

Abstract
Muscle contractures are common in patients with central motor lesions, but the mechanisms responsible for the development of contractures are still unclear. Increased or decreased neural activation, protracted placement of a joint with the muscle in a short position and muscle atrophy have been suggested to be involved, but none of these mechanisms are sufficient to explain the development of muscle contractures alone. Here we propose that changes in tissue homeostasis in the neuromuscular–tendon–connective tissue complex is at the heart of the development of contractures, and that an integrated physiological understanding of the interaction between neural, mechanical and metabolic factors, as well as genetic and epigenetic factors, is necessary in order to unravel the mechanisms that result in muscle contractures. We hope thereby to contribute to a reconsideration of how and why muscle contractures develop in a way which will open a window towards new insight in this area in the future.
Keywords: contracture, motor control, skeletal muscle
Abbreviations
- COL
collagen
- CP
cerebral palsy
- DMD
Duchenne muscular dystrophy
- ECM
extracellular matrix
- IGF‐1
insulin‐like growth factor 1
- miRNA
microRNA
- mTOR
rapamycin
- p70S6K
ribosomal protein S6 kinase β‐1
- TRP
transient receptor potential channel
Introduction
Muscle contractures are defined as unique muscle changes which increase the passive stiffness of the muscle and limit the mobility of the joints without any active force production of the muscles (Smith et al. 2011). Muscle contractures are a common complication to lesions of the central motor pathways such as cerebral palsy (CP), stroke and spinal cord injury. The prevalence after stroke has been reported to be 60% of all patients (Sackley et al. 2008), 36% in CP patients with upper limb involvement (Makki et al. 2014), and 48% following spinal cord injury (Diong et al. 2012). Contractures cause the joints of these patients to gradually become fixated in awkward positions, which obstruct normal physical activity. The exact pathology underlying contractures is not clarified and it is still debated whether the increased stiffness involves elastic elements within the muscle fibres, in the extracellular matrix or both. It is also not clarified to what extent ultrastructural changes in the muscle fibres, such as changes in the number and length of sarcomeres, contribute to the alterations in gross muscle anatomy and joint position. This lack of clarity is probably explained by shortcomings of current techniques for measuring tissue stiffness and muscle fibre lengths in vivo (Smith et al. 2011; Mathewson et al. 2014).
It is generally assumed that the development of muscle contractures is related to an abnormally high muscle activity due to spasticity (Gracies, 2005). However, antispastic medication has no effect on contractures in many patients with lesions of the central motor pathways (Tedroff et al. 2011). Additionally, contractures appear to develop in both stroke and CP without prior evidence of spasticity and, although selective dorsal rhizotomy effectively reduces spasticity, it does not prevent the development of muscle contractures (Tedroff et al. 2011). These findings indicate that spasticity cannot be the deciding cause for the development of muscle contractures.
In contrast, both human and animal studies have shown that immobilization, especially in a shortened position, can cause muscle contractures (Trudel et al. 1999). A study by Trudel et al. showed that hind limb immobilization for various periods ranging from 3 to 32 weeks caused the range of motion (ROM) to decrease simultaneously with an increase in muscle stiffness (Trudel et al. 1999). If immobilization alone can lead to development of muscle contractures, then all spinal cord injury patients should in principle develop muscle contractures in all their paralysed muscles. However, this is not the case. Spinal cord injury patients do sometimes develop contractures (Nas et al. 2015), but usually in one joint only, depending on the location of their injury.
We consequently have to realize that none of the existing theories adequately explain the development of muscle contractures in all patients and under all circumstances. This raises two possibilities: Contractures may be more heterogeneous than we have so far realized and may therefore be caused by different mechanisms in different patients and under different circumstances. Alternatively, or complimentarily, contractures may be caused by an interaction between many different factors where no single factor determines their development. Here we will propose that contractures should be seen as an adaptation in tissue homeostasis in the neuromuscular–tendon–connective tissue complex induced by the lesion of the central motor pathways. In the complex network of interaction between different tissues (neural, muscle, connective tissue etc.), different stimuli (neural activity, mechanical factors, growth factors, nutrients, metabolic substances) and different regulatory factors (genetic, epigenetic, metabolic signalling factors), changes in one factor (neural activity) quickly result in adaptive changes in the rest of the network, leading to a new set value of homeostasis (Fig. 1). It is this complex interaction we need to understand in order to unravel the mechanisms that result in contractures in the individual patients and eventually find ways of preventing and treating contractures. In the following we will discuss the mechanisms that have been proposed to be involved in the development of muscle contractures with a focus on the interaction and signalling between different tissues and compartments in the nerve–muscle–tendon–bone complex. It should be noted that we will not address limitations in joint movements caused by alterations of joint flexibility (joint contractures), although similar mechanisms may be involved.
Figure 1. Tissue homeostasis in the neuromuscular–tendon–connective tissue complex.
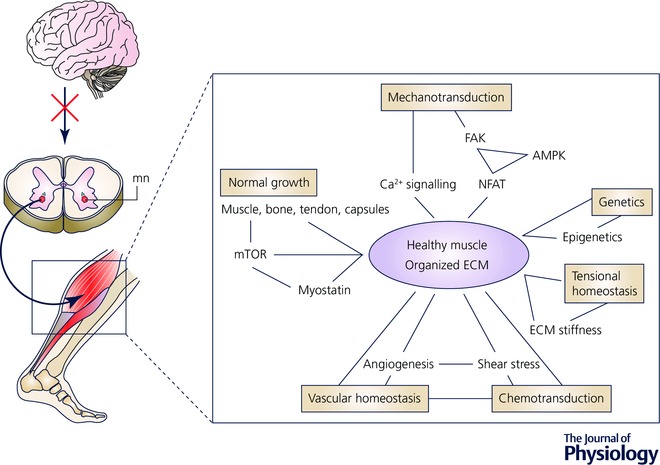
A simplified overview of the different factors involved in the short‐ and long‐term regulation of the triceps surae muscle, tendon and connective tissue. When we use the muscles, neural activity not only signals contraction of the muscle, but also initiates a cascade of signalling factors, which determines the growth of the muscle fibres, the phenotype of the muscle fibres and the properties of the connective tissue, as well as increasing the capillary network in the muscle. As a result of the mechanical stimuli during muscle activity, for instance from the impact of heel strike when we walk, a myriad of different cells are activated, including mechanosensitive fibroblasts, which results in connective and muscle tissue changes. Concomitantly, vascular and metabolic factors are activated in order to meet the energy and metabolic needs of the muscle and connective tissue, when activated. Finally, muscle, tendon and connective tissue elicit signals to the nervous system regarding the present state of the tissue, which is incorporated into the immediate and future neural signalling. The different tissues are thus part of an integrated network that regulates the environment of the cells and maintains a number of key factors (the contractile properties of the muscle, the stiffness of the tissue) within relatively strict limits. Additionally, genetic and epigenetic factors affect the composition of the tissues, thus influencing the capability of the tissue to react and adapt to changes in the tissue homeostasis. This homeostasis is what we observe as a normal and healthy tissue. Alteration in one of the factors involved in the network will inevitably result in adaptive changes throughout the network, in order to maintain homeostasis as far as possible. However, if the alteration is too big the network cannot maintain normal homeostasis and will settle on a new (unhealthy, pathological) set value of homeostasis. This is the state where pathological contractures have developed. In this review we will discuss the possible key contributors to the development of contractures in this homeostatic network. AMPK, 5‐AMP‐activated protein kinase; FAK, focal adhesion kinase; mn, motor neuron; NFAT, nuclear factor of activated T‐cells.
