

Abstract
Key points
Intrauterine growth restriction (IUGR) is associated with vascular dysfunction, oxidative stress and signs of endothelial epigenetic programming of the umbilical vessels.
There is no evidence that this epigenetic programming is occurring on systemic fetal arteries.
In IUGR guinea pigs we studied the functional and epigenetic programming of endothelial nitric oxide synthase (eNOS) (Nos3 gene) in umbilical and systemic fetal arteries, addressing the role of oxidative stress in this process by maternal treatment with N‐acetylcysteine (NAC) during the second half of gestation.
The present study suggests that IUGR endothelial cells have common molecular markers of programming in umbilical and systemic arteries. Notably, maternal treatment with NAC restores fetal growth by increasing placental efficiency and reverting the functional and epigenetic programming of eNOS in arterial endothelium in IUGR guinea pigs.
Abstract
In humans, intrauterine growth restriction (IUGR) is associated with vascular dysfunction, oxidative stress and signs of endothelial programming in umbilical vessels. We aimed to determine the effects of maternal antioxidant treatment with N‐acetylcysteine (NAC) on fetal endothelial function and endothelial nitric oxide synthase (eNOS) programming in IUGR guinea pigs. IUGR was induced by implanting ameroid constrictors on uterine arteries of pregnant guinea pigs at mid gestation, half of the sows receiving NAC in the drinking water (from day 34 until term). Fetal biometry and placental vascular resistance were followed by ultrasound throughout gestation. At term, umbilical arteries and fetal aortae were isolated to assess endothelial function by wire‐myography. Primary cultures of endothelial cells (ECs) from fetal aorta, femoral and umbilical arteries were used to determine eNOS mRNA levels by quantitative PCR and analyse DNA methylation in the Nos3 promoter by pyrosequencing. Doppler ultrasound measurements showed that NAC reduced placental vascular resistance in IUGR (P < 0.05) and recovered fetal weight (P < 0.05), increasing fetal‐to‐placental ratio at term (∼40%) (P < 0.001). In IUGR, NAC treatment restored eNOS‐dependent relaxation in aorta and umbilical arteries (P < 0.05), normalizing eNOS mRNA levels in EC fetal and umbilical arteries (P < 0.05). IUGR‐derived ECs had a decreased DNA methylation (∼30%) at CpG −170 (from the transcription start site) and this epigenetic signature was absent in NAC‐treated fetuses (P < 0.001). These data show that IUGR‐ECs have common molecular markers of eNOS programming in umbilical and systemic arteries and this effect is prevented by maternal treatment with antioxidants.
Keywords: antioxidant, endothelial dysfunction, endothelial nitric oxide synthase, epigenetics, fetal programming, intrauterine growth restriction
Key points
Intrauterine growth restriction (IUGR) is associated with vascular dysfunction, oxidative stress and signs of endothelial epigenetic programming of the umbilical vessels.
There is no evidence that this epigenetic programming is occurring on systemic fetal arteries.
In IUGR guinea pigs we studied the functional and epigenetic programming of endothelial nitric oxide synthase (eNOS) (Nos3 gene) in umbilical and systemic fetal arteries, addressing the role of oxidative stress in this process by maternal treatment with N‐acetylcysteine (NAC) during the second half of gestation.
The present study suggests that IUGR endothelial cells have common molecular markers of programming in umbilical and systemic arteries. Notably, maternal treatment with NAC restores fetal growth by increasing placental efficiency and reverting the functional and epigenetic programming of eNOS in arterial endothelium in IUGR guinea pigs.
Abbreviations
- APD
abdominal anteroposterior diameter
- ECs
endothelial cells
- eNOS
endothelial nitric oxide synthase
- DOHaD
developmental origins of health and disease
- HC
head circumference
- IUGR
intrauterine growth restriction
- l‐NAME
N G‐nitro‐l‐arginine‐methylester
- NAC
N‐acetylcysteine
- Nos3
endothelial nitric oxide synthase gene
- PI
pulsatility index
- qPCR
quantitative PCR
- ROS
reactive oxygen species
- RI
resistance index
- SNP
sodium nitroprusside
Introduction
Compelling evidence shows that adverse intrauterine conditions leading to intrauterine growth restriction (IUGR) increase the risk of developing cardiovascular and metabolic diseases in adulthood (Cohen et al. 2016; Devaskar & Chu, 2016). This has led to the formulation of the ‘Developmental Origins of Health and Disease’ (DOHaD) hypothesis, which relies on the activation of mechanisms ‘sensing and signalling’ diverse stimuli during early development that later lead to higher risk of adult‐onset chronic diseases (diabetes, hypertension, stroke and myocardial infarcts) (Hanson & Gluckman, 2014). Epigenetic modifications in key genes that ‘record’ normal and abnormal perinatal stimuli (Gluckman et al. 2009) are proposed as mechanisms involved in these processes.
IUGR is clinically defined by a fetal weight below the 10th percentile of a distribution obtained in ‘healthy mothers’. In a more comprehensive manner, IUGR constitutes a condition in which the potential growth of the fetus is negatively influenced by maternal nutritional and health status, placental function and other factors (Zhang et al. 2010; Cohen et al. 2016). Placental dysfunction is a common characteristic of IUGR, which is evidenced by an increased placental vascular resistance throughout gestation (Pardi et al. 2002) and an increased placental to fetal weight ratio at term (Macdonald et al. 2014). It has been proposed that the impaired placental vascular function in IUGR results from an augmented synthesis and response to vasoconstrictors (Mills et al. 2005) and a limited action of vasodilatators, which is in part due to the increased effect of pro‐oxidants on endothelial‐dependent vessel relaxation (Herrera et al. 2014). These findings suggest a potential role for antioxidant preventive therapies. However, there is as yet a lack of solid evidence showing the benefits of antioxidants in the prevention of IUGR or improving placental blood flow. Studies in animals have suggested the potential use of vitamins C or E as reactive oxygen species (ROS) scavengers. Nonetheless, results from human clinical trials using these agents have shown limited effects (Hovdenak & Haram, 2012). Conversely there are no data in IUGR models addressing the effects of N‐acetylcysteine (NAC), which has an effective antioxidant capacity via glutathione reposition (Samuni et al. 2013; Lasram et al. 2015) coupled to an efficient transfer from maternal to fetal circulation (Wiest et al. 2014).
The long‐term effects of impaired fetal growth on vascular function could be evidenced at early stages of life (Cohen et al. 2016). It has been reported that endothelial function is impaired in neonates that are small for gestational age (Martin et al. 2000), an effect also observed in umbilical and chorionic arteries derived from IUGR placentae (Krause et al. 2013 a). Interestingly, primary cultures of placental and umbilical endothelium derived from pregnancies complicated by IUGR show abnormal phenotypes which persist under culture conditions (Krause et al. 2013 b), characterized by altered expression of proteins involved in NO‐dependent vasodilatation [i.e. endothelial nitric oxide synthase (eNOS) and arginase], as well as changes in the proteome profile (Caniuguir et al. 2016), suggesting an early programming of endothelial dysfunction. Furthermore, in vitro experiments show that altered expression of eNOS in human IUGR placenta‐derived endothelial cells (ECs) is accompanied by epigenetic alterations in the promoter of its gene (Krause et al. 2013 b). Notably, these cells can be reprogrammed to a ‘normal type’ by interfering the molecular machinery that preserves the DNA methylation pattern. So far, no studies have addressed the role of the antioxidant on the epigenetic programming of placental endothelial dysfunction in IUGR, and whether these epigenetic changes reflect those present in other fetal vascular beds.
We hypothesized that maternal antioxidant treatment prevents the altered endothelial function and eNOS programming in IUGR, as well as improves placental vascular function and fetal growth in a guinea pig model of IUGR (Herrera et al. 2016). To address these effects, pregnant guinea pigs with either control or IUGR fetuses were randomly assigned to receive NAC in drinking water during the second half of gestation. Fetal growth and placental vascular function were followed throughout gestation by ultrasound examinations. NAC effects on the endothelial‐dependent relaxation in isolated aorta and umbilical arteries was determined by wire myography in near‐term fetuses. Finally, the effect of NAC treatment on eNOS epigenetic programming was determined by analysing the mRNA levels of eNOS by quantitative PCR (qPCR), as well as the DNA methylation pattern at the Nos3 promoter in primary cultures of fetal systemic and umbilical artery ECs.
