

Abstract
Key points
Fibrosis occurs secondary to many skeletal muscle diseases and injuries, and can alter muscle function.
It is unknown how collagen, the most abundant extracellular structural protein, alters its organization during fibrosis.
Quantitative and qualitative high‐magnification electron microscopy shows that collagen is organized into perimysial cables which increase in number in a model of fibrosis, and cables have unique interactions with collagen‐producing cells.
Fibrotic muscles are stiffer and have a higher concentration of collagen‐producing cells.
These results improve our understanding of the organization of fibrotic skeletal muscle extracellular matrix and identify novel structures that might be targeted by antifibrotic therapy.
Abstract
Skeletal muscle extracellular matrix (ECM) structure and organization are not well understood, yet the ECM plays an important role in normal tissue homeostasis and disease processes. Fibrosis is common to many muscle diseases and is typically quantified based on an increase in ECM collagen. Through the use of multiple imaging modalities and quantitative stereology, we describe the structure and composition of wild‐type and fibrotic ECM, we show that collagen in the ECM is organized into large bundles of fibrils, or collagen cables, and the number of these cables (but not their size) increases in desmin knockout muscle (a fibrosis model). The increase in cable number is accompanied by increased muscle stiffness and an increase in the number of collagen producing cells. Unique interactions between ECM cells and collagen cables were also observed and reconstructed by serial block face scanning electron microscopy. These results demonstrate that the muscle ECM is more highly organized than previously reported. Therapeutic strategies for skeletal muscle fibrosis should consider the organization of the ECM to target the structures and cells contributing to fibrotic muscle function.
Keywords: collagen, extracellular matrix, fibrosis, skeletal muscle
Key points
Fibrosis occurs secondary to many skeletal muscle diseases and injuries, and can alter muscle function.
It is unknown how collagen, the most abundant extracellular structural protein, alters its organization during fibrosis.
Quantitative and qualitative high‐magnification electron microscopy shows that collagen is organized into perimysial cables which increase in number in a model of fibrosis, and cables have unique interactions with collagen‐producing cells.
Fibrotic muscles are stiffer and have a higher concentration of collagen‐producing cells.
These results improve our understanding of the organization of fibrotic skeletal muscle extracellular matrix and identify novel structures that might be targeted by antifibrotic therapy.
Abbreviations
- des−/−
desmin knockout
- ECM
extracellular matrix
- EDL
extensor digitorum longus
- FACS
fluorescence activated cell sorting
- FAPs
fibro/adipogenic progenitors
- GFP
green fluorescent protein
- PCSA
physiological cross‐sectional area
- SBEM
serial block face scanning electron microscopy
- SEM
scanning electron microscopy
- TEM
transmission electron microscopy
- wt
wild‐type
Introduction
The skeletal muscle extracellular matrix (ECM), once thought to be relatively inert and provide only mechanical support to muscle cells, is now known to play an active role in muscle development, regeneration and disease. It is recognized as a dynamic reservoir for stem cells, growth factors and remodelling proteins, and provides several types of microenvironments that enhance cellular function. Despite its importance, there are limited data available regarding the detailed structure of ECM and how it is organized (beyond the classic transmission electron microscopy (TEM) descriptions of skeletal muscle). While traditional TEM has been invaluable for determining the structure and organization of muscle fibres, its usefulness has been limited in the study of ECM structures due to the inability to isolate and image these structures in a single thin section. With the recent development of serial block face scanning electron microscopy (SBEM), it is now possible to collect two‐dimensional serial images from a tissue which can create a three‐dimensional reconstruction of structures within the tissue (Denk & Horstmann, 2004). Each tissue type presents its own challenges for SBEM, and we optimized SBEM for imaging and reconstructing skeletal muscle ECM (Gillies et al. 2014).
While understanding the normal structure and organization of skeletal muscle ECM is important, determining alterations in ECM organization due to disease or injury is also clinically relevant since these alterations may have functional consequences. Skeletal muscle fibrosis commonly develops in response to skeletal muscle disease and injury and is characterized by increased area of the ECM in muscle cross‐sections (Cullen & Mastaglia, 1980; Lieber et al. 2003). Collagen is the most abundant structural protein found in ECM and is upregulated at the transcriptional level (Haslett et al. 2002; Smith et al. 2009; Meyer & Lieber, 2012) and increased at the protein level (Smith et al. 2011; Meyer & Lieber, 2012) with fibrosis. Secondary to fibrosis, increased muscle stiffness and/or contracture progressively occurs thereby limiting function and mobility (Cornu et al. 1998; Kerr Graham & Selber, 2003). In a model of progressive skeletal muscle fibrosis (desmin knockout, des−/−), bundles of muscle fibres are stiffer than control bundles (Meyer & Lieber, 2012). This increase in stiffness was not observed in single fibres, demonstrating that increased stiffness was due to the presence of passive elements, such as collagen, between muscle fibres. However, collagen content alone did not correlate with the increased stiffness observed. We thus hypothesized that changes in collagen organization, not necessarily content, may be responsible for mechanical changes in fibrotic muscle. We determined whether collagen would reorganize in response to des−/− induced fibrosis and measured accompanying changes in muscle function and cellular profile. Thus, the purpose of this paper was to describe the three‐dimensional nature of the skeletal muscle ECM and to quantify alterations in collagen distribution secondary to fibrosis.
Methods
Animals
Ethical approval for this study was granted by the University of California San Diego Institutional Animal Care and Use Committee and all procedures were performed in accordance with the National Institutes of Health's Guide for the Use and Care of Laboratory Animals. All experiments were performed on muscle from wild‐type 129/Sv (Taconic Farms, Germantown, NY, USA) and desmin knockout 129/Sv mice (Milner et al. 1996). Animal subjects for scanning electron microscopy (SEM), TEM and SBEM analysis were anaesthetized by intraperitoneal injection of either pentobarbital (100 mg kg−1) or rodent cocktail (100 mg kg−1 ketamine; 10 mg kg−1 xylazine; 3 mg kg−1 acepromazine) and transcardially perfusion fixed. Animals for confocal microscopy, whole muscle functional analysis, and fluorescence activated cell sorting (FACS) were killed by CO2 asphyxiation and cervical dislocation.
