

Abstract
Cell surface engineering is an expanding field and whilst extensive research has been performed decorating cell surfaces with biomolecules, the engineering of cell surfaces with particles has been a largely unexploited area. This study reports on the assembly of cell-particle hybrids where irradiated tumor cells were surface engineered with adjuvant-loaded, biodegradable, biocompatible, polymeric particles, with the aim of generating a construct capable of functioning as a therapeutic cancer vaccine. Successfully assembled cell-particle hybrids presented here comprised either melanoma cells or prostate cancer cells stably adorned with Toll-like receptor-9 ligand-loaded particles using streptavidin–biotin cross-linking. Both cell-particle assemblies were tested in vivo for their potential as therapeutic cancer vaccines yielding promising therapeutic results for the prostate cancer model. The ramifications of results obtained for both tumor models are openly discussed.
Keywords: cell surface engineering, cancer vaccines, streptavidin-biotin cross-linking, PLGA particles
Graphical Abstract
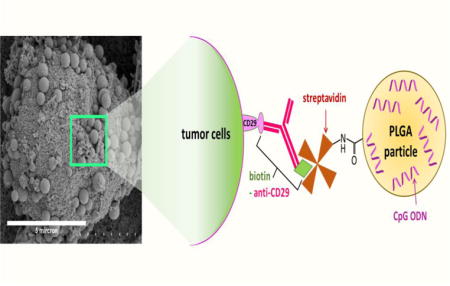
1. Introduction
The use of cells as therapeutic agents is well established, examples of which include adoptive T cell therapy for the eradication of cancers and the use of multipotent stem cells for tissue regeneration [1–3]. Most cell therapies have involved the use of viable cells, capable of proliferation, differentiation, or performing an inherent functional task. However, the use of irradiated, and therefore dying, whole tumor cells is a cancer vaccine strategy that has shown promising outcomes in preclinical and clinical settings [4, 5]. The aim of such a strategy is to generate effective adaptive immune responses to a wide range of tumor-associated, and tumor-specific, antigens and therefore reduce the possibility of immune evasion by tumor cells as a result of antigen loss variants [6]. Cancer accounts for 1 in 4 deaths in the United States [7]. A number of these malignancies, which are often resistant to conventional therapies, have shown promise in terms of responses to immune therapies including irradiated whole tumor cell vaccines which can act as a source of tumor antigens [8]. Examples of such malignancies include melanoma, prostate and pancreatic cancer[9–11]. Irradiation is used to ensure such cell-based vaccines are incapable of proliferation, however, sometimes irradiation has the additional benefit of increasing the antigenicity of tumor cells by promoting an immunogenic form of apoptosis [12, 13]. Whilst genetically modified versions of these cells, often involving transfection with GM-CSF, have delivered encouraging results in clinical trials, it is still apparent that further enhancement of the immunogenicity of these vaccines is required [14]. Cell surface engineering has been the focus of many studies where various cell types have been surface engineered with different macromolecules or nanomaterials for a variety of applications [15–20]. Cell surface engineering of tumor cells with an immune adjuvant can enhance the immunogenicity of the vaccine compared to providing the adjuvant in soluble form. The importance of co-delivering tumor antigen and adjuvant to the same dendritic cell has been illustrated in vitro [21] and has been further confirmed in vivo using a whole cell tumor vaccine where vaccination with the Toll-like receptor 9 ligand, CpG oligodinucleotide (CpG ODN), chemically linked to apoptotic tumor cells resulted in slower tumor growth of established TRAMP C-1 tumors [22]. A limitation of direct chemical linking of immune adjuvants is that one is restricted to using only those adjuvants that are not functionally compromised by the chemical coupling process. By packaging adjuvants into polymeric particles and chemically linking the particles to cells it is possible to circumvent the potential problem of adjuvant inactivation. The use of synthetic particles to tune immune responses is well established [23,24]. However, there have been very few reports in the literature of attempts to engineer cell surfaces with discrete particles [25–27]. These studies have involved either chemical modification of cell surface residues or simple adsorption of particles to cells. The work presented here focuses on the design of a cancer vaccine formulation where the immune adjuvant is loaded into poly(lactic-co-glycolic acid) (PLGA) particles that are then anchored to the tumor cell surface. The particles were anchored onto the cell surface using the streptavidin-biotin cross link that is commonly applied in tissue engineering [28–30]. The method expounded upon here is relatively simple, resulting in a product that should be capable of clinical translation into therapy due to the already established track records of the constituents. For instance, PLGA has FDA approval for clinical use, whilst CpG ODN has been shown to have a good safety profile in clinical trials [31]. The engineered cell-particle assemblies reported here impart a level of versatility to the vaccine formulation where the polymeric particles can be loaded with different immune adjuvants, or even a combination of immune adjuvants as required, and can be tailored to different tumor types.
2. Materials and Methods
2.1. Cell Lines
The murine melanoma cell line, B16.F10, was obtained from ATCC (Manassas, VA). The murine prostate cancer cell line, RM11, was a generous donation from Professor David Lubaroff, (University of Iowa, Iowa city, IA). Cells were maintained in DMEM complete media supplemented with 10% fetal bovine serum, 10 mM HEPES buffer, 1 mM sodium pyruvate, 2 mM Glutamax™, and 50 ng/ml gentamicin sulfate. Cells were incubated in a 5% CO2 humidified incubator at 37°C. For vaccinations, a B16.F10 GM-CSF clone expressing 220 ng GM-CSF/106 cells/day was derived by transducing B16.F10 cells with a lentiviral vector encoding murine GM-CSF (AMSBIO, Cambridge, MA).
