

Abstract
DNA methylation is an essential epigenetic marker associated with the silencing of gene expression. Although various genome-wide studies revealed aberrantly methylated gene targets as molecular biomarkers for early detection, the survival rate of lung cancer patients is still poor. In order to identify methylation-driven biomarkers, genome-wide changes in DNA methylation and differential expression in 32 pairs of lung adenocarcinoma and adjacent normal lung tissue in non-smoking women were examined. This concurrent analysis identified 21 negatively correlated probes (r ≤ −0.5), corresponding to 17 genes. Examining the endogenous expression in lung cancer cell lines, five of the genes were found to be significantly down-regulated. Furthermore, in tumor cells alone, 5-aza-2′-deoxycytidine treatment increased the expression levels of STXBP6 in a dose dependent manner and pyrosequencing showed higher percentage of methylation in STXBP6 promoter. Functional analysis revealed that overexpressed STXBP6 in A549 and H1299 cells significantly decreased cell proliferation, colony formation, and migration, and increased apoptosis. Finally, significantly lower survival rates (P < 0.05) were observed when expression levels of STXBP6 were low. Our results provide a basis for the genetic etiology of lung adenocarcinoma by demonstrating the possible role of hypermethylation of STXBP6 in poor clinical outcomes in lung cancer patients.
Lung carcinoma is one of the most commonly diagnosed cancer types, and it is characterized by poor survival rates. According to recent global cancer statistics, it accounts for 18% of all cancer-related deaths worldwide1. Despite decades of research efforts to improve the clinical outcomes of lung cancer patients, the overall survival rates remain dismal. The mortality rates of lung cancer were the highest in Taiwan2,3.
A wide variety of risk factors, such as genetic, epigenetic, and environmental factors, may cause lung cancer. Nearly 70–90% of lung cancers in Western countries are caused due to cigarette smoking, but only 7% of female lung cancer cases are associated with smoking4,5,6. Furthermore, adenocarcinoma is much more common than other subtypes of non-small cell lung carcinoma seen in non-smokers. These statistics emphasize the necessity of a better understanding of the molecular mechanisms that mediate the development of lung cancer in non-smoking female patients.
Advanced high-throughput technologies have had an important role in identifying the genetic abnormalities that drive the development and growth of various cancers7,8. In addition to genetic changes, epigenetic changes, such as hyper- or hypomethylation, lead to the aberrant expression of tumor suppressor genes or oncogenes9. Hypermethylation within the promoter region was responsible for the inactivation of approximately half of the classical tumor suppressor genes10,11. Tumor suppressor genes undergoing aberrant hypermethylation were expressed in a non-random, tumor-specific pattern in many cancer types12. Therefore, epigenetically disrupted gene expression was able to alter various cancer-related processes, such as cell cycle checkpoints, cell proliferation, apoptosis, signal transduction, regulation of transcription factors, cell adhesion, and angiogenesis13,14. Also, various molecular genetics investigations revealed the impact of methylation on either resistance or sensitivity to chemotherapy or radiation15,16,17,18,19.
The possible role of DNA methylation in lung cancer was identified based on analysis of sputum20 and in prognosis of early-stage lung cancer21,22. Unlike other genetic alterations, methylation-based epigenetic modification is an inherently reversible change, due to which it has gained much attention as an active target of drug development. Therefore, over the past few decades, several research groups have been focused on finding the epigenetic markers (e.g., APC23 and SHOX224,25) or a group of gene set, such as (APC, RASSF1A, CDH13, KLK10 and DLEC1)26 and (AGTR1, GALR1, SLC5A8, ZMYND10 and NTSR1)27, for detection or diagnosis of lung cancer. However, the role of methylation in the tumorigenesis of lung adenocarcinoma and association with prognosis in Taiwan remains largely unknown. For this reason, we performed an integrated analysis of gene expression and DNA methylation status to find novel epigenetic markers of lung cancer. With this approach, we identified STXBP6, whose expression was significantly repressed by methylation, affected cellular function in cancer cell lines, and was associated with overall survival.
Syntaxin binding protein 6, encoded by STXBP6, was initially identified in regulating the formation of the SNARE complex28 and cytogenesis29. The regulatory role of STXBP6 in exocytosis and fusion pore stability was performed by both syntaxin-dependent and syntaxin-independent mechanisms30. It has been reported to be associated with many diseases, such as diabetes31, autism32,33, and systemic lupus erythematosus34. However, there are no studies revealing its biological role in association with lung cancer and its epigenetic regulation.
Therefore, in this study we explored the epigenetic inactivation of STXBP6 expression using lung adenocarcinoma patients. Pyrosequencing analysis using in vitro cellular models revealed the specific CpG sites that are responsible for the hypermethylation of STXBP6. Functional analysis revealed the tumor-suppressive role of STXBP6 in in vitro lung cancer cellular models. Finally, poor survival rates were observed in patients with low expression levels of STXBP6. Thus, methylation-driven, differentially expressed STXBP6 may be used as a novel biomarker to predict clinical outcomes of lung adenocarcinoma patients.
Results
Differential expression and methylation profiling in lung adenocarcinoma
In this genome-wide study, we sought to identify the genes whose expression was differentially regulated by DNA methylation in lung cancer cells. Genome-wide expression (41,789 probes) and DNA methylation profiling (27,578 probes) in 32 pairs of tumor and adjacent normal tissues were analyzed in non-smoking women with lung adenocarcinoma (Table S2). The average age of patients was 62 years old and 78% of them were in Stage I or II. To visualize the distribution of tumor and normal samples based on expression or methylation levels, principal component analyses was executed using differentially expressed probes of gene expression (Fig. 1A) and DNA methylation (Fig. 1B). Black dots denote tumor tissues, gray dots denote normal tissues, and each line indicates the paired samples from the same individual. Each dot represents the expression (Fig. 1A) or methylation values (Fig. 1B) of the significant probes that were summarized at the first two principal component coordinates. The results showed the distinct separation of tumor samples from their corresponding normal samples, indicating distinct patterns of gene expression and methylation levels in normal and tumor tissues.
Figure 1. Identification of differentially expressed genes driven by methylation in non-smoking women with lung adenocarcinoma.