Methods
Ethic statement
All animal care, procedures and experimentation were approved by the Ethics Committee of the Faculty of Medicine, from the Pontificia Universidad Católica de Chile (1130801) and Universidad de Chile (protocol CBA# 0694 FMUCH), and were conducted in accordance with the ARRIVE guidelines and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1996).
Animals
Eighteen adult female Pirbright White guinea pigs (Cavia porcellus) were used for this study. All animals were housed in individual cages under standard conditions (30–35% humidity, 20–21°C and a 12:12 h light–dark cycle), with controlled food‐by‐body weight intake with a commercial diet (LabDiet 5025, Guinea Pigs, 20–30 g day–1). Pregnancies were confirmed by ultrasonography at day 20–25, where the first day with the male was considered day 0 of pregnancy (term ∼67 days).
Experimental design
After confirming pregnancy, the pregnant guinea pigs were randomly assigned to the control (n = 10) or IUGR (n = 8) group. Additionally, starting on day 34 of pregnancy half of the sows from the control and IUGR groups were treated with 500 mg kg–1 day–1 NAC (Evans et al. 2012) (Sigma‐Aldrich, St Louis, MO, USA; cat. no. A7250) in the drinking water, a dose that was adjusted daily to maternal body mass. All pregnant sows were subjected to aseptic surgery on gestation day 35, either sham‐operated (control) or to progressive uterine artery occlusion (IUGR) (Herrera et al. 2016).
Fetal biometry and umbilical artery Doppler ultrasound
Once gestation was confirmed, the sows were examined twice a week and head circumference (HC) and abdominal anteroposterior diameter (APD) were determined by ultrasound (Sonovet R3, Samsung Medison, Seoul, South Korea). From day 30 of gestation, the umbilical artery blood flow was assessed by Doppler ultrasound‐based wave analysis, establishing the resistance index (RI) and pulsatility index (PI). Ultrasound measurements were performed for every fetus, individually identified throughout pregnancy depending on the side and position in the womb (Herrera et al. 2016).
Near‐term processing
At 60–62 days of gestation (∼90% of pregnancy), the pregnant guinea pigs and their fetuses were killed with a maternal anaesthetic overdose (sodium thiopentone 200 mg kg−1 i.p., Opet, Laboratorio Chile, Santiago, Chile). Once cardio‐respiratory arrest was confirmed, the fetuses and their placentae were dissected and weighed.
Aorta and umbilical artery vascular function
At dissection, aorta and umbilical arteries were rapidly excised and 2 mm proximal segments were mounted on a wire myograph (610 M System Myograph multiwire, DMT) to determine vasoactive responses as previously reported (Krause et al. 2015). To determine the NOS‐dependent vasodilatation, vessels were pre‐constricted with half maximal KCl concentration (40.8 mmol l−1) and the isometric force in response to cumulative concentrations of acetylcholine for aortae (10−8 to 10−5 mol l−1) and insulin for umbilical arteries (10−12 to 10−7 mol l−1). Insulin was used as an eNOS‐dependent agent based on preliminary assays in which no responses to other agents was observed (i.e. CGRP, bradykinin and acetylcholine). The relaxing effects of insulin and acetylcholine were determined as the difference between the response in the presence and absence of the NOS inhibitor N G‐nitro‐l‐arginine‐methylester (l‐NAME, 100 μmol l−1). The NOS‐independent response to NO was determined with sodium nitroprusside (SNP, 10−9 to 10−5 mol l−1) in pre‐constricted vessels (Krause et al. 2015).
Primary cultures of ECs from aorta, femoral and umbilical artery
Primary cultures of ECs from umbilical and fetal systemic arteries were obtained according to the method proposed by Chen et al. (1995) with some modifications. Briefly, vessel samples (1–2 cm long) were cut into pieces of ∼1 mm2 and seeded and maintained at 37°C in a 25 cm2 plate with Medium 131 and 20 % Microvascular Growth supplement (MVGS, Thermo Fisher Scientific Inc, MA, USA; cat. no. S00525). After 60 h the tissue was gently removed and the plate was washed with PBS and fresh culture medium was added. The endothelial phenotype was confirmed by immunocytochemistry, using vWF (SAB1402960, Sigma) and CD31 (P8590, Sigma) antibodies. Previous to RNA and DNA extraction, cells were starved overnight in Medium 131 and 2% MVGS. All these determinations were carried out in cells at third passage.
Quantitative PCR
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and PCRs were performed as described (Krause et al. 2016). Aliquots of 1 μg of total RNA were reverse transcribed using IMPROM II RT kit (Promega, Madison, WI, USA). Three different RNAs, the rRNA 18S (sense, 5′‐TGCATGGCCGTTCTTAGTTG; antisense, 5′‐AGTTAGCATGCCAGAGTCTCGTT‐3′), the messenger RNA for β‐actin (sense, 5′‐AACGATGCCGTGCTCAATG‐3′; antisense, ATATCGCTGCGCTCGTTGTC) and RPLP2 (ribosomal protein lateral stalk subunit P2) (sense, 5′‐GCGCCAAGGACATCAAGAAG‐3′; antisense, 5′‐CCAGCAGGTACACTGGCAA‐3′), were used as reference genes for eNOS (sense, 5′‐AGCCAACGCGGTGAAGATC‐3′; antisense, 5′‐TTAGCCATCACCGTGCCC‐3′) mRNA quantification by SYBR®‐green real time PCR. Quantification was carried out using the geometrical average of the 2‐ΔΔCT method (Livak & Schmittgen, 2001) for eNOS relative to the three different reference genes.
Quantification of eNOS protein and p‐eNOS Ser1177
Levels of eNOS protein and its activating phosphorylation at serine 1177 (p‐eNOSSer1177) were quantified by ELISA. Briefly, 20 μg of protein extract was used for determining total eNOS protein levels and p‐eNOSSer1177 using an ‘eNOS Quantikine’ (R&D Systems, Minneapolis, MN, USA; cat. no. DEN00) and a ‘PathScan Phospho‐eNOS (Ser1177)’ (Cell Signaling, Danvers, MA, USA; cat. no. 7980) ELISA Kits, respectively, following the protocol suggested by the provider.
Levels of 3‐nitrotyrosine in vascular tissue
Levels of the oxidative stress marker 3‐nitrotyrosine were determined by dot blot in whole protein extracts from fetal aorta samples. Briefly, 30 μg of protein extract was loaded onto a 0.45 μm nitrocellulose membrane (BioRad, Hercules, CA, USA), blocked with 5% fat‐free milk in Tris‐buffered saline (0.1% Tween) and probed with primary monoclonal anti‐Nitrotyrosine (1:500, sc‐65385, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Proteins were detected by enhanced chemiluminescence and quantified by densitometry using Image J (NIH, Bethesda, MD, USA).
Analysis of DNA methylation
Methylation status of the promoter region of the Nos3 guinea pig gene was determined using a DNA bisulphite modification coupled to DNA sequencing (Krause et al. 2013 b). Briefly, DNA was isolated from ∼10−6 ECs using DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) and 500 ng of total DNA extracts were treated with sodium bisulphite using an EpiTect Bisulfite Kit (Qiagen). Promoter regions were amplified by PCR using specific primers for Nos3 (Table 1). Site‐specific CpG methylations were determined as a percentage using a PyroMark Q96 MD (Qiagen).
Table 1.
Primers for pyrosequencing of Nos3 promoter
| Sequence | ||
|---|---|---|
| * CpGs –220 to –170 | Forward | 5’‐GTTTTTTATATAATGGGATAGGAATAAGGT‐3’ |
| Reverse | biotin5’‐CCCAACCAACCTTATTCCTAT‐3’ | |
| Sequencing | 5’‐GTTGGGAGGTTTTGAA‐3’ | |
| * CpGs –111 to –93 | Forward | 5’‐AAGTAGGGAGGGGGTTGAG‐3’ |
| Reverse | biotin5’‐CCCAACCAACCTTATTCCTAT‐3’ | |
| Sequencing | 5’‐GAGGGGAGGGGTATT‐3’ | |
| CpGs –27 to –11 | Forward | 5’‐AAGGAAAAGGTTAGGGTTTTGT‐3’ |
| Reverse | biotin5’‐TCCTAACCCACACTCTTAAAATTAC‐3’ | |
| Sequencing | 5’‐GTTAGGGTTTTGTTGGA‐3’ | |
| CpGs +34 to +46 | Forward | 5’‐AGAGTTGAAGGGAGGTTGATATGG‐3’ |
| Reverse | biotin5’‐AACCCTACTTACCACACAATCC‐3’ | |
| Sequencing | 5’‐TTAAGAGTGTGGGTTAG‐3’ |
*
This set of primers was designed on the complementary strand.
