

Abstract
Key points
Neuromuscular acetylcholine (ACh) receptors have a high affinity for the neurotransmitter ACh and a low affinity for its metabolic product choline.
At each transmitter binding site three aromatic groups determine affinity, and together provide ∼50% more binding energy for ACh than for choline.
Deprotonation of αY190 by a nearby lysine strengthens the interaction between this aromatic ring and both ACh and choline.
H‐bonds position ACh and choline differently in the aromatic cage to generate the different affinities.
Abstract
Acetylcholine (ACh) released at the vertebrate nerve‐muscle synapse is hydrolysed rapidly to choline (Cho), so endplate receptors (AChRs) are exposed to high concentrations of both of these structurally related ligands. To understand how these receptors distinguish ACh and Cho, we used single‐channel electrophysiology to measure resting affinities (binding free energies) of these and other agonists in adult‐type mouse AChRs having a mutation(s) at the transmitter‐binding sites. The aromatic rings of αY190, αW149 and αY198 each provide ∼50% less binding energy for Cho compared to ACh. At αY198 a phenylalanine substitution had no effect, but at αY190 this substitution caused a large, agonist‐independent loss in binding energy that depended on the presence of αK145. The results suggest that (1) αY190 is deprotonated by αK145 to strengthen the interaction between this benzene ring and the agonist's quaternary ammonium (QA) and (2) AChRs respond strongly to ACh because an H‐bond positions the QA to interact optimally with the rings, and weakly to Cho because a different H‐bond tethers the ligand to misalign the QA and form weaker interactions with the aromatic groups. The results suggest that the difference in ACh versus Cho binding energies is determined by different ligand positions within a fixed protein structure.
Keywords: agonist, cation‐pi, ion channel, neuromuscular, receptor
Key points
Neuromuscular acetylcholine (ACh) receptors have a high affinity for the neurotransmitter ACh and a low affinity for its metabolic product choline.
At each transmitter binding site three aromatic groups determine affinity, and together provide ∼50% more binding energy for ACh than for choline.
Deprotonation of αY190 by a nearby lysine strengthens the interaction between this aromatic ring and both ACh and choline.
H‐bonds position ACh and choline differently in the aromatic cage to generate the different affinities.
Abbreviations
- 3OH‐PTMA
3‐hydroxypropyl‐trimethylammonium tosylate
- 4OH‐BTMA
4‐hydroxybutyltrimethylammonium tosylate
- ACh
acetylcholine
- AChBP
acetylcholine binding protein
- AChR
acetylcholine receptor
- Cho
choline
- E0/E1/E2
unliganded/monoliganded/diliganded gating equilibrium constants
- Kd/Jd
resting/open equilibrium dissociation constants
- PTMA
propyl‐trimethylammonium
- QA
quaternary ammonium
- TMA
tetramethylammonium
Introduction
Agonists produce transient membrane currents by binding to acetylcholine receptors (AChRs) and increasing the rate and probability of channel‐opening over the basal level. For proper signalling at the neuromuscular synapse, AChRs must respond to acetylcholine (ACh) but ignore choline (Cho). Although both of these ligands have a positively charged quaternary ammonium (QA) group, the efficacy (peak open probability; P O max) and resting affinity are ∼20 times greater for ACh than for Cho (Purohit & Grosman, 2006; Lape et al. 2009; Jadey et al. 2011). In adult‐type AChRs the molecular determinants of the resting equilibrium dissociation constant for the neurotransmitter have been localized mainly to the functional groups of three transmitter‐binding site aromatic residues, αW149, αY190 and αY198 (Middleton & Cohen, 1991; Tomaselli et al. 1991; Sine et al. 1994; Nowak et al. 1995; Kearney et al. 1996; Purohit et al. 2012) (Fig. 1), but the roles of these groups in setting affinity for Cho have not yet been determined.
Figure 1. AChR and the aromatic cage.