(A) Principal component analysis of probes with differential gene expression in tumor and normal tissues. Differentially expressed probes were identified by paired t tests (P ≤ 10−6) and fold change (log2 ≥ 1 or ≤ −1) in tumor/normal lung tissues. Connecting lines indicate paired samples from the same individual. (B) Principal component analysis of probes with differential methylation status in tumor and normal tissues. The intensities of methylated probes versus unmethylated probes were converted to M values and examined by paired t tests (P ≤ 10−6). (C) Volcano plot of differentially expressed genes. Green dots indicate down-regulated probes (n = 901) and red dots denote up-regulated probes (n = 307). (D) Volcano plot of probes with differential DNA methylation status. Yellow dots denote hypomethylated probes (n = 863) and blue dots indicate hypermethylated probes (n = 894). (E) Heat map of the probes (n = 273) showing negative correlation (r < 0) between differential expression and methylation status. Hierarchical cluster analysis was performed using Pearson correlation distance. (F) Starburst plot of probes showing negative correlation between gene expression (y-axis) and DNA methylation (x-axis).
Differential expression of probes was filtered by fold change ( ≥ 2-fold) and statistical significance (P ≤ 10−6) in pairs of tumor and normal lung tissues. As shown in Fig. 1C, 901 probes were found to be down-regulated (log2 ≤ −1), whereas 307 probes were up-regulated (log2 ≥ 1). After the intensities of methylated probes were converted to M values and examined by paired-t tests (P ≤ 10−6), 863 probes were found to be hypomethylated, whereas 894 probes were hypermethylated (Fig. 1D). To correlate genome-wide methylation changes with concomitant changes in expression, we integrated the gene expression and DNA methylation probe pairs. In this analysis, we identified 273 negatively correlated probe pairs from 50,948 combined gene expression and methylation probe pairs, meaning that gene expression and methylation changed in opposite directions. Heat maps were used to represent the negative correlation (r < 0) between gene expression and methylation status for these 273 probe pairs (Fig. 1E). Hierarchical cluster analysis identified a distinct cluster for the up-regulated (red) and hypomethylated (yellow) genes. Similarly, down-regulated (green) genes with hypermethylation (blue) status were represented as another cluster. The results depicted in the heat map indicate a clear negative correlation between DNA methylation and genes expression profiles in tumor samples (Fig. 1E; Supplementary Dataset 1).
In order to screen candidate genes for validation, we then narrowed down the probes by increasing the stringency of negative correlation (r ≤ −0.5) between gene expression and DNA methylation (Fig. 1F). As shown in Table 1, 21 probes corresponding to 17 genes met this criterion. These results emphasize that abnormal DNA methylation might play a role in the regulation of 17 genes showing significant differential expression in lung adenocarcinoma.
Table 1. Genes with negative correlation between gene expression and methylation.
| Gene | Exp. Probe IDa | Methylation Probe IDb | CpG Target No. | Corr.c | Exp. Diff. (log2)d | P-value for Exp.e | Beta Diff. (T vs N) | Tumor Beta value | Normal Beta value | Methyla-tion Diff. (M value)f | P-value for Methyla-tiong |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AQP1 | 209047_at | 4600 | cg04551925 | −0.63 | −1.83 | 2.10E-08 | 0.16 | 0.73 | 0.57 | 1.03 | 1.52E-07 |
| HABP2 | 206010_at | 26621 | cg26656452 | −0.61 | 1.89 | 3.91E-07 | −0.23 | 0.58 | 0.81 | −1.76 | 1.33E-08 |
| SPDEF | 220192_x_at | 17272 | cg17240454 | −0.59 | 0.81 | 1.88E-08 | −0.19 | 0.55 | 0.74 | −1.12 | 2.64E-08 |
| EHF | 232361_s_at | 18421 | cg18414381 | −0.59 | 1.29 | 7.14E-07 | −0.17 | 0.28 | 0.45 | −0.93 | 1.52E-09 |
| HMGN1 | 200944_s_at | 18826 | cg18829411 | −0.57 | 0.58 | 1.60E-08 | −0.16 | 0.31 | 0.47 | −1.01 | 4.89E-09 |
| SLC22A18 | 204981_at | 16913 | cg16873863 | −0.57 | 1.07 | 1.79E-07 | −0.23 | 0.40 | 0.63 | −1.35 | 1.90E-09 |
| ARHGEF19 | 226857_at | 18670 | cg18669381 | −0.56 | 1.20 | 7.74E-09 | −0.11 | 0.29 | 0.40 | −0.69 | 2.32E-07 |
| IL11RA | 204773_at | 3676 | cg03662459 | −0.56 | −0.74 | 8.98E-08 | 0.09 | 0.68 | 0.59 | 0.59 | 2.00E-07 |
| GSTM5 | 205752_s_at | 5031 | cg04987894 | −0.55 | −0.69 | 1.57E-07 | 0.21 | 0.52 | 0.31 | 1.04 | 3.51E-11 |
| NIPSNAP1 | 201709_s_at | 13745 | cg13797031 | −0.55 | 0.70 | 2.14E-08 | −0.26 | 0.49 | 0.75 | −1.79 | 3.35E-09 |
| MESP1 | 224476_s_at | 1105 | cg01091565 | −0.54 | 1.24 | 9.77E-07 | −0.18 | 0.62 | 0.80 | −1.28 | 1.72E-08 |
| EHF | 232361_s_at | 13011 | cg13084525 | −0.54 | 1.29 | 7.14E-07 | −0.08 | 0.20 | 0.28 | −0.71 | 1.36E-08 |
| CP | 228143_at | 17465 | cg17439694 | −0.53 | 2.57 | 2.65E-07 | −0.13 | 0.54 | 0.67 | −0.91 | 2.26E-07 |
| SPDEF | 220192_x_at | 7679 | cg07705908 | −0.53 | 0.81 | 1.88E-08 | −0.21 | 0.36 | 0.57 | −1.13 | 1.90E-08 |
| HDHD3 | 221256_s_at | 23967 | cg24012708 | −0.53 | 0.59 | 3.26E-07 | −0.15 | 0.44 | 0.59 | −0.95 | 1.53E-07 |
| DCC | 238914_at | 4292 | cg04272086 | −0.52 | −0.98 | 2.90E-09 | 0.13 | 0.20 | 0.07 | 1.48 | 1.21E-10 |
| STXBP6 | 220994_s_at | 6937 | cg06948294 | −0.52 | −2.07 | 8.79E-12 | 0.11 | 0.15 | 0.04 | 1.62 | 1.68E-08 |
| COX7A1 | 204570_at | 24277 | cg24335895 | −0.52 | −1.27 | 7.66E-10 | 0.22 | 0.52 | 0.30 | 1.44 | 2.71E-11 |
| SPDEF | 213441_x_at | 17272 | cg17240454 | −0.52 | 0.53 | 5.98E-08 | −0.19 | 0.55 | 0.74 | −1.12 | 2.64E-08 |
| RHOJ | 243481_at | 18774 | cg18771300 | −0.52 | −1.25 | 1.19E-10 | 0.21 | 0.52 | 0.31 | 1.05 | 1.20E-07 |
| CP | 1558034_s_at | 17465 | cg17439694 | −0.51 | 2.52 | 3.28E-09 | −0.13 | 0.54 | 0.67 | −0.91 | 2.26E-07 |
| PECAM1 | 208981_at | 3928 | cg03886110 | −0.50 | −1.49 | 1.19E-10 | 0.20 | 0.72 | 0.52 | 1.07 | 2.39E-11 |
aProbes used for the gene expression profiling.
bProbes used for the methylation analysis.
cCorrelation between gene expression and methylation status.
dDifferential median values of gene in tumor/normal tissue samples (log2 ratio).
eStatistical significance of gene expression by paired t test.
fDifferential median values of methylated probes versus unmethylated probes (M value).
gStatistical significance of methylation examined by paired-t test.