Neural activation: hyper‐ or hypo‐activity?
When contractures develop in neurological disorders, altered neural drive to the muscles is a logical suspect as one of the initial steps in the development of contractures and spasticity is also generally assumed in the clinic to be the main cause of contractures (Hagglund & Wagner, 2011). However, there is little solid scientific evidence to support this idea. One main reason for this is that there is considerable confusion regarding the term spasticity in the literature (Malhotra et al. 2009). Spasticity is most commonly defined as a velocity dependent increase of muscle resistance to passive stretch, which implies that hyperactive stretch reflexes have a central pathophysiological role (Malhotra et al. 2009). However, a much broader definition, which emphasizes the presence of involuntary muscle activity and which may better reflect the clinical understanding of spasticity, has also been introduced (Malhotra et al. 2009). Whereas hyperactive stretch reflexes are unlikely by themselves to result in contractures, the same cannot be said of continuous involuntary muscle activity, which may cause the muscle and joint to be fixed in an undesirable position. This so‐called spastic dystonia, which is often seen following stroke and here assumed to interfere with movement ability and lead to abnormal postures, appears to be caused by central mechanisms and is thus unrelated to increased reflex excitability, i.e. spasticity in the original definition (Sheean & McGuire, 2009). Without a clear distinction between these two definitions of spasticity, it is difficult to determine the significance of studies on the pathophysiological role of spasticity for development of contractures. One example is the finding that dorsal rhizotomy has been shown not to have any effect on the development of contractures in children with CP (Tedroff et al. 2011). Rhizotomy will certainly reduce sensory input and diminish stretch reflexes, but would have little effect on spastic dystonia if present.
Recently it has been suggested that reduced rather than increased neural drive is more likely to be the key pathophysiological mechanism in the development of contractures (Gough & Shortland, 2012). Reduced neural activation of the muscle may also in its own right cause changes in connective tissue and other elastic structures in the muscle and tendon through its effect on gene expression of the muscle fibres (Smith et al. 2012). Although contractures do develop in relation to both denervation (Nikolaou et al. 2015) and immobilization (Lake et al. 2016), it should be emphasized that there is still no conclusive evidence of the role of atrophy or reduced neural activity in the development of contractures. This may be mainly due to the problems in disentangling the role of neural activity from the role of secondary changes in muscle loading, muscle structure and muscle signalling. Since techniques which allow dissociation between these interacting components are now available and have been validated, there is a great need to apply these to quantify the effect of each component in the contracture process (Bar‐On et al. 2015). One way of obtaining some information about the role of neural hypo‐activity versus hyper‐activity would be to study a possible relation between contractures and the previous history of muscle activity. To our knowledge no systematic study has so far been performed to document such a relationship in either neurological patients or in animal models. Inactivity models (such as nerve section, limb suspension and Botox injection) are certainly associated with development of contractures (Nikolaou et al. 2015; Pingel et al. 2016), but it is unclear whether this is due to reduced neural activation, the changes in mechanical loading or the altered joint positions, which are unavoidable consequences of the inactivity.
A mismatch between muscle and bone length?
Muscle growth and atrophy
Impaired muscle growth and muscle atrophy have been suggested in a number of studies in later years to be a key to the development of muscle contractures, at least in children with CP, but possibly also in adults with stroke and spinal cord injury (Gough & Shortland, 2012; Aze et al. 2016). Individuals with CP have smaller muscles than healthy individuals, and both the muscle belly length and the cross sectional area are reduced (Gough & Shortland, 2012). This appears to be related to reduced growth of the muscle, possibly caused by reduced neural activation and disuse, since reduced muscle volume is not observed until children are older than 15 months (Herskind et al. 2016). Muscle atrophy and associated structural muscle changes also develop quickly in adults following stroke (Aze et al. 2016) and spinal cord injury (Qin et al. 2010). Some studies have suggested that the shortening of the muscle belly observed in individuals with CP is the result of shorter fascicle length in the muscle (Matthiasdottir et al. 2014), but other studies have failed to confirm this (Mathewson et al. 2015). Muscle growth and atrophy mainly relate to the diameter of the muscle fibres (number of sarcomeres in parallel), but because of the pennate structure of most muscles, the diameter of the fibres contributes significantly to the total muscle length – in the case of the soleus muscle, increase of fibre diameter contributes up to 80% of the growth in muscle length (Gough & Shortland, 2012). Muscle atrophy may therefore cause muscles to be too short in relation to the bone length, and the consequent stretching and stress on the sarcomeres may trigger the development of contractures (Gough & Shortland, 2012).
Increases of muscle mass (hypertrophy) and decreases in muscle mass (atrophy) are controlled by anabolic and catabolic responses, respectively. One key regulator of muscle mass and metabolism that stimulates protein synthesis is rapamycin (mTOR). mTOR modulates protein synthesis in muscle through two distinct pathways, the PHAS‐1 pathway and the ribosomal protein S6 kinase β‐1 (p70S6K) pathway (Glass, 2005). Knockout models for p70S6K have shown that a lack of this gene results in a significantly smaller cross sectional area of muscle cells (Ohanna et al. 2005). Furthermore, insulin like growth factor 1 (IGF‐1) is of crucial importance in muscle growth. IGF‐1 induces an increase of muscle mass by stimulating the phosphatidylinositol‐3 kinase (PI3K)/AKT (protein kinase B) pathway. This activation results in downstream activation of targets required for protein synthesis (Glass, 2005). The downstream signalling of IGF‐I through the AKT pathway is antagonized by myostatin. A blockage of myostatin results in a tremendous increase of muscle mass (13–30%) (Whittemore et al. 2003). Interestingly, children with CP have shown an upregulation of both IGF‐1 and myostatin, indicating an increased turnover in muscles from children with CP compared to muscle tissue from two normally developing children (Smith et al. 2009). Increased muscle turnover has been observed in several conditions presenting muscle atrophy. Thus, the increased turnover in CP muscles observed by Smith et al. is consistent with muscle atrophy in children with CP. However, atrophy alone is not likely to explain the development of the passive stiffness observed in muscle contractures, since other conditions causing muscle atrophy such as sarcopenia do not develop increased muscle stiffness despite of tremendous loss of muscle mass.