2.2. Fabrication and Characterization of Streptavidin-Coated Particles
Particles were prepared using a double emulsion solvent evaporation technique [32]. In brief, 50 μl 1% polyvinyl alcohol (PVA) solution (water phase 1) was emulsified in 1.25 mL dichloromethane (DCM) containing 100 mg PLGA (75:25 m.wt 68 KD, with uncapped carboxyl end groups) (oil phase) using a sonic probe for 30 seconds at 40% amplitude, generating a primary emulsion. This was then emulsified in 8 mL 2.5 % PVA (MW:67 KD) in 0.1 M MES (2-(N-Morphino) ethanesulfonic acid) (water phase 2) using the same probe settings, generating a secondary emulsion. The secondary emulsion was then rapidly poured into 20 mL 1% PVA in 0.1 M MES buffer and stirred in fume hood. EDC and NHS, dissolved in MES buffer, were added sequentially at a ratio of 2 mg EDC (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride) to 3 mg NHS (N-hydroxysuccinimide) per 1 mg PLGA polymer (the amounts of EDC/NHS were empirically determined). The particle suspension was then stirred in a fume hood for 2 hours to allow DCM evaporation and carboxyl end group activation. Particles were collected by sequential centrifugation to ensure narrow size distribution where particle suspension was first centrifuged at 115 × g for 5 minutes and the pellet was discarded. Particles used in the study were collected from the supernatant of the previous step by centrifugation at 10 000 × g for 10 minutes and washed 3 times using nanopure water. Particles suspension was frozen and lyophilized overnight. CpG loaded particles were prepared using 2 mg CpG ODN 1826 in the 50 μl 1% PVA-water phase 1. Rhodamine B-loaded particles were prepared by dissolving 1 mg rhodamine B in the PLGA polymer/DCM solution. Particles were characterized for size and zeta potential using a Zetasizer Nano ZS (Malvern). For rhodamine B-loaded particles (which could not be measured using a Zetasizer), size was measured from SEM images using ImageJ software (n = 100). This size measurement method was validated by measuring the sizes of blank particles using imageJ software and comparing results to their sizes determined using the Zetasizer Nano ZS. To estimate CpG loading, 5–10 mg of particles were degraded in 1 ml 0.3N NaOH until a clear solution was obtained. This solution was neutralized with 1 N HCl. The CpG ODN concentration in the neutralized solution was estimated using an OliGreen® assay kit according to product instructions.
Loading was calculated as follows:
Lyophilized particles were coated with streptavidin by incubating surface activated particles with streptavidin in phosphate buffer saline (PBS), pH 7.4 at a ratio of 8 μg streptavidin per 1 mg particles for 30 minutes at room temperature. Excess streptavidin was removed by washing particles with PBS 3 times. To confirm successful particle coating, as proof of principle, fluorescently labeled streptavidin (streptavidin-PE) was used and particles were analyzed by flow cytometry (FACScan, Becton-Dickinson) (n = 3). Streptavidin-coated particles (unlabeled) were incubated with fluorescently labeled biotin to ensure availability of surface streptavidin for biotin binding. Samples were analyzed using flow cytometry (n = 3).
2.3. Surface Engineering Tumor Cells with PLGA Particles
B16.F10 or RM11 tumor cells were coated with biotin using biotinylated antibody targeting the β1 integrin (anti-mouse/rat CD29). Surface expression of CD29 by tumor cells and successful tumor cell biotinylation were confirmed by immunofluorescent assay using streptavidin-PE and samples were analyzed by flow cytometry (n = 3). To surface functionalize tumor cells with particles, biotinylated tumor cells were incubated with streptavidin-coated particles at a ratio of 1 mg particles per 5 × 105 cells in complete cell culture media for 15 minutes on ice followed by 15 minutes at 37°C. Successful cell surface engineering was confirmed using flow cytometry and microscopic imaging. Initially, the following two negative controls were developed to ensure cell-particle hybrids were formed as a result of streptavidin-biotin chemistry: biotinylated cells incubated with uncoated particles and non-biotinylated cells incubated with streptavidin-coated particles. As both controls showed similar results (see Suppl. Figure 1), only non-biotinylated cells + streptavidin-coated particles was used as the negative control for all upcoming experiments. For scanning electron microscopy (SEM) imaging, cell-particle mixtures were fixed in glutaraldehyde and mounted on poly-L-lysine treated silica wafers. Samples were stained with osmium tetraoxide and gradually dehydrated with ethanol and hexamethyldisilazane (HMDS). Samples were sputter coated with gold/palladium for 3 minutes prior to imaging on SEM at 2 kV accelerating voltage (Hitachi S-4800). For laser scanning confocal microscopy, cell-particle hybrids were assembled using rhodamine loaded particles. Cell-particle mixtures were fixed in 4% paraformaldehyde and deposited on slides using cytospin centrifuge at 700 rpm for 7 minutes. Cover slides were then mounted using Vectasheild mounting media with DAPI. Samples were imaged using differential interference contrast (DIC)/fluorescence mode (Zeiss 710 confocal microscope).
To estimate the percentage of particles binding to the cell surface, an indirect assay was developed (see Suppl. Figure 2). Briefly, cell-particle hybrids were assembled as described above. Aliquots were added to wells of a 96 well tray at a cell density of 1 × 105 cells/well and incubated with biotin-linked alkaline phosphatase (7.5 μg/ml) for 30 minutes. Unbound particles and enzyme were simultaneously removed through washing prior to adding an enzyme substrate (p-nitrophenyl phosphate) in Tris buffer. The plate was centrifuged at 230 × g for 5 minutes and an aliquot of the supernatant was transferred to a new plate to record the absorbance at 450 nm using SpectraMax® Plus384 microplate reader. Blanks involved hybrid samples where the p-nitrophenyl phosphate was added to samples without prior addition of biotinylated enzyme (see Suppl. Figure 2B). A calibration curve was generated by serially diluting known amounts of streptavidin-coated particles that were incubated with biotinylated enzyme prior to mixing with cells and subsequently adding the enzyme substrate. Blanks for calibration curve involved mixing known amounts of particles with cells without the incorporation of the enzyme. All samples were carried out in triplicate. Assays were performed independently for cell-particle hybrids assembled using the B16.F10 and the RM11 tumor cells.
2.4. In Vitro BMDCs Uptake and Activation Studies
Bone marrow derived dendritic cells (BMDCs) were generated as described previously [33]. BMDCs were maintained in RPMI media supplemented with 10% fetal bovine serum, 10 mM HEPES buffer, 1 mM sodium pyruvate, 0.1 mM minimal essential medium non-essential amino acids, 2 mM GlutaMAX, 50 nM 2-mercaptoethanol, and 50 ng/ml gentamicin sulfate and used at day 9 post culture. For uptake experiments, cell-particle hybrids were assembled as described above using rhodamine B-labeled particles and B16.F10-GM-CSF cells stained with carboxyfluorescein succinimidyl ester (CFSE, eBioscience). BMDCs were cultured in 12-well cell culture trays containing poly L lysine-coated cover slips. BMDC were seeded at 4 × 105 cells per well and allowed to adhere for 4 hours before adding the cell-particle hybrids (1 × 105 cell per well). Following a 24 hour incubation, BMDC were stained with anti-mouse CD11c antibody tagged with alexafluor®700. Test and control samples were fixed in 4% paraformaldehyde and imaged using laser scanning confocal microscopy.
