

Abstract
Key points
It is controversial whether glutamate can leak out of vesicles in the nerve terminal.
To address this issue, we abolished vesicular glutamate uptake by washing out presynaptic cytosolic glutamate or by blocking vacuolar ATPase activity using bafilomycin A1.
In the absence of vesicular glutamate uptake, both spontaneous and nerve‐evoked EPSCs underwent a rundown, suggesting that vesicular glutamate can leak out of vesicles.
However, the rundown of evoked EPSCs was caused mainly by accumulation of unfilled vesicles after exocytic release of glutamate, suggesting a minor influence of glutamate leakage on synaptic transmission.
Abstract
Glutamate leaks out of synaptic vesicles when the transvesicular proton gradient is dissipated in isolated vesicle preparations. In the nerve terminal, however, it is controversial whether glutamate can leak out of vesicles. To address this issue, we abolished vesicular glutamate uptake by washing out presynaptic cytosolic glutamate in whole‐cell dialysis or by blocking vacuolar ATPase using bafilomycin A1 (Baf) at the calyx of Held in mouse brainstem slices. Presynaptic glutamate washout or Baf application reduced the mean amplitude and frequency of spontaneous miniature (m)EPSCs and the mean amplitude of EPSCs evoked every 10 min. The percentage reduction of mEPSC amplitude was much less than that of EPSC amplitude or mEPSC frequency, and tended to reach a plateau. The mean amplitude of mEPSCs after glutamate washout or Baf application remained high above the detection limit, deduced from the reduction of mEPSC amplitude by the AMPA receptor blocker 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione. Membrane capacitance measurements from presynaptic terminals indicated no effect of glutamate washout on exocytosis or endocytosis of synaptic vesicles. We conclude that glutamate can leak out of vesicles unless it is continuously taken up from presynaptic cytosol. However, the magnitude of glutamate leakage was small and had only a minor effect on synaptic responses. In contrast, prominent rundowns of EPSC amplitude and mEPSC frequency observed after glutamate washout or Baf application are likely to be caused by accumulation of unfilled vesicles in presynaptic terminals retrieved after spontaneous and evoked glutamate release.
Keywords: calyx of held, glutamate, presynaptic terminal, synaptic transmission
Key points
It is controversial whether glutamate can leak out of vesicles in the nerve terminal.
To address this issue, we abolished vesicular glutamate uptake by washing out presynaptic cytosolic glutamate or by blocking vacuolar ATPase activity using bafilomycin A1.
In the absence of vesicular glutamate uptake, both spontaneous and nerve‐evoked EPSCs underwent a rundown, suggesting that vesicular glutamate can leak out of vesicles.
However, the rundown of evoked EPSCs was caused mainly by accumulation of unfilled vesicles after exocytic release of glutamate, suggesting a minor influence of glutamate leakage on synaptic transmission.
Abbreviations
- Baf
bafilomycin A1
- Cm
membrane capacitance
- CNQX
6‐cyano‐7‐nitroquinoxaline‐2,3‐dione
- MNTB
medial nucleus of the trapezoid body
- P
postnatal day
- RT
room temperature
- VGLUTs
vesicular glutamate transporters
Introduction
Del Castillo & Katz (1954) described synaptic responses as integer multiples of the spontaneous miniature synaptic response, or quanta, at the frog neuromuscular junction. This quantal nature of neurotransmission was confirmed at the calyx of Held glutamatergic synapse in rodent brainstem slices (Sahara & Takahashi, 2001). After exocytic release of neurotransmitters, synaptic vesicles are retrieved by endocytosis, refilled with neurotransmitter and reused for another round of synaptic transmission (Heuser & Reese, 1973; Rizzoli & Betz, 2005). At the calyx of Held, the magnitude of refilling of synaptic vesicles with glutamate depends upon presynaptic cytosolic glutamate concentrations (Ishikawa et al. 2002; Yamashita et al. 2009; Hori & Takahashi, 2012), and washout of glutamate by whole‐cell dialysis gradually reduces the amplitudes of evoked EPSCs and spontaneous miniature (m)EPSCs (Ishikawa et al. 2002; Hori & Takahashi, 2012). During glutamate washout, the magnitude of rundown differs between EPSCs and mEPSCs, with much steeper decline in the EPSC amplitude (Ishikawa et al. 2002; Wu et al. 2007). At the neuromuscular junction (Parsons et al. 1999) or at the hippocampal synapse in culture (Zhou et al. 2000; Ikeda & Bekkers, 2009), pharmacological block of vesicular neurotransmitter transporters slightly reduces (Zhou et al. 2000) or does not reduce at all (Parsons et al. 1999; Ikeda & Bekkers, 2009) the amplitude of spontaneous synaptic responses. Thus, it is controversial whether neurotransmitter can leak out of synaptic vesicles in the nerve terminal. By blocking vesicular glutamate uptake at the calyx of Held, we investigated whether, and to what extent, glutamate can leak out of vesicles.