Bone growth
Although reduced muscle growth and muscle atrophy may lead to a mismatch between muscle length and bone length in children with cerebral palsy this does not necessarily indicate that bone growth is not also affected. The bone density is strongly dependent on weight bearing, and individuals with cerebral palsy show significant abnormalities in bone growth, including delayed maturation, diminished linear growth and low bone density (Henderson et al. 2005 b). Similar changes occur in stroke patients and in subjects with spinal cord injury (Sato et al. 2004; Liu et al. 2008). In children with spinal cord injury, regional osteopenia was furthermore associated with loss of muscle mass (Liu et al. 2008). Children with severe CP develop clinically significant osteopenia throughout their lives, and it has been shown that the reduction of bone minerals is related to decreased growth rate of the bone, rather than to genuine gross losses of bone minerals, as seen with ageing (Henderson et al. 2005 b). Furthermore, the delayed skeletal maturation in CP was found to be correlated with a diminished linear growth (height) and decreased bone density (Henderson et al. 2005 a). Another interesting observation by Stevenson et al. was a diminished linear bone growth, decreased bone density, and delayed skeletal maturation in the affected leg of hemiplegic CP persons when compared to the unaffected leg (Stevenson et al. 1995). This indicates that the impaired bone growth is a local response.
Role of muscle micro‐architecture
Evidence from animal experiments has suggested that the number of sarcomeres in a muscle is regulated according to the joint position (Tamai et al. 1989). Immobilization in elongated position thus increases the number of sarcomeres in series (Goldspink et al. 1974), whereas a decrease of the number of sarcomeres is seen with immobilization in shortened position (Williams & Goldspink, 1984). The addition/decrease of sarcomeres appears to be independent of neural activation (Goldspink et al. 1974). This adaptation of the sarcomeres may take place in order to maintain the most optimal muscle function in a given position (Tamai et al. 1989). There is, furthermore, accumulating evidence that suggests that sarcomeres appear to be stretched and therefore longer in CP muscles compared to typically developed muscles (Smith et al. 2011; Mathewson et al. 2015), although there is not general agreement about this (Smeulders et al. 2004). An unresolved, but important, question is whether other circumstances in the muscle are responsible for an inadequate adaptation of sarcomere length in muscle contractures.
Is it all a question about the right tension in the cell?
Cells exist in a three‐dimensional environment in which they are continuously exposed to mechanical and physical stimuli. Tensional homeostasis is a pervasive concept in mechano‐biology and it has received compelling attention as a way of defining the interplay between the external and internal mechanical state of cells (Banes et al. 1995). Tensional homeostasis describes the state in which cells maintain defined levels of tension within their surroundings despite mechanical perturbations that could change tension (Banes et al. 1995).
Lamin A and myosin II have previously been described as prime candidates for biological tension sensors (Dingal & Discher, 2014). These coiled‐coil proteins assemble into structural networks and transduce mechanical signals between the extracellular matrix (ECM) and the nucleus (Dingal & Discher, 2014). Lamin A is an intermediate filament protein that is found in various cell nuclei. It contributes to nuclear stiffness and nuclear stability (Dingal & Discher, 2014). When culturing cells in soft to stiff environments the expression of lamin A increases tremendously (30‐fold) in stiff environments, while lamin B is only affected to a minor degree (Broers et al. 1997). Myosin II is associated with the extracellular matrix stiffness. High matrix stiffness increases the tension (stress) in the cell, and myosin II responds to the matrix stiffness by increasing in amount and assembling into stress fibres (Rehfeldt et al. 2012). When disrupting the tensional homeostasis by inhibiting myosin, the cells lose the ability to connect with and sense focal adhesions. Elevated tension within the focal adhesions have shown to increase integrin clustering and focal adhesion kinase (FAK) phosphorylation, and these molecular changes induce subsequently a cascade of signalling activation of the Rho pathway (Fig. 1) (Vogel & Sheetz, 2009). Several pathologies, including arteriosclerosis, muscular dystrophies and potential central nervous system disorders, can either be a result of a disruption of the tensional homeostasis, caused by altered forces at the cell or tissue level, a perturbed response to the mechanical stimuli, or altered material properties of the ECM (Jaalouk & Lammerding, 2009). Under static conditions, the gene expression response varies dramatically depending on shear stress and the viscosity of the extracellular matrix in osteoblast cultures (Sikavitsas et al. 2003). Furthermore, ECM‐generated mechanical tension can trigger the synthesis of ECM proteins in fibroblasts (Kessler et al. 2001). On the other hand, reduced mechanical tension in the ECM decreases the expression of ECM building proteins in aged skin (Rittie & Fisher, 2015). It seems that tensional homeostasis is a very sensitive system, and that small alterations of the tension homeostasis can lead to severe consequences both at cell and tissue level. It has previously been proposed that muscle contractures are a result of increased stiffness of the extracellular matrix (Smith et al. 2011). A disruption of tension homeostasis due to progressive extracellular matrix stiffening in the muscle cells could be involved in the development of muscle contractures. However, no research has yet been carried out to elaborate whether the tensional homeostasis is disturbed in muscle cells of patient groups that are predisposed for developing muscle contractures. Future studies will hopefully clarify whether tensional homeostasis due to either alterations of the forces within the cell, a perturbed response to the mechanical stimuli, or stiffening of the extracellular matrix is involved in the development of muscle contractures.
Is the calcium signalling of the muscle cell impaired in muscle contractures?
Intracellular calcium concentration regulates signalling mechanisms, which control various biological processes crucial in the development and regeneration of the muscle (Fig. 1). The calcium signal is unique in a spatiotemporal pattern, and the responses to the calcium signal can either cause short‐term effects such as gene transcription, signal transduction, contraction (Fig. 2) and secretion, or long term regulation of fertilization, proliferation, migration, differentiation, apoptosis and necrosis (Benavides Damm & Egli, 2014). A continuous increase of the calcium concentration can lead to cellular damage and an activation of proteases (Batchelor & Winder, 2006). Consequently, it is necessary that the calcium homeostasis is maintained to ensure a proper function of the cell needed in the development, repair and regeneration of healthy muscle tissue (Benavides Damm & Egli, 2014). The calcium homeostasis is distorted in both CP (Smith et al. 2009, 2012) and muscular dystrophy (Vallejo‐Illarramendi et al. 2014). Smith et al. observed an upregulation of parvalbumin, a calcium binding protein and the voltage‐dependent L‐type calcium channel subunit β‐1 (CACNB1) in wrist muscles of individuals with CP, indicating pathological activation of the ryanodine receptors and increased calcium levels in CP (Smith et al. 2009). Furthermore, a transcriptional analysis of hamstring muscles of individuals with CP showed that several targets involved in calcium handling were significantly changed in CP, with the exception of parvalbumin, which was not significantly altered (Smith et al. 2012). In muscular dystrophy, changes in calcium signalling have been reviewed thoroughly (Vallejo‐Illarramendi et al. 2014). In brief, the lack of the structural protein dystrophin causes membrane fragility of the sarcolemma and increases basal intracellular Ca2+ levels. These increased calcium levels induce activation of the protease calpain, protein degradation, mitochondrial permeability transition pore opening, and subsequently fibre death and necrosis. A blockage of transient receptor potential channels (TRP) reduces the extracellular Ca2+ influx (Formigli et al. 2009). Whether subjects with CP suffering from muscle contractures would benefit from therapeutic approaches towards decreasing intracellular calcium levels has to our knowledge never been investigated.