For BMDC activation studies, BMDC were cultured into 12-well cell culture trays at a density of 3.5 × 105 cells/well. Cells were allowed to adhere for 4 hours prior to adding the different tumor cell/CpG ODN combinations. Treatments included: tumor cells alone, CpG ODN alone, CpG ODN + tumor cells, and untreated control. Tumor cells (B16.F10-GM-CSF, B16.F10 and RM11) were irradiated (35 Gy) and then added at a density of 3.5 × 105 cell/well to the BMDC. Treatments with CpG ODN were at 6 μg/mL. After 24 hour incubation, BMDC were harvested and co-stained for CD11c and CD80 or CD86 using specific fluorescently tagged antibodies and analyzed by flow cytometry. All treatments were performed in triplicate.
2.5. Tumor Studies
Six to eight week old female C57BL/6 and male BALB/c mice were purchased form Jackson laboratories. For the melanoma vaccine study, mice were vaccinated subcutaneously with indicated irradiated (35 Gy) B16.F10-GM-CSF cell/particle formulations on days 0 and 7 on the left dorsal flank and then challenged subcutaneously with 105 live wild type B16.F10 cells in the right shaved flank on day 14. Vaccination groups tested were: untreated (naïve), tumor cells, tumor cells mixed with, but not surface engineered with, CpG ODN-loaded particles (cell-particle mixture), tumor cells surface engineered with CpG ODN-loaded particles (cell-particle hybrid). Vaccinations involved a dose of 3 × 106 irradiated B16.F10-GM-CSF and, when indicated, 2.5 μg CpG ODN. For the prostate cancer tumor model, mice were challenged subcutaneously with 2 × 105 live RM11 cells in the right shaved flank at day 0 and subsequently vaccinated subcutaneously with the indicated irradiated (35 Gy) RM11 cell/particle formulations on days 1 and 8 on the left dorsal flank. Vaccination groups tested were: untreated (naïve), tumor cells + soluble CpG ODN, tumor cells mixed with but not surface engineered with CpG ODN-loaded particles (cell-particle mixture), irradiated tumor cell surface engineered with CpG ODN-loaded particles (cell-particle hybrid). Vaccinations involved a dose of 1.5 × 106 and 3 × 106 irradiated RM11 cells for the prime and boost, respectively. When indicated 5.4 and 10.8 μg CpG ODN was delivered with the vaccine as prime and boost, respectively, in soluble or particle form. Mice were monitored for tumor growth and survival. Tumor volumes were calculated from the following equation for an ellipsoid: Tumor volume = (W*L*H*π/6). Mice were euthanized when tumors reached 20 mm diameter in any direction. All animal handling was carried according to the University of Iowa Institutional Animal Care and Use Committee guidelines. When needed, animals were anesthetized by injecting, intraperitoneally, 87.5mg/kg ketamine and 12.5mg/kg xylazine.
2.6. Statistical Analysis
Data were analyzed by one way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test, Kruskal-Wallis non-parametric test followed by Dunn’s multiple comparison, or student t test as data allowed. Differences were considered significant at p-values < 0.05. Survival data were analyzed using the log-rank test followed by Tukey’s multiple comparison test. Statistical analyses were performed using GraphPad Prism 6 software except for survival data Tukey-Kramer post-test analyses that were performed using Statistical Analysis System 9 (SAS) software.
3. Results and Discussion
3.1. Fabrication and Characterization of Streptavidin-Coated Particles
Here we demonstrate the fabrication of cell-particle hybrids for use as cancer vaccines. The assembly of cell-particle hybrids involved the mixing together of independently prepared streptavidin-coated PLGA particles and biotin-coated cells. Preparation of the particles involved forming the particles (≈ 500 nm in diameter) from PLGA (75:25, free carboxylic end group), using a double emulsion solvent evaporation method, whilst simultaneously activating terminal carboxyl groups of PLGA chains using carbodiimide chemistry (Figure 1A). This approach was chosen over formation and surface activation of particles in a sequential fashion as it resulted in a 5-fold improvement in loading of the cargo, CpG ODN, Table 1. These activated particles were lyophilized and then coated with streptavidin. The sizes, zeta potentials and loadings (with CpG ODN) of variously formulated particles are summarized in Table 2. Confirmation that streptavidin was binding to the particles was performed using PE-labeled streptavidin in a proof of principle study (Figure 1B). That streptavidin (unlabeled), once bound to the particle surface, was still capable of binding to biotin was confirmed using biotinylated fluorescein (Figure 1C).
Figure 1. Particle functionalization and characterization.
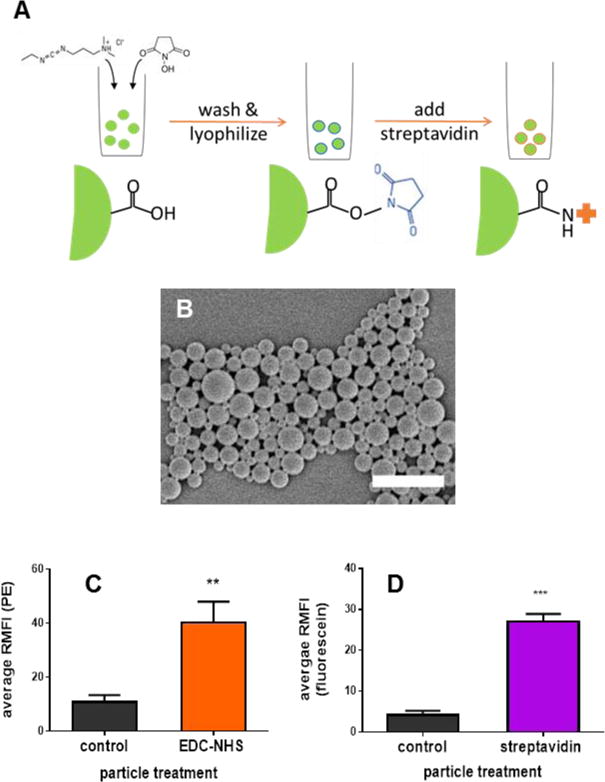
(A): Schematic of preparation of streptavidin-coated particles. PLGA particles were formed using a double emulsion solvent evaporation method with simultaneous activation of terminal carboxyl groups as described in the methods section. Surface activated particles were lyophilized and coated with streptavidin immediately prior to use. (B) Representative SEM image of PLGA particles, scale bar = 2 microns. (C) EDC/NHS-activated and non-activated (control) particles incubated with streptavidin-PE. (D) EDC/NHS-activated particles treated or untreated (control) with streptavidin and then incubated with biotinylated fluorescein. When applicable, error bars = SD. ** p < 0.01, *** p < 0.001, n = 3.
