percentage of non‐fenestrated endothelium, while OVE‐JTMT mice exhibited significant protection against non‐fenestration. B. Mean glomerular basement membrane thickness ± S.D: FVB vs. OVE, JTMT vs. OVE, and OVE‐JTMT vs. OVE all show statistically significant differences. OVE mice show the thickest glomerular basement membrane, while OVE‐JTMT mice show complete elimination of basement membrane thickening. C. Mean podocyte foot process width ± S.D: FVB vs. OVE, JTMT vs. OVE, and OVE‐JTMT vs. OVE all show statistically significant differences. OVE mice show the widest foot processes (greatest foot process effacement) while OVE‐JTMT mice show significant protection against increased foot process width.
Non‐fenestrated endothelial area (NFEA)
In the current study, OVE NFEA was significantly increased (∼14%, P <0.05) compared to FVB animals (Fig. 6A). Importantly, however NFEA was significantly (∼11%, P < 0.05) protected/decreased in OVE‐JTMT mice compared to OVE animals.
Glomerular basement membrane (GBM) thickness
Diabetes‐induced GBM thickening is a hallmark of the disease and the significant (∼30%, P <0.05) GBM thickening in OVE mice was not unexpected (Fig. 6B). However, the similar (∼29%) (P <0.05) protection/decrease in OVE‐JTMT GBM thickness (compared to OVE mice) was less predictable. In this regard, FVB and OVE‐JTMT GBM thicknesses were virtually identical (FVB: 153.72 ± 16.98 nm vs. OVE‐JTMT: 154.79 nm ± 16.99 nm), and GBM thickness protection (29%) in OVE‐JTMT mice matched the diabetes‐driven increase (30%) seen in OVE mice.
Podocyte foot process width (FPw)
In FVB and JTMT control mice, FPW were nearly identical (FVB: 0.593 ± 0.18 µm vs. JTMT: 0.553 mm ± 0.13 µm.) OVE mice, however, showed significantly increased (23%, P < 0.05) FPW (effacement) compared to FVB mice. Furthermore, like the NFEA and GBM thickness parameters, OVE‐JTMT mice exhibited a modest, but statistically significant (∼9%, P < 0.05) protection/decrease in FPW relative to age‐matched OVE animals (Fig. 6C).
Glomerular Cell Morphometry
Glomerular volume (VG)
Since glomerular hypertrophy is a nearly constant feature of DN, VG is an important renal value. It also carries a formidable practical value, because glomerular cell numbers (N) can be calculated as the product of VG and nuclear cell density (Nv). In the current study, OVE mice exhibited significantly increased VG (165%, P < 0.05) compared to FVB controls (Fig. 7). However, glomerulomegaly was significantly protected/reduced (∼27%, P < 0.05) in MT targeted bi‐transgenic OVE‐JTMT mice compared to their age‐matched OVE counterparts.
Figure 7.
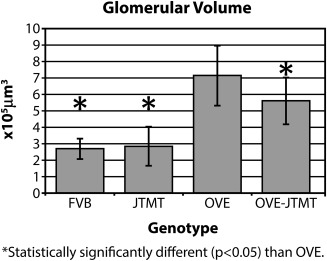
Mean glomerular volume in FVB, JTMT, OVE and OVE‐JTMT mice ± S.D. FVB vs. OVE, OVE‐JTMT, and OVE‐JTMT vs. OVE all show statistically significant difference. OVE mice show the greatest glomerular volume, while OVE‐JTMT mice show significant renoprotection against glomerulomegaly, and though glomerular volume is not reduced to control levels, it is mitigated significantly (∼29%).
Mean endothelial cell density (eNV), mesangial cell density, (mNV), podocyte density (pNV) and total glomerular (gNV) nuclear (cellular) density
Compared to FVB animals, OVE mice demonstrated significant decreases (∼142%) in pNV (Fig. 8A). Podocyte density was least in OVE mice (2.56 × 10 − 4/µm3), which was consistent with OVE mice exhibiting the greatest VG (7.14 × 105 µm3, Fig. 7). Likewise eN v decreased significantly in OVE mice (82%, P < 0.05, Fig. 8B.). Like the pNV, OVE mice exhibited the least eN v, and the greatest VG (Figs. 7 and 8B). Mesangial cell density was not significantly different in FVB, JTMT, OVE or OVE‐JTMT mice (Fig. 8C). In non‐diabetic (FVB and JTMT) mice, total glomerular cell density (gN v) were similar to each other (23.22 × 10− 5 cells/µm3 and 27.57 × 10 − 5 cells/µm3 ∼19%, Fig. 8D), and remarkably, diabetic (OVE and OVE‐JTMT) animals showed similar gN v values (14.58 × 10 − 5 cells/µm3 and 17.35 × 10 − 5 cells/µm3 ∼19%. Fig. 8D.)