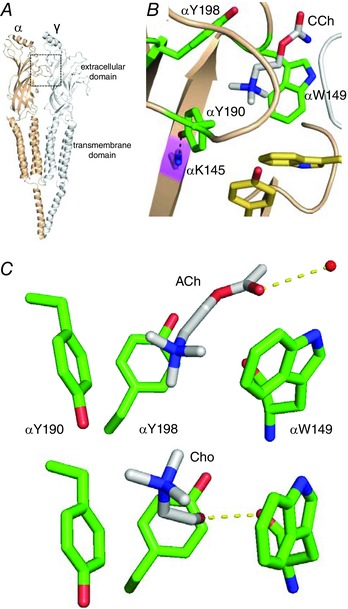
A, low‐resolution view of the Torpedo AChR (PDB accession number 2BG9). Each transmitter binding site is at a subunit interface (box). B, close‐up of the ligand binding site of an acetylcholine binding protein (AChBP; PDB accession number 1UV6). Three α‐subunit aromatics (green) form a cage around carbamylcholine (CCh, white); these determine affinity in adult AChRs but in fetal‐type αY93 and γW55 (yellow) also make a contribution. Black dashed line, interaction between αK145 and αY190. Numbers are mouse AChRs. C, close‐up of the α‐subunit aromatic cage. Top, X‐ray structure of AChBP bound with ACh (PDB accession number 3WIP). Dashed line, H‐bond to a structural water (red sphere). Bottom, homology model of Cho in the mouse α–δ binding site. Dashed line, H‐bond to the αW149 backbone carbonyl. [Colour figure can be viewed at wileyonlinelibrary.com]
The adult‐type neuromuscular AChR is a heteropentamer having two α1 subunits and one each of β, δ and ε. The extracellular domain contains two transmitter‐binding sites at α–δ and α–ε subunit interfaces that are ∼60 Å from a gate in the transmembrane domain that regulates channel conductance. The aromatic amino acids at the transmitter sites stabilize ACh by cation–π interactions (Zhong et al. 1998; Brejc et al. 2001; Dougherty, 2013). In ACh binding proteins, ACh forms an H‐bond with a structural water that H‐bonds with the complementary subunit, and nicotine's tertiary amine forms an H‐bond with the backbone of W149 (Celie et al. 2004; Olsen et al. 2014). Unnatural amino acid substitutions suggest that these interactions are present in AChRs (Xiu et al. 2009; Blum et al. 2010).
In adult AChRs, four functional groups are the main sources of ACh binding energy: αW149 (ring), αY190 (ring and OH) and αY198 (ring) (Purohit et al. 2012). In mouse AChRs the two transmitter sites act independently (Jha & Auerbach, 2010; Nayak et al. 2014), are approximately equivalent for ACh, tetramethylammonium (TMA) and Cho, and together generate binding energies of −10.2, −9.0 and −6.6 kcal mol−1, respectively (Jadey et al. 2011). The per‐site resting equilibrium dissociation constants for these agonists are 175, 490 and 3700 μm, and the P O max values are 0.96, 0.67 and 0.05. At the fetal α–γ site, γW55 in the complementary subunit and αY93 make additional contributions, to increase the affinity for these agonists by ∼30‐fold (Nayak et al. 2014).
Previously, estimates of binding energies for a series of Cho derivatives in adult AChRs showed that the removal or relocation of the ligand's OH group led to higher affinity, whereas an H‐bonding ability at the ethyl tail produced lower affinity (Bruhova et al. 2013). Molecular models and simulations suggested that an H‐bond between the OH of Cho and the αW149 backbone carbonyl led to suboptimal positioning of the QA within the aromatic pocket and less binding energy from αW149.
To better understand molecular recognition in endplate AChRs, and in particular the reasons for the low affinity and efficacy of Cho compared to ACh, we mutated the above three aromatics and the nearby, basic amino acid αK145 and measured binding energies for ACh, TMA, Cho and other agonists. Y‐to‐F mutations of the tyrosines allowed us to estimate the contribution of each OH, and W/F‐to‐A mutations allowed us to estimate the contribution from each aromatic ring.
Methods
Mutagenesis and electrophysiology
Mouse subunit AChR cDNAs were mutated using the QuikChange site‐directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) and verified by nucleotide sequencing. Human embryonic kidney (HEK) 293 cells grown on 35 mm culture dishes were transfected with 3 ± 1 μg cDNA using calcium phosphate precipitation. The cDNA cocktail included α, β, δ and ε subunits in a ratio of 2:1:1:1.
Single‐channel currents were recorded in the cell‐attached patch configuration at 23°C within ∼48 h after transfection. The bath solution was (in mm) 142 KCl, 5.4 NaCl, 1.8 CaCl2, 1.7 MgCl2 and 10 Hepes/KOH (pH 7.4), and the patch pipette solution was Dulbecco's phosphate‐buffered saline (in mm): 137 NaCl, 0.9 CaCl2, 2.7 KCl, 1.5 KH2PO4, 0.5 MgCl2 and 8.1 Na2HPO4 (pH 7.3 with NaOH).
Agonist was added to the pipette at a concentration that was high enough to ensure full occupancy (‘saturation’) of the binding sites (50–140 mm). The agonists (see Fig. 4 for structures) were ACh, Cho, TMA, propyltrimethylammonium bromide (PTMA), 3‐hydroxypropyl‐trimethylammonium tosylate (3OH‐PTMA) and 4‐hydroxybutyltrimethylammonium tosylate (4OH‐BTMA). PTMA, 3OH‐PTMA and 4OH‐BTMA were synthesized as described elsewhere (Bruhova et al. 2013). High concentrations of agonist cause fast open‐channel block at hyperpolarized membrane potentials, so the membrane was depolarized to +70 mV. Depolarization changes the unliganded gating equilibrium constant (E 0) but has no effect on affinity (Nayak et al. 2012).