Table 1.
CpG loading in streptavidin coated particles achieved using the simultaneous versus seqential particle fabrication and surface carboxyl group activation
| Method of carboxylic group surface activation | loading μg CpG/ mg particles (SDa) | |
|---|---|---|
| During particle fabrication (simultaneous) | Initial loading | 4.6 (0.9) |
| Final loading in streptavidin coated particles | 2.3 (0.4) | |
| Post fabrication (sequential) | Initial loading | 2.1 (0.2) |
| Final loading in streptavidin coated particles | 0.4 (0.1) |
SD: standard deviation, n=3
Table 2.
Summary of particles characterized for size and zetapotential.
| Formulation | Size (SDb) d, nm |
PDIc (SD) | Zetapotential (SD) mV |
|---|---|---|---|
| Blank particles | 533 (19) |
0.10 (0.02) |
−16.7 (0.7) |
| CpG loaded particles (before streptavidin coating) | 510 (31) |
0.07 (0.02) |
−18.4 (0.4) |
| Streptavidin-coated CpG loaded particles | 658 (23) |
0.16 (0.03) |
−3.8 (0.1) |
| Rhodamine loaded particles | 592 (174) |
/ | / |
SD: standard deviation, n=3
PDI: polydispersity index
3.2. Surface Engineering Tumor Cells with PLGA Particles
The cells (either B16.F10, a murine melanoma cell line, or RM11, a murine prostate cancer cell line) were coated with biotin using a biotinylated antibody specific for the β1 integrin (CD29). To our knowledge, this is the first report involving CD29 (β1 integrin) in cell-surface engineering. CD29 was chosen as it is ubiquitously expressed and therefore most tumor cell types can be considered as candidates for cell:particle hybrid vaccine formulations [34]. This biotinylation method can also bypass the drawbacks of cell surface chemical modifications using N-hydroxysuccinamide [35] or biotin hydrazide [25] which can result in loss of cell viability as we encountered upon attempting to biotinylate the cell surface of B16.F10 cells by chemical coupling. Confirmation that the cells were coated with biotin was performed using streptavidin-PE (Figure 2A1 and 2A2). Activated particles (initially loaded with rhodamine B to assist with their detection in a proof of principle assay) and biotinylated cells were mixed and incubated together in complete cell culture media for 15 minutes on ice and then for an additional 15 minutes at 37°C to allow for cell-particle hybrid formation. Excess unbound particles were washed away and validation that cell-particle hybrids had formed was performed using flow cytometry (Figure 2B1 and 2B2 (B16.F10) & Figure 2E1 and 2E2 (RM11)), confocal microscopy (Figure 2C1 and 2C2 for B16.F10 & Figure 2F1 and 2F2 for RM11) and scanning electron microscopy (SEM) (Figure 2D1 and 2D2 (B16.F10)). Using flow cytometry, a significant increase in rhodamine B mean fluorescence intensity (MFI) was observed for biotinylated cells over non-biotinylated cells that were independently mixed with streptavidin-coated particles (p < 0.001 and p < 0.01 for the melanoma and prostate cancer cells, respectively), thereby implicating the formation of cell-particle hybrids. These findings confirm the crucial role of avidin-biotin cross-linking in surface engineering tumor cells with particles compared to non-specific adsorption of particles to the cell surface as has been employed previously [27]. Complete shifts in the symmetrical peaks for rhodamine B fluorescence following cell-particle hybrid formation for both B16.F10 (Figure 2B1) and RM11 (Figure 2E1) indicated that most cells, subsequent to washing of unbound particles, were surface-engineered with particles. Confirming the results obtained using flow cytometry, both confocal microscopy and SEM demonstrated successful formation of cell-particle hybrids, therefore eliminating the possibility that the increases in fluorescence observed with flow cytometry were non-specific or that the particles had been mostly endocytosed. Confocal microscopy revealed that the majority of particles were surface bound to tumor cells as opposed to being internalized. These results confirmed that cell-particle hybrids had been successfully assembled for both cell lines tested. In principle this method should work for any cell type expressing sufficient levels of CD29. The proportion of particles that remained bound to cells subsequent to washing the cell-particle mixture was estimated using an indirect assay and was found to be 30% and 25% for hybrids assembled using B16.F10 and RM11 cells, respectively. These results were comparable to the particle binding efficiency of 33.5% reported using a completely different cell-particle conjugation system [26].
Figure 2. Fabrication and characterization of cell-particle assemblies.