Figure 8.
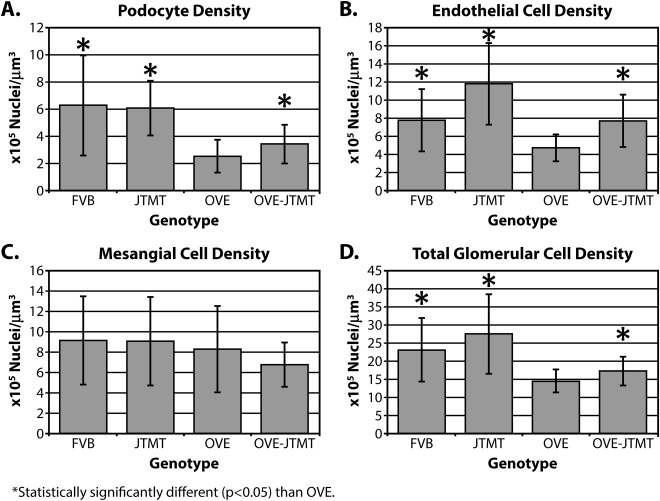
Mean podocyte, endothelial cell, mesangial cell, and total glomerular nuclear density in FVB, JTMT, OVE and OVE‐JTMT mice ± S.D.: While mesangial cells showed no significant differences in cell density in any of the four genotypes studied, podocyte, endothelial cell and total glomerular glomerular cell density exhibited significant differences in OVE vs. FVB, and OVE‐JTMT vs. OVE comparisons. FVB mice showed the greatest density in mesangial cells and podocytes, while JTMT mice exhibited the greatest density in endothelial cells and in total glomerular density. On the contrary, OVE diabetics exhibited the least cell density in all cell types except mesangial cells. Interestingly, when the endothelial cell‐targeted MT transgene was present, both diabetic (OVE‐JTMT) and non‐diabetic (JTMT) control mice showed strong tendencies toward increased endothelial cell density.
Mean endothelial cell number (eN), mesangial cell number (mN), podocyte number (pN) and total glomerular nuclear (cellular) number (gN) per glomerulus (N)
It is widely accepted that podocytes do not undergo mitosis in vivo (Kriz et al., 1998, Carlson et al., 2013). This is consistent with data in the current investigation, which show no significant pN differences in FVB, JTMT, OVE, or OVE‐JTMT mice (Fig. 9A). Contrariwise, eN clearly increases in diabetes, and OVE eN exceeds FVB eN by 127 cells (∼64%; P <0.05; Fig. 9B).
Figure 9.
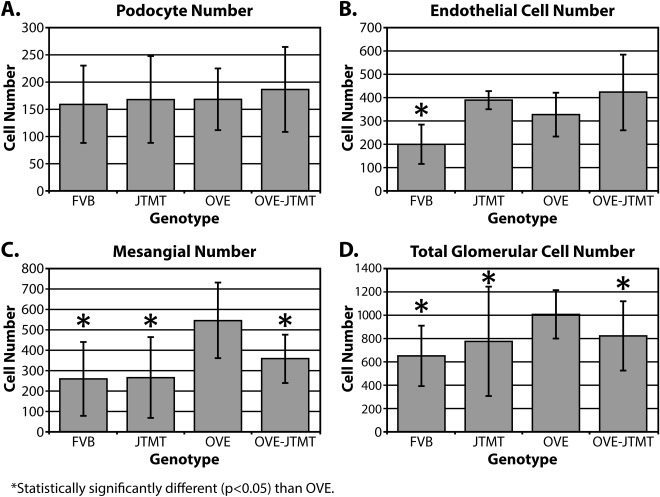
Mean podocyte, endothelial cell, mesangial cell, and total glomerular cell numbers per glomerulus in FVB, JTMT, OVE and OVE‐JTMT mice ± S.D. Podocyte cell number was not significantly different in any of the four genotypes studied. Endothelial cell proliferation increased significantly in diabetic mice, with or without the presence of the MT transgene. Mesangial cell number (i.e., 547), was significantly greater in OVE mice than any other genotype and were sufficient to drive the total glomerular cell number in OVE mice significantly greater than in any other genotype. OVE‐JTMT mice had an intermediate total glomerular cell number, and total glomerular cells was least in FVB mice.