Transcription factors binding site prediction
Prediction of binding sites for transcription factors in the Nos3 promoter was performed with the online software MatInspector, with selected transcription factors chosen considering a cut‐off of 0.900 for matrix similarity (Krause et al. 2013 b). The data generated were validated considering the presence of conserved sequences for transcription factors that regulate eNOS expression in humans (Karantzoulis‐Fegaras et al. 1999) and mice (Teichert et al. 1998).
Data and statistical analyses
Biometry growth curves were analysed with a Pearson test to assess correlations. Thereafter, data were analysed with linear regression, and functions were compared by ANCOVA. Data for scalar units were expressed as median and interquartile range, whilst ratios, percentages and indices were expressed as mean ± SEM for normal distributions. All other concentration–response curves were analysed using an agonist‐response best‐fit line, where the maximal vasomotor response was expressed as a percentage of the contraction induced by 40.8 mm K+ (%Kmax for relaxation) and the vascular sensitivity was expressed as pD2 (−logEC50) (Krause et al. 2013 a; Schneider et al. 2015). Differences were considered significant at P ≤ 0.05 (Prism 5.0; GraphPad Software, La Jolla, CA, USA).
Results
Fetal biometry and umbilical artery Doppler during gestation
HC and APDgrowth curves were represented by linear regressions with high correlation in all groups for both parameters (r 2 > 0.7). The linear regression slope for HC was similar between control and IUGR animals, but the maternal treatment with NAC increased (∼1.2‐fold) this slope in control without changes in IUGR animals (Fig. 1 A and C). Conversely, the APD linear regression slope was markedly diminished in IUGR animals (∼30%) compared to control, an effect that was prevented by NAC in IUGR fetuses (Fig. 1 B and D).
Figure 1. Fetal biometry during gestation.
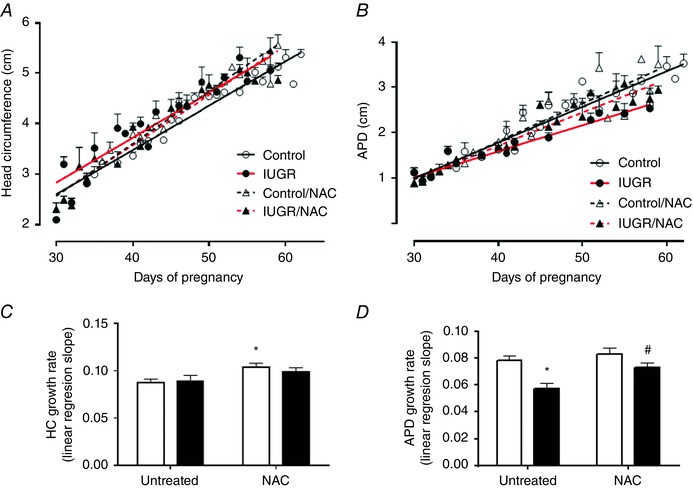
Ecographic determinations of head circumference (HC, A) and antero‐posterior diameter (APD, B) growth trajectory during gestation in control (open circles, continous black lines), control treated with NAC (open triangles, dashed black lines), IUGR (solid circles, continous red lines) and IUGR treated with NAC (solid triangles, dashed red lines) fetal guinea pigs. HC (C) and APD (D) growth rate slope in control (open bars) and IUGR (solid bars) with (treated) or without (untreated) NAC treatment. Values are expressed as mean ± SEM, * P < 0.05 vs untreated control, # P < 0.05 vs untreated IUGR, ANCOVA. [Colour figure can be viewed at wileyonlinelibrary.com]
Umbilical artery Doppler variables showed a progressive decrease in PI and RI indexes throughout gestation in all groups (Fig. 2). In untreated IUGR animals the decreases in PI (Fig. 2 A) and RI (Fig. 2 B) were slower compared with untreated controls, whilst NAC prevented the IUGR effects, where fetuses showed similar PI and RI curves relative to controls. At term, the levels of a pro‐oxidant marker, 3‐nitrotyrosine, were increased in IUGR aorta (∼2.2‐fold) compared to controls and this effect was prevented by maternal treatment with NAC (Fig. 3).
Figure 2. Resistance indices in the umbilical artery during gestation.

Umbilical artery pulsatility (A) and resistance (B) indices derived from waveforms acquired before and after the surgery in control (open circles, continous black lines), control treated with NAC (open triangles, dashed black lines), IUGR (solid circles, continous red lines) and IUGR treated with NAC (solid triangles, dashed red lines) fetal guinea pigs. Slope quantification of PI (C) and RI (D) changes along gestation in control (open bars) and IUGR (solid bars) with (treated) or without (untreated) anitoxidant treatment. Values expressed as mean ± SEM, * P < 0.05 vs untreated control, # P < 0.05 vs untreated‐IUGR, ANCOVA. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 3. Levels of protein peroxynitration in fetal aorta.
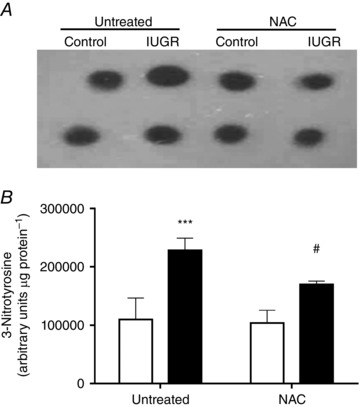
Representative blot (A) and relative levels (B) of 3‐nitrotyrosine determined by dot blot in whole protein extract from fetal aorta of control (open bars, n = 6) and IUGR (solid bars, n = 7) guinea pigs without (untreated) or with (NAC) maternal antioxidant supplementation. Values expressed as mean ± SEM, *** P < 0.001 vs untreated‐control, # P < 0.05 vs untreated‐IUGR, two‐way ANOVA, Newman–Keuls multiple comparison test.
Fetal and placental weights at near term
Fetal weights were similar among untreated (83.7 ± 3.1 g, n = 16) and NAC‐treated (79.1 ± 5.5 g, n = 13) controls (Fig. 4 A). However, there was a ∼ 31% reduction in fetal weight in untreated IUGR animals (57.9 ± 5.1 g, n = 9), which was reverted with NAC (74.1 ± 4.5 g, n = 9). Conversely, placental weight was comparable between untreated (6.86 ± 0.50 g) and NAC‐treated (6.25 ± 0.37 g) controls, but reduced at comparable levels in untreated (4.09 ± 0.39 g) and NAC‐treated (4.78 ± 0.37 g) IUGR groups (Fig. 4 B). Further, fetal to placental weight ratio, as an index of placental sufficiency, was decreased in untreated IUGR animals (∼30%) and was fully prevented by NAC treatment (∼1.1‐fold) (Fig. 4 C).
Figure 4. Placental and fetal weight at near term.
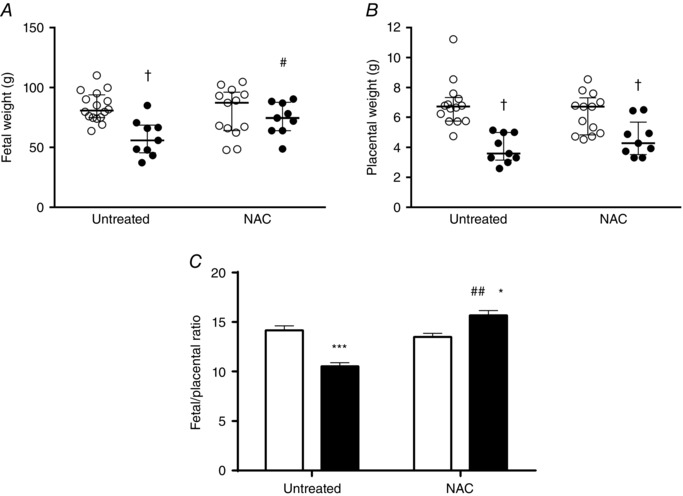
A and B, fetal (A) and placental (B) weight of near‐term fetuses from control (open circles) and IUGR (solid circles) groups whose mothers received (NAC) or did not receive (untreated) antioxidant treatment. C, fetal to placental weight ratio as an index of placental efficiency in control (open bar) and IUGR (solid bar) groups. Values expressed as median and interquartile range for weights and as mean ± SEM for weight ratio, † P < 0.05, *P < 0.05, ***P < 0.001 vs untreated control, #P < 0.05 vs untreated IUGR, two‐way ANOVA, Newman–Keuls multiple comparison test.