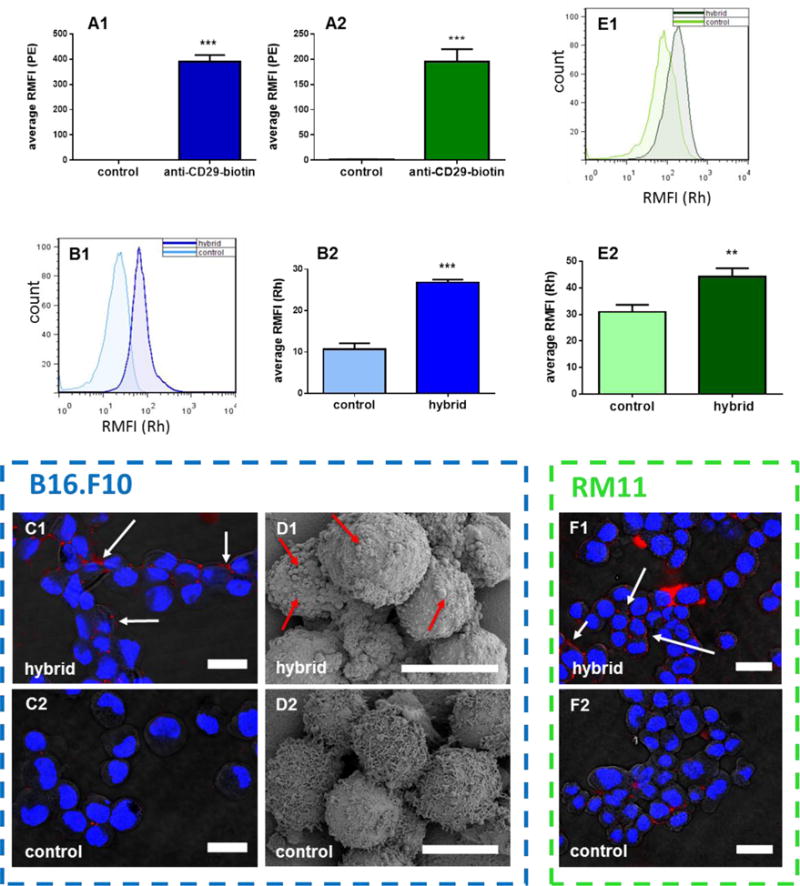
(A1) B16.F10 cells and (A2) RM11 cells treated or not treated (control) with biotinylated anti-CD29 antibodies (anti-CD29-biotin) followed by strepavidin-PE. (B–F) Validation of cell-particle hybrid assembly from B16.F10 or RM11 cells subsequent to surface biotinylation using anti-CD29-biotin and mixed with streptavidin-coated particles loaded with rhodamine B and washed to remove unbound particles (hybrid). Control involved the same conditions except cells were not treated with anti-CD29-biotin. Validation was performed using: (B1–2 and E1–2): flow cytometry where (B1 and E1) representive (n = 1) and (B2 and E2) mean (n = 3) results from measuring relative mean rhodamine fluorescence intensity (RMFI(Rh)) of (B1 and B2) B16.F10 and (E1 and E2) RM11 cells; (C1–2 and F1–2): laser scanning confocal microscopy showing (C1 and F1) hybrid and (C2 and F2) control cell-particle mixtures for (C1 and C2) B16.F10 and (F1 and F2) RM11 cells (blue = DAPI stained cell nuclei, red = rhodamine-labeled PLGA particles); (D): scanning electron microscopy showing (D1) B16.F10 hybrid and (D2) control cell-particle mixtures (arrows in C1, D1, and F1 indicate particles bound to cell surface). Scale bar= 20 micron for C1–2 and F1–2, 10 microns for D1–2. When applicable, error bars = SD. ** p < 0.01, *** p < 0.001, n = 3.
3.3. In Vivo Anti-Tumor Efficacy of Cell-Particle Hybrids
Between 10% and 20% of prostate cancer patients have their disease progress to castration resistant prostate cancer for which there is no cure [36]. Prostate cancer is considered a good candidate for immune therapy [37] and it has been the focus of many cancer vaccine formulations [38, 39] culminating recently in the introduction of the FDA approved dendritic cell based vaccine, Sipuleucel T [40]. Despite this, the survival prognosis of patients seems to be only marginally improved for patients treated with Sipuleucel T and, as such, improved or alternative cancer vaccines are sought. Thus, having established that it is feasible to manufacture cell-particle hybrids (above) we proceeded to generate a prostate cancer vaccine that we could test in an in vivo mouse prostate cancer model. Cell-particle hybrids were prepared as described above except that the PLGA particles used were loaded with the Toll-like receptor-9 agonist, CpG ODN, prior to mixing with biotinylated RM11 cells. Upon mixing the cells with CpG ODN-loaded particles the mixture was treated with 35 Gy of gamma-irradiation to render the cells non-viable. In addition, the irradiation of tumor cells has been reported to cause an immunogenic form of cell death [12], however, whether such a phenomenon occurred for the tumor cells used here was not investigated. Thus, the aim of this in vivo study was to determine if the cell-particle hybrid vaccine had greater therapeutic efficacy than cells and particles mixed (non-hybrid) or cells plus soluble CpG and, as such, could significantly improve the survival of tumor-challenged mice. Immunocompetent BALB/c mice were challenged with viable RM11 cells and then administered with the cell-particle hybrid vaccine as a prime/boost on day 1/day 8 post-tumor challenge, as described in the methods, and tumor volumes and survival were subsequently determined (Figure 3A and 3B). The cell-particle hybrid vaccinated mice was the only group to demonstrate statistically enhanced survival over the untreated (naïve) group (Figure 3B: p = 0.01). Whilst the other treatment groups displayed a trend towards improved therapy, the improvement was not significantly different to the untreated group (Figure 3B). However, the survival of mice vaccinated with the cell-particle hybrid was not statistically significant from those vaccinated with the cell-particle mixture, p = 0.8. Parallel results were observed with the tumor volume data (Figure 3A). These results highlight the potential advantage of delivering tumor antigens (irradiated tumor cells) and the immune adjuvant as one package, thereby enabling enhanced immune activation by dendritic cells as has been reported [22]. The method we propose here adds an extra level of versatility to the vaccine formulation where the polymeric particles have the potential to be loaded with a variety, or combination, of immune adjuvants as required.
Figure 3. Irradiated murine prostate cancer cells, RM11, are effective therapeutic cancer vaccines when conjugated to adjuvant-loaded particles.
