

ABSTRACT
Viruses in the family Coronaviridae, within the order Nidovirales, are etiologic agents of a range of human and animal diseases, including both mild and severe respiratory diseases in humans. These viruses encode conserved replicase and structural proteins as well as more diverse accessory proteins, encoded in the 3′ ends of their genomes, that often act as host cell antagonists. We previously showed that 2′,5′-phosphodiesterases (2′,5′-PDEs) encoded by the prototypical Betacoronavirus, mouse hepatitis virus (MHV), and by Middle East respiratory syndrome-associated coronavirus antagonize the oligoadenylate-RNase L (OAS-RNase L) pathway. Here we report that additional coronavirus superfamily members, including lineage A betacoronaviruses and toroviruses infecting both humans and animals, encode 2′,5′-PDEs capable of antagonizing RNase L. We used a chimeric MHV system (MHVMut) in which exogenous PDEs were expressed from an MHV backbone lacking the gene for a functional NS2 protein, the endogenous RNase L antagonist. With this system, we found that 2′,5′-PDEs encoded by the human coronavirus HCoV-OC43 (OC43; an agent of the common cold), human enteric coronavirus (HECoV), equine coronavirus (ECoV), and equine torovirus Berne (BEV) are enzymatically active, rescue replication of MHVMut in bone marrow-derived macrophages, and inhibit RNase L-mediated rRNA degradation in these cells. Additionally, PDEs encoded by OC43 and BEV rescue MHVMut replication and restore pathogenesis in wild-type (WT) B6 mice. This finding expands the range of viruses known to encode antagonists of the potent OAS-RNase L antiviral pathway, highlighting its importance in a range of species as well as the selective pressures exerted on viruses to antagonize it.
IMPORTANCE Viruses in the family Coronaviridae include important human and animal pathogens, including the recently emerged viruses severe acute respiratory syndrome-associated coronavirus (SARS-CoV) and Middle East respiratory syndrome-associated coronavirus (MERS-CoV). We showed previously that two viruses within the genus Betacoronavirus, mouse hepatitis virus (MHV) and MERS-CoV, encode 2′,5′-phosphodiesterases (2′,5′-PDEs) that antagonize the OAS-RNase L pathway, and we report here that these proteins are furthermore conserved among additional coronavirus superfamily members, including lineage A betacoronaviruses and toroviruses, suggesting that they may play critical roles in pathogenesis. As there are no licensed vaccines or effective antivirals against human coronaviruses and few against those infecting animals, identifying viral proteins contributing to virulence can inform therapeutic development. Thus, this work demonstrates that a potent antagonist of host antiviral defenses is encoded by multiple and diverse viruses within the family Coronaviridae, presenting a possible broad-spectrum therapeutic target.
KEYWORDS: lineage A coronavirus, torovirus, RNase L, phosphodiesterase, interferon antagonist, oligoadenylate synthetase
INTRODUCTION
Coronaviruses (CoVs) and the closely related toroviruses (ToVs) are well-known agents of disease in mammals, including humans. Coronaviruses and toroviruses, which are members of the family Coronaviridae within the order Nidovirales, contain positive-sense single-stranded RNA (ssRNA) genomes that are the longest known RNA genomes, ranging from 28 to 31 kb (1). The first two-thirds of the genome encodes the replicase proteins, which include the viral RNA-dependent RNA polymerase, and numerous nonstructural proteins (NSPs) which are required for replication and in some cases have host immune antagonist activities (2).
The structural proteins are encoded in the 3′ third of the genome and consist of spike (S), small membrane (E), membrane (M), nucleocapsid (N), and sometimes hemagglutinin-esterase (HE) proteins. Interspersed among the structural genes are diverse genes encoding accessory proteins that are not essential for replication but are believed to be required for virulence in vivo (1).