On the other hand, fetal organ weights (i.e. brain, heart, lungs, liver, spleen and kidneys) (Table 2) were reduced in untreated IUGR animals compared to controls. These effects were partially reverted in kidneys and liver weights, and fully reverted in brain and heart weights with NAC treatment. Furthermore, brain to liver weight ratio, as an index of fetal symmetry, was increased (∼2‐fold) in untreated IUGR animals with a partial reversion in IUGR animals treated with NAC.
Table 2.
Weight of fetal organs at term
| Untreated | NAC‐treated | |||
|---|---|---|---|---|
| Control | IUGR | Control | IUGR | |
| Brain (g) | 2.33 ± 0.05 | 2.03 ± 0.06 a | 2.25 ± 0.07 | 2.25 ± 0.03 b |
| Heart (g) | 0.59 ± 0.03 | 0.36 ± 0.03 a | 0.51 ± 0.05 | 0.51 ± 0.05 b |
| Lungs (g) | 1.88 ± 0.05 | 1.45 ± 0.20 a | 1.83 ± 0.13 | 1.67 ± 0.14 |
| Liver (g) | 5.07 ± 0.23 | 2.21 ± 0.18 a | 4.72 ± 0.56 | 3.99 ± 0.66 a,b |
| Spleen (g) | 0.10 ± 0.01 | 0.09 ± 0.01 | 0.12 ± 0.01 | 0.10 ± 0.02 |
| Kidney (g) | 0.39 ± 0.01 | 0.24 ± 0.02 a | 0.36 ± 0.02 | 0.31 ± 0.01 a,b |
| Brain/liver ratio | 0.46 ± 0.01 | 0.92 ± 0.05 a | 0.48 ± 0.03 | 0.56 ± 0.03 a,b |
Values expressed as mean ± SEM. a P < 0.05 vs untreated control; b P < 0.05 vs untreated IUGR, ANOVA.
Ex vivo relaxation in aorta and umbilical arteries at near term
Umbilical arteries from fetal guinea pigs showed a concentration‐dependent relaxation to insulin (Fig. 5 A) which was totally blocked by the NOS inhibitor l‐NAME (data not shown). The maximal relaxation response to insulin was decreased in IUGR (13.9 ± 6.7 %Kmax) compared to both control groups (untreated, 36.9 ± 5.8 %Kmax; treated 28.9 ± 4.1 %Kmax), an effect that was reverted with NAC treatment in IUGR subjects (35.5 ± 9.3 %Kmax) (Fig. 5 A and B). Sensitivity for insulin, expressed as pD2, was similar among the four groups (Fig. 5 C). Conversely, the maximal relaxation to SNP (NO donor) was comparable among controls and IUGR (∼80 %Kmax). However, NAC treatment increased the maximal relaxation to SNP in IUGR animals (101.8 ± 5.0 %Kmax) (Fig. 5 D and E), whilst pD2 (Fig. 5 F) was increased in untreated (7.76 ± 0.25) and NAC‐treated (7.03 ± 0.13) IUGR animals compared to controls (untreated, 6.48 ± 0.09; NAC treated, 6.37 ± 0.22).
Figure 5. Endothelial‐dependent and independent relaxation of isolated umbilical arteries.
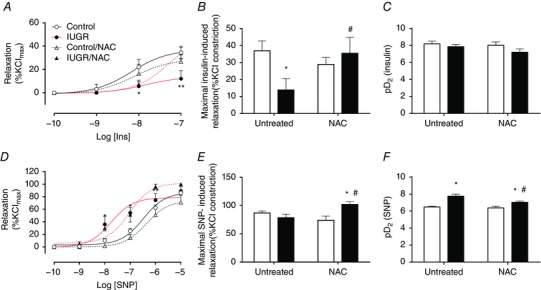
A–C, cumulative concentration relaxation curve (A), maximal response (B) and sensitivity (C) to insulin in isolated umbilical arteries. D–F, cumulative concentration relaxation curve (D), maximal response (E) and sensitivity (F) to the NO donor SNP in isolated umbilical arteries. Groups are control (open circles, continuous black lines), control treated with NAC (open triangles, dash black lines), IUGR (solid circles, continous red lines) and IUGR treated with NAC (solid triangles, dash red lines) fetal guinea pigs. Values expressed as mean ± SEM, *P < 0.05, **P < 0.01 vs untreated control, #P < 0.05 vs untreated IUGR, two‐way ANOVA, Newman–Keuls multiple comparison test. [Colour figure can be viewed at wileyonlinelibrary.com]
Similarly, in isolated aorta the maximal relaxation response to acetylcholine was decreased in IUGR animals (11.7 ± 1.7 %Kmax) compared to both control groups (untreated, 27.3 ± 0.8 %Kmax; treated, 23.5 ± 0.9 %Kmax), whilst NAC treatment in IUGR subjects led to a normal relaxing response (30.4 ± 0.8 %Kmax) (Fig. 6 A and B). Sensitivity for acetylcholine was similar among the four groups (Fig. 6 C). Maximal relaxation to the NO donor SNP was comparable among controls and untreated IUGR animals (∼95 %Kmax), but reduced in aorta from IUGR animals treated with NAC (69.8 ± 3.5 %Kmax) (Fig. 6 D and E), with no differences in pD2 (∼ 6.7) (Fig. 6 F).
Figure 6. Endothelial‐dependent and independent relaxation of isolated fetal aorta.
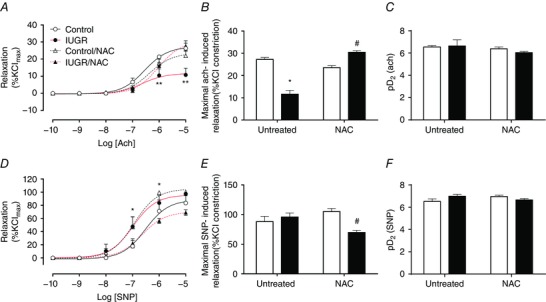
A–C, cumulative concentration relaxation curve (A), maximal response (B) and sensitivity (C) to acetylcholine in isolated fetal aortae. D–F, cumulative concentration relaxation curve (D), maximal response (E) and sensitivity (F) to the NO donor SNP in isolated umbilical arteries. Groups are control (open circles, continuous black lines), control treated with NAC (open triangles, dash black lines), IUGR (solid circles, continous red lines) and IUGR treated with NAC (solid triangles, dash red lines) fetal guinea pigs. Values expressed as mean ± SEM, *P < 0.05, **P < 0.01 vs untreated control, #P < 0.05 vs untreated IUGR, two‐way ANOVA, Newman–Keuls multiple comparison test. [Colour figure can be viewed at wileyonlinelibrary.com]
Levels of eNOS mRNA in fetal ECs
Levels of eNOS mRNA in primary cultures of endothelium from fetal aorta, femoral and umbilical arteries were determined by qPCR. In untreated control fetuses there were no differences in the levels of eNOS transcript among ECs from the different vascular beds studied (data not shown). In marked contrast, eNOS levels were increased (∼5‐fold) in aorta, femoral and umbilical artery ECs derived from untreated IUGR fetuses (Fig. 7) compared to untreated controls. IUGR umbilical ECs showed increased eNOS protein levels (∼2.5‐fold) but decreased levels of its activating phosphorylation at serine 1177 (Fig. 8). In IUGR fetuses treated with NAC, levels of eNOS mRNA were comparable to controls and lower than untreated IUGR animals in aorta and femoral arteries, and this effect was partially reverted in umbilical artery ECs (∼2.5‐fold increase relative to untreated control) (Fig. 7).