Methods
Ethical approval
All experiments were performed in accordance with the guidelines of the Physiological Society of Japan and Doshisha University and were approved by the local committee for handling experimental animals in Doshisha University.
Slice preparation and solutions
Transverse brainstem slices (175 μm thick) containing the medial nucleus of the trapezoid body (MNTB) were prepared from postnatal day (P) 12–15 C57BL6 mice. Mice were deeply anaesthetized by inhalation of isoflurane and killed by decapitation prior to rapid dissection of the brainstem. Slices were incubated for 1 h at 36–37℃ in artificial cerebrospinal fluid containing (mm): 125 NaCl2, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 glucose, 3 myo‐inositol, 2 sodium pyruvate and 0.5 ascorbic acid (pH adjusted to 7.3 with 95% O2–5% CO2; 310 mosmol kg−1) and maintained thereafter at room temperature (RT, 24–28℃), at which all recordings were made. The artificial cerebrospinal fluid routinely contained picrotoxin (100 μm) and strychnine hydrochloride (0.5 μm) to block GABAA receptors and glycine receptors, respectively. It also contained d(−)‐2‐amino‐5‐phosphonopentanoic acid (50 μm) to block NMDA receptors. For recording presynaptic Ca2+ currents, the artificial cerebrospinal fluid contained 10 mm tetraethylammonium chloride, 0.5 mm 4‐aminopyridine, 1 μm tetrodotoxin, 10 μm bicuculline methiodide and 0.5 μm strychnine hydrochloride. Principal neurons in the MNTB and the calyx of Held presynaptic terminal were visualized under an upright microscope (Axioskop, Carl Zeiss; BX51WI, Olympus) using a ×60 water‐immersion objective (Olympus). Simultaneous whole‐cell recordings were made from the calyx of Held and MNTB principal neurons, using Axopatch 700A amplifiers. The postsynaptic pipette solution contained (mm): 140 CsCl, 40 Hepes, 5 EGTA, 1 MgCl2 and 5 QX‐314 (pH 7.3; 320 mosmol kg−1). The presynaptic pipette solution contained (mm): 100 potassium methanesulfonate, 30 KCl, 40 Hepes, 0.5 EGTA, 12 phosphocreatine (sodium salt), 3 ATP (magnesium salt), 0.5 GTP (sodium salt) and 0 or 3 potassium glutamate (pH 7.3; 310–320 mosmol kg−1). The postsynaptic pipette was pulled to 5–7 MΩ and had a series resistance of 10–25 MΩ, which was compensated by up to 75% for a final value of 7 MΩ. The resistance of the presynaptic pipette was 7–10 MΩ, and series resistance was typically 14–20 MΩ, which was not compensated. When the postsynaptic series resistance increased by >15 MΩ during recording, data were discarded. The postsynaptic holding potential was −70 mV. The liquid junction potential between the pipette and the external solution was not corrected. Bafilomycin A1 (Baf; Wako Pure Chemical Industries, Tokyo, Japan) was dissolved in DMSO and bath applied at 5 μm (final DMSO, 0.5%) for 100 s.
Membrane capacitance (C m) measurements
Membrane capacitance measurements were made from calyx of Held presynaptic terminals in the whole‐cell configuration at RT (Yamashita et al. 2010; Eguchi et al. 2012). Calyceal terminals were voltage clamped at a holding potential of −80 mV, and a sinusoidal voltage command with a peak‐to‐peak voltage of 60 mV was applied at 1 kHz. The presynaptic pipette solution contained (mm): 125 caesium methanesulfonate, 30 CsCl, 10 Hepes, 0.5 EGTA, 12 disodium phosphocreatine, 3 MgATP, 1 MgCl2, 0.3 Na2GTP and 0 or 3 caesium glutamate (pH 7.3; 315–320 mosmol kg−1). Single‐pulse step depolarization (to +10 mV, 10 ms) was used to induce presynaptic I Ca. Membrane capacitance changes within 450 ms of square‐pulse stimulation were excluded from analysis to avoid contamination by conductance‐dependent capacitance artifacts (Yamashita et al. 2005). To avoid the influence of capacitance drift on baseline, we discarded data when the baseline drift measured 0–10 s before stimulation was >5 fF s−1. When the capacitance baseline drift was 1–5 fF s−1, we subtracted a linear regression line of the baseline from the data for the baseline correction.
Data were acquired at a sampling rate of 100 kHz, using an EPC‐10 patch‐clamp amplifier controlled by PatchMaster software (HEKA Elektronik, Lambrecht, Germany) after online filtering at 5 kHz. The recording pipette was pulled to 5–7 MΩ and had a series resistance of 9–14 MΩ, which was compensated by up to 50% for a final value of 7 MΩ. Care was taken to maintain series resistance <14 MΩ to allow dialysis of the terminal with pipette solution.
Data analysis
Synaptic currents were low‐pass filtered at 10 kHz. Data were digitized at 50 kHz using Digidata 1320A (Axon Instruments) and analysed using IGOR Pro 6 (WaveMetrics Inc., Lake Oswego, OR, USA). Spontaneous mEPSCs were detected using a sliding template method implemented in IGOR Pro 6. The template was made by averaging eye‐selected 50–100 mEPSCs.