Identification of methylation-driven down-regulated genes in lung cancer cell lines
To validate differentially expressed genes driven by methylation in non-smoking women with lung adenocarcinoma and select candidate genes for functional analysis, endogenous expression levels of the 17 genes were examined in lung cancer cell lines (A549 and H1299). Five genes, including IL11RA, GSTM5, STXBP6, RHOJ, and PECAM1, were significantly down-regulated (P ≤ 0.0001) in A549 and H1299 cells as compared to normal BEAS-2B cells (Figs 2A and S1A–D).
Figure 2. Validation of STXBP6 regulated by methylation in lung cancer cells.

(A) Endogenous levels of STXBP6 in lung cancer cells (A549 and H1299) and BEAS-2B normal control cells. The mRNAs were measured by quantitative RT-PCR. GAPDH was the internal control. Relative expression levels were normalized against BEAS-2B cells. Bars represent the means ± SDs of 3 independent experiments. (B) Relative expression of STXBP6 in cells treated with 5-aza-2-deoxycytidine (5-aza). Cells were treated with 5 and 10 μM of methylation inhibitor 5-aza for 3 days. Relative expression levels of STXBP6 were normalized against the untreated group of each cell line. (C) Scheme of CpG sites in the 5′-untranslated region of STXBP6 for pyrosequencing. Top: CpG sites for pyrosequencing were highlighted in light blue area. Bottom: CpG sites were numbered as shown in parentheses according to genome assembly version (GRCh37/hg19). (D) Quantification of DNA methylation of STXBP6 using pyrosequencing analysis. Genomic DNA was extracted from A549 and BEAS-2B cells and treated with bisulfide to examine the methylation percentage of CpG sites by pyrosequencing. **P ≤ 0.001, ***P ≤ 0.0001.
To validate the role of methylation in the regulation of the expression of these 5 genes, we treated A549, H1299, and BEAS-2B cell lines with 5-aza. Interestingly, only STXBP6, which expression was significantly up-regulated in a dose-dependent manner, was found when the A549 and H1299 cell lines were treated with 5-aza (Fig. 2B). In contrast, non-significant differences were observed in BEAS-2B normal cells (Fig. 2B). Furthermore, pyrosequencing analysis was performed to identify the specific CpG sites of methylation in STXBP6. Five CpG sites in the 5′ untranslated region of STXBP6 were examined (Fig. 2C). Four of the five CpG sites (the CpG site at Chr14:25518748 was undetected) showed a significantly higher methylation percentage in A549 cells compared to normal BEAS-2B cells (Fig. 2D). Lastly, as shown in Table 2, the expression levels of STXBP6 were significantly down-regulated in adenocarcinoma samples in eight data set. Also, the methylation levels of STXBP6 were significantly hypermethylated in six data set (Table 2). While datasets containing both expression and methylation data, the converse relationship between methylation and gene expression of STXBP6 was not only observed in our data set (GSE19804/GSE49996) but also in three publicly available data set (Table 2; Supplementary Dataset 2). These findings suggested a possible role of methylation in down-regulating the expression of STXBP6 in lung cancer cells.
Table 2. Expression values and methylation status of STXBP6 in lung adenocarcinoma among public data set.
| Data Type | GSE No. (reference) | Sample Size | Correlationa | Expression Difference (log2)b | P-value for Expressionc | Methylation Difference (M value)d | P-value for Methylatione |
|---|---|---|---|---|---|---|---|
| Expression | |||||||
| GSE1918847 | 110 | −4.22** | 3.72E-20 | ||||
| GSE767049 | 56 | −1.37** | 1.40E-13 | ||||
| GSE3121048 | 246 | −3.77** | 2.47E-12 | ||||
| GSE1007250 | 107 | −1.52** | 1.06E-26 | ||||
| Methylation | |||||||
| GSE19034 | 60 | 2.24* | 2.48E-03 | ||||
| GSE3286644 | 50 | 2.04** | 3.58E-08 | ||||
| Expression & Methylation | |||||||
| GSE19804/GSE49996 | 120/64 | −0.52 | −4.38** | 9.57E-22 | 1.73** | 8.61E-08 | |
| GSE62949/GSE62948 | 56/56 | −0.60 | −0.24** | 3.84E-11 | 1.08** | 5.98E-08 | |
| GSE32863/GSE32861(44) | 114/118 | −0.68 | −1.83** | 9.29E-22 | 2.54** | 2.49E-18 | |
| GSE63459/GSE6338451 | 62/70 | −0.53 | −1.79* | 4.20E-02 | 1.34** | 2.21E-08 | |
aCorrelation between gene expression and methylation status.
bDifferential median values of gene in tumor/normal tissue samples (log2 ratio).
cStatistical significance of gene expression by t test or paired-t test.
dDifferential median values of methylated probes versus unmethylated probes (M value).
eStatistical significance of methylation examined by paired-t test.
*P < 1.00E-2; **P < 1.00E-07.
Functional investigation of STXBP6 in lung cancer cell lines
Since STXBP6 was epigenetically down-regulated in lung cancer cells, we investigated the functional roles of STXBP6 by transiently transfecting a STXBP6 expression plasmid into A549 and H1299 cells. As shown in Fig. 3A, the mRNA levels of STXBP6 in A549 and H1299 cells after transfection were significantly increased (P ≤ 0.0001). Western blot analysis also validated the increased protein amounts of STXBP6 in both A549 and H1299 cells upon transfection of STXBP6 plasmid (Fig. 3B).
Figure 3. STXBP6 suppresses cell proliferation and colony formation in lung cancer cells.