Scanning electron microscopy
After anaesthesia administration, animals (n = 4 wild‐type (wt)) were perfused transcardially with mammalian Ringer solution warmed to 35°C containing heparin (20 units ml−1) and 0.2% dextrose for 2 min followed by fixative solution containing 2% paraformaldehyde, 2.5% glutaraldehyde and 0.2% tannic acid in 0.15 m sodium cacodylate buffer containing 2 mm calcium chloride at 35°C for 5 min. All reagents were obtained from Electron Microscopy Sciences (Hartfield, PA, USA) unless otherwise noted. After fixation, 5th toe extensor digitorum longus (EDL) muscles were incubated in fixative solution overnight at 4°C and stained following the OTOTO method. Briefly, muscles were washed with 0.15 m sodium cacodylate buffer followed by incubation with 2% osmium tetroxide containing 1.5% potassium ferrocyanide in 0.15 m sodium cacodylate buffer for 30 min on ice. After washing in distilled water, muscles were incubated in 1% thiocarbohydrazide for 20 min then washed with distilled water followed by a second incubation in 2% osmium tetroxide for 30 min. After washing in distilled water, muscles were again incubated in 1% thiocarbohydrazide for 20 min, washed with distilled water, and finally incubated a third time in 2% osmium tetroxide for 30 min. Muscles were washed with distilled water and dehydrated in 50%, 70%, 90%, 100% and 100% ethanol for 5 min each. Muscles were then cut longitudinally or into cross‐sections, critical point dried, sputter coated with gold, and viewed with a Zeiss Sigma SEM at 2 kV.
Confocal microscopy of muscle fibre bundles
After dissection, 5th toe EDL muscles (n = 4 wt) were pinned to cork at slack length and stored at −20°C in glycerinated storage solution containing (in mm): potassium propionate (170), K3EGTA (5), MgCl2 (5.3), imidazole (10), Na2ATP (21.2), NaN3 (1), glutathione (2.5); and 50 μm leupeptin and 50% (v/v) glycerol. Mouse anti‐type I collagen antibodies (Chondrex, Redmond, WA, USA) were labelled with Alexa Fluor 488 using an antibody labelling kit (Thermo Fisher Scientific, Waltham, MA, USA). Antibody conjugates were purified prior to use to remove any unlabelled antibody or dye. Muscles for confocal analysis were pinned to Sylgard coated dishes in a relaxing solution containing (in mm): imidazole (59.4), KCH4O3S (86), Ca(KCH4O3S)2 (0.13), Mg(KCH4O3S)2 (10.8), K3EGTA (5.5), KH2PO4 (1), Na2ATP (5.1); and 50.0 μm leupeptin, at pCa 8.0 and pH 7.1. Bundles of muscle fibres (5–10 fibres) were isolated and placed in a custom stretching chamber with a cover glass bottom designed for confocal imaging of skeletal muscle bundles (Shah & Lieber, 2003). Bundles were attached using 10‐0 monofilament nylon suture on one end to a stationary titanium pin and on the other end to a titanium wire attached to a micrometer. Bundles were blocked in 1% bovine serum albumin (BSA) in relaxing solution for 30 min, followed by blocking in 15 μl ml−1 goat serum and 10% mouse serum in 0.1% BSA in relaxing solution for 15 min. Bundles were incubated with Alexa Fluor conjugated anti‐type I collagen (1:250) overnight at 4°C. After removing primary antibodies, DRAQ5 (1:1000) was added to label nuclei.
Confocal imaging was performed at 21°C with a Leica True Confocal Scanner SP5 microscope (Buffalo Grove, IL, USA) using a 63× planapochromat oil immersion lens (1.3 numerical aperture). Muscle bundle length was adjusted to slack length, measured as the suture knot‐to‐knot distance. Image stacks of muscle bundles were obtained at slack length and at 10% strain increments. Bundles that slipped on the pin with strain or were damaged were not included in image analyses. X–Y image stacks (512 × 512 raster, 400 Hz) were acquired at 0.99 μm optical section thickness in Leica Application Suite Advanced Fluorescence (LAS AF) software. Three‐dimensional projections of image stacks were created in LAS AF software using the 3D Projection tool in the x–y plane. Because fluorescence intensity decreased with imaging time (photobleaching) and distance from the cover glass, black and white levels were adjusted in Adobe Photoshop CS6 (San Jose, CA, USA) when necessary to extend the image histogram to the full range of 0–255, without adjusting midtone.
Transmission electron microscopy
After anaesthesia administration, animals (n = 8, aged >12 months) were perfused transcardially as described for SEM with the addition of 0.05% ruthenium red to the fixative solution. After complete fixation, both 5th toe EDL muscles were dissected from each animal with one EDL prepared for transmission electron microscopy and the other for SBEM analysis. After removal, EDL muscles were cut into five pieces of equal length and incubated in fixative solution overnight at 4°C. Tissues were washed with 0.15 m sodium cacodylate buffer followed by incubation with 2% osmium tetroxide containing 1.5% potassium ferrocyanide and 0.05% ruthenium red in 0.15 m sodium cacodylate buffer for 30 min on ice. Tissues were washed in distilled water and placed in 2% uranyl acetate at 4°C overnight, followed by washing in distilled water and dehydration in 50%, 70%, 90%, 100% and 100% ethanol for 5 min each. Dehydrated tissues were placed in anhydrous acetone and then embedded in Durcupan ACM resin (Sigma‐Aldrich, St Louis, MO, USA). Cross‐sections of tissue blocks were sectioned at 70 nm thickness on a Leica Ultracut UCT ultramicrotome with a DiATOME diamond knife (Hatfield, PA, USA), and collected on 300 hexagonal mesh thin bar copper grids. Sections were post‐stained for 10 min in 2% uranyl acetate, washed with distilled water, and stained for 1 min in Sato's lead solution. Sections were observed at 80 kV using a FEI Tecnai Spirit TEM (Hillsboro, OR, USA) with objective aperture size 40 μm and condenser aperture size 100 μm. Images were acquired with a 2k × 2k Gatan CCD camera (Pleasanton, CA, USA) and SerialEM Software (Mastronarde, 2005).
Volume fraction estimation by stereology
Transmission electron micrographs for stereological analysis were acquired after a multi‐stage sampling scheme. One section from each tissue block prepared during fixation and embedding was imaged (n = 5 blocks per muscle, n = 8 muscles). The number of test points required to calculate volume fraction with a desired 95% confidence level was calculated based on pilot studies (Weibel, 1979). Eight low magnification (1650×) and five high magnification (11,000×) micrographs were acquired from each section by uniform random systematic sampling, with high magnification images acquired only from areas containing extracellular space. If the randomly selected 11,000× field of view contained no extracellular space it was excluded from analysis and the next field of view was analysed. Sections were also excluded from analysis if they contained tendon tissue (n = 2 out of 80 total).