Statistical analysis
All statistical analyses were performed using IBM‐SPSS 24‐sofware (SPSS Inc., Chicago, IL, USA). The effects of glutamate washout were analysed by a repeated‐measures ANOVA, with glutamate concentrations ([Glu], 3 or 0 mm) as a between‐subjects factor and time (0, 10, 20 and 30 min after glutamate washout) as a within‐subjects factor. The effects of Baf were also analysed by a repeated‐measures ANOVA, with drug (DMSO or Baf) as a between‐subjects factor and time (before drug application, 0, 10, 20 and 30 min after drug application) as a within‐subjects factor. Bonferroni tests were used for post hoc comparisons. All data were expressed as means ± SEM.
Results
Washout of presynaptic cytosolic glutamate and block of vacuolar ATPase with bafilomycin A1
Glutamate is concentrated in synaptic vesicles at 60–150 mm (Burger et al. 1989) by vesicular glutamate transporters (VGLUTs; Bellocchio et al. 2000; Takamori et al. 2000) using the transvesicular proton gradient produced by vacuolar ATPase (Naito & Ueda, 1985). The endogenous cytosolic glutamate concentration in presynaptic terminals is 1–10 mm at the calyx of Held in different postnatal periods (Ishikawa et al. 2002; Yamashita et al. 2009). In simultaneous whole‐cell recordings from a calyx of Held presynaptic terminal and a postsynaptic MNTB neuron in slices of mouse brainstem, we dialysed presynaptic terminals with a glutamate‐free pipette solution (Ishikawa et al. 2002; Yamashita et al. 2009; Hori & Takahashi, 2012). We evoked EPSCs every 10 min (Fig. 1) and recorded spontaneous mEPSCs, which represent quantal, or single vesicular, EPSCs at this synapse (Sahara & Takahashi, 2001). After glutamate washout, EPSCs decreased in amplitude with a time constant of 22 min, whereas mEPSC amplitude underwent a less significant decline, with a tendency to reach a plateau (Fig. 1 B). These results are similar to those reported by Ishikawa et al. (2002), who evoked EPSCs at 0.1 Hz. During glutamate washout, the mEPSC frequency decreased from 33 ± 12 to 3.6 ± 2.0 Hz (n = 5 pairs) in 30 min (Fig. 1 C). In control experiments, when glutamate was included in presynaptic pipettes at 3 mm, no significant change was observed in the mean amplitude of EPSCs, mEPSCs or the mean frequency of mEPSCs. The number of vesicles undergoing spontaneous exocytosis during glutamate washout for 30 min was estimated as 24,700 ± 6700 (n = 5 pairs). Thus, fast rundown of EPSCs can be explained, at least in part, by an increased number of empty vesicles involved in EPSCs.
Figure 1. Rundowns of EPSCs and miniature (m)EPSCs after whole‐cell washout of cytosolic glutamate in the presynaptic terminal.
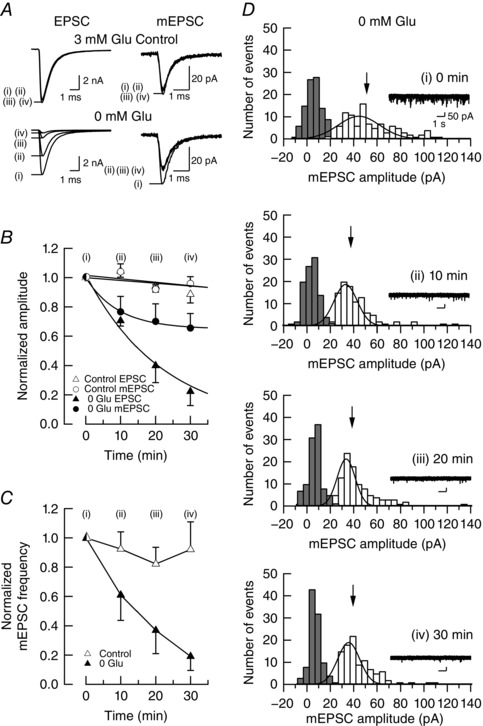
A, sample traces of EPSCs and mEPSCs, before and 10–30 min after glutamate washout (lower traces, superimposed), and controls with 3 mm glutamate in presynaptic pipettes (upper traces). B, mean amplitudes of EPSCs (triangles) and mEPSCs (circles) at different time periods after glutamate washout (filled symbols) or controls with 3 mm glutamate (open symbols). Each data point was derived from five experiments and normalized to the mean amplitudes at time 0 immediately after rupturing presynaptic membrane (ordinate). Error bars indicate ±SEMs in this and the following figures. Exponential curves were best fitted to data points with an equation of , where the time constant (τ) and I 0 were 22 min and 0, respectively, for EPSCs, whereas they were 9.4 min and 0.65, respectively, for mEPSCs. At time 