Figure 4. Effects of OH removal at αY190 and αY198 are agonist‐independent.
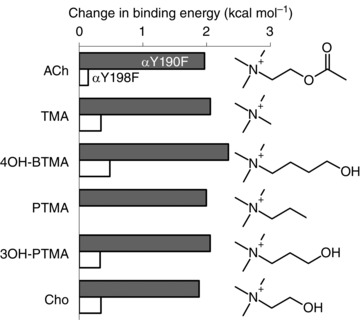
Removal of the hydroxyl group of αY190 causes a ∼+2.0 kcal mol−1 reduction in binding energy for six different agonists, but removal of a hydroxyl group of αY198 has little effect.
Analyses of current interval durations were performed by using QUB software (Nicolai & Sachs, 2013). Single‐channel currents were analog‐filtered at 20 kHz and digitized at a sampling frequency of 50 kHz. Clusters of shut↔open gating activity (Fig. 2 A) were selected and idealized into noise‐free interval durations using the segmental k‐means algorithm (Qin, 2004) after digitally low‐pass filtering at 12 kHz. Because of the high [agonist], gating was only A2C↔A2O. The idealized interval durations were fitted to a simple, two‐state shut↔open model, with an additional shut state sometimes added to account for an approximately millisecond lifetime, non‐conducting state outside of gating (Salamone et al. 1999; Elenes & Auerbach, 2002). The forward and backward gating rate constants were estimated by using a maximum‐interval likelihood algorithm after imposing a dead time of 25–50 μs (Qin et al. 1997) (Table S2). The diliganded or monoliganded gating equilibrium constant (E 2 or E 1) is the forward/backward rate constant ratio.
Figure 2. Single‐channel currents activated by Cho or TMA.
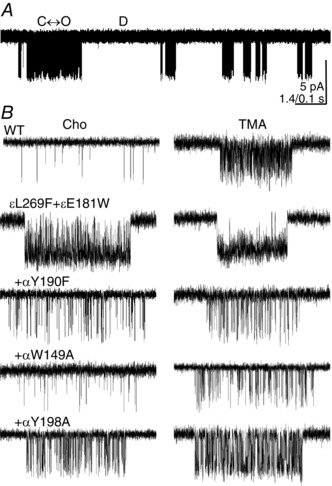
A, at low time resolution, αW149A openings are clustered (100 mm Cho, +70 mV, openings are down). Within a cluster a single AChR oscillates between C(losed) and O(pen), and between clusters all AChRs in the patch are D(esensitized). B, higher time resolution views of clusters. The background was either WT or ε(L269F+E181W), which increased the unliganded gating equilibrium constant 1492‐fold. The binding site mutations all reduce PO.
For measurements of unliganded gating equilibrium constant (E 0), the pipette solution was agonist‐free and currents were measured at −100 mV. Background mutations were used to enforce a high, constitutive P O (to allow cluster formation). After adding the binding site mutation(s), an unliganded gating equilibrium constant was estimated from the intra‐cluster interval durations using a C↔O model. The fold‐change in this constant relative to the background is the fold‐change relative to the WT (7.4 × 10−7; Nayak et al. 2012), establishing E 0 for the background+mutation(s) combination. The changes in E 0 for the aromatic mutants are published elsewhere (Purohit & Auerbach, 2010).
Affinity estimation
In adult mouse AChRs the two transmitter binding sites are approximately equivalent and independent for ACh and choline (Salamone et al. 1999; Jha & Auerbach, 2010; Nayak et al. 2016). We estimated K d from E 2 (measured using a single, saturating agonist concentration), as follows. The first component of the method is based on two facts: (1) AChRs switch between resting and active conformations (C↔O) with or without bound agonists (Jackson, 1986; Purohit & Auerbach, 2009; Auerbach, 2012), and (2) agonists can bind to C and to O (Grosman & Auerbach, 2001; Purohit & Auerbach, 2013). Hence, agonist activation can occur by either of two pathways. In WT AChRs the predominant path is C↔AC↔A2C↔A2O (where A is the agonist). The per‐site equilibrium dissociation constant for binding to C is K d and the diliganded gating equilibrium constant is E 2. The alternative, rarely taken path is C↔O↔AO↔A2O, where the unliganded gating equilibrium constant is E 0 and the per‐site equilibrium dissociation constant for binding to O is J d.
The two pathways can be connected to form a square (a closed cycle) (Monod et al. 1965). Without external energy, the product of equilibrium constants between any two states (here, C and A2O) is independent of the connecting path, so
or
| (1) |
The Kd/J d ratio is called the ‘coupling’ constant and is > 1 for agonists because C has a lower affinity than O. This ratio gives the extent to which agonists increase the gating equilibrium constant over the basal level.