Figure 2. Mechanotransduction in muscle tissue.
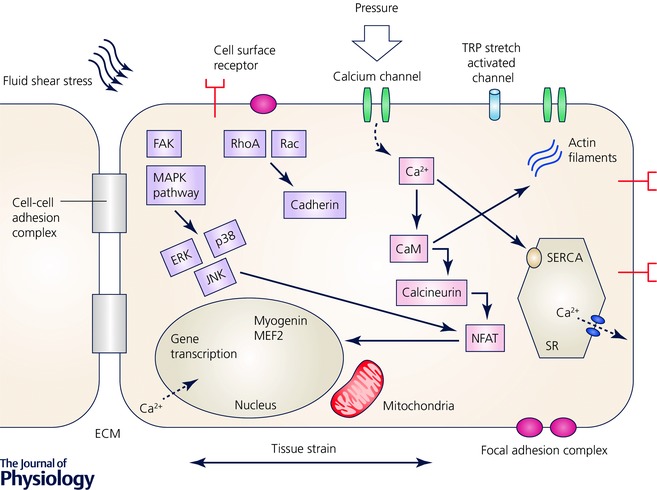
Mechanical loading is sensed through a diverse group of membrane‐anchored mechano‐sensors, including stretch‐activated ion channels, focal adhesion complexes and cell surface receptors. This mechanical sensation is then converted into biochemical signals by triggering several downstream signalling cascades in the cytoplasm. Elevated tension increases integrin clustering and phosphorylation of the focal adhesion kinase FAK, which triggers the mitogen‐activated protein kinase (MAPK) sub‐pathways and the Rho pathways: the extracellular signal‐regulated kinases (ERK), c‐Jun NH‐2‐terminal protein kinases (JNKs) and p38 MAPKs. Elevated tension within the focal adhesions increases integrin clustering and FAK phosphorylation, and these molecular changes induce subsequently a cascade of signalling activation of the Rho pathway including Rac and cadherin. Calcium binds to camodulin and activates calcineurin which dephosphorylates the nuclear factor of activated T‐cells (NFAT). This initiates gene transcription of muscle remodelling transcription factors including myogenin and MEF2. The sarcoplasmic reticulum represents the internal calcium stores. The release of calcium is controlled by inositol‐1,4,5‐trisphosphate and the reuptake of calcium is controlled by the sarco‐endoplasmic reticulum ATPase pumps (SERCA).
Figure 2 shows a schematic overview including several factors affecting calcium homeostasis in the cell. A dysregulation of any of these factors shown in Fig. 2 can lead to disturbances in the muscle remodelling, cell cycle progression, growth, differentiation, proliferation and apoptosis, which can have fatal consequences for the muscle tissue.
Cellular mechano‐sensing
The cells sense physical forces through cell–cell interactions and cell substrate interactions and translate the mechanical input into biochemical signals. Myosin and the giant protein titin are believed to be the main players in mechano‐sensing through the sarcomere itself (Gautel, 2011), and the kinase in the M‐band is believed to play a role in this (Gautel, 2011). In spastic CP patients, a reduced resting length of the sarcomeres has been observed. This was accompanied by a doubling of the elastic modulus compared to normal muscle cells (Friden & Lieber, 2003). The same observation was made in spastic spinal cord injury patients (Olsson et al. 2006). Friden and Lieber suggested that titin isoforms may be critical to the structural changes in the muscles of spastic patients (Friden & Lieber, 2003). Furthermore, titin also binds calcium, and the protein charge on titin is dependent on the calcium concentration, showing a sharp transition at a pCa of 6.8. This indicates that titin could be the component controlling the calcium dependence along the total length of a sarcomere (Coomber et al. 2011). One can therefore speculate if titin is involved in the development of contractures.
Whether any of these various processes with calcium regulation and mechano‐sensing are impaired or affected in muscle contractures is yet to be discovered. We propose that this particular area should achieve meticulous attention in future research into the origin of muscle contractures. If the calcium signalling either downstream or upstream is involved in the initiation of muscle contractures, or if binding of calcium to titin is impaired, this would open completely new opportunities for the treatment of muscle contractures.
Is impaired micro‐vascularization involved in development of contractures?
Blood flow to the skeletal muscle is determined by mechanical, neural and metabolic forces (Uchida et al. 2015). The micro‐vascularization of skeletal muscles is crucial for tissue maintenance, repair and remodelling (Fig. 1) (Uchida et al. 2015). Even a short‐term blockade of the blood supply to a skeletal muscle causes significant injuries to the muscle cell. Furthermore, denervated skeletal muscles have been shown to express devascularization of the tissue accompanied by degeneration and necrosis of muscle cells (Borisov et al. 2000). Why denervation causes capillary necrosis is still unclear. Possible reasons for this development could be either vascular degeneration or increased shear stress.
Vascular degeneration
Patients suffering from amyopathic lateral sclerosis and axonal demyelinative neuropathies suffer from poor vascular perfusion of the muscle tissue caused by necrotic capillaries (Carpenter & Karpati, 1982). An investigation of biceps brachii muscle of seven spastic patients and seven healthy control participants demonstrated the same in muscle contractures. Individuals with CP had 38% lower capillary density than the healthy control subjects (Ponten & Stal, 2007). The authors suggest that the low proportion of oxidative fibres and the low oxidative capacity and low capillary supply could explain that muscles with contractures show increased fatigue (Ponten & Stal, 2007). Stroke patients also show reduced capillarization in the paretic side when compared to the non‐paretic side (Prior et al. 2009). Reduced capillarization affects various physiological processes, including oxygen uptake and glucose metabolism (Ponten & Stal, 2007; Prior et al. 2009). However, the does not correlate with reduced capillarization in stroke patients, which indicates that the restricted mobility limits the stroke patients more than the reduced capillarization of their muscles (Prior et al. 2009).