Figure 7. Levels of mRNA for eNOS in primary cultures of guinea pig fetal artery endothelial cells.
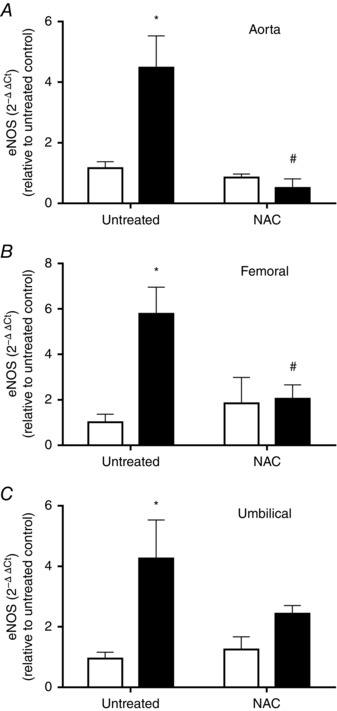
Levels of eNOS mRNA in primary cultures of fetal ECs from aorta (A), femoral (B) and umbilical (C) arteries in control (open bars) and IUGR (solid bars) guinea pig groups whose mothers received (NAC) or did not receive (untreated) antioxidant treatment. Values expressed as mean ± SEM, * P < 0.05 vs untreated control, # P < 0.05 vs untreated IUGR, two‐way ANOVA, Newman–Keuls multiple comparison test.
Figure 8. Levels of eNOS protein in umbilical artery endothelial cells.
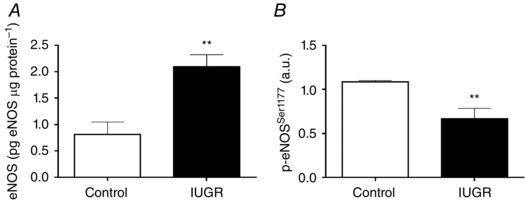
Total eNOS protein levels (A) and relative levels of eNOS (B) activating phosphorylation in ECs from control (n = 5) and IUGR (n = 4) fetuses quantified by ELISA. Values expressed as mean ± SEM, ** P < 0.01 vs control, Mann–Whitney U test.
DNA methylation of Nos3 promoter in primary cultures of fetal ECs
The methylation status of CpGs located in the proximal and core Nos3 gene promoter (−220 to +46) was evaluated in primary cultures of fetal ECs from aorta, femoral and umbilical arteries by pyrosequencing. In silico analysis of the proximal promoter of the Cavia porcellus Nos3 gene showed the presence of 12 CpGs dinucleotides between −500 and +100 bp, relative to the transcription start site, which were numbered according to their position. Seven of these 12 CpGs were located in conserved binding sites for transcription factors which have been reported that regulate basal eNOS gene expression (Figs 9 and 10). Fetal aorta and umbilical artery ECs from all the guinea pig groups showed a comparable methylation profile in most of the CpGs studied (Fig. 10 B and D). However, there was a substantial decrease (∼ 30%) in the methylation status of CpG −170 in IUGR fetuses relative to controls and this change was absent in fetuses whose mother received NAC. Notably, femoral artery ECs from IUGR animals showed two CpGs with significant decrease in the methylation status (CpG −170, ∼20%; CpG +34, ∼25%) as well as an increase in the methylation of CpG −27 (∼ 25%) relative to all the other groups studied (Fig. 10 C). Similar to aorta and umbilical arteries, all these changes in IUGR femoral ECs were prevented by maternal treatment with NAC.
Figure 9. Cavia porcellus Nos3 gene promoter and first exon sequence.
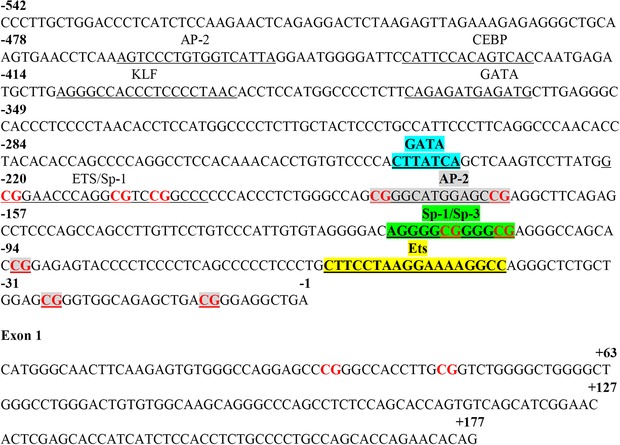
Sequence of Nos3 gene obtained from Ensembl genome browser. Transcription factor (TF) binding sites are indicated as underlined sequences with the cognate TF above. Colours highlight binding sites for TFs that have been previously reported in mice and humans which are conserved in guinea pigs. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 10. Level of DNA methylation in the Nos3 promoter of guinea pig fetal artery endothelial cells.
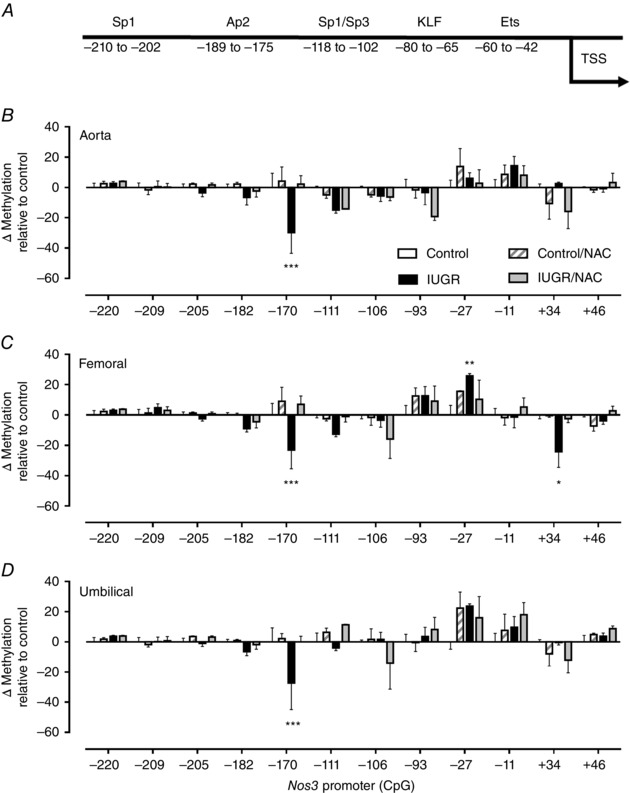
A, schematic representation of guinea pig Nos3 promoter and cognate binding sites for transcription factors predicted with MatInspector. B–D, change in DNA methylation levels relative to control in CpGs present in the Nos3 promoter in primary cultures of fetal ECs from aorta (B), femoral (C) and umbilical (D) arteries in untreated control (open bars) and IUGR (solid bars), as well as treated control (dashed bars) and IUGR (grey bars) guinea pigs. Values expressed as mean ± SEM, * P < 0.05, ** P < 0.01, *** P < 0.001 vs untreated control, two‐way ANOVA, Newman–Keuls multiple comparison test.
Discussion
This study demonstrates that maternal treatment with NAC during the second half of gestation prevents specific IUGR signatures, improving umbilical artery flow indicators and preserving vascular function leading to an enhancement in placental efficiency in a guinea pig model of progressive uterine artery occlusion. Furthermore, maternal NAC treatment was associated with an improvement in the ex vivo aortic and umbilical artery endothelial function, as well as a normalization in the expression of eNOS and Nos3 promoter DNA methylation profile in primary cultures of fetal ECs from IUGR animals. Together these data serve to demonstrate that, in pregnant guinea pigs, maternal treatment with NAC improves fetal endothelial function and prevents the epigenetic programming in an animal model of placental insufficiency.