0, mean amplitudes of evoked EPSCs were 7.5 ± 0.5 nA (3 mm Glu control, n = 5 pairs) and 7.8 ± 0.7 nA (0 mm Glu, n = 5 pairs) and those of mEPSCs were 35 ± 1.4 pA (3 mm Glu control, n = 5 pairs) and 43 ± 7.2 pA (0 mm Glu, n = 5 pairs). Glutamate concentrations ([Glu]) had significant effects on the amplitude of mEPSCs (repeated‐measures ANOVA: main effect of [Glu], F 1,8 = 5.03, P > 0.05; main effect of time, F 3,24 = 8.3, P < 0.001; [Glu] × time interaction, F 3,24 = 4.2, P < 0.05) and that of EPSCs (repeated‐measures ANOVA: main effect of [Glu], F 1,7 = 31, P < 0.001; main effect of time, F 1.4,10 = 35, P < 0.001; [Glu] × time interaction, F 1.4,10 = 18, P < 0.001). Miniature EPSC amplitudes between 0 and 30 min without glutamate in presynaptic pipettes were significantly different (Bonferroni tests, P < 0.05). The difference in rundown magnitude between controls with 3 mm glutamate and those without glutamate was statistically significant for EPSCs at 10, 20 and 30 min (Bonferroni tests, P < 0.01). C, mean frequency of mEPSCs in different time periods after glutamate washout, normalized to the mean frequency at time 0. The mean frequency of mEPSCs at time 0 was 17 ± 1.3 Hz (3 mm Glu control, n = 5 pairs) and 15 ± 4.2 Hz (0 mm Glu, n = 5 pairs). The glutamate concentration had significant effects on the frequency of mEPSCs (repeated‐measures ANOVA: main effect of [Glu], F 1,8 = 7.4, P < 0.05; main effect of time, F 3,24 = 9.7, P < 0.001; [Glu] × time interaction, F 3,24 = 5.4, P < 0.05). The statistical difference between controls and 0 mm glutamate was significant at 20 min (Bonferroni tests, P < 0.05) and 30 min (Bonferroni tests, P < 0.01). D, representative amplitude histograms of mEPSCs (open bars) in different time periods after glutamate washout. Inset traces show mEPSCs at a slow time scale in this figure and in Figs 2 D and 3E. The total number of events is 100 for each histogram. Background noise distributions (filled bars) were obtained from the baselines of records with no clear mEPSC events. Arrows indicate the mean amplitude of mEPSCs in this figure and in Figs 2 D and 3E. Gaussian curves are fitted to the mEPSC amplitude histograms using the least‐squares method. The coefficient of variation of mEPSC amplitudes was 0.47, 0.52, 0.45 and 0.40, respectively, for 0, 10, 20 and 30 min after glutamate washout.
In intact synapses without presynaptic whole‐cell recording, we bath applied the vacuolar ATPase blocker bafilomycin A1 (Baf in DMSO, 5 μm for 100 s; Fig. 2), as previously reported at cultured hippocampal synapses (Ikeda & Bekkers, 2009). In control experiments, we bath applied DMSO alone (0.5%). Within 5 min after Baf application (100 s), the mEPSC amplitude started to decline and remained similar thereafter, whereas the EPSC amplitude and mEPSC frequency declined continuously. These results are consistent with those of Zhou et al. (2000) but inconsistent with those of Ikeda & Bekkers (2009), who reported that the mean amplitude of mEPSCs does not change after Baf application. Immediately after Baf application, there was a transient increase in mEPSC frequency (Fig. 2 C). Together with a transient increase in EPSC amplitude and a decrease in the paired‐pulse ratio, seen immediately after Baf application, at cultured hippocampal synapses (Ikeda & Bekkers, 2009), Baf probably has a side‐effect causing a transient increase in release probability.
Figure 2. Rundowns of EPSCs and mEPSCs after blocking glutamate uptake with bafilomycin A1 (Baf).