(A) Relative expression levels of STXBP6 in A549 and H1299 cells overexpressing STXBP6 by quantitative RT-PCR. GAPDH was the internal control. Bars represent the means ± SDs of 3 independent experiments. (B) Western blot analysis of STXBP6 in A549 and H1299 cells overexpressing STXBP6. GAPDH was used as the loading control. (C) MTT assays of A549 cells overexpressing STXBP6. Proliferation assays were executed by adding MTT at different time points after seeding 4,000 cells/well. (D) MTT assays of H1299 cells overexpressing STXBP6. (E) Colony formation assays of A549 cells overexpressing STXBP6. A representative image is shown above and a quantitative graph below. (F) Colony formation assays of H1299 cells overexpressing STXBP6. A representative image is shown above and a quantitative graph below. *P < 0. 05, **P < 0. 001, ***P < 0.0001.
After successfully overexpressing STXBP6 in lung cancer cells, we first examined the effect of STXBP6 on cell growth by MTT assays. The results showed a significant decrement in proliferation in both A549 and H1299 cells overexpressing STXBP6 (P ≤ 0.05) (Fig. 3C,D). Furthermore, STXBP6 overexpression markedly reduced colony formation in both A549 and H1299 cells (Fig. 3E,F). Next, the role of STXBP6 in migration of lung cancer cell lines was investigated by transwell migration assays. The results revealed that STXBP6 significantly suppressed the migration abilities of both A549 and H1299 cells (P ≤ 0.001) (Fig. 4A–D).
Figure 4. STXBP6 decreases cell migration in lung cancer cells.

(A) Representative image of the bottom surface of a transwell migration assay in A549 cells. (B) Quantification of migrated cells in (A). (C) Representative image of the bottom surface of a transwell migration assay in H1299 cells. (D) Quantification of migrated cells in (C). The absorbance values of migratory cells were normalized against a mock transfection group. Bars represent the means ± SDs of 3 independent experiments. **P < 0.001.
To evaluate the possible significance of STXBP6 expression in the modulation of apoptosis, annexin V-FITC and PI staining were carried out. The results showed a noticeable increment in apoptotic percentage in both A549 and H1299 cells overexpressing STXBP6 (P ≤ 0.05) (Fig. 5A–D). Furthermore, cell cycle analysis was performed on day 3 of transfection with the STXBP6 plasmid. The percentage of apoptotic cells in G1 phase was significantly increased in both A549 and H1299 cells overexpressing STXBP6 (P ≤ 0.001) (Fig. 5E–H). These results demonstrated the suppressive effects of STXBP6 on lung carcinogenesis.
Figure 5. STXBP6 increases apoptosis in lung cancer cells.

(A) A representative diagram of annexin V-FITC and propidium iodide (PI) staining assays in A549 cells overexpressing STXBP6. (B) Quantification of annexin V results in (A). The percentage of apoptotic cells was derived from the sum of the percentages of late and early apoptotic cells. (C) A representative diagram of annexin V-FITC and PI staining assays in H1299 cells overexpressing STXBP6. (D) Quantification of annexin V-FITC results in (C). (E) Flow cytometry analysis for apoptosis in A549 cells overexpressing STXBP6. (F) Quantification of apoptotic cells in (E). The percentage of cells in sub-G1 phase was used to identify the apoptotic cells by PI staining. Bars represent the means ± SDs of 3 independent experiments. (G) Flow cytometry analysis for apoptosis in H1299 cells overexpressing STXBP6. (H) Quantification of apoptotic cells in (G). *P < 0. 05, **P < 0. 001.
Expression levels of STXBP6 in tumor tissues correlate with patients’ overall survival
Lastly, Kaplan–Meier analysis was used to examine overall survival in relation to the expression values of STXBP6 in adenocarcinoma patients from Shedden’s study35 (Fig. 6A) and Tomida’s study36 (Fig. 6B). Patients were divided into “high expression” or “low expression” groups based on the median value of all samples. The results showed that patients with lower expression of STXBP6 had poorer survival than those with high expression (Fig. 6A,B). These findings indicate that epigenetic changes in STXBP6 may be useful for predicting the prognosis of patients with lung adenocarcinoma.
Figure 6. Down-regulation of STXBP6 in lung cancer tissues was associated with poor overall survival.

Patients were divided into “high expression” or “low expression” groups based on the median value of all samples. Kaplan–Meier analysis was used to examine overall survival in relation to the expression values of STXBP6 in adenocarcinoma patients from (A) Shedden’s study35 and (B) Tomida’s study36. P-values were calculated by a log-rank test.
Discussion
In this study, we sought to uncover epigenetic-based molecular targets by analyzing the association between genome-wide DNA methylation and gene expression patterns in tumor and adjacent normal tissues from non-smoking Taiwanese female lung adenocarcinoma patients. First, differential expression levels and methylation status between tumor and normal tissues were identified using both gene expression and methylation microarrays. Second, selected genes with negative correlations between expression levels and methylation status were then validated by examining their endogenous levels in lung cancer cell lines. Third, pyrosequencing and 5-aza-2′-deoxycytidine treatment showed the regulatory role of methylation of STXBP6 in tumor cells. Fourth, functional analysis revealed that STXBP6 suppressed tumor growth in lung cancer cell lines. Finally, lower expression of STXBP6 was found to be associated with poor clinical outcomes in lung cancer patients.
Because of the development of targeted therapy resistance or the absence of targetable mutations in lung cancer patients, developing alternative therapeutic strategies for lung cancer in early diagnosis, prognosis prediction, and treatment are urgent and important. Epigenetics approaches, including DNA methylation, histone modification, and miRNA regulation, may solve these problems by affecting multiple pathways that regulate major properties of the cancer cells. Among them, DNA methylation at CpG sites is the most characterized epigenetic modification described in lung cancer. Therefore, targeting DNA methylation of tumor suppressor genes or oncogenes may hold promise in lung cancer therapy.
For the last several years, more and more evidence has accumulated to emphasize the hypermethylation status of CpG islands located in the promoter regions of tumor suppressor genes37. Several groups have performed epigenetic analyses of methylation in types of cancer other than lung adenocarcinoma. A recent genome-wide analysis of DNA methylation and gene expression changes in lung squamous cell carcinoma identified several methylation-driven genes, including CCDC37, CYTL1, CDO1, SLIT2, LMO3 and SERPINB538. Another genomic analysis of idiopathic pulmonary fibrosis identified methylation–gene expression relationships within genes that were either involved in fibroproliferation or were feasible candidates in this process39. Suzuki et al. performed an integrative multi-omics analysis to understand how cancers harbor various types of aberrations at the genomic, epigenomic, and transcriptional levels40. Additional investigations using larger numbers of samples with varied clinical features can help to reveal novel gene targets associated with methylation changes in tumorigenesis. Further research direction may also include the timing of methylation and the difference in methylation levels between epithelial and stromal tissues. In addition, more studies should focus on finding markers for epigenetic priming agents that render lung cancer more susceptible to cytotoxic chemotherapy and immunotherapy.