Interestingly, MT overexpression stimulates but does not mitigate EC proliferation in both non‐diabetic (JTMT) and diabetic (OVE‐JTMT) mice. In JTMT, the average increase (vs. FVB) is ∼95%; ∼188 cells), and in OVE‐JTMT eN shows an average increase (vs. OVE) of (29%; ∼95 cells; Fig. 9B).
Mesangial cell number also increases in OVE mice (vs. FVB mice), and the average mN increase is ∼287 cells. Unlike the EC population, mN is decreased ∼189 cells, (or ∼53%) when MT targeted EC in OVE‐JTMT mice are present (Fig. 9C). With a 63% increase in eN, and a concomitant 113% increase in mN, (no change in pN), a total glomerular cell increase in OVE is not unexpected. Our data confirm this and show that in OVE mice, gN is ∼1007 cells compared with ∼651 in age‐matched FVB mice, an increase of ∼356 cells (∼55%; P <0.05; Fig. 9D). Moreover, data in the current investigation verified the expected eN increase and mN decrease in OVE‐JTMT mice (relative to OVE animals). OVE‐JTMT gN was ∼823 cells, an increase of ∼172 cells (47%) compared to FVB mice, and a reduction of 184 (∼9%, P <0.05) relative to OVE animals.
DISCUSSION
Data in the current study demonstrate that targeted MT overexpression in vascular EC results in significant renoprotection against several highly refractory complications of DN. These include GFB injury, glomerulomegaly, and altered glomerular cell densities and numbers.
Although the specific sequence of diabetic pathogenic events leading ultimately to end stage renal disease (ESRD) is not well understood, several molecular mechanisms are believed to play major roles in the pathogenesis of DN (Viberti et al., 1994). An interesting “unifying” hypothesis (Brownlee, 2005) has been proposed, that implicates hyperglycemia‐driven increases in ROS as the focal point of a final common pathway leading to chronic renal complications, renal decompensation, and finally ESRD.
Our findings provide rigorous TEM stereometric evidence for molecular intervention in the overproduction of ROS by EC and demonstrate that in several specific experimental diabetic paradigms, targeted MT overexpression in EC results in significant mitigation of an array of structural diabetic pathobiological consequences including GFB injury, glomerulomegaly and altered glomerular cell densities and numbers.
Body Features and Fluids
Although body weights were relatively similar in all genotypes in the present study, significant diabetic nephromegaly (enlarged kidneys) was a consistent DN feature in OVE and OVE‐JTMT animals. Similar enlargement is reported in human diabetics where their increased size and weight most often are attributed to hyperfiltration, or otherwise unknown causes (Hughson et al., 2002). Interestingly, however, this condition often accompanies increased glomerular volume (V G), a common chronic complication of DN (Bilous et al., 1989). However, the demonstrated attenuation of both by MT overexpression in glomerular cells is not expected (Zheng et al., 2008; Carlson et al., 2013). Although nephromegaly is not generally considered a result of oxidative stress, its parallel mitigation in animals overexpressing MT in glomerular cells, favors such an interpretation.
Importantly, blood glucose and HbA1c were not reduced in animals carrying the MT transgene. This confirmed the sustained maintenance of diabetic conditions in transgenic animal progeny (OVE‐JTMT) in the present study, and showed that any mitigation of DN complications in OVE‐JTMT animals could not be the result of reduced diabetes.
Glomerular Filtration Barrier (GFB) Damage
Non‐fenestrated endothelial area, (NFEA)
In DN, injury to GFB structural components including NFEA, GBM thickening, and increased podocyte foot process width (FPW) typically are manifested following the onset of albuminuria. Although no direct relationship has been established, albumin is known to damage renal tubules (Kralik et al., 2009) and it seems likely that leaking albumin or other nephropathic agents may result in GFB injury.