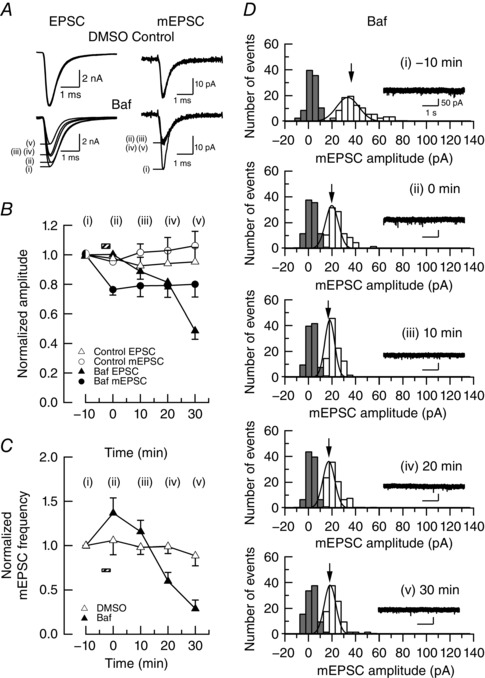
A, sample traces of EPSCs and mEPSCs before and 0–30 min after 100 s bath application (hatched boxes in B and C) of Baf (5 μm with 0.5% DMSO, lower traces, superimposed) or DMSO alone (controls, upper traces). Presynaptic terminals were kept intact without whole‐cell recording. B, mean amplitudes of EPSCs (triangles) and mEPSCs (circles) in different time periods after application of Baf (filled symbols) or DMSO alone (open symbols). Each data point was derived from five experiments and normalized to the amplitudes before application of Baf or DMSO. The mean amplitude of evoked EPSCs before drug application was 7.3 ± 0.8 nA (DMSO, n = 5 cells) and 7.5 ± 0.6 nA (Baf, n = 8 cells) and that of mEPSCs was 38 ± 5.5 pA (DMSO, n = 5 cells) and 38 ± 3.7 pA (Baf, n = 8 cells). Drug application had a significant effect on the amplitude of mEPSCs (repeated‐measures ANOVA: main effect of drug, F 1,11 = 6.0, P < 0.05; main effect of time, F 2,24 = 2.2, P > 0.05; [Glu] × time interaction, F 2,24 = 2.3, P > 0.05) and that of EPSCs (repeated‐measures ANOVA: main effect of drug, F 1,8 = 8.1, P < 0.05; main effect of time, F 4,32 = 16, P < 0.001; [Glu] × time interaction, F 4,32 = 13, P < 0.001). Differences in the magnitude of amplitude reduction between DMSO controls and Baf application data were statistically significant for mEPSCs at 0 min (Bonferroni tests, P < 0.05) and EPSCs at 30 min (Bonferroni tests, P < 0.01). C, mean frequency of mEPSCs in different time periods after application of Baf (filled triangles) or DMSO alone (open symbols) normalized to the initial values before drug application. The mean frequency of mEPSCs before drug application was 5.6 ± 1.4 Hz (DMSO, n = 5 cells) and 8.7 ± 2.0 Hz (Baf, n = 8 cells). Drug application had a significant effect on the frequency of mEPSCs (repeated‐measures ANOVA: main effect of drug, F 1,11 = 0.8, P > 0.05; main effect of time, F 4,44 = 13, P < 0.001; [Glu] × time interaction, F 4,44 = 8.0, P < 0.001). The mEPSC frequency was significantly reduced at 0, 10, 20 and 30 min after Baf application (Bonferroni tests, P < 0.05). D, representative amplitude histograms of mEPSCs (open bars) in different time periods after Baf application. The total number of events is 100 for each histogram. The coefficient of variation of mEPSC amplitudes was 0.28, 0.25, 0.27, 0.23 and 0.35, respectively, for before and 0, 10, 20 and 30 min after application of Baf.
Quantal size is reduced by 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX)
The reduction of mEPSC amplitude after glutamate washout or Baf application suggests that glutamate leaks out of vesicles when glutamate uptake is blocked. However, the small rundown with a plateau of mEPSC amplitude after Baf application in cultured hippocampal synapses has been ascribed to a low detectability of mEPSCs (Zhou et al. 2000). To re‐examine this possibility, we tested the effect of postsynaptic AMPA receptor block on mEPSC amplitudes by applying CNQX at incremental concentrations (0.2–1.5 μm; Fig. 3). As the CNQX concentration increased, mean amplitudes of EPSCs and mEPSCs declined in parallel (Fig. 3 B). At 1.5 μm CNQX, mEPSCs were still detectable above noise level (Fig. 3 E), with a mean amplitude of 19 ± 0.8 pA (n = 5 pairs), that is 40% of control size. This mean amplitude was significantly smaller than that measured after glutamate washout for 30 min (26 ± 3.0 pA, n = 5 pairs, P < 0.05) or after Baf application (27 ± 3.5 pA, n = 5 cells), indicating that mEPSCs after rundown were well above the detection limit of mEPSCs, unlike those at hippocampal synapses (Zhou et al. 2000).
Figure 3. Reductions in the amplitudes of EPSCs and mEPSCs by 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX) titration.
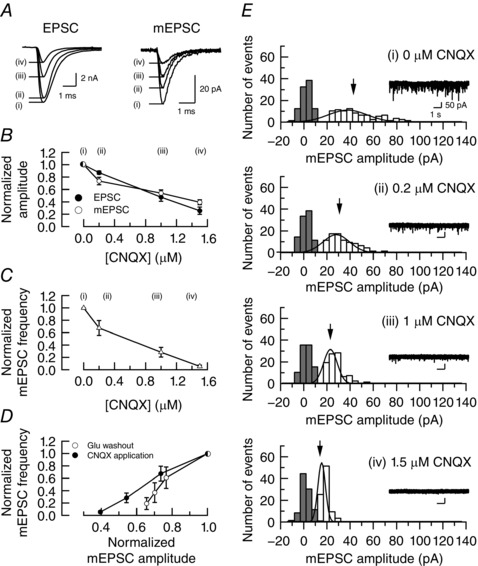
A, sample traces of EPSCs and mEPSCs without or with CNQX at different concentrations (0.2–1.5 μm, superimposed). B, mean amplitudes of EPSCs (filled circles) and mEPSCs (open circles) plotted against CNQX concentrations. Each data point was derived from five experiments and normalized to control values without CNQX (ordinate). In the presence of 1.5 μm CNQX, the mean amplitude of evoked EPSCs was 2.1± 0.5 nA and that of mEPSCs was 19 ± 0.8 pA. C, mean frequency of mEPSCs in the presence of CNQX at different concentrations, normalized to control values. D, relative mEPSC frequency (ordinate) plotted against relative mEPSC amplitude during glutamate washout (open circles) and in the presence of CNQX at different concentrations (filled circles). E, amplitude histograms of mEPSCs recorded from a postsynaptic neuron in the presence of CNQX at different concentrations. The coefficient of variation of mEPSC amplitudes was 0.44, 0.43, 0.32 and 0.23, respectively, in the presence of 0, 0.2, 1.0 and 1.5 μm CNQX.