Images were analysed with IMOD software using the stereology plug‐in (Kremer et al. 1996). A 20 × 20 square grid (grid spacing 0.79 μm, 400 points per grid) was overlaid on each low magnification micrograph and each point was classified as myofibril, mitochondria, myonucleus, satellite cell, fibroblast, extracellular space, nerve, capillary, or other. Cell classifications were based on histology atlases (Carpenter & Karpati, 2001; Gartner & Hiatt, 2009). Fibroblasts were identified by their location within the extracellular matrix, long cellular processes, and lack of a basal lamina. Perivascular cells were identified by their close proximity to capillaries and the presence of a basal lamina. Immune cells were identified by the presence of densely stained granules, convoluted plasma membrane and lack of a basal lamina. Nerves were identified by densely stained Schwann cells surrounding the axon or by the characteristic motor end plate morphology. Satellite cells were characterized based on their location under the basal lamina of muscle fibres. To quantify objects within the extracellular space, 27 × 27 square grids (grid spacing 85.6 nm, 729 points per grid) were overlaid on high magnification micrographs and points within the extracellular space were classified as interstitial space (no visible object), single collagen fibril, collagen fibril in cable, or basal lamina. Points lying on objects not within the extracellular space were left unclassified and not counted. Object point counts were summed for each animal and volume fractions of myofibrils, mitochondria, myonuclei, satellite cells, fibroblasts, extracellular space, nerves and capillaries were defined as the total object point count divided by the total number of points counted at low magnification. Volume fractions for single collagen fibril, collagen fibrils in cable, and basement membrane in the muscle, V col/V M, were calculated using the following equation:
| (1) |
where is the fraction of collagen (single fibril, fibril in cable, or basement membrane) in the extracellular space calculated at high magnification and is the fraction of extracellular space in the muscle calculated at low magnification. Overall, this sampling approach measured a total of 1014 images at 1650× and 11,000× magnifications.
Whole muscle functional analysis
Hindlimbs were skinned and placed in muscle relaxing solution (n = 8, aged >12 months). Second to fourth toe EDL muscles were removed and sutures were tied at the proximal and distal ends of the 5th toe muscle. In vivo muscle length was measured using calipers with the knee in full extension and ankle at maximum dorsiflexion. Proximal and distal tendons were cut and the muscle was placed in a custom mechanical testing chamber at room temperature (20°C) (Friden & Lieber, 2003). The distal tendon was attached to a force transducer (Model 300B, sensitivity 0.1879 g V−1, Aurora Scientific, Ontario, Canada) and the proximal tendon was tied to a micrometer arm. Muscles were set to slack length (L 0), measured as the suture‐to‐suture distance at which passive tension was just measurable (∼0.3 mN). Slack sarcomere length (L s) was measured using laser diffraction (Lieber et al. 1984). Muscles were lengthened in increments of 0.5 mm with peak force and sarcomere length measured at each step. Muscles were allowed to stress relax between steps for 5 min during which stress relaxation was measured. Muscles were strained to a sarcomere length of 4.0 μm or until the micrometer maximum was reached. Muscles were then removed from the chamber, sutures were removed, and the muscle was blotted and weighed. Physiological cross‐sectional area (PCSA) was calculated with the following equation (Sacks & Roy, 1982):
| (2) |
where pennation angle θ = 11.3 deg (Chleboun et al. 1997) and 1.056 = muscle density (g cm−3) (Mendez & Keys, 1960). Stress was defined as muscle tension divided by PCSA.
Muscle passive force data were acquired using custom LabView software (National Instruments, Austin, TX, USA) and analysed in Microsoft Excel (Redmond, WA, USA). Stress was plotted against muscle strain and fitted with a quadratic regression. Tangent modulus was calculated as the slope of the regression line at a sarcomere length of 3.5 μm.
Estimation of collagen cable size and number
Custom MATLAB (Mathworks, Natick, MA, USA) code was written to estimate the size and measure the number of collagen cables from stereology data. Classified stereology grids from high magnification images in IMOD were output to WLMP format containing coordinates for each crosshair and its classification. The coordinates for crosshairs classified as ‘collagen fibril in cable’ were input into MATLAB where adjacent coordinates were grouped based on a connectivity of eight neighbours. This yielded the number of crosshairs contained in each identified collagen cable. The cross‐sectional area of each cable was estimated by multiplying the number of crosshairs in each cable by the area of a circle with radius equal to half the distance between crosshairs.
Generation of type I collagen reporter mice and FACS analysis
Identification of collagen I producing cells was achieved by breeding wt and des−/− mice with collagen I reporter mice expressing green fluorescent protein (GFP) under the control of the collagen α1(I) promoter (Yata et al. 2003). FACS was used to determine the amount of collagen producing cells in wt (n = 16) and des−/− (n = 4) skeletal muscle (Chapman et al. 2016). Briefly, the tibialis anterior, quadriceps and gastrocnemius muscles were dissected and cleaned of all visible tendons and placed in a collagenase/dispase digestive solution (0.27% type I collagenase, 0.06 units ml−1 dispase II, 50 units ml−1 streptomycin, 50 units ml−1 penicillin) for 50 min at 37°C. Muscles were mechanically broken down and then strained through a 70 μm filter followed by a 40 μm filter. Cells were pelleted and resuspended in FACS buffer (2.5% normal goat serum and 1 mm EDTA in PBS). Cells were stained with antibodies against CD31 (102422; Biolegend, San Diego, CA, USA) and CD45 (103126; Biolegend) to identify endothelial and haematopoietic cells, respectively. Cells were then sorted using the BD FACS Aria II Special Order Research Project (BD Biosciences, San Jose, CA, USA) with four lasers (405 nm, 100 mW; 488 nm, 50 mW). Cells were first gated based on size to eliminate cellular debris (side vs. forward scatter) and clumped cells (side scatter area vs. forward scatter area). Endothelial and haematopoietic cells were then eliminated based on staining for CD31 and CD45 antibodies conjugated to Pacific Blue. The remaining cells were gated on GFP expression and the number of cells in each sample was determined. The number of collagen producing cells (GFP+) was normalized to muscle mass to control for differences among muscle preparations and size differences among animals.
Serial block face scanning electron microscopy
Tissue preparation methods for SBEM were previously published (Gillies et al. 2014). Briefly, perfusion fixed EDL muscles (n = 5, aged >12 months) were incubated in fixative solution containing 0.05% ruthenium red overnight at 4°C. Tissues were washed with 0.15 m sodium cacodylate buffer followed by incubation with 2% osmium tetroxide containing 1.5% potassium ferrocyanide and 0.05% ruthenium red in 0.15 m sodium cacodylate buffer for 30 min on ice. After washing in distilled water, tissues were incubated in 1% thiocarbohydrazide for 20 min then washed with distilled water followed by a final incubation in 2% osmium tetroxide for 30 min. Tissues were washed with distilled water and placed in 2% uranyl acetate overnight at 4°C. Tissues were then stained en bloc with Warton's lead aspartate for 15 min at 60°C followed by washing with distilled water and dehydration in 50%, 70%, 90%, 100% and 100% ethanol for 5 min each. Tissues were embedded in Durcupan ACM resin. Embedded tissues were trimmed to ∼1 mm3 and mounted on aluminum pins.