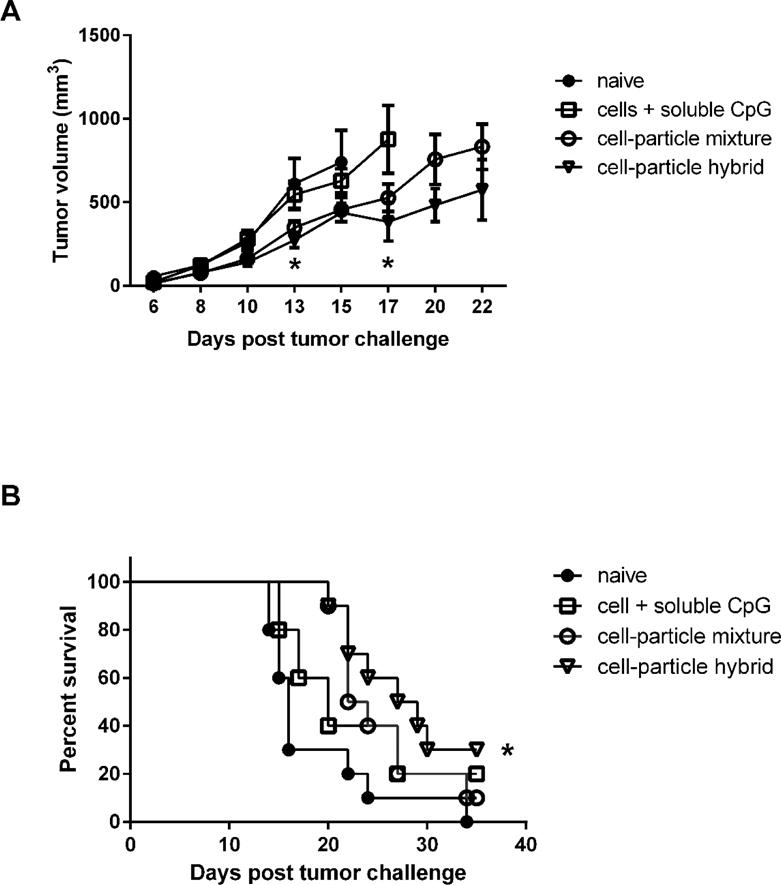
(A–B): Irradiated RM11 cells surface engineered with CpG ODN-loaded particles were effective as a therapeutic vaccine (see methods for tumor challenge and vaccination details) as shown by: (A) significantly reducing prostate cancer tumor burden compared to naïve mice (Day 13, * p < 0.05) and mice vaccinated with irradiated RM11 plus soluble CpG ODN (Day 17, * p < 0.05), and (B) significantly extending the survival of mice compared to naïve mice as shown in Kaplan-Meier survival curve (* p < 0.05). Median survival = 28 days for cell-particle hybrid group, 23 days for cell-particle mixture group, 20 days for cells + soluble CpG group, and 16 days for naïve mice. See methods for tumor challenge and vaccination details. Samples are presented as mean ± SEM, n= 5 – 10 mice per group.
We also tested a cell-particle hybrid formulation in a murine melanoma tumor model where we used B16.F10 cells that had been transfected to express GM-CSF. It has been previously demonstrated that such cells, once irradiated, are capable of therapeutic and prophylactic protection against B16.F10 tumor challenge, whilst irradiated B16.F10 cells that do not express GM-CSF are ineffective as vaccines [41]. In contrast to the therapeutic approach used for the RM11 model above we chose a prophylactic approach because pilot therapeutic studies using B16.F10-GMCSF cells alone (non-hybrid) revealed no therapeutic efficacy, nor did any of our preliminary B16.F10-particle hybrid constructs (data not shown). Thus we switched to the more permissive prophylactic setting in order to maximize the possibility of observing changes in immune protection against tumor challenge. We established that the cell-particle constructs could be taken up by dendritic cells in vitro (Figure 4). However, we found that neither the cell-particle hybrid nor the irradiated B16.F10-GM-CSF cells improved survival of mice challenged with a lethal dose of B16.F10 in a prophylactic setting (Suppl. Figure 3). This finding was unexpected since Dranoff et al (1993) had demonstrated therapeutic efficacy of irradiated B16 cells (transduced with GMCSF) [41]. A possible reason for this discrepancy may have been genetic drift resulting from a difference in the passage number status of the B16.F10 cells. Thus genetic drift may have been responsible for differences observed between our group and Dranoff et al. Further investigation in vitro revealed that the irradiated B16.F10-GM-CSF cells, when co-cultured with dendritic cells, had a significant dampening of the response to soluble CpG ODN in terms of CD80 and CD86 expression by dendritic cells (Figure 5A and 5B). CD80/CD86, expressed on the surface of dendritic cells, are important in promoting T lymphocyte activation and therefore sub-optimal levels of expression would be a great disadvantage in the context of vaccine formulations. Thus, we surmise that the B16.F10-GM-CSF cells used in the hybrid construct were possibly interfering with the potential immunogenic properties of the hybrid vaccine, in particular, the adjuvant role of CpG ODN. This attenuated response by dendritic cells to CpG ODN did not appear to be mediated by TGFβ1 (Suppl. Figure 4) and was also evident, albeit to a lesser extent, with the parent, non-GM-CSF secreting, B16.F10 cells (Figure 5A and 5B). In contrast, the RM11 tumor cells used to construct the cell-particle hybrid prostate cancer vaccine described above and shown to work therapeutically did not have a negative impact on the up-regulation of CD80/CD86 expression when co-cultured in vitro with dendritic cells in the presence of CpG ODN (Figure 5A and 5B).
Figure 4. Cell-particle hybrid uptake by BMDC in vitro.
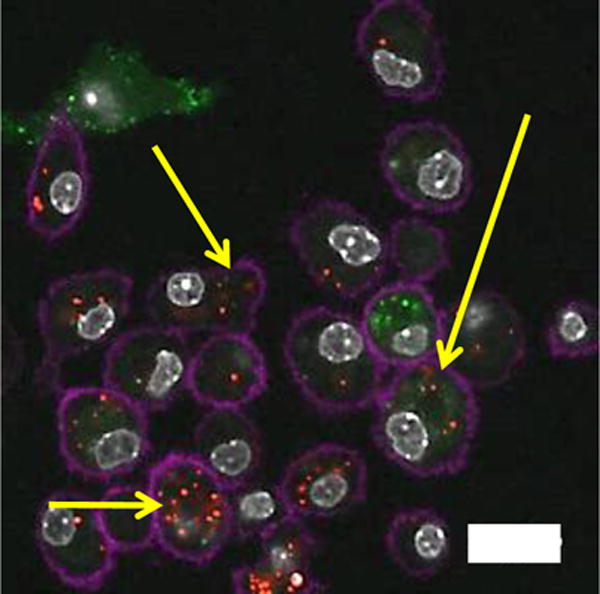
Laser scanning confocal microscopy imaging of cell-particle hybrid uptake by BMDC. Cell-particle hybrids were incubated with BMDC Yellow arrows indicating colocalization of B16.F10 cells (green) and particles (red) inside BMDC (magenta). Magenta: Alexa flour®700 (CD11c) stained BMDC, green: CFSE labeled B16.F10 melanoma cells, red: rhodamine B-labeled PLGA particles, gray: DAPI stained cell nuclei. Scale bar: 20 micron.