Notably, the reduction of mEPSC amplitude by CNQX was associated with a decline of mEPSC frequency (Fig. 3 C). This is likely to be a secondary effect of the reduction in amplitude, with small events merging into the noise level. Although AMPA can depolarize calyceal presynaptic terminals (Takago et al. 2005), CNQX by itself at 1.5 μm had no effect on presynaptic membrane potential (n = 4 cells; data not shown), excluding its presynaptic effect. For a given reduction of mEPSC amplitude, the percentage reduction of mEPSC frequency was less for CNQX application than for glutamate washout (Fig. 3 D), suggesting that the reduction of mEPSC frequency after glutamate washout was caused primarily by accumulation of empty vesicles and secondarily by an increase of undetectable events associated with a reduction of mEPSC amplitude.
Influence of vesicle filling state on exocytosis
In hippocampal autaptic culture preparations, the vesicular neurotransmitter filling state is proposed to regulate exocytosis, with poorly filled vesicles having a low release probability (Herman et al. 2014). To examine whether greater reductions of EPSC amplitude than mEPSC amplitude after glutamate washout (Fig. 1 B) might be caused by a reduction of release probability, we measured the C m of calyceal presynaptic terminals (Sun et al. 2002; Yamashita et al. 2005) during glutamate washout. If release probability declines, exocytic capacitance change (ΔC m) is expected to decrease. However, ΔC m remained unchanged during glutamate washout (Fig. 4). Furthermore, no change was discerned for the presynaptic Ca2+ currents or the time course of endocytic capacitance change (Fig. 4 B), suggesting that presynaptic glutamate washout has no effect on exocytosis or endocytosis of synaptic vesicles. These results are consistent with those of unchanged FM dye destaining after Baf application in cultured hippocampal synapses (Zhou et al. 2000), as well as with those of similar C m changes, with or without intracellular glutamate, in salamander retinal cells (Bartoletti & Thorenson, 2011).
Figure 4. Exo‐endocytic membrane capacitance changes in presynaptic terminals after glutamate washout.
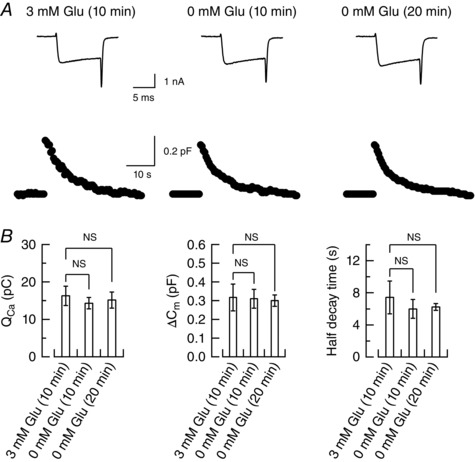
A, presynaptic membrane capacitance changes (lower traces) induced by Ca2+ currents (upper traces) elicited by a depolarizing command pulse (10 ms, from −80 to 0 mV), with 3 mm glutamate in whole‐cell pipettes or 10 or 20 min after washing out glutamate with glutamate‐free pipette solution (middle and right traces). B, bar graphs summarize Ca2+ charges (QCa, left panel), magnitudes of exocytic capacitance changes (ΔC m, middle panel) and endocytic capacitance half‐decay time (right panel). There was no significant difference (NS) in these parameters between controls (3 mm glutamate) and glutamate washout.