Mounted specimens were viewed in a Zeiss Sigma SEM outfitted with a specialized specimen chamber for serial block face imaging (Gatan 3View). Specimens were imaged at 3 kV under variable pressure (∼32 Pa), high current mode, at 2300× magnification with aperture size 60 mm. Images were acquired with a Gatan backscattered electron detector (1 mm aperture) and visualized with Gatan Digital Micrograph software at 8k × 8k raster, 1 μs dwell time, at 70 nm cutting thickness. Images were converted to 8 bit for analysis and manual segmentation of cells and collagen cables was performed with IMOD software.
Data deposition
A sample of transmission electron micrographs used for stereological analysis, the high magnification stereology analysis grid, and examples of cell types that were identified via stereology can be accessed through the Figshare website https://figshare.com/s/6cb9bf94bef5b166e388.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA). Data are presented as means ± standard error of the mean and significance level (α) was set to P = 0.05 for all data except collagen cable size and number. These data were non‐normally distributed and therefore plotted as a histogram of pooled data and analysed by a Mann–Whitney test (non‐parametric). Stereology and FACS data were analysed by two‐tailed unpaired t tests and data from whole muscle functional analysis were analysed by repeated measures two‐way analysis of variance (two‐way ANOVA) with Bonferroni post hoc paired comparisons.
Results
Identification of bundles of collagen fibrils in skeletal muscle ECM
To provide an overview of the structure and organization of ECM, scanning electron microscopy (SEM) was used to image wild‐type (wt) muscle. We observed muscle fibres enveloped in a mesh of connective tissue, but, importantly, in addition to the mesh we observed discrete cable‐like structures that appeared to be bundles of collagen fibrils. These ‘cables’ were approximately 1 μm in diameter, had a wavy appearance, and were present on the surface of muscle fibres and spanned between fibres (Fig. 1). Due to their location, morphology, and composition we postulated that these cables might be load‐bearing structures, but SEM analysis did not allow investigation of the mechanics of the cables or their composition. Since type I collagen is the primary structural protein found in skeletal muscle ECM and the size of the observed cables was within the resolving capability of confocal microscopy, bundles of muscle fibres containing their surrounding ECM were immunolabelled for type I collagen and viewed using confocal microscopy. Image stacks revealed cable‐like structures labelled positively for type I collagen surrounding muscle fibres (Fig. 2 A and D). To test the response of these cables to uniaxial strain, fibre bundle preparations were stretched in a custom stretching device (Shah & Lieber, 2003) and imaged at 20% (Fig. 2 B and E) and 40% muscle fibre strain (Fig. 2 C and F). Collagen cables clearly reorganized with increased strain becoming less wavy in appearance and aligning with the direction of strain, and appeared to form discrete connections with muscle fibres.
Figure 1. Collagen fibrils are organized into cables in skeletal muscle ECM.
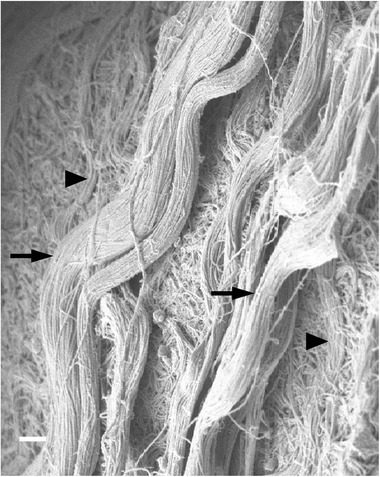
Large bundles of collagen fibrils, or collagen cables, were observed by scanning electron microscopy in wild‐type EDL muscle (n = 4). Cables had a wavy appearance and were present on the surface of muscle fibres (arrowheads) as well as in the space between fibres (arrows). Scale bar = 1 μm.
Figure 2. Collagen cables are composed of type I collagen and realign with increased strain.
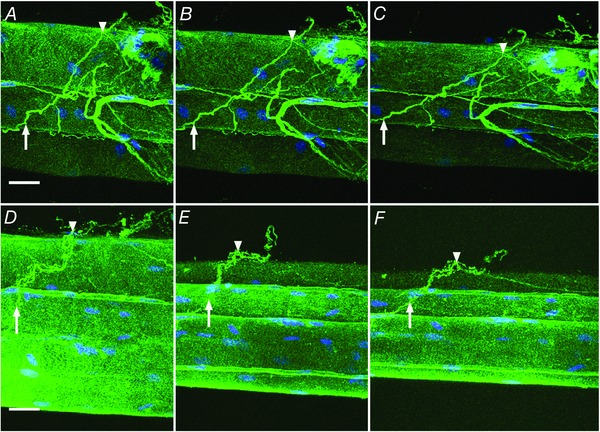
Bundles of wild‐type muscle fibres were observed by confocal microscopy in a custom stretching chamber (n = 4). Bundle preparations (two examples shown in A–C and D–F) were immunolabelled for type I collagen (green) and nuclei (blue). At slack length (0% strain; A and D) intensely stained type I collagen positive structures that traverse multiple fibres are defined as collagen cables. Two points on a cable are identified in each preparation (arrow and arrowhead) and these points are again identified at 20% strain (B and E) and 40% strain (C and F) showing reorganization of cables with strain. Scale bar = 25 μm. [Colour figure can be viewed at wileyonlinelibrary.com]
des−/− muscle has an increased volume fraction of collagen in cables
To observe differences in ECM ultrastructure in fibrotic muscle, we used the des−/− model that was previously shown to progressively develop skeletal muscle fibrosis (Milner et al. 1996; Meyer & Lieber, 2012). To emphasize the fibrosis, aged (>12 months) animals were studied. Differences in volume fraction of muscle components (both intracellular and extracellular) were quantified in both wt and des−/− muscle using rigorous stereological principles (Weibel, 1979). A total of 421,041 locations on 1014 transmission electron micrographs were classified from areas selected using multi‐stage uniform random systematic sampling. The extracellular space accounted for 26.8 ± 3.6% volume of des−/− muscle and 24.1 ± 3.0% of wt muscle (not significantly different). While no change was detected in the volume of isolated collagen fibrils, importantly, the volume fraction of collagen fibrils in cables was significantly increased by ∼60% in des−/− muscle (Fig. 3 A and B). The decreased volume fraction of satellite cells in des−/− muscle was not significant (Fig. 3 C), even though fibroblast volume fraction showed a ∼60% increase in des−/− muscle (Fig. 3 D, not significant, P = 0.055). No significant differences were observed in the volume fraction of any other muscle component (Table 1).