Figure 5. In vitro effect of irradiated tumor cells on the immunostimulatory properties of CpG ODN.
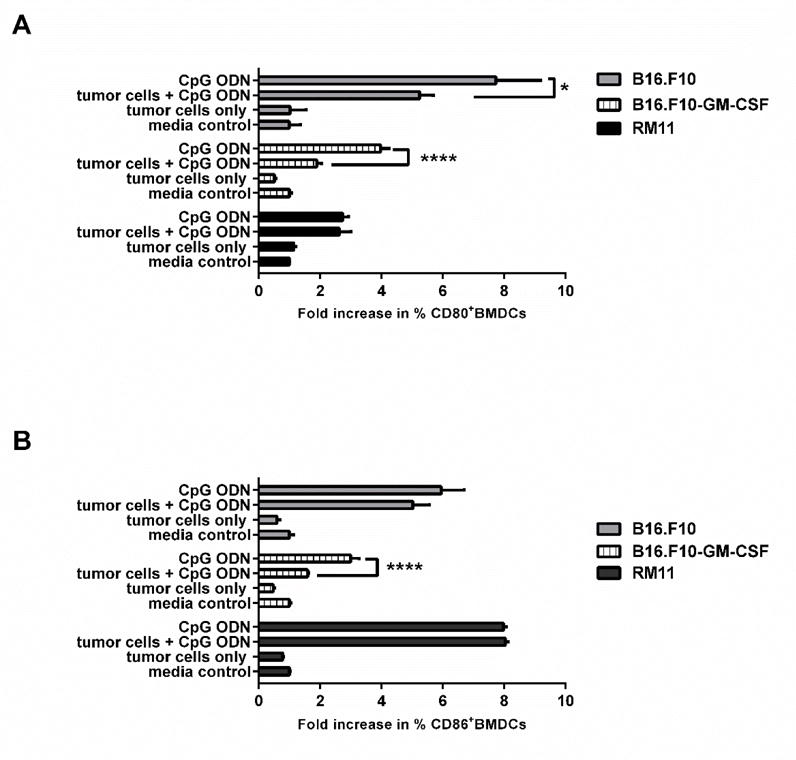
(A) CD80 and (B) CD86 expression by BMDC after 24 hour incubation in vitro with media alone (control), CpG ODN alone, CpG ODN + indicated irradiated tumor cells, or indicated irradiated tumors cells alone. Samples are presented mean ± SD. *p< 0.05, ****p<0.0001, n = 3. These experiments did not involve any cell-particle hybrids.
The results obtained for the prostate cancer and melanoma tumor models presented here highlight the need for careful selection of the cellular component of the cell-particle hybrid vaccine construct prior to its introduction into the clinic. In order for the cell:hybrid vaccine to be effective in the clinic it would ideally be applied to those tumor cell vaccines (non-hybrid) that have already shown promise in clinical trials in terms of generating antitumor immunity. In this way the use of an inappropriate tumor cell type lacking immunogenicity will be avoided and the addition of adjuvant loaded particles would be predicted to further enhance tumor immunity, tumor regression and patient survival.
4. Conclusions
We have demonstrated the successful fabrication of cell-particle hybrids where the particle component has the potential to be loaded with a variety of agents including immune adjuvants, such as CpG ODN, as was used here. The versatility of the system was further demonstrated by the successful fabrication of hybrids comprising different cell types. Using irradiated tumor cells as the cell component engender the hybrid constructs with the potential to be used as safe cancer vaccines, the immunogenicity of which can be further potentiated by the presence of adjuvant(s) in the particle component. The cell-particle hybrids were capable of co-delivering the cells (antigen) and particles (containing adjuvant) to the same dendritic cells (Figure 4) which is an essential rate limiting step toward the initiation of anti-tumor immune responses [42]. Promisingly, the hybrid construct comprising irradiated prostate cancer cells conjugated to PLGA particles loaded with CpG ODN had a significant therapeutic effect in a prostate tumor model. Further manipulation of the vaccination regime and/or adjuvant composition may result in further improvements of the therapeutic effects. Although the melanoma hybrid construct did not result in protection from tumor challenge in a melanoma model, it highlighted the need to choose carefully the cell component of the cell-particle hybrid construct in terms of its ability to affect the immunopotency of dendritic cells and consequently the efficacy of the vaccine.
Supplementary Material
Acknowledgments
The flow cytometry data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine / Holden Comprehensive Cancer Center core research facility at the University of Iowa (NIH grant number of 1S10RR025439-01). We also acknowledge the Radiation and Free Radical Research Core supported in part by the Holden Comprehensive Cancer Center Grant P30 CA086862 and the University of Iowa, Carver College of Medicine. We thank Nattawut Leelakanok for his support in performing statistical analysis using the SAS software.