Discussion
At the calyx of Held presynaptic terminal, whole‐cell washout of cytosolic glutamate or blocking glutamate uptake into vesicles with the vacuolar ATPase inhibitor bafilomycin A1 slightly reduced the spontaneous mEPSC amplitude and strongly reduced the evoked EPSC amplitude. The relatively small rundown of mEPSCs after Baf treatment has previously been attributed to an artifact owing to limited detectability of mEPSCs (Zhou et al. 2000). In the present study, we have excluded this possibility by demonstrating that the mEPSC amplitude after rundown is high above the detection limit. The number of vesicles undergoing exocytosis during a 30 min period of glutamate washout was estimated to be 513 ± 195 (n = 5 pairs) from evoked EPSCs and 24,700 ± 6700 (n = 5 pairs) from a spontaneous mEPSC frequency that was 15 ± 4.2 Hz (n = 5 pairs) on average during the period. Given that the total number of vesicles is estimated as 190,000 at the calyx of Held (De Lange et al. 2003), 13% of the total vesicles underwent spontaneous exocytosis and recycled without being refilled with glutamate. Given that a fraction of the total vesicles undergoes recycling in physiological conditions (Rizzoli & Betz, 2005; but see Xue et al. 2013), relatively stronger rundown of evoked EPSCs after blocking glutamate uptake can be explained by accumulation of empty vesicles recycled following spontaneous and evoked release of glutamate. It is controversial whether the recycling pool of vesicles undergoing spontaneous exocytosis is distinct from that undergoing evoked exocytosis (Chung et al. 2010; Wilhelm et al. 2010). At the calyx of Held, marked reductions of mEPSC frequency and evoked EPSC amplitude after blocking glutamate uptake suggest that their recycling pools overlap.
At hippocampal synapses in culture, Zhou et al. (2000) reported that Baf treatment (1 μm, at 37°C for 1 h) reduces the mEPSC amplitude to 60% of control conditions, with little further reduction after longer treatment, whereas Ikeda & Bekkers (2009) saw no significant reduction in mEPSC amplitude 10 min after local Baf application (5 μm, at RT for 100 s). At the calyx of Held, bath‐applied Baf (5 μm, 100 s) decreased mEPSC amplitude to 70% of the control value, and glutamate washout decreased it to 80% in 30 min. The present study indicates that a small percentage reduction in mEPSC amplitude after glutamate washout or Baf application reflects the magnitude of glutamate leakage out of vesicles. This leakage is normally compensated by uptake of glutamate from the presynaptic cytosol using the proton gradient produced by vacuolar ATPase, as there was no change in mEPSC amplitude at least for 30 min in the presence of 3 mm glutamate in presynaptic pipettes (Fig. 1; see also Ishikawa et al. 2002). Based on an assumption that glutamate does not leak out of vesicles, Ikeda & Bekkers (2009) calculated the number of recycling vesicles by dividing the total EPSC charge over 30–40 min by a single mEPSC charge. Owing to its minor influence on synaptic efficacy, glutamate leakage may not affect such calculations much. However, as the spontaneous mEPSC frequency can affect the magnitude of EPSC rundown after blocking glutamate uptake, estimation of the functional vesicle pool size should take this factor into account, unless vesicle recycling after evoked and spontaneous release are totally independent.
In isolated vesicles, glutamate leaks out of the vesicles when the vesicular proton gradient is dissipated (Carlson & Ueda, 1990; Maycox et al. 1990). GABA, but not glutamate or glycine, can permeate phospholipid liposomes without vesicular protein (Hell et al. 1991; Schenck et al. 2009), whereas glutamate leakage is reportedly caused by the H+–glutamate antiport through VGLUTs (Mackenzie et al, 2008). The calyces of Held in rodents express both VGLUT1 and VGLUT2 (Billups, 2005; Blaesse et al. 2005). Thus, it remains to be seen whether the magnitude of glutamate leakage is different between vesicles expressing different VGLUT subtypes.
We have identified that glutamate leakage is expressed as a reduction of mEPSC amplitude that is continuously compensated by vacuolar ATPase‐dependent glutamate reuptake. The relatively small leakage of vesicular glutamate may economize ATP consumption in the nerve terminal and will minimize rundown of excitatory neurotransmission when synapses are exposed to anoxic conditions.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
T.T. and T.H. designed the study. C.T., K.E. and T.H. performed experiments and data analysis. C.T., T.H. and T.T. wrote the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the Core Research for Evolutional Science and Technology of Japan Science and Technology Agency (JST CREST) to T.T. and by the Okinawa Institute of Science and Technology.
Acknowledgements
We thank Takeshi Sakaba and Shigeo Takamori for comments, and Steve Aird for editing this manuscript.