Figure 3. Volume fraction of muscle components determined by stereology.
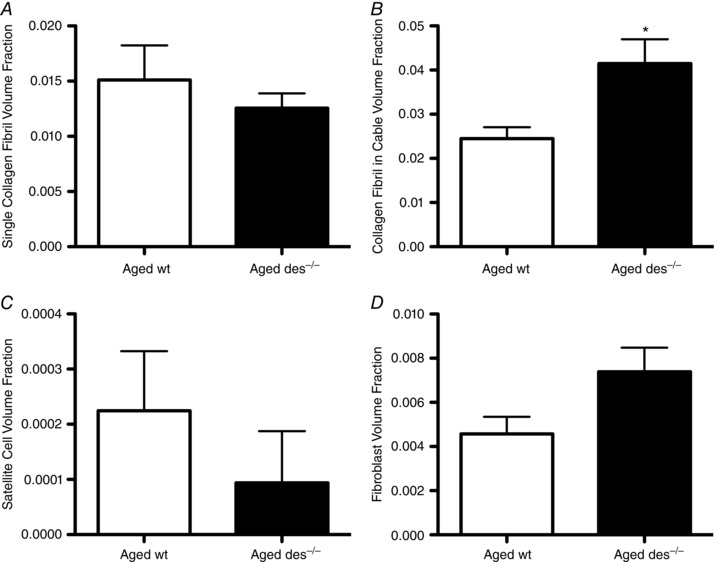
Stereological analysis was performed on aged wild‐type and aged des−/− EDL muscle (n = 8) to quantify the volume fractions of single collagen fibrils (A), collagen fibrils in cables (B), satellite cells (C) and fibroblasts (D). Increased collagen cable volume fraction in des−/− muscle was statistically significant (B), while fibroblast volume fraction increased by about 60% in des−/− muscle (D, not significant, P = 0.055). Means ± standard error of the mean; * P < 0.05 (two‐tailed t tests).
Table 1.
Volume fraction of EDL muscle components
| Muscle component | Aged wt (% EDL volume, n = 8) | Aged des−/− (% EDL volume, n = 8) |
|---|---|---|
| Myofibril | 61.9 ± 2.25 | 59.2 ± 2.78 |
| Mitochondria | 9.96 ± 0.81 | 8.20 ± 1.00 |
| Myonucleus | 0.54 ± 0.06 | 0.78 ± 0.13 |
| Basement membrane | 2.03 ± 0.27 | 1.83 ± 0.08 |
| Interstitial space | 18.1 ± 2.61 | 19.6 ± 3.09 |
| Nerve | 0.50 ± 0.31 | 0.86 ± 0.45 |
| Vessel | 1.88 ± 0.26 | 2.45 ± 0.44 |
| Other | 0.71 ± 0.60 | 0.93 ± 0.36 |
Values are means ± standard error of the mean. No significant differences between groups were detected by two‐tailed t tests (P = 0.05).
des−/− muscle is stiffer at the whole muscle level
Increased stiffness of des−/− muscle has been shown at the muscle bundle level (Meyer & Lieber, 2012), but it was unknown whether this difference would be observed at the whole muscle scale. Functional analysis of 5th toe extensor digitorum longus (EDL) muscles was performed by stepwise lengthening of muscles in a custom stretching chamber while recording passive force and sarcomere length. Muscle stress was determined by dividing passive force by the physiological cross‐sectional area and the stress vs. sarcomere length relationship was fitted with a quadratic regression. des−/− muscle exhibited significantly higher stresses when subjected to passive stretch at sarcomere lengths of 2.9 μm and greater (Fig. 4 A). The tangent stiffness, defined as the derivative of the stress vs. sarcomere length quadratic at 3.5 μm sarcomere length, was ∼4‐fold greater in des−/− muscle compared to wt (Fig. 4 B). Because the stiffness of individual myofibres is not altered in aged des−/− compared to wt (Meyer & Lieber, 2012), composite theory indicates that increased stiffness observed in aged des−/− muscle at the whole muscle level is due to changes in the ECM rather than intrinsic changes in the fibres themselves.
Figure 4. Aged des−/− muscles are stiffer compared to wt.
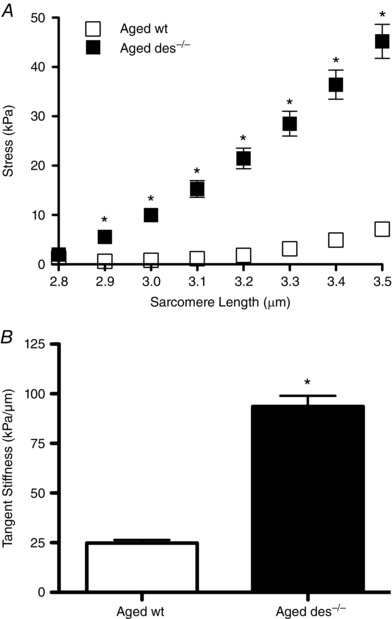
A, average stress after stress relaxation vs. sarcomere length curves from aged wild‐type and des−/− 5th toe EDL muscle (n = 8) were fitted with a quadratic. Aged des−/− muscles generated significantly higher stress than aged wild‐type at sarcomere lengths of 2.9 μm and higher. Means ± standard error of the mean; * P < 0.05 (two‐way ANOVA, Bonferroni post hoc tests). B, tangent stiffness values at 3.5 μm sarcomere length were significantly higher in aged des−/− muscle compared to aged wt. Means ± standard error of the mean; * P < 0.05 (two‐tailed t test).
Increased collagen cable volume fraction is a result of more collagen cables in des−/− muscle
To determine whether increased volume fraction of collagen cables in des−/− muscle was accomplished by cells generally increasing the amount of collagen synthesized or whether the actual cable structure was modified, the number of cables was obtained from high‐magnification micrographs from stereological analysis and the cross‐sectional area of each cable was calculated from classified high‐magnification stereology grids. The total number of collagen cables was 468 (wt) and 686 (des−/−) and the size distribution was skewed right for both genotypes showing a greater number of smaller cables (Fig. 5 A). The shape of the distribution was similar for both genotypes and non‐parametric analysis revealed that the medians were significantly different (0.041 μm2 wt vs. 0.029 μm2 des−/−, P < 0.05) indicating that the des−/− muscle has more collagen cables with smaller cross‐sectional area. The increased volume fraction of collagen cables in des−/− muscle could also theoretically be a result of increased cable length. While it is possible that some cables may be counted twice in our analysis if they are very long, due to section spacing it is extremely unlikely that this would be the case. Therefore, while there are slightly more des−/− cables at larger sizes there are hundreds more small cables, thus the increase in volume fraction is primarily due to an increase in cable number.