Shear stress
The vascular system is continually exposed to stress. The blood flow applies shear stress, blood pressure and muscle contraction apply compression, and finally strains of the surrounding muscles apply tension (Fig. 2) (Egginton, 2009). Intracellular signalling pathways can be activated by any of these haemodynamic forces, and can thereby regulate proliferation, apoptosis, adhesion and matrix degradation (Egginton, 2009). Increased shear stress causes hyperpolarization of the cell membrane, whereas decreased shear stress causes depolarization (Chatterjee & Fisher, 2014). Endothelial cells sense the shear stress associated with blood flow and the resultant mechano‐signalling initiates downstream signalling events to regulate the vascular diameter and vessel tone (Chatterjee & Fisher, 2014). In endothelial cells this concept has been described as chemotransduction (Lopez‐Barneo et al. 1988). A dysregulation of the chemotransduction in endothelial cells can lead to angiogenesis. In other patient groups where the micro‐vascularization is poor, such as in intermittent claudication patients, exercise is the most effective therapy, since it increases the oxidative and vascular capacity of the muscles, indicating an improvement of the micro‐vascularization with exercise (Hiatt et al. 1990). It is crucial for the development and maintenance of the micro‐vascularization that the chemotransduction is highly regulated and shut down or upregulated as required.
Whether chemotransduction is impaired or altered in muscle contractures is still unsolved, but it is very likely that the vascularization of the muscle tissue is affected by muscle contractures, since the muscle function and activity level is reduced in the affected muscles. A better understanding of micro‐vascularization in connection with this particular patient group, and a possible effect of a reduced blood flow, could possibly lead to several new opportunities of treatments for patients with muscle contractures. It would also help to understand one of the connections to exercise therapy, since exercise is known to cause an increase in vascularization.
The impact of genetics and epigenetics on the development of contractures
Genetics
In regard to the cause of muscle contractures it is the general opinion that muscle contractures are not caused by a gene defect, but that the disease mainly develops depending on the muscle activation and/or the lack thereof. However, some genetic diseases develop muscle and joint contractures, for example Ullrich congenital muscular dystrophy (UD) (Yonekawa & Nishino, 2015). UD was first described in 1930 by Otto Ullrich and is a gene mutation of collagen 6 alpha‐1 (COL6A1), collagen 6 alpha‐2 (COL6A2) or collagen 6 alpha‐3 (COL6A3) (Yonekawa & Nishino, 2015). The disease has clinical–pathological features of a muscle disorder and connective tissue disorganization, and causes progressive joint and muscle contractures, preventing normal physical activity (Yonekawa & Nishino, 2015). Collagen VI is an extracellular protein, forming a distinct microfibrillar network in most interstitial connective tissues, and it has a pivotal role in maintaining skeletal muscle integrity and function (Bonnemann, 2011). Since mutations of collagen type 6 can cause contractures, it is tempting to elaborate whether patients with different contractures might show differences in gene variants of either COL6 or other crucial collagen types, or connective tissue components, responsible for the integrity and functionality of the muscle tissue. Furthermore, will these patients show different epigenetic regulations of these genes (Fig. 1)?
Epigenetics
The most extensively studied mechanisms in epigenetics are DNA methylation, histone acetylation and histone methylation. DNA methylation is the most stable modification of the chromatin structure, providing long‐term regulatory directions for transcriptional control and generally instructions for responsiveness (Siegfried et al. 1999). DNA methylation occurs at CpG islands (regions of repeated cytosine‐guanine pairs) at the 5′ position of the cytosine ring (Siegfried et al. 1999). In contrast histone acetylation is a transient and enzymatically controlled process like phosphorylation that generally governs gene activation (Clayton et al. 2006). Histone methylation can activate or suppress (silence) transcription in a context dependent manner (Clayton et al. 2006). There is increasing appreciation for the contributions of genetic and epigenetic regulations to muscle development, regeneration and function (Baar, 2010). Although a detailed description of these epigenetic regulatory mechanisms is beyond the scope of this review, it is opportune to mention that epigenetics could play a significant role in muscle function, integrity and pathology. Regarding muscle function, several studies have investigated the involvement of epigenetics regarding fibre type differentiation within the muscle (reviewed by Baar, 2010). Furthermore, recent reports highlight the influence of epigenetic mechanisms in several diseases including muscular dystrophies (Colussi et al. 2008). The genetic disorder Duchenne muscular dystrophy (DMD), the most severe muscular dystrophy, is caused by mutations of the dystrophin gene. The pathology of the disease is characterized by a rapid progression of muscle degeneration, loss of ambulation and early death (Lopez‐Hernandez et al. 2015). Previous studies have shown an involvement of epigenetic mechanisms in the development of the disease (Cacchiarelli et al. 2011). Studies on monozygotic twins have also shown a different penetrance of facioscapulohumeral muscular dystrophy (FSHD), indicating a strong epigenetic contribution to the pathology of FSHD (Griggs et al. 1995). Whether epigenetic mechanisms are involved in muscle contractures is still unknown, but since there is a very high inter‐individual variability in the severity of the condition, it is very likely that epigenetic mechanisms are involved in the regulation of the severity of contractures, and maybe even in the initiation of the latter. The discovery of epigenetic regulatory mechanisms in muscle contractures could offer new promising targets of pharmacological treatments for patients with contractures.
MicroRNAs
MicroRNAs (miRNAs) are a group of evolutionary conserved nucleotide non‐coding RNAs involved in the regulation of post‐transcriptional gene expression (Liu & Bassel‐Duby, 2015). miRNAs can silence specific downstream target mRNA through an incorporation of miRNA into target mRNAs with imperfect base pairing causing translational inhibition and RNA degradation (Liu & Bassel‐Duby, 2015). This fine‐tuning process of RNA expression causes downregulations of the target mRNAs and protein levels (Liu & Bassel‐Duby, 2015). miRNAs influence various biological events, including cell death, differentiation, proliferation and cell growth (Liu & Bassel‐Duby, 2015). Recent studies have demonstrated that miRNAs may play critical roles in diseases affecting skeletal muscle, including hereditary spastic paraplegia (Henson et al. 2012), muscular dystrophies (Liu & Bassel‐Duby, 2015), and amyotrophic lateral sclerosis (ALS) (Campos‐Melo et al. 2013). Cacchiarelli et al. 2010 observed a higher expression of miR‐31 in muscle biopsies of DMD patients when compared to healthy muscle. The authors conclude that the higher expression of miR‐31 indicates insensitive regeneration and a disability of Duchenne myoblasts to complete differentiation (Cacchiarelli et al. 2010). Furthermore, post mortem spinal cord specimens from ALS patients revealed that the expression of several miRNAs was significantly altered (Campos‐Melo et al. 2013). Dysfunctions of skeletal muscle mitochondria have been suggested to be involved in the progression of severity of ALS. However, no studies have so far investigated whether muscle contractures show alterations of miRNA expression. Since miRNAs are emerging as key players in regulating several muscle diseases, it is of tremendous importance to clarify whether miRNAs are involved in the development of muscle contractures. If alterations of miRNA expression cause skeletal muscle maladaptation during the development of muscle contractures, they could offer potential miRNA‐based therapies to benefit skeletal muscle health.