The second component of the method is based on the empirical observation that in adult AChRs, and for a series of structurally related agonists (including ACh, TMA and choline), K d and J d are correlated (Jadey & Auerbach, 2012; Auerbach, 2016):
Inserting this into eqn (1) and rearranging,
| (2) |
Hence, in molar units, K d = (E 2/E 0)−0.5. E 0 is the same for all agonists and was known for each AChR construct (see above), so K d could be estimated just from the measured value of E 2. For example, E 2 ACh = 25 and E 0 WT = 7.4 × 10−7, so we calculate K d = 170 μm, which is the same as measured by fitting across multiple [ACh] (Chakrapani et al. 2003). Using this shortcut, which allows K d to be estimated from activity recorded at a single agonist concentration, increased the throughput substantially.
We describe the affinity changes as free energy differences. By taking the natural log of K d and multiplying by RT (where R is the gas constant and T is the absolute temperature), the equilibrium dissociation constant is converted into a binding free energy in the units kcal mol−1. In our experiments, RT = 0.59. Hence, the agonist binding energy in kcal mol−1 was +0.59lnK d. Free energy differences of 1, 2 and 3 kcal mol−1 correspond to 5.4‐, 30‐ and 161‐fold changes in affinity, with negative energies indicating larger affinities (smaller K d values). Using energy to describe affinity is no more complicated than using a Richter scale to describe earthquakes.
We estimate conservatively that we are able to measure a 2‐fold change in E 2. Assuming independent errors, by the law of propagation of errors (Taylor, 1997) the net error in the E 2/E 0 ratio is ±√(22 + 22), or 2.8‐fold. Because of the square root in eqn (2), the error in K d is 1.7‐fold, that in binding energy is ± 0.3 kcal mol−1.
The difference in binding energy with a Y‐to‐F substitution was the estimate of the OH contribution. The effect of the W/F‐to‐A substitution was the estimate of the ring contribution (in the case of tyrosine, without the OH). Previously it was shown that Ala and Ser mutations give the same energies for ACh binding, so there appears to be no additional favourable or unfavourable interactions from the Ala group (Purohit et al. 2012).
Protein engineering
To engineer the gating rate constants to be in a range suitable for analysis, we added background mutations that, like depolarization, only changed E 0 and did not influence the coupling constant [eqn (1); Table S3] (Jadey et al., 2011). The observed gating equilibrium constants were corrected for the backgrounds (both observed and background‐corrected values are given in Table S2).
To create a receptor having just one functional transmitter binding site, the mutations δP123R+δW55A/R or εP121R were added to eliminate binding at either α–δ or α–ε (Gupta et al. 2013). These mutations also reduce E 0, and thus both E 1 and E 0 were determined for each ‘crippled’ construct (one WT and one inoperative binding site; Table S4). For example, the effect of the αW149A mutation at the α–δ site with Cho was measured on a εP121R+εL269F+εE181T+βL262M background to be E 1 = 0.7 (after correction for depolarization). E 0 for this construct without agonist was 0.02. As described above [eqn (2) without the square root, because there was only one binding site], K d Cho = 28.6 mm, which corresponds to a binding energy of −2.1 kcal mol−1. At the WT α‐site this energy is −3.5 kcal mol−1, so we estimate that the αW149A mutation caused a +1.4 kcal mol−1 loss in Cho binding energy, or a ∼10‐fold decrease in affinity.
Results
Agonists activate AChRs to produce clusters of single‐channel openings that represent C(losed)↔O(pen) ‘gating’ separated by silent, D(esensitized) periods (Fig. 2). For each agonist–receptor combination, the diliganded gating equilibrium constant E 2 was measured from the intra‐cluster interval durations using a saturating [agonist], and the resting equilibrium dissociation constant (K d) was calculated from this value as described in the Methods. The agonist binding free energy (kcal mol−1) was +0.59lnK d.
F and A mutations of the α‐subunit aromatics reduced the affinity for ACh, TMA and Cho (Fig. 3 and Table S1). For all three agonists the reduction in favourable binding energy was αY190A>αW149A≈αY198A. Our objective was to estimate the binding energy contributions from five functional groups of these three aromatic amino acids (two OH and three rings) for different agonists.
Figure 3. Effects of mutations on binding free energy.