References
- Bartoletti TM & Thorensen WB (2011). Quantal amplitude at the cone ribbon synapse can be adjusted by changes in cytosolic glutamate. Mol Vis 17, 920–931. [PMC free article] [PubMed] [Google Scholar]
- Bellocchio EE, Reimer RJ, Fremeau RT Jr & Edwards RH (2000). Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289, 957–960. [DOI] [PubMed] [Google Scholar]
- Billups B (2005). Colocalization of vesicular glutamate transporters in the rat superior olivary complex. Neurosci Lett 382, 66–70. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Ehrhardt S, Friauf E & Nothwang HG (2005). Developmental pattern of three vesicular glutamate transporters in the rat superior olivary complex. Cell Tissue Res 320, 33–50. [DOI] [PubMed] [Google Scholar]
- Burger PM, Mehl E, Cameron PL, Maycox PR, Baumert M, Lottspeich F, De Camilli P & Jahn R (1989). Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron 3, 715–720. [DOI] [PubMed] [Google Scholar]
- Carlson MD & Ueda T (1990). Accumulated glutamate levels in the synaptic vesicle are not maintained in the absence of active transport. Neurosci Lett 110, 325–330. [DOI] [PubMed] [Google Scholar]
- Chung C, Barylko B, Leitz J, Liu X & Kavalali ET (2010). Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J Neurosci 30, 1363–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lange RP, de Roos AD & Borst JG (2003). Two modes of vesicle recycling in the rat calyx of Held. J Neurosci 23, 10164–10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J & Katz B (1954). Quantal components of the end‐plate potential. J Physiol 124, 560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K, Nakanishi S, Takagi H, Taoufiq Z & Takahashi T (2012). Maturation of a PKG‐dependent retrograde mechanism for exoendocytic coupling of synaptic vesicles. Neuron 74, 517–529. [DOI] [PubMed] [Google Scholar]
- Hell JW, Edelmann L, Hartinger J & Jahn R (1991). Functional reconstitution of the γ‐aminobutyric acid transporter from synaptic vesicles using artificial ion gradients. Biochemistry 30, 11795–11800. [DOI] [PubMed] [Google Scholar]
- Herman MA, Ackermann F, Trimbuch T & Rosenmund C (2014). Vesicular glutamate transporter expression level affects synaptic vesicle release probability at hippocampal synapses in culture. J Neurosci 34, 11781–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE & Reese TS (1973). Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol 57, 315–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T & Takahashi T (2012). Kinetics of synaptic vesicle refilling with neurotransmitter glutamate. Neuron 76, 511–517. [DOI] [PubMed] [Google Scholar]
- Ikeda K & Bekkers JM (2009). Counting the number of releasable synaptic vesicles in a presynaptic terminal. Proc Natl Acad Sci USA 106, 2945–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Sahara Y & Takahashi T (2002). A single packet of transmitter does not saturate postsynaptic glutamate receptors. Neuron 34, 613–621. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Illing AC, Morris MEK, Varoqui H & Erickson JD (2008). Analysis of a vesicular glutamate transporter (VGLUT2) supports a cell‐leakage mode in addition to vesicular packaging. Neurochem Res 33, 238–247. [DOI] [PubMed] [Google Scholar]
- Maycox PR, Deckwerth T & Jahn R (1990). Bacteriorhodopsin drives the glutamate transporter of synaptic vesicles after co‐reconstitution. EMBO J 9, 1465–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito S & Ueda T (1985). Characterization of glutamate uptake into synaptic vesicles. J Neurochem 44, 99–109. [DOI] [PubMed] [Google Scholar]
- Parsons RL, Calupca MA, Merriam LA & Prior C (1999). Empty synaptic vesicles recycle and undergo exocytosis at vesamicol‐treated motor nerve terminals. J Neurophysiol 81, 2696–2700. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO & Betz WJ (2005). Synaptic vesicle pools. Nat Rev Neurosci 6, 57–69. [DOI] [PubMed] [Google Scholar]
- Sahara Y & Takahashi T (2001). Quantal components of the excitatory postsynaptic currents at a rat central auditory synapse. J Physiol 536, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck S, Wojcik SM, Brose N & Takamori S (2009). A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat Neurosi 12, 156–162. [DOI] [PubMed] [Google Scholar]
- Sun JY, Wu XS & Wu LG (2002). Single and multiple vesicle fusion induce different rates of endocytosis at a central synapse. Nature 417, 555–559. [DOI] [PubMed] [Google Scholar]
- Takago H, Nakamura Y & Takahashi T (2005). G protein‐dependent presynaptic inhibition mediated by AMPA receptors at the calyx of Held. Proc Natl Acad Sci USA 102, 7368–7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C & Jahn R (2000). Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407, 189–194. [DOI] [PubMed] [Google Scholar]
- Wilhelm BG, Groemer TW & Rizzoli SO (2010). The same synaptic vesicles drive active and spontaneous release. Nat Neurosci 13, 1454–1456. [DOI] [PubMed] [Google Scholar]
- Wu XS, Xue L, Mohan R, Paradiso K, Gillis KD & Wu LG (2007). The origin of quantal size variation: vesicular glutamate concentration plays a significant role. J Neurosci 27, 3046–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Sheng J, Wu XS, Wu W, Luo F, Shin W, Chiang HC & Wu LG (2013). Most vesicles in a central nerve terminal participate in recycling. J Neurosci 33, 8820–8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Eguchi K, Saitoh N, von Gersdorff H & Takahashi T (2010). Developmental shift to a mechanism of synaptic vesicle endocytosis requiring nanodomain Ca2+ . Nat Neurosci 13, 838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Hige T & Takahashi T (2005). Vesicle endocytosis requires dynamin‐dependent GTP hydrolysis at a fast CNS synapse. Science 307, 124–127. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Kanda T, Eguchi K & Takahashi T (2009). Vesicular glutamate filling and AMPA receptor occupancy at the calyx of Held synapse of immature rats. J Physiol 587, 2327–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Petersen CC & Nicoll RA (2000). Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J Physiol 525, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]