Figure 5. The number of collagen cables is increased in aged des−/− muscle and aged des−/− muscle contains more collagen producing cells.
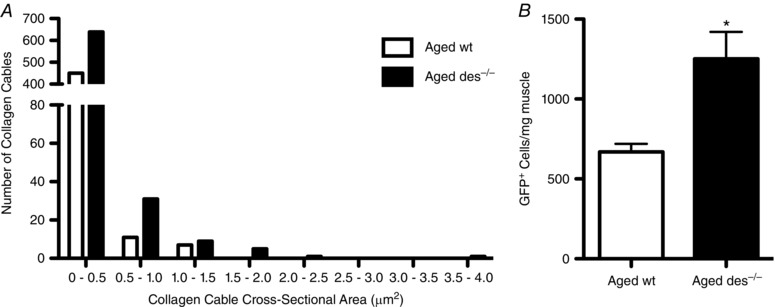
A, the number of collagen cables was counted and the cross‐sectional area of each cable estimated for collagen cables identified on high magnification transmission electron micrographs used for stereological analysis. The total number of cables was 468 (aged wild‐type) and 686 (aged des−/−) and the cross‐sectional area distribution shape was similar between genotypes (n = 8). Medians were found to be significantly different by a Mann–Whitney test (non‐parametric) at 0.041 μm2 for wild‐type vs. 0.029 μm2 for des−/− (P < 0.05). Therefore, the increased volume fraction of collagen cables in des−/− muscle is due to an increase in cable number, not size. B, type I collagen reporter mice were generated in which cells that produce collagen are positively labelled with GFP. GFP expressing cells from aged wt (n = 16) and aged des−/− (n = 4) were sorted and counted by FACS analysis and normalized to muscle mass (Chapman et al. 2016). GFP+ cells were significantly increased in aged des−/− muscle. Means ± standard error of the mean; * P < 0.05 (two‐tailed t test).
Collagen producing cells are more abundant in des−/− muscle
The increased number of collagen cables and increased volume fraction of fibroblasts in des−/− muscle suggests that more cables are present in des−/− muscle due to increased cellular production of collagen. However, it is also possible that the increase in fibroblast volume fraction is due to larger, not more, collagen producing cells. Fluorescence activated cell sorting (FACS) was used to resolve this question by quantifying the number of collagen producing cells. Type I collagen reporter mice were developed that express green fluorescent protein (GFP) under the control of the collagen α1(I) promoter (Yata et al. 2003). GFP expressing (collagen producing) cells were counted and normalized to muscle mass. The number of GFP+ cells was significantly increased in des−/− muscle, indicating an ∼90% increase in the number of collagen producing cells compared to wt (Fig. 5 B) (Chapman et al. 2016).
Unique interactions occur between ECM cells and collagen cables
Through stereology and FACS analysis we showed increased collagen cables and an increase in collagen producing cells, but these data do not provide information about the organization or interaction between these two structures. SBEM was performed to identify and define these interactions in three dimensions. Most fibroblasts observed by SBEM analysis were either relatively aligned with capillaries or isolated in the space between muscle fibres, occasionally sending out very long finger‐like processes. In some cases, fibroblasts were observed that were intimately associated with collagen cables (see Movie S1 in the online Supporting information). In one case, a fibroblast wrapped itself around a collagen cable (Fig. 6 A–D). This extremely long fibroblast (57.5 μm from top to bottom) was aligned with the collagen cable and even followed it after a branching point. A process of the fibroblast extends into the branch fork (Fig. 6 A). Fibroblasts were frequently observed interacting with cables and are known to specifically adhere to type I collagen through integrin binding, but it is unknown whether fibroblasts interact with collagen cables simply because they are abundant in the ECM or whether they also interact with other structural proteins present in smaller amounts (e.g. elastin) and we have not observed these interactions. The reconstruction shown is from a des−/− muscle; however, cables have also been observed interacting with fibroblasts in wt muscle (Gillies et al. 2014). It is not possible to determine based on the data acquired whether interactions between fibroblasts and collagen cables are quantitatively different between des−/− and wt muscle. Another ECM cell, presumed to be an eosinophil, also tightly wrapped itself around a collagen cable (Fig. 6 E, F and G).
Figure 6. Cellular interactions with collagen cables in aged des−/− muscle.
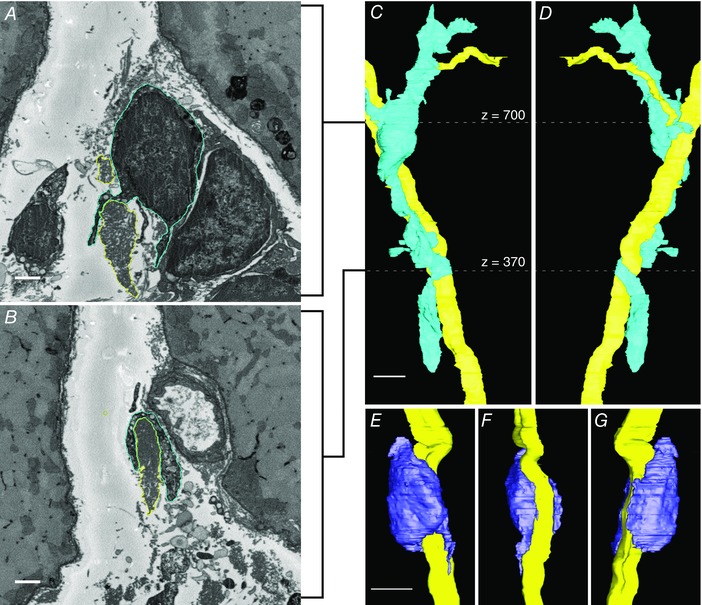
Collagen cables and ECM cells were imaged by SBEM, segmented, and reconstructed in IMOD. Panels A and B are x–y slices (cropped to show detail) from the reconstruction shown in C and D. A, x–y slice at z = 700 showing contours for a fibroblast (blue outline) and collagen cable (yellow outline). B, x–y slice at z = 370 showing contours of the same fibroblast (blue) and collagen cable (yellow). C, reconstruction of a fibroblast (blue) that forms a long, flat process that wraps around a collagen cable (yellow). The cable is branched and the fibroblast appears to follow the branches. The white dotted lines indicate the z‐location of x–y slices shown in A and B. D, 180 deg rotation of C around the z‐axis. E, a suspected eosinophil (purple) wraps around a collagen cable (yellow). F, 90 deg rotation of E around the z‐axis. G, 180 deg rotation of E around the z‐axis. Scale bars in A and B = 1 μm, in C–G = 5 μm. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
This study demonstrates the complex combination of cells and structural support proteins that occupy the skeletal muscle ECM. SBEM reveals that collagen fibrils are organized into large bundles, or cables, that traverse hundreds of micrometres. They have a wavy structure that reorients with strain, becoming aligned with the muscle fibre axis with increasing strain. Similar collagen cables have previously been observed in skeletal muscle by SEM (Passerieux et al. 2006; Gillies & Lieber, 2011). These discrete collagen cables do not fit with the traditional definition of skeletal muscle perimysium as generic, poorly organized connective tissue that surrounds only muscle fascicles. This is probably due to the lack of high resolution images available (such as were generated in this study) where the size and complexity of such structures cannot be appreciated on a single TEM section. We describe these cables as perimysial since the most obvious difference between endomysium and perimysium at the electron microscopy level is the organization of collagen fibrils into cables. The collagen cables observed in this study appear to be primarily composed of type I collagen based on their morphology and antibody labelling with confocal imaging. Other structural proteins known to be present in skeletal muscle ECM in lesser amounts (e.g. fibronectin, proteoglycans, laminin, elastin) were not identified.