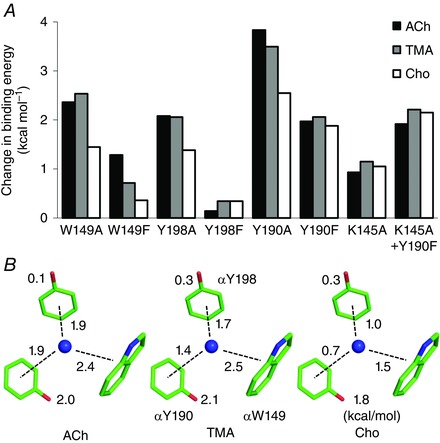
A, all mutations result in a positive binding free energy change (decrease affinity) for all agonists. αY190A has the largest effect. The αK145A mutation has no effect when combined with αY190F. B, graphical summary. Numbers are free energy losses (kcal mol−1) resulting from the removal of an OH (Y‐to‐F) or a ring (W/F‐to‐A). For all agonists, indole>benzene; for all rings, ACh>Cho. For clarify, only the agonist's quaternary nitrogen is visualized as a blue sphere. [Colour figure can be viewed at wileyonlinelibrary.com]
The contributions of the OH groups were estimated from the effects of an F substitution at either of the two tyrosines (both in loop C). αY190F caused a large decrease in affinity (∼30‐fold; ∼+2 kcal mol−1) that was about the same for ACh, TMA and Cho. In contrast, αY198F had a negligible effect for all three agonists. To explore this further, we measured affinity of αY190F and αY198F AChRs for three additional Cho derivatives (Fig. 4). The OH contribution is large only at αY190, where it is agonist‐independent and in all cases ∼+2 kcal mol−1. We consider the mechanism for the large contribution of the αY190 OH group, below.
The αW149F substitution replaces the indole ring (in loop B) with a benzene ring. The effect of this mutation was agonist‐dependent, with the binding energy loss being ACh>TMA>Cho. This mutation reduced favourable ACh binding energy by +1.3 kcal mol−1 but the effect for Cho was minimal. However, having an aromatic ring here is important for affinity, because the αW149A mutation decreased affinity substantially and to the same extent for ACh and TMA (70‐fold or +2.5 kcal mol−1), but less so for Cho (13‐fold or +1.5 kcal mol−1) (Fig. 3 B).
The contributions of the loop C benzenes were estimated by comparing the energy losses caused by F versus A mutations of the tyrosines. The results show that without an OH each of these rings stabilizes ACh by ∼−1.9 kcal mol−1, or slightly less than the loop B indole. For TMA, the benzene stabilization was slightly less favourable, even if the indole interaction was the same as for ACh. For Cho, the benzene rings were each ∼+1 kcal mol−1 less favourable than for ACh, which is about the same difference as for the indole. The lower binding energy for Cho from the three less favourable ring interactions combined is more than enough to account for its ∼+1.8 kcal mol−1 (∼20‐fold) lower resting affinity compared to the neurotransmitter.
In the above experiments, affinities were estimated from AChRs having two agonist‐binding sites and the binding energy estimates were averages. In the next experiments, one of the two binding sites was rendered inoperative by a mutation so that the effects of αY198A and αW149A mutations could be studied at each site, separately (Fig. 5). For all agonists, the single‐site energy losses were approximately equal to the averages from two‐site AChRs.
Figure 5. Single‐site analyses.
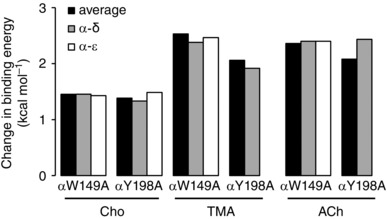
The change in binding energy with αW149A and αY198A is approximately equal at both adult‐type binding sites.
We now return to the observation that αY190F (but not αY198F) results in a large, agonist‐independent loss of binding energy. Others have pointed out the importance of αK145, in strand β7. In an ACh binding protein, this terminal amino‐group can approach (2.6 Å) and interact with the OH group of αY190 (Celie et al. 2004). In human adult AChRs, it was proposed that upon agonist binding αK145 disrupts its contact with αD200 to form a salt bridge with αY190 and launch channel‐opening (Mukhtasimova et al. 2005). We hypothesized that deprotonation of the αY190 hydroxyl by αK145 increases the negative charge of the ring (Mecozzi et al. 1996), to convert a cation–π interaction with the agonist's QA into a stronger, cation–anion interaction.
We measured the effects of αK145A, either alone or in combination with αY190F (Fig. 3 A). The mutations αK145A and αY190F, and the mutation pair αK145A+αY190F, had little effect on the unliganded gating equilibrium constant (Tables S1 and S2). With agonists, αK145A reduced the ACh, TMA and Cho binding energies to about the same extent, ∼+1.2 kcal mol−1, which is ∼60% of the αY190F energy loss. The mutation pair reduced ACh, TMA and Cho binding energies by +1.9, +2.2 and +2.1 kcal mol−1, which is about the same as observed with αY190F alone. Hence, the αK145A mutation has no effect when the OH of αY190 is absent.
Discussion
The two main experimental findings were (1) deprotonation of αY190 by αK145 accounts for the unusually strong interaction of this aromatic with all agonists, and (2) weaker interactions with all of the aromatic rings account for the lower affinity of Cho compared to ACh. We hypothesize that the ACh versus choline affinity difference arises from different H‐bonds that influence the position of each agonist within a fixed aromatic cage (Fig. 1 C).