Several studies have demonstrated that oxidative stress during gestation leads to reduced fetal growth and altered placental vascular function (Friedman & Cleary, 2014; Roberts, 2014). Increased levels of pro‐oxidants in maternal urine during the first trimester of pregnancy correlate negatively with birth weight (Potdar et al. 2009). Furthermore, women with altered uterine artery and adverse neonatal outcomes (i.e. IUGR and/or pre‐eclampsia) (Stepan et al. 2004) have a reduced plasma antioxidant capacity. In addition, fetal and maternal plasma antioxidant capacity is decreased at birth in IUGR animals (Bar‐Or et al. 2005; Saker et al. 2008; Mert et al. 2012). Evidence from IUGR animal models demonstrates that maternal antioxidant treatment with vitamins C and E can prevent the adverse perinatal outcomes, although these treatments have failed to show clear benefits in human clinical studies (Hovdenak & Haram, 2012). A possible explanation for this is the slower reaction rate of these vitamins with pro‐oxidants in comparison with endogenous antioxidant defences and cellular oxidative stress sensors (Winterbourn & Hampton, 2008), and the deficient metabolism of vitamin C in humans in contrast to rodents (mice and rats) (Yu & Schellhorn, 2013).
The data presented show that maternal treatment with NAC prevents the altered abdominal growth pattern and normalized umbilical artery parameters (Figs 1 and 2), suggesting the potential use of NAC in preventing IUGR under conditions of impaired utero‐placental perfusion. Note that NAC treatment in controls was associated with an increase in head circumference growth rate but this did not result in an increased brain weight at term. In humans, NAC has a high transfer rate from the maternal to fetal circulation (Wiest et al. 2014) and it has been used during pregnancy as a treatment for maternal acetaminophen overdose (Kozer & Koren, 2001) and preterm labour (Shahin et al. 2009). NAC has a weak antioxidant capacity against superoxide and hydrogen peroxide, but has a very effective action on hypoclorous acid, hydroxyl, alkoxy and peroxy radicals (Samuni et al. 2013). It is proposed that NAC is a potent antioxidant acting as an effective glutathione precursor due to its high stability and good absorption by the digestive track, and also serves as a precursor for cysteine at the hepatic level (Lasram et al. 2015). Notably, in this study treatment of pregnant sows with NAC was associated with a reduction in levels of 3‐nitrotyrosine in IUGR fetal aorta (Fig. 2), an effect that has been also observed in fetal heart (Evans et al. 2012) and liver (Hashimoto et al. 2012). In this context, glutathione is a key molecule involved in the redox cellular signalling regulating the oxidation of cysteines from oxidative stress sensors (Winterbourn & Hampton, 2008, 2015). Together these data support a potential therapeutic use of this agent during pregnancy to prevent altered placental vascular function and thus IUGR.
Other studies have addressed the effects of maternal treatments with NAC on IUGR. Studies in rats in which uterine artery perfusion is reduced during late gestation showed that maternal treatment with NAC does not prevent the reduction in fetal weight but contributes to the recovery in brain development (Chang et al. 2005). Similarly, NAC treatment in a model of IUGR in guinea pigs by exposure to hypoxia in the last quarter of gestation revealed a protection from oxidative stress at the systemic level and a partial recovery of placenta efficiency but did not prevent fetal growth restriction (Evans et al. 2012; Al‐Hasan et al. 2013). In contrast, in the present study, NAC treatment reverted the effect of an impaired utero‐placental perfusion on fetal growth restriction not only at the brain level but also by improving fetal weight as well as lower body organs (Table 2). These differences might be explained by the different timing of the antioxidant treatment and/or the origin of placental insufficiency. Human IUGR is frequently characterized by a progressive impairment of placental vascular function that has its origins in the first half of gestation rather than sudden changes in perfusion, a condition that resembles our model. Our data show that the glutathione precursor NAC prevents placental vascular dysfunction by improving NO‐dependent relaxation and increases placental efficiency in IUGR guinea pigs with a compensatory effect on fetal growth (Figs 2 and 5). Pioneering studies by Myatt et al. (1996) demonstrated that placentae from IUGR pregnancies show increased levels of eNOS and also augmented levels of peroxynitrite derivative. In agreement with this, it has been suggested that the endothelial dysfunction present in IUGR pregnancies occurs by a reduced NO bioavailability or impaired eNOS activity (Krause et al. 2011). Our results show that umbilical arteries from IUGR guinea pigs have a decreased endothelial‐dependent relaxation with an enhanced response to exogenous NO. Notably similar characteristics are present in umbilical and chorionic arteries from IUGR human samples (Mills et al. 2005; Krause et al. 2013 a).
Further studies on our model will focus on the NO‐dependent mechanisms involved in the vascular effects of NAC. Conversely, treatment with NAC in IUGR fetuses led to normal umbilical artery RI and PI (Fig. 2), normal endothelial‐dependent relaxation as well as an improved response to NO (Fig. 5) and placental efficiency (Fig. 4). These data are in agreement with previous reports in which acute treatment with NAC restored the endothelial function in isolated placental arteries from pregnancies with IUGR (Schneider et al. 2015) and pre‐eclampsia (Bisseling et al. 2004). Moreover, the increase in placental efficiency induced by NAC could also reflect a better antioxidant capacity, preventing the impairment in maternal‐to‐fetal amino acid transport by the syncytiotrophoblast, which is negatively affected by oxidative stress (Khullar et al. 2004). Together, these data support the concept that NAC prevents and may restore placental vascular dysfunction associated with a reduced utero‐placental blood flow in IUGR pregnancies.
A clear association between IUGR and an impaired endothelial function in adulthood has been extensively reported, although the evidence showing the direct effects of IUGR on fetal endothelial function as well as markers of epigenetic programming in the endothelium at birth is limited. In this study we have provided novel data addressing the effects of IUGR on fetal systemic and umbilical endothelial function and cellular programming. We found an impaired endothelial function in the aorta of IUGR fetuses, which was reverted by maternal NAC treatment (Fig. 6). Comparable changes in endothelial NO‐mediated relaxation in the aorta have been reported in a transgenic mouse model of IUGR, with a sex‐specific decrease in the maximal relaxation in male fetuses (Renshall et al. 2014). We found no sex‐specific effect of IUGR, but this discrepancy on the effects of IUGR on endothelial function could result from the different normalization procedure used for determining the basal tone of the vessels. Nevertheless, signs of endothelial dysfunction at birth have been reported in human neonates small for gestational age (Martin et al. 2000). Similarly, the presence of endothelial dysfunction has been consistently reported in placental and umbilical arteries from IUGR subjects, suggesting a common ‘IUGR signature’ in systemic and placental endothelium (Krause et al. 2013 a). Notably in the present study, changes in endothelial‐dependent relaxation followed the same direction in IUGR aorta and umbilical arteries, with an important decrease in NOS‐mediated reactivity (Figs 5 and 6).
Compelling data have led to the notion that in the endothelium, eNOS expression and function are mainly determined by transcriptional and post‐translational mechanisms (Fish & Marsden, 2006; Balligand et al. 2009; Eisenreich, 2013). In this context, studies in primary cultures of human umbilical and placental arteries show that endothelial dysfunction in IUGR (Krause et al. 2013 a; Jones et al. 2015) and pre‐term (Postberg et al. 2015) is characterized by an increased expression of eNOS mRNA with a parallel increase in eNOS protein levels, but a significant decrease in activating post‐translational modifications (Krause et al. 2013 a). Similarly, we found that eNOS activation (p‐eNOSser1177) was substantially reduced in IUGR umbilical artery endothelia cells (Fig. 8). This counterintuitive increase in eNOS mRNA and protein in conditions of endothelial dysfunction could result from an eNOS gene induction in the early stages of hypoxia (Krause et al. 2012) whose activity would be finely tuned by post‐translational mechanisms in the long term. Based on the limited quantity of endothelium available in freshly isolated vessels of fetal guinea pigs, we aimed to determine the mRNA expression of eNOS in primary cultures of ECs. We observed a comparable increase in eNOS mRNA levels in cultured arterial ECs (aorta, femoral and umbilical) derived from IUGR guinea pig fetuses, an effect reverted by maternal treatment with NAC (Fig. 7). Together, these data suggest a common programming of eNOS expression in umbilical and systemic ECs which would be mediated by oxidative stress.