Worsening lung cancer statistics (e.g., increasing global incidence), particularly in women41,42, invoke researchers to develop accurate and highly sensitive markers for the early detection of disease. Several biomarkers for the diagnosis of lung cancer have been identified43; however, sensitivities of these biomarkers differ for each subtype of lung cancer. Thus, it is also highly challenging to find specific biomarkers for each subtype, as the various lung cancers are known to have diverse pathological features. Hence, we set out to find suitable biomarker genes for adenocarcinoma, to distinguish it from normal samples. To meet this expectation, we processed our data using a stringent cutoff for the negative correlation between gene expression and DNA methylation in non-smoking women with lung adenocarcinoma. From the genome-wide analysis, 167 methylation-driven, differentially expressed genes (273 probes) were identified in lung adenocarcinoma. In spite of differences in clinical features of tumor samples and selection criteria, previous genome-wide methylation studies showed results similar to our study44,45. For instance, our hierarchical clustering analysis also resulted in a large cluster for most of the probes, which are hypermethylated and down-regulated. The current findings along with previous findings indicate that a larger number of genes may undergo hypermethylation in the case of lung carcinogenesis.
Interestingly, differential expression of some of the candidate genes in our study was in agreement with previous studies. For example, CDKN3 was found to be overexpressed in hepatocellular carcinoma and to promote cell proliferation by affecting cell cycle progression46, and was also found be overexpressed in this study. To the best of our knowledge, this is the first study reporting the regulatory role of methylation in the control of STXBP6 in lung cancer with different validation approaches, including microarrays and 5-aza and pyrosequencing analyses. Moreover, STXBP6 was significantly down-regulated or hypermethylated in many public data set (Table 2)44,47,48,49,50. Furthermore, negative correlation between gene expression and methylation status for STXBP6 was observed in other three publicly available datasets (Table 2)44,51, indicating the possibility of epigenetic inactivation of STXBP6 even in different races. The possible role of methylation in the control of STXBP6 expression was also suggested by a human cell model of breast cancer52,53. These results indicate that epigenetic silencing of STXBP6 could occur in different cancer types.
Administration of methylation inhibitors, such as 5-aza, is one of the most commonly used strategies to uncover the role of aberrant methylation changes in gene inactivation54,55. When cells were treated with 5-aza, up-regulation of STXBP6 was observed only in cancerous cell lines. Pyrosequencing results further identified the methylated CpG sites modulating the expression of STXBP6 in these cell lines.
To investigate the functional roles of STXBP6, it was overexpressed in A549 and H1299 cancer cells. Ectopic expression of STXBP6 resulted in slower cell proliferation, less colony formation, slower migration ability, and a greater percentage of apoptosis in lung cancer cells. These results suggested that STXBP6 could function as a tumor suppressor, although further experiments are warranted using in vivo studies.
Lastly, survival analysis using two publicly available datasets35,36 indicated that the survival probability of lung cancer patients increased with higher expression of STXBP6 in tumor tissues. This result suggested an avenue for developing a novel therapeutic regimen for treating lung cancer.
In conclusion, our results indicate that the pathogenesis of lung adenocarcinoma may result from epigenetically regulated expression levels of STXBP6. Before this biomarker can be translated into clinical utility, further studies using larger sample sizes will help to reveal the importance of STXBP6 as novel potential biomarker for the prognosis of lung adenocarcinoma56. Multicenter studies are also needed to validate the tests and analyze the reproducibility of promising results derived from limited samples.
Methods
Clinical tissue samples
Thirty-two pairs of lung adenocarcinoma and adjacent normal lung tissue samples were acquired from non-smoking female patients admitted to National Taiwan University Hospital or Taichung Veterans General Hospital. Written informed consent was obtained from all subjects and/or guardians for the use of their tissue samples. Acquisition and subsequent use of all the clinical samples were in accordance with the Declaration of Helsinki, and were approved by the Institutional Review Board of National Taiwan University Hospital (IRB approval Number: 200610015 R) and the Institutional Review Board of Taichung Veterans General Hospital (IRB approval Number: C09204). Lung tissue samples were quickly immersed in RNAlater® solution (Life Technologies, Gaithersburg, MD, USA), snap-frozen in liquid nitrogen, and stored at −80 °C. Before the extraction of RNA and DNA, frozen tumor tissue blocks were sectioned.
RNA extraction and cDNA synthesis
Total RNA from sectioned tissue samples was isolated using TRIzol reagent (Life Technologies, Gaithersburg, MD, USA) and purified with the RNeasy mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA integrity was confirmed by agarose gel electrophoresis and the Agilent 2100 Bioanalyzer RNA 6000 LabChip kit (Agilent Technologies, Santa Clara, CA, USA). The purified total RNAs were then used as templates to synthesize the labeled double-stranded cDNA and cRNA according to the Affymetrix standard synthesis protocols.
Genomic DNA isolation, bisulfite treatment, and methylation profiling
Genomic DNA from tumor and adjacent normal tissue samples was extracted using TRIzol reagent (Life Technologies, Gaithersburg, MD, USA). The DNA was then subjected to bisulfite conversion using an EZ DNA methylation kit (Zymo Research, Orange, CA, USA). In the bisulfite reaction, the samples were cycled 16 times for 30 sec at 95 °C and 1 h at 50 °C. Then, bisulfite-converted DNA was used for methylation microarrays.
Gene expression and methylation profiling
mRNA expression profiling was performed by Human Genome U133 plus 2.0 arrays (Affymetrix, Inc., Santa Clara, CA, USA) based on reverse transcription and probe hybridization. This platform contains 41,789 probes. Gene expression levels were detected by relative fluorescence intensity. The expression array data of this study have been submitted to the Gene Expression Omnibus database (accession number GSE19804).
To identify the DNA methylation status of 27,578 CpG sites, the Illumina Infinium Human Methylation27 beadchip (Illumina, San Diego, CA, USA) was used. The accession number for the methylation array data set in the Gene Expression Omnibus database is GSE49996. The methylation levels (beta values) of a given gene were determined by ratio of the methylated probe intensity to the overall probe intensity of that gene. Methylation beta values were then converted to an M-value through a logistic transformation and expressed as the log2 ratio of the intensities of methylated probe versus unmethylated probe. The M-value for the ith interrogated CpG site is defined as:
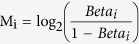 |
Furthermore, two-dimensional principal component analyses were used for a visual representation of differential expression patterns between tumor and normal samples. Next, hierarchical cluster analysis using Pearson correlation distances was executed to group the probes with similar expression and methylation profiles. Then, differentially expressed and methylated probes were analyzed by paired t tests (P ≤ 10−6). Genes with differential expression between tumor and adjacent normal tissues were further filtered by at least 2-fold changes. A negative correlation coefficient (r < 0) was used to identify a converse relationship between gene expression and methylation status. A stringent correlation coefficient, defined as r ≤ −0.5, was used for selecting genes for further validation in in vitro cell models.