OH groups
For all three agonists, mutations of αY190 had the largest effects on affinity. Removal of this OH group (αY190F) produced about the same, large reduction in binding energy for ACh, TMA and Cho. This one‐atom (per‐site) perturbation costs ∼+2.0 kcal mol−1 for all of the tested ligands, which is of similar magnitude to the removal of the entire αY198 or αW149 side chain. At the fetal α–γ site removal of the αY190 OH group mutation has the same, substantial effect on ACh affinity (Nayak et al. 2014). For all three agonists, the combined contribution from the αY190 OH and benzene ring represents ∼75% of the total binding free energy at each adult site.
αK145A alone reduced binding energies for ACh and Cho but was without effect when αY190F was added. This result is consistent with the hypothesis that the α‐amino group of αK145 deprotonates the αY190 hydroxyl to create a more electronegative ring and stronger bond with the agonist's QA. However, the αK145A effect alone was smaller (+1.2 kcal mol−1) than that of αY190F alone (+2 kcal mol−1). We speculate this quantitative difference can be attributed to water. In two high resolution (<1.8 Å) structures of ACh binding protein (AChBP) (PDB code 4ZK1 and 4ZJT), a water molecule approaches the OH of this tyrosine in ∼65% of the structures. We hypothesize that with αK145A, a water molecule can substitute and polarize partially the αY190 ring.
Unliganded gating was hardly affected by the αK145A and αY190F mutations. This observation argues against the idea that the transfer of the αY190 proton to αK145 is an essential event in channel opening, even if this may occur when agonists are present.
In contrast to αY190F, the αY198F mutation has little effect on affinity for any agonist. This suggests that αY198 is fully protonated. Although the OH group of αY198 is not important for affinity it has other roles, because αY198F reduces the probability of long‐opening events that are characteristic of unliganded gating (Purohit & Auerbach, 2010, 2013).
Rings
The energy contributions of the OH groups, substantial for αY190 and nil for αY198, are the same for all agonists, so the ability to distinguish between agonists (molecular recognition) lies with the aromatic rings. Without deprotonation, for each agonist the αY198 and αY190 rings make similar contributions to affinity (all agonists) at α–ε, α–δ and α–γ binding sites (Nayak et al. 2014). There was some indication that the αY198 ring interaction is slightly stronger than αY190, but the two loop C benzenes (without the OH group) are nearly equivalent.
For all three agonists, the αW149 indole contributes more to binding energy compared to the benzenes. αW149A results in an even larger reduction in ACh binding energy at the fetal site compared to the adult sites (+3.0 vs. +2.5 kcal mol−1) (Nayak et al. 2014). The greater effect of αW149A can probably be attributed to the fact that indoles make stronger cation–π bonds compared to benzene (Mecozzi et al. 1996).
At each adult site the sum of the energy contributions from the three aromatic residues is larger by ∼160% than the WT binding energy. This suggests that the functional groups at the binding site interact (share binding energy).
There was a clear agonist‐dependence to the αY198 and αY190 benzene binding energies, ACh>TMA>>Cho. The combined energy from the two loop C rings (per‐site) was −3.8, −3.1 and −1.7 kcal mol−1, respectively, which roughly parallels the total binding free energies (–5.1, –4.5 and –3.3 kcal mol−1). The indole binding energy was the same for ACh and TMA but, as with the benzenes, substantially smaller for Cho. The relative Cho binding energy relative to that for ACh was 62% from αW149, 53% from αY198 and 64% from αY190. The reduction in Cho affinity appears to be a general effect involving all of the binding site aromatics rather than from any particular side chain.
K d is the A+C↔AC equilibrium dissociation constant, so in order to interpret structurally the binding energies we would need high‐resolution structures of both C and AC binding sites. However, there are no published structures of an AC binding site in any pentameric ligand‐gated ion channel. AChBP is an excellent model for the fetal, α–γ AC site, but less so for the adult sites (Sixma & Smit, 2003; Nayak et al. 2016). However, there are no AChBP structures with Cho or TMA. Therefore, we are limited to considering functional results and simulations of homology models based on AChBP in the following discussion of the structural basis of the ACh versus Cho affinity difference.
The balance between the cation–π energies suggests that the three rings maintain the same symmetry with respect to the QA with different agonists, with a ∼+1 kcal mol−1 weaker interaction of Cho with each ring compared to ACh being the basis for its low affinity. Two additional observations are that ACh (and TMA) is stabilized more than Cho by the αW149 indole, and that replacing the indole with a benzene reduces affinity significantly with ACh but has almost no effect with Cho.