Although a quantitative correlation was not attempted, collagen cable volume fraction and muscle stiffness significantly increased in des−/− muscle. It should be noted that a relatively small change in collagen cable volume fraction (+1.7% muscle volume) corresponded to a 4‐fold increase in muscle stiffness. We have shown in both mouse and human muscle that collagen content alone does not predict muscle stiffness (Smith et al. 2011; Chapman et al. 2015) so we suggest that collagen organization may be a better predictor of muscle stiffness. As muscle fibres were stretched, perimysial collagen cables straightened and realigned with the muscle axis and passive tension increased non‐linearly with strain suggesting that the wavy orientation of collagen cables may provide a strain relief function. Given the very high tensile strength of collagen fibrils (Burton, 1954), it is feasible that the increased collagen cable volume fraction and the actual increase in the number of collagen cables in des−/− muscle caused increased stiffness. However, the increase in stiffness would have to increase non‐linearly with fibril number for stiffness to change as described.
The significant increase in collagen producing cells is consistent with the stereological finding of a trend of increased fibroblast volume fraction. However, the increase in collagen producing cells was greater than the increase in fibroblast volume fraction, which could be due to the heterogeneous population of GFP+ cells. GFP expressing cells included fibroblasts, but also skeletal muscle progenitor cells and fibro/adipogenic progenitors (FAPs) (Chapman et al. 2016). Additionally, cells of haematopoietic and endothelial origin that exhibit fibrogenic plasticity and have low or no expression of CD45 and CD31, respectively, could be present in the pool of GFP+ cells measured (Pessina et al. 2015). FAPs have been shown to contribute to skeletal muscle fibrosis (Uezumi et al. 2011), but the extent of their involvement relative to fibroblasts or other collagen producing cells is not yet clear. The localization of FAPs and fibroblasts in muscle are similar and it is possible that some fibroblasts classified via stereology were actually FAPs.
The deletion of desmin initiates an inflammatory response in skeletal muscle (Meyer & Lieber, 2012), which may lead to the observed increase in the number of collagen producing cells and potentially dysregulate fibroblast activity eventually leading to fibrosis. It was previously shown that des−/− muscle contains more collagen (measured by hydroxyproline assay) and an increase in inflammatory cells (Meyer & Lieber, 2012). It is interesting that only the number of collagen cables and not the volume fraction of single collagen fibrils or basement membrane were increased in des−/− muscle by collagen producing cells. This may not necessarily be a result of fibrosis, but perhaps collagen fibrils preferentially organize into cables in the ECM. We now know that the fibrotic thickening of the ECM observed in histological samples from des−/− muscle (Meyer & Lieber, 2012) is due to the presence of an increased number of collagen cables. Such cables have not previously been appreciated as cellular organelles but the current study suggests that they are synthesized and regulated much as any other organelle. Future studies are required to determine the factors that regulate cable size and number.
Conclusion
Fibrosis occurs in skeletal muscle secondary to injury and disease and is generally defined as an increase in ECM. Here we have shown, in the des−/− model of fibrosis, that increased ECM is due to an increase in the volume fraction and number of collagen cables, and that the stiffness of these muscles is also increased. Fibroblasts often appear to interact with collagen cables, although it is unknown whether these interactions differ between wt and des−/− muscle. Taken together, these data suggest that collagen cables and the cells/factors regulating their size and number are functionally significant and may be targets for antifibrotic therapy. The data also illustrate that measurement of collagen content alone may not be useful to provide a mechanistic understanding of the biomechanical properties of fibrotic muscle tissue.
Additional information
Competing interests
None declared.
Author contributions
This work was completed at the University of California San Diego. Study conception and design: A.R.G., M.H.E. and R.L.L. Data acquisition and analysis: A.R.G., M.A.C., E.A.B. and T.J.D. Manuscript preparation: A.R.G., M.A.C. and R.L.L. Manuscript editing: A.R.G., E.A.B., T.J.D., M.H.E. and R.L.L. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health under award numbers R01AR40050 and R01AR57393 and by the National Institute of Child Health and Human Development (NICHD) of the National Institutes of Health under award number R24HD05083. The National Centre for Microscopy and Imaging Research is supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number P41GM103412. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Translational perspective.
Fibrosis of skeletal muscle occurs secondary to muscle diseases such as muscular dystrophy, neuromuscular diseases such as cerebral palsy, and mechanical injury such as occurs during intense exercise. This causes muscle stiffening (resistance to elongation) and may even deform the joint requiring corrective surgery. Until now, the cellular physiological and structural basis of fibrosis has not been known. In this study, the authors use a combination of biomechanical, microscopy and biological tools to understand this basis in a mouse fibrosis model. Interestingly, they show that while the collagen content of these muscles does not change significantly, the organization of collagen changes with fibrosis. Specifically, a greater fraction of the collagen fibrils (the basic building block of connective tissue) organizes into what the authors term ‘perimysial cables’ that traverse hundreds of micrometres of tissue and may be several micrometres in diameter. In addition, they show that an increase in the number of collagen producing cells occurs after fibrosis, which also suggests regulation of this process at the cellular level. Thus, this study has demonstrated a higher degree of organization of skeletal muscle extracellular matrix than previously appreciated and may provide new strategies for treating fibrotic muscle. Such strategies may specifically target the collagen‐producing cells or may target the perimysial cables themselves as a way to relieve the deforming and often painful effects of fibrosis.