Mouse hepatitis virus (MHV) is a lineage A Betacoronavirus and the prototypical CoV. MHV encodes the accessory protein NS2, which was previously identified as a 2-His phosphoesterase (2H-PE) superfamily member (3) that we have demonstrated has 2′,5′-phosphodiesterase (2′,5′-PDE) activity that antagonizes host interferon (IFN) signaling via antagonism of the 2′,5′-oligoadenylate synthetase (OAS)–RNase L pathway (4). Upon sensing double-stranded RNA (dsRNA), OAS synthesizes 2′,5′-oligoadenylates (2-5A), which catalyze the activation of RNase L via homodimerization. RNase L subsequently cleaves host and viral ssRNA, leading to termination of protein synthesis and subsequent apoptosis (5). NS2 cleaves 2-5A, thus preventing the activation of RNase L. NS2 is a critical determinant of MHV strain A59 liver tropism in C57BL/6 (B6) mice and is required for the virus to cause hepatitis. A mutant A59 strain (NS2H126R; referred to herein as MHVMut) expressing an inactive phosphodiesterase is unable to antagonize the OAS-RNase L pathway in the mouse liver. Infection with this virus does not result in hepatitis, and MHVMut replication is reduced at least 10,000-fold compared to that of wild-type (WT) A59. However, in mice genetically deficient for RNase L (RNase L−/−), MHVMut replicates to wild-type levels and causes hepatitis (4).
As might be expected of antagonists of a potent innate antiviral pathway, 2′,5′-PDEs are not a host evasion mechanism unique to MHV. We recently showed that Middle East respiratory syndrome-associated coronavirus (MERS-CoV) and related bat coronaviruses, all lineage C betacoronaviruses, encode NS4b accessory proteins with 2′,5′-PDE activity (6). Additionally, unrelated group A rotaviruses (RVA) encode a PDE located in the C-terminal domain of the VP3 structural protein (7). We show here that lineage A betacoronaviruses closely related to MHV, including the human respiratory coronavirus HCoV-OC43 (OC43), the human enteric coronavirus CoV-4408 (HECoV), the equine coronavirus ECoV-NC99 (ECoV), and porcine hemagglutinating encephalomyelitis virus (PHEV), as well as the more distantly related equine torovirus (EToV) Berne (BEV), also encode NS2 homologs with predicted PDE activity. We found that these proteins (with the exception of PHEV NS2) do possess enzymatic 2′,5′-PDE activity that is capable of antagonizing RNase L and thus countering a potent host antiviral response, suggesting that PDE-mediated OAS-RNase L antagonism is an important virulence strategy for lineage A betacoronaviruses and toroviruses.
RESULTS
Alignment and modeling of coronavirus and torovirus NS2 proteins.
To determine whether the MHV-related betacoronaviruses encode proteins with 2′,5′-PDE activity, we first analyzed the primary amino acid sequences of the NS2 proteins from OC43, HECoV, ECoV, and PHEV and the pp1a C-terminal domain (pp1a-CTD) of BEV. While the NS2 homologs are encoded within ORF2a, the PDE of BEV is encoded at the 3′ end of ORF1a and processed from the pp1a polyprotein (2). All of these proteins contain two conserved HxS/Tx motifs spaced ∼80 residues apart, where “x” is any hydrophobic residue, which is characteristic of 2H-PE superfamily proteins (3, 4, 8) (Fig. 1). Interestingly, the carboxy termini of the PHEV and BEV PDEs are truncated relative to those of the other NS2 proteins, similarly to rotavirus VP3-CTD PDEs (8, 9). We further entered the primary amino acid sequences of these proteins into Phyre2 to predict their tertiary structures (Fig. 2). All of these proteins scored highly for homology with the published structure of the A-kinase anchoring protein 7 (AKAP7) central domain (CD) (10), a previously identified host-encoded 2H-PE with 2′,5′-PDE activity (7). We previously showed that the MHV NS2 and RVA VP3-CTD proteins, which are also structural homologs of AKAP7 CD, exhibit 2′,5′-PDE activity and can antagonize RNase L (4, 7, 8).