Cell culture
Cancerous lung cells (A549 and H1299) and normal lung cells (BEAS-2B) were cultured in RPMI medium 1640 (GIBCO, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS; Biological Industries, Beit-Haemek, Israel) and 1% antibiotics, including puromycin and streptomycin (Biological Industries, Beit-Haemek, Israel), at 37 °C in a humidified atmosphere with 5% CO2. To validate the role of methylation in regulating the expression of target genes, A549, H1299, and BEAS-2B cells were seeded on a 6-well plate. After 24 h, the cell lines were treated with 5 and 10 μM of 5-aza-2′-deoxycytidine (5-aza) (Sigma Chemical Company, St. Louis, MO, USA). Expression values of target genes were analyzed 3 days after treatment.
Quantitative reverse transcription PCR
The quality and quantity of the RNA were measured by NanoDrop™ 2000 (Thermo Scientific™, USA). One μg of total RNA from each cell line was reverse transcribed by the High Capacity cDNA Reverse Transcription Kit (Life Technologies, Gaithersburg, MD, USA). The final cDNA products were used as the templates for subsequent real time PCR (RT-PCR). RT-PCR was performed with SYBR Green (Roche, Germany) on an ABI 7900 system (Life Technologies, Gaithersburg, MD, USA) according to standard protocols. The primers used in this study are listed in Table S1. All individual experiments were carried out in triplicate, and data were normalized using GAPDH as the loading control. The statistical significance of gene expression in different samples was identified by the t-test calculator in GraphPad Prism 5 (GraphPad Software, Inc., CA, USA).
Pyrosequencing assay
To quantify the methylation levels of multiple CpG sites in the 5′ untranslated region of STXBP6, a predesigned PyroMark CpG Assay was used (Hs_STXBP6_01_PM PyroMark CpG assay, PM00057414, Qiagen, Venlo, Netherlands). Specific CpG sites including Chr14: 25518720, 25518735, 25518737, 25518743, and 25518748 were examined.
Overexpression of STXBP6 in lung cancer cells
STXBP6 was overexpressed in A549 and H1299 cells to evaluate its functional significance. Full-length STXBP6 cDNA with a C-terminal Myc-DDK tag was inserted into the pCMV6-Entry mammalian vector (OriGene Technologies, Rockville, MD, USA). The pCMV6-Entry-Myc-STXBP6 vector and an empty vector were transiently transfected into A549 and H1299 cell lines using TransIT-2020 transfection reagent (MirusBio, Madison, WI, USA) according to the manufacturer’s instruction. All sequences, including STXBP6 in the vector, were verified by Sanger sequencing (the first core laboratory, College of Medicine, National Taiwan University). mRNA levels were quantified by quantitative RT-PCR using STXBP6-specific primers (F-5′-GTCTATACTTACTGCCAGCG-3′ and R-5′-GTTAAATGCCTTGATGGCCTC-3′), and protein levels were examined by western blotting.
Western blot
Total cell lysates were prepared and proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins in the gel were then electrotransferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked with 5% milk and were incubated with monoclonal anti-FLAG antibody (Sigma-Aldrich, St. Louis, MO, USA) or anti-GAPDH antibody (Sigma-Aldrich, St. Louis, MO, USA) overnight. After washing, the bound primary antibodies on the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit IgG or rabbit anti-mouse IgG (GeneTex, Irvine, CA, USA). Finally, the blots were developed with a chemiluminescent western blotting system (Millipore, Billerica, MA, USA).
Cell proliferation assay
A549 and H1299 cells were seeded into 96-well plates in triplicate and incubated for 12 h at 37 °C in a CO2 incubator. Next, all cells in 96-well plates were divided into groups and transfected with STXBP6 plasmid or mock vectors. At different time points (24, 48, and 72 h) of transfection, proliferative activity was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (EMD Biosciences, La Jolla, CA, USA) assay using a microtiter plate reader (BioTek, Winooski, VT, USA) at 570 nm. The absorbance of A549 and H1299 cells was measured.
Colony formation assay
Cells were seeded in 6-well plates and incubated overnight. The adherent cells were transfected with STXBP6 plasmid or mock vector. After two weeks of incubation, cells were fixed using 3:1 methanol-acetic acid and stained using 0.1% crystal violet. Finally, the dried plates were used for image acquisition with a digital camera.
Cell migration assay
Migration assays were carried out using 24-well transwell units (Corning, NY, USA). The upper chamber of each transwell unit was loaded with 4 × 104 cells/well in 0.2 mL serum-free RPMI medium and the lower chambers contained 0.6 mL of RPMI with 10% FBS as chemoattractant. Cells were then incubated for 24 h at 37 °C. Then a methanol-acetic acid (3:1) mixture was added into the lower chambers to fix the cells for 20 min at room temperature, followed by staining with 0.1% crystal violet for another 20 min. Cells on the upper side of the membrane surface were removed by scraping with a cotton swab, and the cells that passed through the filter were destained using 10% acetic acid. The absorbance was measured at 570 nm with an ELISA reader (BioTek, Winooski, VT, USA). Images of the bottom surface of the transwell migration chambers were captured at 10X magnification before destaining.
Apoptosis assay
In order to perform the annexin V-FITC and propidium iodide (PI) double staining assay, cells were trypsinized, washed with phosphate-buffered saline (PBS), and resuspended in 500 μL of 1X binding buffer (Becton Dickinson, NJ, USA). Thereafter, cells were stained using 10 μL of Annexin V (5 μL) and PI (5 μL) mix (Becton Dickinson, NJ, USA) for 15 min. The suspension was passed through a nylon mesh filter and analyzed using a Beckman Coulter FC500 (Beckman, Brea, CA, USA) and CXP analysis software.
Cell cycle analysis
Initially, cells were trypsinized, washed with PBS, and fixed with cold 100% ethanol at −20 °C overnight. Thereafter, cells were washed twice and resuspended in PBS containing 20 μg/mL PI (Life Technologies, NY, USA), 0.1% triton-X-100 (Sigma, St. Louis, MO, United States), and 100 μg/mL RNase A (Sigma, St. Louis, MO, United States) for 30 min. The suspension was passed through a nylon mesh filter and analyzed using a Beckman Coulter FC500 (Beckman, Brea, CA, USA) and CXP analysis software.