Glomerular endothelial damage is difficult to measure, but quantification of its most distinctive feature (glycocalyx‐filled glomerular capillary endothelial fenestrations) could offer some useful information. Our previous TEM stereometric investigation (Teiken et al., 2008) compared GFB features (including NFEA), in aging FVB and diabetic OVE mice and the data showed that in diabetic mice, significant DN injury was age‐related and generally occurred in all GFB components after five months, and well after substantial albuminuria had been established. Likewise, data in the current study shows that by 150 days, NFEA is significantly increased in OVE mice (relative to FVB controls), which is believed to indicate increased DN severity in patients (Toyoda et al., 2008). Although increased NFEA cannot be attributed exclusively to DN, as similar changes have been reported in serum sickness [as well as other conditions (Satchell and Braet, 2009)], our data show that age‐matched OVE‐JTMT mice exhibit significantly decreased (∼11%) NFEA relative to OVE diabetics. Thus, ROS reduced EC targeted MT scavenging may be at least partially responsible for recovery from increased percent NFEA.
Glomerular basement membrane (GBM)
GBM is compositionally similar to other basement membranes (Tsilibary, 2003, Miner, 2011) but its histoarchitectural position (interposed between two cell types), thickness, and other attributes including isoforms of major GBM proteins, (Miner, 2011) sets them apart.
GBM thickening in diabetic human (Reddi, 1978) and a variety of animal models (Romen et al., 1980) has been thoroughly investigated, and though the precise metabolic processes by which basement membranes increase in width have not been clearly uncovered, a number of pathogenetic mechanisms are implicated, most of which include hyperglycemia‐induced paradigms (Tsilibary, 2003)
Utilizing the “gold standard” orthogonal intercept technique, we carried out a number of GBM thickness studies in OVE diabetics and age‐matched FVB controls (Carlson et al., 2003; Teiken et al., 2008, 2011; Carlson et al., 2013), all of which demonstrated that diabetic GBMs were ubiquitously wider than control mice. Importantly, however, in a recent paper (Carlson et al., 2013), we showed for the first time, that increased GBM thickness was completely eliminated, and in mice with targeted MT overexpression in podocytes, widths were equal to controls.
An identical experimental design was chosen in the current study in an effort to demonstrate similar protection by targeted MT overexpression in EC. Accordingly, GBMs were compared in age‐matched genotypes. As expected, OVE GBMs were significantly thicker than FVB controls. But more importantly (as shown in Fig. 6), in OVE‐JTMT mice GBM thickening was completely eliminated, and GBM widths were basically identical with controls.
These GBM studies, as well as an analyses of myocardial capillary basement membrane thickness (Velic et al., 2013), which was the first to demonstrate complete elimination of increased diabetic basement membrane thickness in transgenic animals (OVE‐MT), strongly suggest that GBM thickening may be the result of hyperglycemia‐driven ROS production and subsequent decreased extracellular matrix turnover (Tsilibary, 2003). Furthermore, it affirms the concept that both EC and podocyte targeted MT scavenging of ROS contribute to renoprotection against increased GBM thickness.
Podocyte foot process width, FPw
Together with their intervening slit membranes, podocytes foot processes are the final major structural element in the GFB. (Tsilibary, 2003).
Early in clinical DN, FPW is enlarged and with time, it continues to increase (Binder et al., 1999) until intervening slit membranes are displaced and a continuous layer replaces discrete foot processes (effacement).
Podocytes are a target for oxidative damage (Arif et al., 2014). Moreover, they can act as a source of free radical production (Greiber et al., 1998). It is likely, therefore, that increased OVE FPW may be the result of hyperglycemia‐induced ROS production. Data in our previous report (Carlson et al., 2013), support this paradigm and show a significant FPW protection in animals overexpressing MT specifically in podoctyes relative to OVE diabetics.
Also, the current investigation shows that FPW is mitigated 9% in OVE‐JTMT mice vs. OVE diabetics (P < 0.05). It seems possible therefore, that specific MT overexpression in both podocytes and EC may be partially responsible for scavenging ROS and could lead to protection against increased podocyte effacement.
Furthermore, since it is widely accepted that ROS play roles in increased NEFA, increased GBM width, and increased FPW (Vijayalingam et al., 2004; Zheng et al., 2008; Binder et al., 1999), it seems reasonable to suggest that failure of a combined “endothelio‐podocytic” response to hyperglycemia‐driven oxidative stress may lead to inadequate free radical scavenging, as a possible global cause of diabetic GFB injury.