FIG 1.

Alignment of lineage A betacoronavirus and Berne torovirus PDE sequences. The PDEs examined (accession no.) were MHV NS2 (P19738) (26), OC43 NS2 (AAT84352.1) (27), HECoV NS2 (ACJ35484.1), ECoV NS2 (ABP87988.1) (28), PHEV NS2 (AAY68295.1) (29), and BEV pp1a (CAA36600.1) (30). Conserved catalytic HxS/Tx motifs are indicated by boxes.
FIG 2.
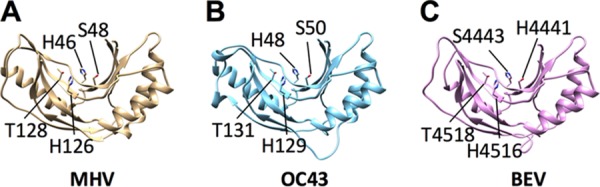
Known and predicted structures of nidovirus PDEs. The crystal structure of MHV NS2 (PDB entry 4Z5V) and predicted structures of OC43 NS2 (B) and BEV pp1a-CTD (C) are shown. Predicted structures were generated using Phyre2 and then visualized and annotated using UCSF Chimera 1.8. Catalytic His and conserved Ser/Thr residues are indicated.
Putative coronavirus and torovirus 2′,5′-PDEs are enzymatically active and cleave 2-5A.
To determine whether the predicted nidovirus PDEs (from OC43, BEV, and PHEV) are enzymatically active, the genes encoding them as well as their corresponding mutants with an Arg substitution of the second predicted catalytic His residue were expressed in Escherichia coli as maltose binding protein (MBP) fusion proteins and purified by affinity chromatography followed by ion-exchange chromatography and size exclusion chromatography as described in Materials and Methods (4). Purified wild-type or catalytic mutant proteins were incubated with the 2-5A substrate, and an indirect fluorescence resonance energy transfer (FRET) assay in which higher relative light unit (RLU) values represent active RNase L, as described in Materials and Methods and in detail previously (11), was used to assess the activation of RNase L. MHV NS2 was utilized as a positive control for inhibition of RNase L (Fig. 3). The OC43 and BEV proteins reduced RNase L activation to a degree similar to that with MHV NS2, while PHEV NS2 was significantly less active. The mutant proteins containing a His → Arg mutation in the second catalytic motif did not inhibit RNase L, as expected and consistent with previous results for MHV NS2 (4).
FIG 3.
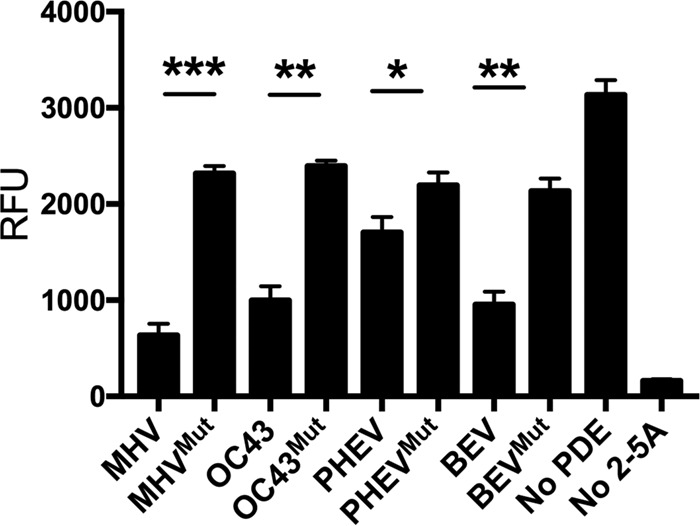
Assay of PDE activities of coronavirus and torovirus PDEs. Recombinant WT and mutant PDEs were incubated with 2-5A for 60 min, and the remaining substrate was quantified using an indirect FRET-based assay as described in Materials and Methods. The number of relative fluorescence units (RFU) is proportional to the amount of 2-5A remaining. Data shown are from one representative experiment of three independent experiments, each carried out in triplicate with separate enzyme preparations, and are expressed as means and standard errors of the means (SEM). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Coronavirus and torovirus PDEs inhibit RNase L when expressed from a chimeric MHV NS2 mutant backbone.