Conclusion
In this review we have summarized several of the factors which are likely to contribute to the development of contractures in patients with central motor lesions, including neural activation, mismatch between bone and muscle growth, mechanotransduction, tensional homeostasis, micro‐vascularization, genetics and epigenetics. A framework of factors triggering contracture is appearing, but all the factors and processes involved need to be examined further in relation to the triggering of muscle contractures, and we are convinced that a comprehensive investigation of these topics will open the door to a better understanding of why muscle contractures develop. This may then hopefully pave the way for a more optimal, patient‐focused treatment of this heterogeneous patient group.
Additional information
Competing interests
None declared.
Funding
This project was funded by the Danish Research Council (DFF‐1333‐00197) and the Elsass Foundation.
Acknowledgements
We thank Dr Adrian Harrison for his help and great scientific discussions about this work.
Biographies
Jessica Pingel did her PhD at the Institute of Sports Medicine Copenhagen, and postdoctoral fellowship at the Department of Exercise, Nutrition and Sports at the University of Copenhagen Denmark. She has been working with connective tissue research for the last 10 years.
Else Marie Bartels PhD, DSc, started her career at Biophysics at University of Copenhagen and went from here to The Oxford Research Unit, The Open University Oxford, and later to the Department of Physiology and the Nuffield Orthopedic Centre at Oxford University, UK. She is at present in Copenhagen as head of the Biochemistry and Physiology Laboratory at the Parker Institute ‐ a clinical research institute studying musculoskeletal diseases.
Jens Bo Nielsen is Professor of Human Motor Control at the Department of Neuroscience and Pharmacology, University of Copenhagen, Denmark. He is also Research Director at the Helene Elsass center, which aims to transfer knowledge from basic science into new ways of helping people with Cerebral palsy. Professor Nielsen has been conducting research in how the nervous system controls movement in health and disease for the past 20 years.

References
- Aze O, Odjardias E, Devillard X, Akplogan B, Calmels P & Giraux P (2016). Structural and physiological muscle changes after post‐stroke hemiplegia: A systematic review. Ann Phys Rehabil Med 59S, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baar K (2010). Epigenetic control of skeletal muscle fibre type. Acta Physiol (Oxf) 199, 477–487. [DOI] [PubMed] [Google Scholar]
- Banes AJ, Tsuzaki M, Yamamoto J, Fischer T, Brigman B, Brown T & Miller L (1995). Mechanoreception at the cellular level: the detection, interpretation, and diversity of responses to mechanical signals. Biochem Cell Biol 73, 349–365. [DOI] [PubMed] [Google Scholar]
- Bar‐On L, Molenaers G, Aertbelien E, Van Campenhout A, Feys H, Nuttin B & Desloovere K (2015). Spasticity and its contribution to hypertonia in cerebral palsy. Biomed Res Int 2015, 317047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor CL & Winder SJ (2006). Sparks, signals and shock absorbers: how dystrophin loss causes muscular dystrophy. Trends Cell Biol 16, 198–205. [DOI] [PubMed] [Google Scholar]
- Benavides Damm T & Egli M (2014). Calcium's role in mechanotransduction during muscle development. Cell Physiol Biochem 33, 249–272. [DOI] [PubMed] [Google Scholar]
- Bonnemann CG (2011). The collagen VI‐related myopathies: muscle meets its matrix. Nat Rev Neurol 7, 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisov AB, Huang SK & Carlson BM (2000). Remodeling of the vascular bed and progressive loss of capillaries in denervated skeletal muscle. Anat Rec 258, 292–304. [DOI] [PubMed] [Google Scholar]
- Broers JL, Machiels BM, Kuijpers HJ, Smedts F, van den Kieboom R, Raymond Y & Ramaekers FC (1997). A‐ and B‐type lamins are differentially expressed in normal human tissues. Histochem Cell Biol 107, 505–517. [DOI] [PubMed] [Google Scholar]
- Cacchiarelli D, Incitti T, Martone J, Cesana M, Cazzella V, Santini T, Sthandier O & Bozzoni I (2011). miR‐31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep 12, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M, Nicoletti C, Santini T, Sthandier O, Barberi L, Auricchio A, Musaro A & Bozzoni I (2010). MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab 12, 341–351. [DOI] [PubMed] [Google Scholar]
- Campos‐Melo D, Droppelmann CA, He Z, Volkening K & Strong MJ (2013). Altered microRNA expression profile in amyotrophic lateral sclerosis: a role in the regulation of NFL mRNA levels. Mol Brain 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S & Karpati G (1982). Necrosis of capillaries in denervation atrophy of human skeletal muscle. Muscle Nerve 5, 250–254. [DOI] [PubMed] [Google Scholar]
- Chatterjee S & Fisher AB (2014). Mechanotransduction: forces, sensors, and redox signalling. Antioxid Redox Signal 20, 868–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton AL, Hazzalin CA & Mahadevan LC (2006). Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell 23, 289–296. [DOI] [PubMed] [Google Scholar]
- Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S, Ragone G, Pescatori M, Zaccagnini G, Antonini A, Minetti G, Martelli F, Piaggio G, Gallinari P, Steinkuhler C, Clementi E, Dell'Aversana C, Altucci L, Mai A, Capogrossi MC, Puri PL & Gaetano C (2008). HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci USA 105, 19183–19187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coomber SJ, Bartels EM & Elliott GF (2011). Calcium‐dependence of Donnan potentials in glycerinated rabbit psoas muscle in rigor, at and beyond filament overlap; a role for titin in the contractile process. Cell Calcium 50, 91–97. [DOI] [PubMed] [Google Scholar]
- Dingal PC & Discher DE (2014). Systems mechanobiology: tension‐inhibited protein turnover is sufficient to physically control gene circuits. Biophys J 107, 2734–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diong J, Harvey LA, Kwah LK, Eyles J, Ling MJ, Ben M & Herbert RD (2012). Incidence and predictors of contracture after spinal cord injury–a prospective cohort study. Spinal Cord 50, 579–584. [DOI] [PubMed] [Google Scholar]