Glomerular Cell Morphometry
Mean glomerular volume, VG
Glomerular volume, (V G) is a significant benchmark that is often inversely related to N. Moreover, increased V G is a widely accepted complication of human DN (Viberti et al., 1994; Zheng et al., 2004; Zheng et al., 2008 Teiken et al., 2008, 2011), and we have shown that it is a consistent feature of OVE mice at ∼150 days of age (Teiken et al., 2008, 2011, Carlson et al., 2013)
However, increased V G often is accompanied by renal enlargement (nephromegaly, and unexpectedly, both conditions are mitigated by MT overexpression in glomerular cells. In this regard, Zheng and co‐workers (2008) reported that a significant (∼165%) increase in V G in severely diabetic OVE mice V G was reduced by 28% (vs. OVE mice) in OVE‐Nmt transgenics. Remarkably, in the current study, OVE V G also exceeded FVB control mice by ∼165% and was significantly mitigated (27%) in OVE‐JTMT animals.
Mean endothelial cell (eNV), mesangial cell (mNV), podocyte (pNV), and total glomerular (gNV) nuclear (cellular) density
Our data show that pNV in 150 day‐old OVE mice is significantly reduced (∼142%) relative to age‐matched FVB controls, which is consistent with findings in other investigations (Teiken et al., 2008; Carlson et al., 2013) of pN V in the same age and genotypes. Although in recent investigations (Teiken et al. 2008; Carlson et al., 2013; and current study), pN V was significantly reduced in diabetic mice, pN was nearly identical in diabetics and controls. This was somewhat surprising, as pN is generally reduced (Dalla Vestra et al., 2003). both in diabetic patients (Pagtalunan et al., 1997; Dalla Vestra et al., 2003; Susztak et al., 2006) and diabetic animal models (Kriz et al., 1998; Petermann et al., 2004). Also comparing current cell density findings with data in our previous study (Teiken et al., 2011; Carlson et al., 2013) showed remarkably similar significant reductions in eN v (82% vs. 67%) and gN v (61% vs. 63%) which are consistent with findings of a recent aging analysis (Teiken et al., 2011). In the current study mN v is not significantly different in the four genotypes and pN v, eN v, mN v, and gN v were not significantly different in diabetic (OVE and OVE‐JTMT) animals despite significantly increased V G in these mice. In our most recent work (Teiken et al., 2011, Carlson et al., 2013, and current), all cell types in diabetic animals trend toward decreased density (relative to FVB and JTMT controls), and are inversely related to their respective V G.
Mean endothelial cell (eN), mesangial cell (mN), podocyte (pN) and total glomerular (gN) nuclear (cellular) number per glomerulus (N)
Glomerular cell number is highly dynamic and could be perhaps the most important indicator of renal tissue injury and decompensation. Our data indicate that non‐diabetic control (FVB and JTMT) animals show similar podocyte and mesangial cell numbers, while endothelial cells carrying the MT transgene (JTMT, OVE‐JTMT) show significantly greater (∼96%, ∼111% respectively) numbers than their FVB counterparts.
On the contrary, however, mesangial cells from diabetic (OVE) mice are highly prolific (∼113%), much as might be expected from their known in vivo activity in the human diabetic mesangium. Mesangial cells carrying the MT transgene are considerably less prolific (∼38%), possibly indicating “molecular recognition” of conditions simulating reduced mesangial oxidative stress in human DN. Interestingly, these conditions (i.e., presence of the MT transgene and increased mesangial proliferation) are EC prolific (∼60% in animals overexpressing MT), and may simulate conditions of increased extracellular matrix production in late stages of human chronic DN.
The fruitful utility of excellent imaging and precise methodologies
For many years, precise dimensions of microscopic cellular and extracellular renal glomerular structures, and absolute numbers of cells per glomerulus have been sought in human and various animal models in health and disease (Viberti et al., 1994, Drummond and Mauer, 2002; Petermann et al., 2004; Basgen et al., 2006; Susztak et al., 2006 Teiken et al., 2011). Though many possible cytopathic “trends” have been established, it is difficult rigorously to compare the data from disparate investigators, due primarily to variability in the methods used to collect and calculate reliable numbers.
On the contrary, TEM morphometric techniques, including those used in the current investigation generate highly verifiable data that are “uncharacteristically reliable.” Our laboratory has been particularly fortunate in this regard as we have had the luxury of comparing “new” measurements with scrupulously maintained data derived from a series of published studies (Zheng et al., 2008, Tieken et al., 2008; Carlson et al., 2013; Velic et al., 2013; Carlson et al., Current study) in which identical methods were applied to the same animal genotypes, and ages.