Funding
This work was supported by National Cancer Institute at the National Institutes of Health (P50 CA97274/ UI Mayo Clinic Lymphoma SPORE grant and P30 CA086862 Cancer Center support grant) and the Lyle and Sharon Bighley Professorship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Syed BA, Evans JB. Stem cell therapy market. Nat Rev Drug Discov. 2013;12(3):185–186. doi: 10.1038/nrd3953. [DOI] [PubMed] [Google Scholar]
- 2.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotech. 2013;31(11):999–1008. doi: 10.1038/nbt.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee HL, et al. Hypoxia-specific, VEGF-expressing neural stem cell therapy for safe and effective treatment of neuropathic pain. J Control Release. 2016;226:21–34. doi: 10.1016/j.jconrel.2016.01.047. [DOI] [PubMed] [Google Scholar]
- 4.Fu J, et al. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res. 2014;74(15):4042–52. doi: 10.1158/0008-5472.CAN-13-2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le DT, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. Journal of immunotherapy (Hagerstown, Md: 1997) 2013;36(7):382–389. doi: 10.1097/CJI.0b013e31829fb7a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson BM, McNeel DG. Antigen loss and tumor-mediated immunosuppression facilitate tumor recurrence. Expert review of vaccines. 2012;11(11):1315–1317. doi: 10.1586/erv.12.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer facts and figures,[cited March, 2016]; Available from: http://www.cancer.org/research/cancerfactsstatistics/
- 8.Goldman B, DeFrancesco L. The cancer vaccine roller coaster. Nat Biotech. 2009;27(2):129–139. doi: 10.1038/nbt0209-129. [DOI] [PubMed] [Google Scholar]
- 9.Drake CG. Update on prostate cancer vaccines. Cancer J. 2011;17(5):294–9. doi: 10.1097/PPO.0b013e3182325e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dranoff G. GM-CSF-secreting melanoma vaccines. Oncogene. 2003;22(20):3188–92. doi: 10.1038/sj.onc.1206459. [DOI] [PubMed] [Google Scholar]
- 11.Springett GM. Novel pancreatic cancer vaccines could unleash the army within. Cancer Control. 2014;21(3):242–6. doi: 10.1177/107327481402100311. [DOI] [PubMed] [Google Scholar]
- 12.Kroemer G, et al. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 13.Vandenberk L, et al. Exploiting the Immunogenic Potential of Cancer Cells for Improved Dendritic Cell Vaccines. Front Immunol. 2015;6:663. doi: 10.3389/fimmu.2015.00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Gruijl TD, et al. Whole-cell cancer vaccination: from autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol Immunother. 2008;57(10):1569–77. doi: 10.1007/s00262-008-0536-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen W, et al. Improving cell-based therapies by nanomodification. J Control Release. 2015;219:560–575. doi: 10.1016/j.jconrel.2015.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo CY, et al. Cell surface glycoengineering improves selectin-mediated adhesion of mesenchymal stem cells (MSCs) and cardiosphere-derived cells (CDCs): Pilot validation in porcine ischemia-reperfusion model. Biomaterials. 2016;74:19–30. doi: 10.1016/j.biomaterials.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang X, et al. Mesenchymal stem cell-based cell engineering with multifunctional mesoporous silica nanoparticles for tumor delivery. Biomaterials. 2013;34(7):1772–1780. doi: 10.1016/j.biomaterials.2012.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson JT, et al. Cell surface engineering with polyelectrolyte multilayer thin films. J Am Chem Soc. 2011;133(18):7054–64. doi: 10.1021/ja110926s. [DOI] [PubMed] [Google Scholar]
- 19.Medof ME, et al. Cell-surface engineering with GPI-anchored proteins. The FASEB Journal. 1996;10(5):574–86. doi: 10.1096/fasebj.10.5.8621057. [DOI] [PubMed] [Google Scholar]
- 20.Rabuka D, et al. Noncovalent cell surface engineering: incorporation of bioactive synthetic glycopolymers into cellular membranes. J Am Chem Soc. 2008;130(18):5947–5953. doi: 10.1021/ja710644g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nierkens S, et al. In vivo colocalization of antigen and CpG [corrected] within dendritic cells is associated with the efficacy of cancer immunotherapy. Cancer Res. 2008;68(13):5390–6. doi: 10.1158/0008-5472.CAN-07-6023. [DOI] [PubMed] [Google Scholar]
- 22.Shirota H, Klinman DM. CpG-conjugated apoptotic tumor cells elicit potent tumor-specific immunity. Cancer Immunol Immunother. 2011;60(5):659–69. doi: 10.1007/s00262-011-0973-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varypataki EM, et al. Synthetic long peptide-based vaccine formulations for induction of cell mediated immunity: A comparative study of cationic liposomes and PLGA nanoparticles. J Control Release. 2016;226:98–106. doi: 10.1016/j.jconrel.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 24.Geary SM, et al. Diaminosulfide based polymer microparticles as cancer vaccine delivery systems. J Control Release. 2015;220, Part B:682–690. doi: 10.1016/j.jconrel.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krishnamachari Y, et al. Self-assembly of cell–microparticle hybrids. Adv Mater. 2008;20(5):989–993. [Google Scholar]
- 26.Stephan MT, et al. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010;16(9):1035–1041. doi: 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chambers E, Mitragotri S. Long circulating nanoparticles via adhesion on red blood cells: mechanism and extended circulation. Exp Biol Med (Maywood) 2007;232(7):958–66. [PubMed] [Google Scholar]
- 28.Richter JF, et al. A novel method for imaging sites of paracellular passage of macromolecules in epithelial sheets. J Control Release. 2016;229:70–79. doi: 10.1016/j.jconrel.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y, et al. Enhanced in vivo antitumor efficacy of dual-functional peptide-modified docetaxel nanoparticles through tumor targeting and Hsp90 inhibition. J Control Release. 2016;221:26–36. doi: 10.1016/j.jconrel.2015.11.029. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, et al. Enzyme-functionalized vascular grafts catalyze in-situ release of nitric oxide from exogenous NO prodrug. J Control Release. 2015;210:179–188. doi: 10.1016/j.jconrel.2015.05.283. [DOI] [PubMed] [Google Scholar]
- 31.Bode C, et al. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines. 2011;10(4):499–511. doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joshi VB, et al. Biodegradable Particles as Vaccine Delivery Systems: Size Matters. AAPS J. 2013;15(1):85–94. doi: 10.1208/s12248-012-9418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lutz MB, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223(1):77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 34.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J, et al. GM-CSF-surface-modified B16.F10 melanoma cell vaccine. Vaccine. 2006;24(25):5265–8. doi: 10.1016/j.vaccine.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 36.Crawford ED, et al. Treating Patients with Metastatic Castration Resistant Prostate Cancer: A Comprehensive Review of Available Therapies. J Urol. 2015;194(6):1537–1547. doi: 10.1016/j.juro.2015.06.106. [DOI] [PubMed] [Google Scholar]
- 37.Geary SM, et al. Proposed mechanisms of action for prostate cancer vaccines. Nat Rev Urol. 2013;10(3):149–60. doi: 10.1038/nrurol.2013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cole G, et al. DNA vaccination for prostate cancer: key concepts and considerations. Cancer Nanotechnol. 2015;6(1):2. doi: 10.1186/s12645-015-0010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Eertwegh AJ, et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(5):509–17. doi: 10.1016/S1470-2045(12)70007-4. [DOI] [PubMed] [Google Scholar]
- 40.Lubaroff DM. Prostate cancer vaccines in clinical trials. Expert Rev Vaccines. 2012;11(7):857–68. doi: 10.1586/erv.12.54. [DOI] [PubMed] [Google Scholar]
- 41.Dranoff G, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90(8):3539–43. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy KP. Janeway’s immunobiology. Garland Science; New York: 2011. Antigen Presentation to T Lymphocytes; pp. 211–238. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.