Supporting information
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Movie S1. Interaction of a collagen cable and fibroblast in aged des−/− muscle reconstructed from SBEM analysis.
Acknowledgements
We acknowledge Dr Samuel Ward and Dr Ana Rodríguez‐Soto for helpful discussion. Mason Mackey and Andrea Thor from the National Centre for Microscopy and Imaging Research and Michael Villongco, Rachel Meza, Blair Conner and Shannon Bremner from the Muscle Physiology Laboratory are thanked for technical assistance.
References
- Burton AC (1954). Relation of structure to function of the tissues of the wall of blood vessels. Physiol Rev 34, 619–642. [DOI] [PubMed] [Google Scholar]
- Carpenter S & Karpati G (2001). Pathology of Skeletal Muscle. Oxford University Press, Oxford. [Google Scholar]
- Chapman MA, Mukund K, Subramaniam S, Brenner D & Lieber RL (2016). Three distinct cell populations express extracellular matrix proteins and increase in number during skeletal muscle fibrosis. Am J Physiol Cell Physiol DOI: 10.1152/ajpcell.00226.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MA, Pichika R & Lieber RL (2015). Collagen crosslinking does not dictate stiffness in a transgenic mouse model of skeletal muscle fibrosis. J Biomech 48, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chleboun GS, Patel TJ & Lieber RL (1997). Skeletal muscle architecture and fiber‐type distribution with the multiple bellies of the mouse extensor digitorum longus muscle. Acta Anat (Basel) 159, 147–155. [DOI] [PubMed] [Google Scholar]
- Cornu C, Goubel F & Fardeau M (1998). Stiffness of knee extensors in Duchenne muscular dystrophy. Muscle Nerve 21, 1772–1774. [DOI] [PubMed] [Google Scholar]
- Cullen MJ & Mastaglia FL (1980). Morphological changes in dystrophic muscle. Br Med Bull 36, 145–152. [DOI] [PubMed] [Google Scholar]
- Denk W & Horstmann H (2004). Serial block‐face scanning electron microscopy to reconstruct three‐dimensional tissue nanostructure. PLoS Biol 2, e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friden J & Lieber RL (2003). Spastic muscle cells are shorter and stiffer than normal cells. Muscle Nerve 27, 157–164. [DOI] [PubMed] [Google Scholar]
- Gartner LP & Hiatt JL (2009). Color Atlas of Histology. Wolters Kluwer Health/Lippincott William & Wilkins, Alphen aan den Rijn. [Google Scholar]
- Gillies AR, Bushong EA, Deerinck TJ, Ellisman MH & Lieber RL (2014). Three‐dimensional reconstruction of skeletal muscle extracellular matrix ultrastructure. Microsc Microanal 20, 1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies AR & Lieber RL (2011). Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 44, 318–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslett JN, Sanoudou D, Kho AT, Bennett RR, Greenberg SA, Kohane IS, Beggs AH & Kunkel LM (2002). Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc Natl Acad Sci USA 99, 15000–15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr Graham H & Selber P (2003). Musculoskeletal aspects of cerebral palsy. J Bone Joint Surg Br 85, 157–166. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN & McIntosh JR (1996). Computer visualization of three‐dimensional image data using IMOD. J Struct Biol 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Lieber RL, Runesson E, Einarsson F & Friden J (2003). Inferior mechanical properties of spastic muscle bundles due to hypertrophic but compromised extracellular matrix material. Muscle Nerve 28, 464–471. [DOI] [PubMed] [Google Scholar]
- Lieber RL, Yeh Y & Baskin RJ (1984). Sarcomere length determination using laser diffraction. Effect of beam and fiber diameter. Biophys J 45, 1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Mendez J & Keys A (1960). Density and composition of mammalian muscle. Metabolism 9, 184–188. [Google Scholar]
- Meyer GA & Lieber RL (2012). Skeletal muscle fibrosis develops in response to desmin deletion. Am J Physiol Cell Physiol 302, C1609–C1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner DJ, Weitzer G, Tran D, Bradley A & Capetanaki Y (1996). Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol 134, 1255–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passerieux E, Rossignol R, Chopard A, Carnino A, Marini JF, Letellier T & Delage JP (2006). Structural organization of the perimysium in bovine skeletal muscle: Junctional plates and associated intracellular subdomains. J Struct Biol 154, 206–216. [DOI] [PubMed] [Google Scholar]
- Pessina P, Kharraz Y, Jardi M, Fukada S, Serrano AL, Perdiguero E & Munoz‐Canoves P (2015). Fibrogenic cell plasticity blunts tissue regeneration and aggravates muscular dystrophy. Stem Cell Reports 4, 1046–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks RD & Roy RR (1982). Architecture of the hind limb muscles of cats: functional significance. J Morphol 173, 185–195. [DOI] [PubMed] [Google Scholar]
- Shah SB & Lieber RL (2003). Simultaneous imaging and functional assessment of cytoskeletal protein connections in passively loaded single muscle cells. J Histochem Cytochem 51, 19–29. [DOI] [PubMed] [Google Scholar]
- Smith LR, Lee KS, Ward SR, Chambers HG & Lieber RL (2011). Hamstring contractures in children with spastic cerebral palsy result from a stiffer extracellular matrix and increased in vivo sarcomere length. J Physiol 589, 2625–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LR, Ponten E, Hedstrom Y, Ward SR, Chambers HG, Subramaniam S & Lieber RL (2009). Novel transcriptional profile in wrist muscles from cerebral palsy patients. BMC Med Genomics 2, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezumi A, Ito T, Morikawa D, Shimizu N, Yoneda T, Segawa M, Yamaguchi M, Ogawa R, Matev MM, Miyagoe‐Suzuki Y, Takeda S, Tsujikawa K, Tsuchida K, Yamamoto H & Fukada S (2011). Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci 124, 3654–3664. [DOI] [PubMed] [Google Scholar]
- Weibel ER (1979). Stereological Methods, Vol. 1: Practical Methods for Biological Morphometry. Academic Press, London. [Google Scholar]
- Yata Y, Scanga A, Gillan A, Yang L, Reif S, Breindl M, Brenner DA & Rippe RA (2003). DNase I‐hypersensitive sites enhance α1(I) collagen gene expression in hepatic stellate cells. Hepatology 37, 267–276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Movie S1. Interaction of a collagen cable and fibroblast in aged des−/− muscle reconstructed from SBEM analysis.