The remarkable repeatability of TEM stereometric methodology coupled with frequent similarities in data and measurements offer uniquely effective protocols for future studies requiring accurate cellular and extracellular morphometric data.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the superb electron microscopic technical assistance of Laurie Kim Young. They thank Dr. Patrick Carr for expert assistance with statistical analysis. The authors acknowledge use of the Edward C. Carlson Imaging and Image Analysis Core Facility. They extend sincere gratitude to Elizabeth Schaff for clerical assistance and to Dr. John Swinscoe for technical consultation. There are no potential conflicts of interest regarding financial and personal relationships between any of the authors and other individuals that might bias their work. Also, no part of this manuscript has been submitted or is currently being considered for publication elsewhere. The ethical background for this study derives from an ethically neutral hypothesis and all data collection and interpretation have been carried out in the absence of unethical behavior. No ethical committee has reviewed the manuscript.
The copyright line for this article was changed on 14 March 2017 after original online publication
Literature Cited
- Arif E, Rathore Y, Kumari B, Ashish F, Wong H, Holzman L, Nihalani D. 2014. Slit diaphragm protein Neph1 and its signaling: A novel therapeutic target for protection of podocytes against glomerular injury. J Biol Chem 289:9502–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basgen J, Nicholas S, Mauer M, Rozen S, Nyengaard J. 2006. Comparison of methods for counting cells in the mouse glomerulus. Nephron Exp Nephrol 103:e:139–e148. [DOI] [PubMed] [Google Scholar]
- Baur P, Stacey T. 1977. The use of PIPES buffer in the fixation of mammalian and marine tissues for electron microscopy. J Microsc 109:315–327. [DOI] [PubMed] [Google Scholar]
- Baynes J. 1991. Role of oxidative stress in development of complications in diabetes. Diabetes 40:405–412. [DOI] [PubMed] [Google Scholar]
- Bilous R, Mauer M, Sutherland D, Steffes M. 1989. Mean glomerular volume and rate of development of diabetic nephropathy. Diabetes 38:1142–1147. [DOI] [PubMed] [Google Scholar]
- Binder C, Weiher H, Exner M, Kerjaschki D. 1999. Glomerular overproduction of oxygen radicals in Mpv17 gene‐inactivated mice causes podocyte foot process flattening and proteinuria: A model of steroid‐resistant nephrosis sensitive to radical scavenger therapy. Am J Pathol 154:1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M. 2005. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 54:1615–1625. [DOI] [PubMed] [Google Scholar]
- Carlson E, Audette J, Klevay L, Nguyen H, Epstein P. 1997. Ultrastructural and functional analyses of nephropathy in calmodulin‐ induced diabetic transgenic mice. Anat Rec 247:9–19. [DOI] [PubMed] [Google Scholar]
- Carlson E, Audette J, Veitenheimer N, Risan J, Laturnus D, Epstein P. 2003. Ultrastructural morphometry of capillary basement membrane thickness in normal and transgenic diabetic mice. Anat Rec 271:A: 332–341. [DOI] [PubMed] [Google Scholar]
- Carlson E, Chhoun J, Laturnus D, Bikash K, Berg B, Zheng S, Epstein P. 2013. Podocyte‐specific overexpression of metallothionein mitigates diabetic complications in the glomerular filtration barrier and glomerular histoarchitecture: A transmission electron microscopy stereometric analysis. Diabetes Metab Res Rev 29:113–124. [DOI] [PubMed] [Google Scholar]
- Dalla Vestra M, Masiero A, Roiter A, Saller A, Crepaldi G, Fioretto P. 2003. Is podocyte injury relevant in diabetic nephropathy? Studies in patients with type 2 diabetes. Diabetes 52:1031–1035. [DOI] [PubMed] [Google Scholar]
- Dische F. 1992. Measurement of glomerular basement membrane thickness and its application to the diagnosis of thin‐membrane nephropathy. Arch Pathol Lab Med 116:43–49. [PubMed] [Google Scholar]