The results are consistent with the hypothesis that the QA is positioned outside the aromatic cage with Cho, but within the plane of the cage with ACh and TMA (Fig. 1C). Such differential positioning accounts for the symmetrical, but smaller, effects of the aromatic interactions with Cho. Furthermore, we hypothesize that QA position depends on agonist H‐bonds. Nicotine and ACh are probably H‐bonded to the complementary subunit (Celie et al. 2004; Amiri et al. 2007; Xiu et al. 2009; Blum et al. 2010; Olsen et al. 2014), but Cho and TMA are too small to make a cross‐subunit interaction. Simulations suggest that Cho makes an H‐bond with the backbone carbonyl of αW149, and we speculate that this tether prevents the Cho QA from being centred in the cage. TMA cannot H‐bond at all and interacts only with the α‐subunit aromatics. This ligand, which has nearly the same affinity as ACh, may be free to move so as to position its QA optimally within the cage, but perhaps with greater dynamics compared to ACh. It is also possible that the cage has different configurations with different agonists (compactness, ring orientation, dynamics), perhaps because of different degrees of loop C closure (Hansen et al. 2005).
The results suggest that agonist affinity differences arise from agonist H‐bonds that influence the position of the QA relative to the aromatic cage, and that deprotonation of αY190 adds favourable binding energy for all agonists but is not important for molecular recognition. We hypothesize that in AChRs molecular recognition of ACh versus choline is determined by a variable ligand position within a fixed protein structure.
Additional information
Conflict of interest
The authors declare no conflict of interest.
Author contributions
I.B. performed research and analysed data; A.A. and I.B. wrote the paper. The authors declare no conflict of interest.
Funding
This work was funded by National Institutes of Health Grant NS‐64969 to A.A. and a Canadian Institutes of Health Research fellowship to I.B.
Supporting information
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Table S1. Energy estimates for aromatic mutants.
Table S2. Observed and corrected rate/equilibrium constants.
Table S3. Effects of mutations on E0.
Table S4. Single site AChRs with transmitter binding site mutations.
Acknowledgements
We thank M. Merritt, M. Shero and M. Teeling for technical assistance.
References
- Amiri S, Sansom MS & Biggin PC (2007). Molecular dynamics studies of AChBP with nicotine and carbamylcholine: the role of water in the binding pocket. Protein Eng Des Sel 20, 353–359. [DOI] [PubMed] [Google Scholar]
- Auerbach A (2012). Thinking in cycles: MWC is a good model for acetylcholine receptor‐channels. J Physiol 590, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A (2016). Dose‐response analysis when there is a correlation between affinity and efficacy. Mol Pharmacol 89, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum AP, Lester HA & Dougherty DA (2010). Nicotinic pharmacophore: the pyridine N of nicotine and carbonyl of acetylcholine hydrogen bond across a subunit interface to a backbone NH. Proc Natl Acad Sci USA 107, 13206–13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB & Sixma TK (2001). Crystal structure of an ACh‐binding protein reveals the ligand‐binding domain of nicotinic receptors. Nature 411, 269–276. [DOI] [PubMed] [Google Scholar]
- Bruhova I, Gregg T & Auerbach A (2013). Energy for wild‐type acetylcholine receptor channel gating from different choline derivatives. Biophys J 104, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celie PH, van Rossum‐Fikkert SE, van Dijk WJ, Brejc K, Smit AB & Sixma TK (2004). Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41, 907–914. [DOI] [PubMed] [Google Scholar]
- Chakrapani S, Bailey TD & Auerbach A (2003). The role of loop 5 in acetylcholine receptor channel gating. J Gen Physiol 122, 521–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty DA (2013). The cation–pi interaction. Acc Chem Res 46, 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenes S & Auerbach A (2002). Desensitization of diliganded mouse muscle nicotinic acetylcholine receptor channels. J Physiol 541, 367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C & Auerbach A (2001). The dissociation of acetylcholine from open nicotinic receptor channels. Proc Natl Acad Sci USA 98, 14102–14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Purohit P & Auerbach A (2013). Function of interfacial prolines at the transmitter‐binding sites of the neuromuscular acetylcholine receptor. J Biol Chem 288, 12667–12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P & Bourne Y (2005). Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J 24, 3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB (1986). Kinetics of unliganded acetylcholine receptor channel gating. Biophys J 49, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadey S & Auerbach A (2012). An integrated catch‐and‐hold mechanism activates nicotinic acetylcholine receptors. J Gen Physiol 140, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadey SV, Purohit P, Bruhova I, Gregg TM & Auerbach A (2011). Design and control of acetylcholine receptor conformational change. Proc Natl Acad Sci USA 108, 4328–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A & Auerbach A (2010). Acetylcholine receptor channels activated by a single agonist molecule. Biophys J 98, 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney PC, Nowak MW, Zhong W, Silverman SK, Lester HA & Dougherty DA (1996). Dose–response relations for unnatural amino acids at the agonist binding site of the nicotinic acetylcholine receptor: tests with novel side chains and with several agonists. Mol Pharmacol 50, 1401–1412. [PubMed] [Google Scholar]