Considering that the increased eNOS expression in IUGR endothelium potentially results from an epigenetic effect, we aimed to determine the DNA methylation pattern in the Nos3 promoter in fetal systemic (aorta and femoral) and umbilical ECs, as a mechanism mediating the mRNA levels of eNOS (Fish & Marsden, 2006). We found that, similar to reports in human (Chan et al. 2004) and rat (Xu et al. 2010), the guinea pig Nos3 gene had a low density of CpGs in the proximal and core promoter, laying most of them in binding sites for transcription factors that regulate basal eNOS expression (Zhang et al. 1995; Teichert et al. 1998; Karantzoulis‐Fegaras et al. 1999) (Fig. 9). These results showed that in IUGR ECs a specific CpG (−170) had a decreased methylation, suggesting the presence of an epigenetic signature of IUGR that is common for the systemic and umbilical endothelium (Fig. 10). Conversely, IUGR femoral ECs showed two additional changes in methylation, CpG −27 (increased) and CpG +34 (decreased), which were absent in fetuses from the other groups. Notably, the IUGR hallmark (CpG −170) was in the context of a conserved binding site for Ap2, which could potentially regulate basal eNOS transcription and expression (Zhang et al. 1995; Teichert et al. 1998). A previous report has shown that a brief exposure to hypoxia of near‐term pregnant rats, a condition associated with fetal growth restriction, induces an increase in eNOS expression in fetal pulmonary ECs which correlates with a decreased methylation in the Nos3 promoter (Xu et al. 2010). Similarly, we have previously reported that altered eNOS mRNA levels in human umbilical and placental artery ECs from IUGR subjects is associated with a decreased DNA methylation in a single CpG (−352) in the NOS3 promoter and is reverted in vitro by the knock‐down of DNMT1 (Krause et al. 2013 b). Together, these data show that conditions limiting in utero nutrients and oxygen supply affect endothelial programming by changing the methylation profile of the eNOS gene. To the best of our knowledge, this is the first study showing the effects of IUGR on the epigenetic programming of eNOS in fetal systemic endothelium and, moreover, comparing this effect with that observed in umbilical arteries. Nevertheless, further studies are required to determine the participation of additional epigenetic mechanisms that regulate eNOS expression (i.e. histone post‐translational modifications and non‐coding RNAs) (Fish & Marsden 2006), which could contribute to the vascular programming in IUGR. In fact, it has demonstrated that the increased eNOS expression in HUAECs from pre‐term neonates with altered vascular function is associated with changes in the levels of permissive post‐transcriptional histone modifications (Postberg et al. 2015). Nonetheless, considering that maternal treatment with NAC reverts the functional and epigenetic signatures of IUGR it is possible to suggest that these changes are importantly mediated by increased oxidative stress present in IUGR pregnancies (Herrera et al. 2014).
In conclusion, this study suggests that maternal treatment with NAC prevents the effects of a reduced utero‐placental perfusion on fetal growth by increasing placental efficiency and preventing umbilical and systemic endothelial dysfunction. Notably, the endothelial dysfunction observed in IUGR is related to a DNA methylation signature in the promoter region of the Nos3 gene in primary cultures of aorta, femoral and umbilical arteries, which is reverted by maternal NAC treatment.
Additional information
Competing interests
The authors have no competing interests.
Author contributions
EAH, MF, PC and BJK conceived and designed the experiments. EAH, FCZ, EF, CV, CH, RA, VA, MF and BJK collected, analysed and interpreted the experimental data. EAH, RU, PC and BJK drafted the article, and all authors revised it critically and approved the final version.
Funding
This work was funded by grants 1130801 and 115119 from the National Fund for Scientific and Technological Development (FONDECYT‐Chile).
Acknowledgements
We are very grateful to Marta Gonzalez and René Vergara for their excellent technical assistance.
References
- Al‐Hasan YM, Evans LC, Pinkas GA, Dabkowski ER, Stanley WC & Thompson LP (2013). Chronic hypoxia impairs cytochrome oxidase activity via oxidative stress in selected fetal Guinea pig organs. Reprod Sci 20, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balligand JL, Feron O & Dessy C (2009). eNOS activation by physical forces: from short‐term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev 89, 481–534. [DOI] [PubMed] [Google Scholar]
- Bar‐Or D, Heyborne KD, Bar‐Or R, Rael LT, Winkler JV & Navot D (2005). Cysteinylation of maternal plasma albumin and its association with intrauterine growth restriction. Prenat Diagn 25, 245–249. [DOI] [PubMed] [Google Scholar]
- Bisseling TM, Maria Roes E, Raijmakers MT, Steegers EA, Peters WH & Smits P (2004). N‐Acetylcysteine restores nitric oxide‐mediated effects in the fetoplacental circulation of preeclamptic patients. Am J Obstet Gynecol 191, 328–333. [DOI] [PubMed] [Google Scholar]
- Caniuguir A, Krause BJ, Hernandez C, Uauy R & Casanello P (2016). Markers of early endothelial dysfunction in intrauterine growth restriction‐derived human umbilical vein endothelial cells revealed by 2D‐DIGE and mass spectrometry analyses. Placenta 41, 14–26. [DOI] [PubMed] [Google Scholar]
- Cohen E, Wong FY, Horne RS & Yiallourou SR (2016). Intrauterine growth restriction: impact on cardiovascular development and function throughout infancy. Pediatr Res 79, 821–830. [DOI] [PubMed] [Google Scholar]
- Chan Y, Fish JE, D'Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis‐Fegaras F, Keightley A, Steer BM & Marsden PA (2004). The cell‐specific expression of endothelial nitric‐oxide synthase: a role for DNA methylation. J Biol Chem 279, 35087–35100. [DOI] [PubMed] [Google Scholar]
- Chang EY, Barbosa E, Paintlia MK, Singh A & Singh I (2005). The use of N‐acetylcysteine for the prevention of hypertension in the reduced uterine perfusion pressure model for preeclampsia in Sprague‐Dawley rats. Am J Obstet Gynecol 193, 952–956. [DOI] [PubMed] [Google Scholar]
- Chen SF, Fei X & Li SH (1995). A new simple method for isolation of microvascular endothelial cells avoiding both chemical and mechanical injuries. Microvasc Res 50, 119–128. [DOI] [PubMed] [Google Scholar]
- Devaskar SU & Chu A (2016). Intrauterine growth restriction: hungry for an answer. Physiology (Bethesda) 31, 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenreich A (2013). Regulation of vascular function on posttranscriptional level. Thrombosis 2013, 948765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans LC, Liu H, Pinkas GA & Thompson LP (2012). Chronic hypoxia increases peroxynitrite, MMP9 expression, and collagen accumulation in fetal guinea pig hearts. Pediatr Res 71, 25–31. [DOI] [PubMed] [Google Scholar]
- Fish JE & Marsden PA (2006). Endothelial nitric oxide synthase: insight into cell‐specific gene regulation in the vascular endothelium. Cell Mol Life Sci 63, 144–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AM & Cleary KL (2014). Prediction and prevention of ischemic placental disease. Semin Perinatol 38, 177–182. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Buklijas T, Low FM & Beedle AS (2009). Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol 5, 401–408. [DOI] [PubMed] [Google Scholar]
- Hanson MA & Gluckman PD (2014). Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev 94, 1027–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Pinkas G, Evans L, Liu H, Al‐Hasan Y & Thompson LP (2012). Protective effect of N‐acetylcysteine on liver damage during chronic intrauterine hypoxia in fetal guinea pig. Reprod Sci 19, 1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera EA, Alegria R, Farias M, Diaz‐Lopez F, Hernandez C, Uauy R, Regnault TR, Casanello P & Krause BJ (2016). Assessment of in vivo fetal growth and placental vascular function in a novel intrauterine growth restriction model of progressive uterine artery occlusion in guinea pigs. J Physiol 594, 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera EA, Krause B, Ebensperger G, Reyes RV, Casanello P, Parra‐Cordero M & Llanos AJ (2014). The placental pursuit for an adequate oxidant balance between the mother and the fetus. Front Pharmacol 5, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovdenak N & Haram K (2012). Influence of mineral and vitamin supplements on pregnancy outcome. Eur J Obstet Gynecol Reprod Biol 164, 127–132. [DOI] [PubMed] [Google Scholar]