- Drummond K, Mauer M. 2002. International Diabetic Nephropathy Study Group. The early natural history of nephropathy in type 1 diabetes: II. Early renal structural changes in type 1 diabetes. Diabetes 51:1580–1587. [DOI] [PubMed] [Google Scholar]
- Epstein P, Overbeek P, Means A. 1989. Calmodulin‐induced early‐onset diabetes in transgenic mice. Cell 58:1067–1073. [DOI] [PubMed] [Google Scholar]
- Giacco F, Brownlee M. 2010. Oxidative stress and diabetic complications. Circ Res 107:1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiber S, Munzel T, Kastner S, Muller B, Schollmeyer P, Pavenstadt H. 1998. NAD(P)H oxidase activity in cultured human podocytes: Effects of adenosine triphosphate. Kidney Int 53:654–663. [DOI] [PubMed] [Google Scholar]
- Ha H, Yu M, Choi J, Lee B. 2001. Activation of protein kinase C‐delta and C‐epsilon by oxidative stress in early diabetic rat kidney. Am J Kidney Dis 38:S204–S207. [DOI] [PubMed] [Google Scholar]
- Hughson M, Johnson K, Young R, Hoy W, Bertram J. 2002. Glomerular size and glomerulosclerosis: Relationships to disease categories, glomerular solidification, and ischemic obsolescence. Am J Kidney Dis 39:679–688. [DOI] [PubMed] [Google Scholar]
- Jensen E, Gunderson H, Osterby R. 1979. Determination of membrane thickness distribution from orthogonal intercepts. J Microsc 115:19–33. [DOI] [PubMed] [Google Scholar]
- Kashihara N, Haruna Y, Kanwar Y. 2010. Oxidative stress in diabetic nephropathy. Curr Med Chem 17:4256–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi M, Yamada M, Wada H, Okigaki T. 1992. Roles of active oxygen species in glomerular epithelial cell injury in vitro caused by puromycin aminonucleoside. Toxicology 72:329–340. [DOI] [PubMed] [Google Scholar]
- Kralik P, Long Y, Song Y, Yang L, Wei H, Coventry S, Zheng S, Epstein P. 2009. Diabetic albuminuria is due to a small fraction of nephrons distinguished by albumin‐stained tubules and glomerular adhesions. Am J Pathol 2175:500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz W, Gretz N, Lemley K. 1998. Progression of glomerular diseases: Is the podocyte the culprit? Kidney Int 54:687–697. [DOI] [PubMed] [Google Scholar]
- Mifsud S, Allen T, Bertram J, Hulthen U, Kelly D, Cooper M, Wilkinson‐Berka J, Gilbert R. 2001. Podocyte foot process broadening in experimental diabetic nephropathy: Amelioration with renin‐angiotensin blockade. Diabetologia 44:878–882. [DOI] [PubMed] [Google Scholar]
- Miner J. 2011. Glomerular basement membrane composition and the filtration barrier. Pediatr Nephrol. [2001 Feb15.e‐Pub ahead]. [DOI] [PMC free article] [PubMed]
- Pagtalunan M, Miller P, Jumping‐Eagle S, Nelson R, Myers B, Rennke H, Coplon NS, Sun L, Meyer T. 1997. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 99:342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann A, Pippin J, Krofft R, Blonski M, Griffin S, Durvasula R, Shankland S. 2004. Viable podocytes detach in experimental diabetic nephropathy: Potential mechanism underlying glomerulosclerosis. Neph Exp Nephrol 98:E144–E123. [DOI] [PubMed] [Google Scholar]
- Reddi A. 1978. Diabetic microangiopathy. I. Current status of the chemistry and metabolism of the glomerular basement membrane. Metabolism 27:107–124. [DOI] [PubMed] [Google Scholar]
- Romen W, Heck T, Rauscher G, Lange HU, Hempel K. 1980. Glomerular basement membrane turnover in young, old, and streptozotocin‐diabetic rats. Ren Physiol 3:324–329. [DOI] [PubMed] [Google Scholar]
- Satchell S, Braet F. 2009. Glomerular endothelial cell fenestrations: An integral component of the glomerular filtration barrier. Am J Physiol Renal Physiol 296:F947–F956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S. 1995. The role of reactive oxygen metabolites in glomerular disease. Annu Rev Physiol 57:245–262. [DOI] [PubMed] [Google Scholar]
- Susztak K, Raff A, Schiffer M, Bottinger E. 2006. Glucose‐induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55:225–233. [PubMed] [Google Scholar]
- Takemoto M, Asker N, Gerhardt H, Lundkvist A, Johansson B, Saito Y, Betsholtz C. 2002. A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol 161:799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teiken J, Audette J, Laturnus D, Zheng S, Epstein P, Carlson E. 2008. Podocyte loss in aging OVE26 diabetic mice. Anat Rec 291:114–121. [DOI] [PubMed] [Google Scholar]