- Lape R, Krashia P, Colquhoun D & Sivilotti LG (2009). Agonist and blocking actions of choline and tetramethylammonium on human muscle acetylcholine receptors. J Physiol 587, 5045–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecozzi S, West AP, Jr & Dougherty DA (1996). Cation–pi interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide. Proc Natl Acad Sci USA 93, 10566–10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton RE & Cohen JB (1991). Mapping of the acetylcholine binding site of the nicotinic acetylcholine receptor: [3H]nicotine as an agonist photoaffinity label. Biochemistry 30, 6987–6997. [DOI] [PubMed] [Google Scholar]
- Monod J, Wyman J & Changeux JP (1965). On the nature of allosteric transitions: a plausible model. J Mol Biol 12, 88–118. [DOI] [PubMed] [Google Scholar]
- Mukhtasimova N, Free C & Sine SM (2005). Initial coupling of binding to gating mediated by conserved residues in the muscle nicotinic receptor. J Gen Physiol 126, 23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak TK, Bruhova I, Chakraborty S, Gupta S, Zheng W & Auerbach A (2014). Functional differences between neurotransmitter binding sites of muscle acetylcholine receptors. Proc Natl Acad Sci USA 111, 17660–17665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak TK, Chakraborty S, Zheng W & Auerbach A (2016). Structural correlates of affinity in fetal versus adult endplate nicotinic receptors. Nat Commun 7, 11352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak TK, Purohit PG & Auerbach A (2012). The intrinsic energy of the gating isomerization of a neuromuscular acetylcholine receptor channel. J Gen Physiol 139, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai C & Sachs F (2013). Solving ion channel kinetics with the QuB software. Biophys Rev Lett 8, 191–211. [Google Scholar]
- Nowak MW, Kearney PC, Sampson JR, Saks ME, Labarca CG, Silverman SK, Zhong W, Thorson J, Abelson JN, Davidson N, et al (1995). Nicotinic receptor binding site probed with unnatural amino acid incorporation in intact cells. Science 268, 439–442. [DOI] [PubMed] [Google Scholar]
- Olsen JA, Balle T, Gajhede M, Ahring PK & Kastrup JS (2014). Molecular recognition of the neurotransmitter acetylcholine by an acetylcholine binding protein reveals determinants of binding to nicotinic acetylcholine receptors. PLoS One 9, e91232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P & Auerbach A (2009). Unliganded gating of acetylcholine receptor channels. Proc Natl Acad Sci USA 106, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P & Auerbach A (2010). Energetics of gating at the apo‐acetylcholine receptor transmitter binding site. J Gen Physiol 135, 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P & Auerbach A (2013). Loop C and the mechanism of acetylcholine receptor‐channel gating. J Gen Physiol 141, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Bruhova I & Auerbach A (2012). Sources of energy for gating by neurotransmitters in acetylcholine receptor channels. Proc Natl Acad Sci USA 109, 9384–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit Y & Grosman C (2006). Estimating binding affinities of the nicotinic receptor for low‐efficacy ligands using mixtures of agonists and two‐dimensional concentration‐response relationships. J Gen Physiol 127, 719–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F (2004). Restoration of single‐channel currents using the segmental k‐means method based on hidden Markov modeling. Biophys J 86, 1488–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Auerbach A & Sachs F (1997). Maximum likelihood estimation of aggregated Markov processes. Proc Biol Sci 264, 375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone FN, Zhou M & Auerbach A (1999). A re‐examination of adult mouse nicotinic acetylcholine receptor channel activation kinetics. J Physiol 516, 315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ & Taylor P (1994). Conserved tyrosines in the alpha subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. J Biol Chem 269, 8808–8816. [PubMed] [Google Scholar]
- Sixma TK & Smit AB (2003). Acetylcholine binding protein (AChBP): a secreted glial protein that provides a high‐resolution model for the extracellular domain of pentameric ligand‐gated ion channels. Annu Rev Biophys Biomol Struct 32, 311–334. [DOI] [PubMed] [Google Scholar]
- Taylor J (1997). Introduction to error analysis, the study of uncertainties in physical measurements, vol. 1 University Science Books, New York. [Google Scholar]
- Tomaselli GF, McLaughlin JT, Jurman ME, Hawrot E & Yellen G (1991). Mutations affecting agonist sensitivity of the nicotinic acetylcholine receptor. Biophys J 60, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiu X, Puskar NL, Shanata JA, Lester HA & Dougherty DA (2009). Nicotine binding to brain receptors requires a strong cation–pi interaction. Nature 458, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA & Dougherty DA (1998). From ab initio quantum mechanics to molecular neurobiology: a cation–pi binding site in the nicotinic receptor. Proc Natl Acad Sci USA 95, 12088–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Table S1. Energy estimates for aromatic mutants.
Table S2. Observed and corrected rate/equilibrium constants.
Table S3. Effects of mutations on E0.
Table S4. Single site AChRs with transmitter binding site mutations.