Survival analysis
The gene expression signatures from GSE6846535 and GSE1321336 were used to elucidate the prognostic roles of STXBP6 in lung adenocarcinoma patients. Patients were categorized as “STXBP6 High” if their RNA expression levels of STXBP6 were higher than the median expression in all samples, and as “STXBP6 Low” if their RNA expression levels of STXBP6 were lower than the median expression in all samples. The association between gene expression and overall survival (up to 100 months) of lung adenocarcinoma patients was examined using Kaplan-Meier survival analysis. The statistical significance of the relationship between gene expression and survival was examined by a log-rank test.
Statistical analysis
Data were expressed as the means ± SDs from at least three independent experiments. The statistical significance of gene expression in different samples was identified by a t-test calculator in GraphPad Prism 5 (GraphPad Software, Inc., CA, USA). P-values less than 0.05 were considered significant.
Additional Information
How to cite this article: Lenka, G. et al. Identification of Methylation-Driven, Differentially Expressed STXBP6 as a Novel Biomarker in Lung Adenocarcinoma. Sci. Rep. 7, 42573; doi: 10.1038/srep42573 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This research was supported by a grant from the Ministry of Science and Technology (MOST 103-2320-B-002-065-MY3), and also by another grant from the Department of Health (DOH96-TD-G-111-014). We thank Melissa Stauffer for editorial assistance and Tsai-Chun Lai and Wei-Ting Kuo for technical help and timely discussions during data analysis.
Footnotes
The authors declare no competing financial interests.
Author Contributions G.L., M.-H.T., L.-C.L. and E.Y.C. conceived and designed the experiments. G.L. and H.-C.L. performed the experiments. G.L., J.-H.H., Y.-C.L. and T.-P.L. analyzed the data. M.-H.T., E.Y.C., J.-M.L., C.-P.H. and L.-C.L. contributed reagents, materials and/or analysis tools. G.L., H.-C.L. and L.-C.L. wrote the paper. All authors reviewed the manuscript.
References
- Jemal A. et al. Global cancer statistics. CA: a cancer journal for clinicians 61, 69–90, doi: 10.3322/caac.20107 (2011). [DOI] [PubMed] [Google Scholar]
- Chen C. J. et al. Epidemiologic characteristics and multiple risk factors of lung cancer in Taiwan. Anticancer research 10, 971–976 (1990). [PubMed] [Google Scholar]
- Yang S. P., Luh K. T., Kuo S. H. & Lin C. C. Chronological observation of epidemiological characteristics of lung cancer in Taiwan with etiological consideration—a 30-year consecutive study. Japanese journal of clinical oncology 14, 7–19 (1984). [PubMed] [Google Scholar]
- Wood M. E., Kelly K., Mullineaux L. G. & Bunn P. A. Jr. The inherited nature of lung cancer: a pilot study. Lung cancer (Amsterdam, Netherlands) 30, 135–144 (2000). [DOI] [PubMed] [Google Scholar]
- Ger L. P., Liou S. H., Shen C. Y., Kao S. J. & Chen K. T. Risk factors of lung cancer. Journal of the Formosan Medical Association = Taiwan yi zhi 91 Suppl 3, S222–231 (1992). [PubMed] [Google Scholar]
- Hirayasu T. et al. Human papillomavirus DNA in squamous cell carcinoma of the lung. Journal of clinical pathology 49, 810–817 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., So A. Y.-L. & Fan J.-B. Analysis of cancer genomes through microarrays and next-generation sequencing. Translational Cancer Research 4, 212–218 (2015). [Google Scholar]
- Peng L. et al. Large-scale RNA-seq transcriptome analysis of 4043 cancers and 548 normal tissue controls across 12 TCGA cancer types. Scientific reports 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J. et al. Detection and quantification of methylation in DNA using solid-state nanopores. Scientific reports 3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A. P. & Tycko B. The history of cancer epigenetics. Nature reviews. Cancer 4, 143–153, doi: 10.1038/nrc1279 (2004). [DOI] [PubMed] [Google Scholar]
- Jones P. A. & Baylin S. B. The fundamental role of epigenetic events in cancer. Nature reviews. Genetics 3, 415–428, doi: 10.1038/nrg816 (2002). [DOI] [PubMed] [Google Scholar]
- Costello J. F. et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nature genetics 24, 132–138, doi: 10.1038/72785 (2000). [DOI] [PubMed] [Google Scholar]
- Baylin S. B. et al. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Human molecular genetics 10, 687–692 (2001). [DOI] [PubMed] [Google Scholar]
- Lenka G., Tsai M.-H., Hsiao J.-H., Lai L.-C. & Chuang E. Y. Overexpression of methylation-driven DCC suppresses proliferation of lung cancer cells. Translational Cancer Research 5, 169–175, doi: 10.21037/tcr.2016.04.08 (2016). [DOI] [Google Scholar]
- Noro R. et al. PTEN inactivation in lung cancer cells and the effect of its recovery on treatment with epidermal growth factor receptor tyrosine kinase inhibitors. International journal of oncology 31, 1157–1163 (2007). [PubMed] [Google Scholar]
- Hegi M. E., Sciuscio D., Murat A., Levivier M. & Stupp R. Epigenetic deregulation of DNA repair and its potential for therapy. Clinical cancer research: an official journal of the American Association for Cancer Research 15, 5026–5031, doi: 10.1158/1078-0432.ccr-08-1169 (2009). [DOI] [PubMed] [Google Scholar]
- Ibanez de Caceres I. et al. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene 29, 1681–1690, doi: 10.1038/onc.2009.454 (2010). [DOI] [PubMed] [Google Scholar]
- Seng T. J. et al. DLEC1 and MLH1 promoter methylation are associated with poor prognosis in non-small cell lung carcinoma. British journal of cancer 99, 375–382, doi: 10.1038/sj.bjc.6604452 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T. et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nature medicine 9, 568–574, doi: 10.1038/nm852 (2003). [DOI] [PubMed] [Google Scholar]
- Leng S. et al. Defining a gene promoter methylation signature in sputum for lung cancer risk assessment. Clinical cancer research: an official journal of the American Association for Cancer Research 18, 3387–3395, doi: 10.1158/1078-0432.ccr-11-3049 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock M. V. et al. DNA methylation markers and early recurrence in stage I lung cancer. The New England journal of medicine 358, 1118–1128, doi: 10.1056/NEJMoa0706550 (2008). [DOI] [PubMed] [Google Scholar]
- Sandoval J. et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 31, 4140–4147, doi: 10.1200/jco.2012.48.5516 (2013). [DOI] [PubMed] [Google Scholar]
- Guo S. et al. Quantitative assessment of the diagnostic role of APC promoter methylation in non-small cell lung cancer. Clin Epigenetics 6, 5, doi: 10.1186/1868-7083-6-5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneip C. et al. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer 6, 1632–1638, doi: 10.1097/JTO.0b013e318220ef9a (2011). [DOI] [PubMed] [Google Scholar]