- Egginton S (2009). Invited review: activity‐induced angiogenesis. Pflugers Arch 457, 963–977. [DOI] [PubMed] [Google Scholar]
- Formigli L, Sassoli C, Squecco R, Bini F, Martinesi M, Chellini F, Luciani G, Sbrana F, Zecchi‐Orlandini S, Francini F & Meacci E (2009). Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1‐phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J Cell Sci 122, 1322–1333. [DOI] [PubMed] [Google Scholar]
- Friden J & Lieber RL (2003). Spastic muscle cells are shorter and stiffer than normal cells. Muscle Nerve 27, 157–164. [DOI] [PubMed] [Google Scholar]
- Gautel M (2011). Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Arch 462, 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass DJ (2005). Skeletal muscle hypertrophy and atrophy signalling pathways. Int J Biochem Cell Biol 37, 1974–1984. [DOI] [PubMed] [Google Scholar]
- Goldspink G, Tabary C, Tabary JC, Tardieu C & Tardieu G (1974). Effect of denervation on the adaptation of sarcomere number and muscle extensibility to the functional length of the muscle. J Physiol 236, 733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough M & Shortland AP (2012). Could muscle deformity in children with spastic cerebral palsy be related to an impairment of muscle growth and altered adaptation? Dev Med Child Neurol 54, 495–499. [DOI] [PubMed] [Google Scholar]
- Gracies JM (2005). Pathophysiology of spastic paresis. II: Emergence of muscle overactivity. Muscle Nerve 31, 552–571. [DOI] [PubMed] [Google Scholar]
- Griggs RC, Tawil R, McDermott M, Forrester J, Figlewicz D & Weiffenbach B (1995). Monozygotic twins with facioscapulohumeral dystrophy (FSHD): implications for genotype/phenotype correlation. Muscle Nerve Suppl, S50–55. [PubMed] [Google Scholar]
- Hagglund G & Wagner P (2011). Spasticity of the gastrosoleus muscle is related to the development of reduced passive dorsiflexion of the ankle in children with cerebral palsy: a registry analysis of 2,796 examinations in 355 children. Acta Orthop 82, 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson RC, Gilbert SR, Clement ME, Abbas A, Worley G & Stevenson RD (2005. a). Altered skeletal maturation in moderate to severe cerebral palsy. Dev Med Child Neurol 47, 229–236. [DOI] [PubMed] [Google Scholar]
- Henderson RC, Kairalla JA, Barrington JW, Abbas A & Stevenson RD (2005. b). Longitudinal changes in bone density in children and adolescents with moderate to severe cerebral palsy. J Pediatr 146, 769–775. [DOI] [PubMed] [Google Scholar]
- Henson BJ, Zhu W, Hardaway K, Wetzel JL, Stefan M, Albers KM & Nicholls RD (2012). Transcriptional and post‐transcriptional regulation of SPAST, the gene most frequently mutated in hereditary spastic paraplegia. PLoS One 7, e36505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskind A, Ritterband‐Rosenbaum A, Willerslev‐Olsen M, Lorentzen J, Hanson L, Lichtwark G & Nielsen JB (2016). Muscle growth is reduced in 15‐month‐old children with cerebral palsy. Dev Med Child Neurol 58, 485–491. [DOI] [PubMed] [Google Scholar]
- Hiatt WR, Regensteiner JG, Hargarten ME, Wolfel EE & Brass EP (1990). Benefit of exercise conditioning for patients with peripheral arterial disease. Circulation 81, 602–609. [DOI] [PubMed] [Google Scholar]
- Jaalouk DE & Lammerding J (2009). Mechanotransduction gone awry. Nat Rev Mol Cell Biol 10, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D, Dethlefsen S, Haase I, Plomann M, Hirche F, Krieg T & Eckes B (2001). Fibroblasts in mechanically stressed collagen lattices assume a ‘synthetic’ phenotype. J Biol Chem 276, 36575–36585. [DOI] [PubMed] [Google Scholar]
- Lake SP, Castile RM, Borinsky S, Dunham CL, Havlioglu N & Galatz LM (2016). Development and use of an animal model to study post‐traumatic stiffness and contracture of the elbow. J Orthop Res 34, 354–364. [DOI] [PubMed] [Google Scholar]
- Liu AJ, Briody JN, Munns CF & Waugh MC (2008). Regional changes in bone mineral density following spinal cord injury in children. Dev Neurorehabil 11, 51–59. [DOI] [PubMed] [Google Scholar]
- Liu N & Bassel‐Duby R (2015). Regulation of skeletal muscle development and disease by microRNAs. Results Probl Cell Differ 56, 165–190. [DOI] [PubMed] [Google Scholar]
- Lopez‐Barneo J, Lopez‐Lopez JR, Urena J & Gonzalez C (1988). Chemotransduction in the carotid body: K+ current modulated by PO2 in type I chemoreceptor cells. Science 241, 580–582. [DOI] [PubMed] [Google Scholar]
- Lopez‐Hernandez LB, Gomez‐Diaz B, Luna‐Angulo AB, Anaya‐Segura M, Bunyan DJ, Zuniga‐Guzman C, Escobar‐Cedillo RE, Roque‐Ramirez B, Ruano‐Calderon LA, Rangel‐Villalobos H, Lopez‐Hernandez JA, Estrada‐Mena FJ, Garcia S & Coral‐Vazquez RM (2015). Comparison of mutation profiles in the Duchenne muscular dystrophy gene among populations: implications for potential molecular therapies. Int J Mol Sci 16, 5334–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makki D, Duodu J & Nixon M (2014). Prevalence and pattern of upper limb involvement in cerebral palsy. J Child Orthop 8, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S, Pandyan AD, Day CR, Jones PW & Hermens H (2009). Spasticity, an impairment that is poorly defined and poorly measured. Clin Rehabil 23, 651–658. [DOI] [PubMed] [Google Scholar]
- Mathewson MA, Chambers HG, Girard PJ, Tenenhaus M, Schwartz AK & Lieber RL (2014). Stiff muscle fibres in calf muscles of patients with cerebral palsy lead to high passive muscle stiffness. J Orthop Res 32, 1667–1674. [DOI] [PubMed] [Google Scholar]
- Mathewson MA, Ward SR, Chambers HG & Lieber RL (2015). High resolution muscle measurements provide insights into equinus contractures in patients with cerebral palsy. J Orthop Res 33, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthiasdottir S, Hahn M, Yaraskavitch M & Herzog W (2014). Muscle and fascicle excursion in children with cerebral palsy. Clin Biomech (Bristol, Avon) 29, 458–462. [DOI] [PubMed] [Google Scholar]
- Nas K, Yazmalar L, Sah V, Aydin A & Ones K (2015). Rehabilitation of spinal cord injuries. World J Orthop 6, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaou S, Hu L & Cornwall R (2015). Afferent innervation, muscle spindles, and contractures following neonatal brachial plexus injury in a mouse model. J Hand Surg Am 40, 2007–2016. [DOI] [PubMed] [Google Scholar]
- Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, Sonenberg N, Kelly PA, Sotiropoulos A & Pende M (2005). Atrophy of S6K1 −/− skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol 7, 286–294. [DOI] [PubMed] [Google Scholar]
- Olsson MC, Kruger M, Meyer LH, Ahnlund L, Gransberg L, Linke WA & Larsson L (2006). Fibre type‐specific increase in passive muscle tension in spinal cord‐injured subjects with spasticity. J Physiol 577, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingel J, Wienecke J, Lorentzen J & Nielsen JB (2016). Botulinum toxin injection causes hyperreflexia and increased muscle stiffness of the triceps surae muscle in the rat. J Neurophysiol; DOI: 10.1152/jn.00452.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponten EM & Stal PS (2007). Decreased capillarization and a shift to fast myosin heavy chain IIx in the biceps brachii muscle from young adults with spastic paresis. J Neurol Sci 253, 25–33. [DOI] [PubMed] [Google Scholar]
- Prior SJ, McKenzie MJ, Joseph LJ, Ivey FM, Macko RF, Hafer‐Macko CE & Ryan AS (2009). Reduced skeletal muscle capillarization and glucose intolerance. Microcirculation 16, 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Bauman WA & Cardozo C (2010). Bone and muscle loss after spinal cord injury: organ interactions. Ann NY Acad Sci 1211, 66–84. [DOI] [PubMed] [Google Scholar]
- Rehfeldt F, Brown AE, Raab M, Cai S, Zajac AL, Zemel A & Discher DE (2012). Hyaluronic acid matrices show matrix stiffness in 2D and 3D dictates cytoskeletal order and myosin‐II phosphorylation within stem cells. Integr Biol (Camb) 4, 422–430. [DOI] [PubMed] [Google Scholar]
- Rittie L & Fisher GJ (2015). Natural and sun‐induced aging of human skin. Cold Spring Harb Perspect Med 5, a015370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackley C, Brittle N, Patel S, Ellins J, Scott M, Wright C & Dewey ME (2008). The prevalence of joint contractures, pressure sores, painful shoulder, other pain, falls, and depression in the year after a severely disabling stroke. Stroke 39, 3329–3334. [DOI] [PubMed] [Google Scholar]
- Sato Y, Kaji M, Honda Y, Hayashida N, Iwamoto J, Kanoko T & Satoh K (2004). Abnormal calcium homeostasis in disabled stroke patients with low 25‐hydroxyvitamin D. Bone 34, 710–715. [DOI] [PubMed] [Google Scholar]
- Sheean G & McGuire JR (2009). Spastic hypertonia and movement disorders: pathophysiology, clinical presentation, and quantification. PM R 1, 827–833. [DOI] [PubMed] [Google Scholar]
- Siegfried Z, Eden S, Mendelsohn M, Feng X, Tsuberi BZ & Cedar H (1999). DNA methylation represses transcription in vivo . Nat Genet 22, 203–206. [DOI] [PubMed] [Google Scholar]
- Sikavitsas VI, Bancroft GN, Holtorf HL, Jansen JA & Mikos AG (2003). Mineralized matrix deposition by marrow stromal osteoblasts in 3D perfusion culture increases with increasing fluid shear forces. Proc Natl Acad Sci USA 100, 14683–14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeulders MJ, Kreulen M, Hage JJ, Huijing PA & van der Horst CM (2004). Overstretching of sarcomeres may not cause cerebral palsy muscle contracture. J Orthop Res 22, 1331–1335. [DOI] [PubMed] [Google Scholar]
- Smith LR, Chambers HG, Subramaniam S & Lieber RL (2012). Transcriptional abnormalities of hamstring muscle contractures in children with cerebral palsy. PLoS One 7, e40686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LR, Lee KS, Ward SR, Chambers HG & Lieber RL (2011). Hamstring contractures in children with spastic cerebral palsy result from a stiffer extracellular matrix and increased in vivo sarcomere length. J Physiol 589, 2625–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LR, Ponten E, Hedstrom Y, Ward SR, Chambers HG, Subramaniam S & Lieber RL (2009). Novel transcriptional profile in wrist muscles from cerebral palsy patients. BMC Med Genomics 2, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson RD, Roberts CD & Vogtle L (1995). The effects of non‐nutritional factors on growth in cerebral palsy. Dev Med Child Neurol 37, 124–130. [DOI] [PubMed] [Google Scholar]
- Tamai K, Kurokawa T & Matsubara I (1989). In situ observation of adjustment of sarcomere length in skeletal muscle under sustained stretch. Nihon Seikeigeka Gakkai Zasshi 63, 1558–1563. [PubMed] [Google Scholar]
- Tedroff K, Lowing K, Jacobson DN & Astrom E (2011). Does loss of spasticity matter? A 10‐year follow‐up after selective dorsal rhizotomy in cerebral palsy. Dev Med Child Neurol 53, 724–729. [DOI] [PubMed] [Google Scholar]
- Trudel G, Uhthoff HK & Brown M (1999). Extent and direction of joint motion limitation after prolonged immobility: an experimental study in the rat. Arch Phys Med Rehabil 80, 1542–1547. [DOI] [PubMed] [Google Scholar]
- Uchida C, Nwadozi E, Hasanee A, Olenich S, Olfert IM & Haas TL (2015). Muscle‐derived vascular endothelial growth factor regulates microvascular remodelling in response to increased shear stress in mice. Acta Physiol (Oxf) 214, 349–360. [DOI] [PubMed] [Google Scholar]
- Vallejo‐Illarramendi A, Toral‐Ojeda I, Aldanondo G & Lopez de Munain A (2014). Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev Mol Med 16, e16. [DOI] [PubMed] [Google Scholar]
- Vogel V & Sheetz MP (2009). Cell fate regulation by coupling mechanical cycles to biochemical signalling pathways. Curr Opin Cell Biol 21, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittemore LA, Song K, Li X, Aghajanian J, Davies M, Girgenrath S, Hill JJ, Jalenak M, Kelley P, Knight A, Maylor R, O'Hara D, Pearson A, Quazi A, Ryerson S, Tan XY, Tomkinson KN, Veldman GM, Widom A, Wright JF, Wudyka S, Zhao L & Wolfman NM (2003). Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun 300, 965–971. [DOI] [PubMed] [Google Scholar]
- Williams PE & Goldspink G (1984). Connective tissue changes in immobilised muscle. J Anat 138, 343–350. [PMC free article] [PubMed] [Google Scholar]
- Yonekawa T & Nishino I (2015). Ullrich congenital muscular dystrophy: clinicopathological features, natural history and pathomechanism(s). J Neurol Neurosurg Psychiatry 86, 280–287. [DOI] [PubMed] [Google Scholar]