- Teiken J, Grove B, Epstein P, Carlson E. 2011. Production of a new transgenic mouse (Jtmt) that specifically overerxpresses the antioxidant metallothionein in endothelial cells. FASEB J 868:Abstract. [Google Scholar]
- Teiken J, Epstein P, Carlson E. 2011. TEM stereometric analyses of glomeruli in aging OVE26 transgenic diabetic mice. Am J Nephrol 33:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Diabetes Control IINA and Complications Trial Research Group . 1993. The effect of intensive treatment of diabetes on the development and progression of long‐term complications in insulin‐dependent diabetes mellitus. N Engl J Med 329:977–986. [DOI] [PubMed] [Google Scholar]
- Toyoda M, Najafian B, Kim Y, Caramori M, Mauer M. 2007. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes 56:2155–2160. [DOI] [PubMed] [Google Scholar]
- Tsilibary E. 2003. Microvascular basement membranes in diabetes mellitus. J Pathol 200:537–546. [DOI] [PubMed] [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group . 1998. Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853. [PubMed] [Google Scholar]
- Vlassara H, Palace M. 2002. Diabetes and advanced glycation endproducts. J Intern Med 251:87–101. [DOI] [PubMed] [Google Scholar]
- Velic A, Laturnus D, Chhoun J, Zheng S, Epstein P, Carlson E. 2013. Diabetic basement membrane thickening does not occur in myocardial capillaries of transgenic mice when metallothionein is overexpressed in cardiac myocytes. Anat Rec 296:480–487. [DOI] [PubMed] [Google Scholar]
- Venable J, Coggeshall R. 1965. A simplified lead stain for use in electron microscopy. J Cell Biol 25:407–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viberti G, Wiseman M, Pinto J, Messent J. 1994. Diabetic Nephropathy in Joslin's Diabetes Mellitus 13th ed In: Kahn CR, Weir GC, editors. Philadelphia: Lea & Febiger; p 691–737. [Google Scholar]
- Vijayalingam S, Parthiban A, Shanmugasundaram K, Mohan V. 1996. Abnormal antioxidant status in impaired glucose tolerance and non‐ insulin‐dependent diabetes mellitus. Diabet Med 13:715–719. [DOI] [PubMed] [Google Scholar]
- Wallner E, Wada J, Tramonti G, Lin S, Srivastava S, Kanwar Y. 2001. Relevance of aldo‐keto reductase family members to the pathobiology of diabetic nephropathy and renal development. Renal Failure 23:311–320. [DOI] [PubMed] [Google Scholar]
- Wang J, Song Y, Elsherif L, Song Z, Zhou G, Prabhu S, Saari J, Cai L. 2006. Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation 113:544–554. [DOI] [PubMed] [Google Scholar]
- Weibel E, Gomez D. 1962. A principle for counting tissue structures on random sections. J App Physiol 17:343–348. [DOI] [PubMed] [Google Scholar]
- Weibel E. 1979. Stereological methods, Vol. 1: Practical methods for biological morphometry. London: Academic Press; p 44–45. [Google Scholar]
- White K, Bilous R. 2004. Estimation of podocyte number: A comparison of methods. Kidney Int 66:663–667. [DOI] [PubMed] [Google Scholar]
- Wolff S, Jiang Z, Hunt J. 1991. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Radic Biol Med 10:339–352. [DOI] [PubMed] [Google Scholar]
- Xu J, Huang Y, Li F, Zheng S, Epstein P. 2010. FVB mouse genotype confers susceptibility to OVE26 diabetic albuminuria. Am J Physiol Renal Physiol 299:F487–F494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Carlson E, Yang L, Kralik P, Huang Y, Epstein P. 2008. Podocyte‐specific overexpression of the antioxidant metallothionein reduces diabetic nephropathy. J Am Soc Nephrol 19:2077–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Noonan W, Metreveli N, Coventry S, Kralik P, Carlson E, Epstein P. 2004. Development of late‐stage diabetic nephropathy in OVE26 diabetic mice. Diabetes 53:3248–3257. [DOI] [PubMed] [Google Scholar]
- Ziyadeh F. 2004. Mediators of diabetic renal disease: The case for tgf‐Beta as the major mediator. J Am Soc Nephrol 15 Suppl 1:S55–S57. [DOI] [PubMed] [Google Scholar]