- Dietrich D. et al. Performance evaluation of the DNA methylation biomarker SHOX2 for the aid in diagnosis of lung cancer based on the analysis of bronchial aspirates. International journal of oncology 40, 825–832, doi: 10.3892/ijo.2011.1264 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. Methylation of multiple genes as a candidate biomarker in non-small cell lung cancer. Cancer Lett 303, 21–28, doi: 10.1016/j.canlet.2010.12.011 (2011). [DOI] [PubMed] [Google Scholar]
- Guo S. et al. Identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC). Clin Epigenetics 7, 3, doi: 10.1186/s13148-014-0035-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales S. J., Hesser B. A., Masuda E. S. & Scheller R. H. Amisyn, a novel syntaxin-binding protein that may regulate SNARE complex assembly. The Journal of biological chemistry 277, 28271–28279, doi: 10.1074/jbc.M204929200 (2002). [DOI] [PubMed] [Google Scholar]
- Kleppe L. et al. Cortisol treatment of prespawning female cod affects cytogenesis related factors in eggs and embryos. General and comparative endocrinology 189, 84–95, doi: 10.1016/j.ygcen.2013.04.028 (2013). [DOI] [PubMed] [Google Scholar]
- Constable J. R., Graham M. E., Morgan A. & Burgoyne R. D. Amisyn regulates exocytosis and fusion pore stability by both syntaxin-dependent and syntaxin-independent mechanisms. The Journal of biological chemistry 280, 31615–31623, doi: 10.1074/jbc.M505858200 (2005). [DOI] [PubMed] [Google Scholar]
- Collins S. C. et al. Increased expression of the diabetes gene SOX4 reduces insulin secretion by impaired fusion pore expansion. Diabetes, doi: 10.2337/db15-1489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castermans D. et al. The neurobeachin gene is disrupted by a translocation in a patient with idiopathic autism. Journal of medical genetics 40, 352–356 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castermans D. et al. SCAMP5, NBEA and AMISYN: three candidate genes for autism involved in secretion of large dense-core vesicles. Human molecular genetics 19, 1368–1378, doi: 10.1093/hmg/ddq013 (2010). [DOI] [PubMed] [Google Scholar]
- Lessard C. J. et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. American journal of human genetics 90, 648–660, doi: 10.1016/j.ajhg.2012.02.023 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedden K. et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nature medicine 14, 822–827, doi: 10.1038/nm.1790 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida S. et al. Relapse-related molecular signature in lung adenocarcinomas identifies patients with dismal prognosis. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 27, 2793–2799, doi: 10.1200/jco.2008.19.7053 (2009). [DOI] [PubMed] [Google Scholar]
- Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 21, 5427–5440, doi: 10.1038/sj.onc.1205600 (2002). [DOI] [PubMed] [Google Scholar]
- Kwon Y. J. et al. Genome-wide analysis of DNA methylation and the gene expression change in lung cancer. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer 7, 20–33, doi: 10.1097/JTO.0b013e3182307f62 (2012). [DOI] [PubMed] [Google Scholar]
- Yang I. V. et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine 190, 1263–1272, doi: 10.1164/rccm.201408-1452OC (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A. et al. Aberrant transcriptional regulations in cancers: genome, transcriptome and epigenome analysis of lung adenocarcinoma cell lines. Nucleic acids research 42, 13557–13572, doi: 10.1093/nar/gku885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. Y., Chang C. H., Yu C. J., Kuo S. H. & Yang P. C. Distribution according to histologic type and outcome by gender and age group in Taiwanese patients with lung carcinoma. Cancer 103, 2566–2574, doi: 10.1002/cncr.21087 (2005). [DOI] [PubMed] [Google Scholar]
- Yoshimi I., Ohshima A., Ajiki W., Tsukuma H. & Sobue T. A comparison of trends in the incidence rate of lung cancer by histological type in the Osaka Cancer Registry, Japan and in the Surveillance, Epidemiology and End Results Program, USA. Japanese journal of clinical oncology 33, 98–104 (2003). [DOI] [PubMed] [Google Scholar]
- Hirschowitz E. A. Biomarkers for lung cancer screening: interpretation and implications of an early negative advanced validation study. American journal of respiratory and critical care medicine 179, 1–2, doi: 10.1164/rccm.200809-1395ED (2009). [DOI] [PubMed] [Google Scholar]
- Selamat S. A. et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome research 22, 1197–1211, doi: 10.1101/gr.132662.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield A. S. et al. DNA methylation and RNA expression profiles in lung adenocarcinomas of never-smokers. Cancer genetics 208, 253–260, doi: 10.1016/j.cancergen.2014.12.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing C. et al. Cyclin-dependent kinase inhibitor 3 is overexpressed in hepatocellular carcinoma and promotes tumor cell proliferation. Biochemical and biophysical research communications 420, 29–35, doi: 10.1016/j.bbrc.2012.02.107 (2012). [DOI] [PubMed] [Google Scholar]
- Hou J. et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PloS one 5, e10312 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayama H. et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer research 72, 100–111 (2012). [DOI] [PubMed] [Google Scholar]
- Su L.-J. et al. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC genomics 8, 1 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi M. T. et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PloS one 3, e1651 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles A. I. et al. An integrated prognostic classifier for stage I lung adenocarcinoma based on mRNA, microRNA, and DNA methylation biomarkers. Journal of Thoracic Oncology 10, 1037–1048 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez S. V. et al. DNA methylation changes in a human cell model of breast cancer progression. Mutation research 688, 28–35, doi: 10.1016/j.mrfmmm.2010.02.007 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez S. V. & Russo J. Estrogen and xenoestrogens in breast cancer. Toxicologic pathology 38, 110–122, doi: 10.1177/0192623309354108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momparler R. L. Epigenetic therapy of cancer with 5-aza-2′-deoxycytidine (decitabine). Seminars in oncology 32, 443–451, doi: 10.1053/j.seminoncol.2005.07.008 (2005). [DOI] [PubMed] [Google Scholar]
- Lenka G., Weng W. H., Chuang C. K., Ng K. F. & Pang S. T. Aberrant expression of the PRAC gene in prostate cancer. International journal of oncology 43, 1960–1966, doi: 10.3892/ijo.2013.2117 (2013). [DOI] [PubMed] [Google Scholar]
- Wang Y. Development of cancer diagnostics—from biomarkers to clinical tests. Translational Cancer Research 4, 270–279 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.