- Jones S, Bischof H, Lang I, Desoye G, Greenwood SL, Johnstone ED, Wareing M, Sibley CP & Brownbill P (2015). Dysregulated flow‐mediated vasodilatation in the human placenta in fetal growth restriction. J Physiol 593, 3077–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantzoulis‐Fegaras F, Antoniou H, Lai SL, Kulkarni G, D'Abreo C, Wong GK, Miller TL, Chan Y, Atkins J, Wang Y & Marsden PA (1999). Characterization of the human endothelial nitric‐oxide synthase promoter. J Biol Chem 274, 3076–3093. [DOI] [PubMed] [Google Scholar]
- Khullar S, Greenwood SL, McCord N, Glazier JD & Ayuk PT (2004). Nitric oxide and superoxide impair human placental amino acid uptake and increase Na+ permeability: implications for fetal growth. Free Radic Biol Med 36, 271–277. [DOI] [PubMed] [Google Scholar]
- Kozer E & Koren G (2001). Management of paracetamol overdose: current controversies. Drug Safety 24, 503–512. [DOI] [PubMed] [Google Scholar]
- Krause BJ, Carrasco‐Wong I, Caniuguir A, Carvajal J, Farias M & Casanello P (2013. a). Endothelial eNOS/arginase imbalance contributes to vascular dysfunction in IUGR umbilical and placental vessels. Placenta 34, 20–28. [DOI] [PubMed] [Google Scholar]
- Krause BJ, Costello PM, Munoz‐Urrutia E, Lillycrop KA, Hanson MA & Casanello P (2013. b). Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics 8, 944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause BJ, Hanson MA & Casanello P (2011). Role of nitric oxide in placental vascular development and function. Placenta 32, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause BJ, Hernandez C, Caniuguir A, Vasquez‐Devaud P, Carrasco‐Wong I, Uauy R & Casanello P (2016). Arginase‐2 is cooperatively up‐regulated by nitric oxide and histone deacetylase inhibition in human umbilical artery endothelial cells. Biochem Pharmacol 99, 53–59. [DOI] [PubMed] [Google Scholar]
- Krause BJ, Herrera EA, Diaz‐Lopez FA, Farias M, Uauy R & Casanello P (2015). Pre‐gestational overweight in guinea pig sows induces fetal vascular dysfunction and increased rate of large and small fetuses. J Dev Orig Health Dis 7, 237–243. [DOI] [PubMed] [Google Scholar]
- Krause BJ, Prieto CP, Munoz‐Urrutia E, San Martin S, Sobrevia L & Casanello P (2012). Role of arginase‐2 and eNOS in the differential vascular reactivity and hypoxia‐induced endothelial response in umbilical arteries and veins. Placenta 33, 360–366. [DOI] [PubMed] [Google Scholar]
- Lasram MM, Dhouib IB, El Fazaa S & Gharbi N (2015). A review on the possible molecular mechanism of action of N‐acetylcysteine against insulin resistance and type‐2 diabetes development. Clin Biochem 48, 1200–1208. [DOI] [PubMed] [Google Scholar]
- Livak KJ & Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Macdonald EM, Natale R, Regnault TR, Koval JJ & Campbell MK (2014). Obstetric conditions and the placental weight ratio. Placenta 35, 582–586. [DOI] [PubMed] [Google Scholar]
- Martin H, Gazelius B & Norman M (2000). Impaired acetylcholine‐induced vascular relaxation in low birth weight infants: implications for adult hypertension? Pediatr Res 47, 457–462. [DOI] [PubMed] [Google Scholar]
- Mert I, Oruc AS, Yuksel S, Cakar ES, Buyukkagnici U, Karaer A & Danisman N (2012). Role of oxidative stress in preeclampsia and intrauterine growth restriction. J Obstet Gynaecol Res 38, 658–664. [DOI] [PubMed] [Google Scholar]
- Mills TA, Wareing M, Bugg GJ, Greenwood SL & Baker PN (2005). Chorionic plate artery function and Doppler indices in normal pregnancy and intrauterine growth restriction. Eur J Clin Invest 35, 758–764. [DOI] [PubMed] [Google Scholar]
- Myatt L, Rosenfield RB, Eis AL, Brockman DE, Greer I & Lyall F (1996). Nitrotyrosine residues in placenta. Evidence of peroxynitrite formation and action. Hypertension 28, 488–493. [DOI] [PubMed] [Google Scholar]
- Pardi G, Marconi AM & Cetin I (2002). Placental‐fetal interrelationship in IUGR fetuses–a review. Placenta 23 Suppl A, S136–141. [DOI] [PubMed] [Google Scholar]
- Postberg J, Kanders M, Forcob S, Willems R, Orth V, Hensel KO, Weil PP, Wirth S & Jenke AC (2015). CpG signalling, H2A.Z/H3 acetylation and microRNA‐mediated deferred self‐attenuation orchestrate foetal NOS3 expression. Clin Epigenet 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potdar N, Singh R, Mistry V, Evans MD, Farmer PB, Konje JC & Cooke MS (2009). First‐trimester increase in oxidative stress and risk of small‐for‐gestational‐age fetus. BJOG 116, 637–642. [DOI] [PubMed] [Google Scholar]
- Renshall LJ, Dilworth MR, Greenwood SL, Sibley CP & Wareing M (2014). In vitro assessment of mouse fetal abdominal aortic vascular function. Am J Physiol Regul Integr Comp Physiol 307, R746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JM (2014). Pathophysiology of ischemic placental disease. Semin Perinatol 38, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saker M, Soulimane Mokhtari N, Merzouk SA, Merzouk H, Belarbi B & Narce M (2008). Oxidant and antioxidant status in mothers and their newborns according to birthweight. Eur J Obstet Gynecol Reprod Biol 141, 95–99. [DOI] [PubMed] [Google Scholar]
- Samuni Y, Goldstein S, Dean OM & Berk M (2013). The chemistry and biological activities of N‐acetylcysteine. Biochim Biophys Acta 1830, 4117–4129. [DOI] [PubMed] [Google Scholar]
- Schneider D, Hernandez C, Farias M, Uauy R, Krause BJ & Casanello P (2015). Oxidative stress as common trait of endothelial dysfunction in chorionic arteries from fetuses with IUGR and LGA. Placenta 36, 552–558. [DOI] [PubMed] [Google Scholar]
- Shahin AY, Hassanin IM, Ismail AM, Kruessel JS & Hirchenhain J (2009). Effect of oral N‐acetyl cysteine on recurrent preterm labor following treatment for bacterial vaginosis. Int J Gynaecol Obstet 104, 44–48. [DOI] [PubMed] [Google Scholar]
- Stepan H, Heihoff‐Klose A & Faber R (2004). Reduced antioxidant capacity in second‐trimester pregnancies with pathological uterine perfusion. Ultrasound Obstet Gynecol 23, 579–583. [DOI] [PubMed] [Google Scholar]
- Teichert AM, Karantzoulis‐Fegaras F, Wang Y, Mawji IA, Bei X, Gnanapandithen K & Marsden PA (1998). Characterization of the murine endothelial nitric oxide synthase promoter. Biochim Biophys Acta 1443, 352–357. [DOI] [PubMed] [Google Scholar]
- Wiest DB, Chang E, Fanning D, Garner S, Cox T & Jenkins DD (2014). Antenatal pharmacokinetics and placental transfer of N‐acetylcysteine in chorioamnionitis for fetal neuroprotection. J Pediatr 165, 672–677 e672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC & Hampton MB (2008). Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 45, 549–561. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC & Hampton MB (2015). Redox biology: signaling via a peroxiredoxin sensor. Nature Chem Biol 11, 5–6. [DOI] [PubMed] [Google Scholar]
- Xu XF, Ma XL, Shen Z, Wu XL, Cheng F & Du LZ (2010). Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J Hypertens 28, 2227–2235. [DOI] [PubMed] [Google Scholar]
- Yu R & Schellhorn HE (2013). Recent applications of engineered animal antioxidant deficiency models in human nutrition and chronic disease. J Nutr 143, 1–11. [DOI] [PubMed] [Google Scholar]
- Zhang J, Merialdi M, Platt LD & Kramer MS (2010). Defining normal and abnormal fetal growth: promises and challenges. Am J Obstet Gynecol 202, 522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Min W & Sessa WC (1995). Functional analysis of the human endothelial nitric oxide synthase promoter. Sp1 and GATA factors are necessary for basal transcription in endothelial cells. J Biol Chem 270, 15320–15326. [DOI] [PubMed] [Google Scholar]