To investigate whether the NS2 proteins of OC43, HECoV, ECoV, and PHEV and the BEV pp1a-CTD can antagonize RNase L during infection, we constructed chimeric viruses expressing each exogenous PDE from ORF4 (encoding NS4a and NS4b) of an MHV backbone (Fig. 4). The MHV A59 backbone we utilized encodes an H126R substitution in NS2 (MHVMut; referred to in the literature as NS2H126R) that abrogates its enzymatic activity and its ability to antagonize RNase L. MHVMut exhibits minimal replication in primary bone marrow-derived macrophages (BMMs) or in vivo (4). The chimeric viruses we constructed expressed either the exogenous PDE protein or its catalytic mutant from the ORF4 locus of MHV, which is dispensable for MHV replication in vitro and in vivo (12). Each exogenous protein was constructed with a C-terminal Flag tag to allow verification of expression from the chimeric viruses.
FIG 4.
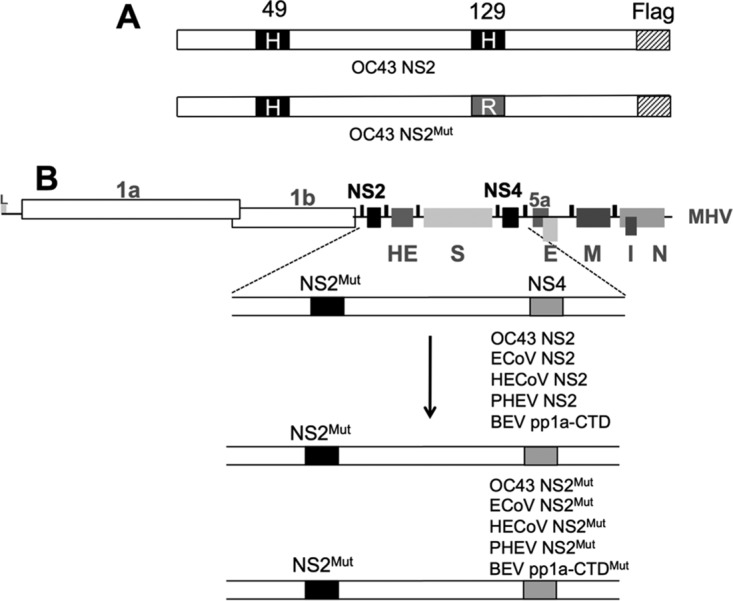
NS2 organization and construction of chimeric viruses. (A) Depiction of the NS2 protein of HCoV-OC43. The catalytic His residues at positions 49 and 129 are shown, with the His → Arg mutation shown below. (B) Genome organization of MHV, with the NS2 and NS4 loci indicated. Also shown are replicase open reading frames 1a and 1b and the genes encoding the structural proteins HE, S, E, M, N, and I as well as nonstructural protein 5a. In chimeric viruses, MHV NS2 residue 126 is mutated from H to R, rendering NS2 catalytically inactive (NS2Mut). The gene encoding the exogenous PDE or its catalytically inactive mutant was inserted in place of MHV NS4.
To assess the expression of PDEs by Western blotting, we infected L2 cells with the chimeric viruses and harvested protein lysates at 10 h postinfection (hpi). We probed for the exogenous PDEs by using a primary antibody directed against the Flag tag, utilizing glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control (Fig. 5). The OC43, HECoV, and ECoV PDEs were detectable at high levels of abundance by Western blotting, while detection of BEV pp1-CTD expression was less robust. PHEV NS2 expression from multiple viral clones as well as the swarm of uncloned recombinant viruses could not be detected by Western blotting.
FIG 5.
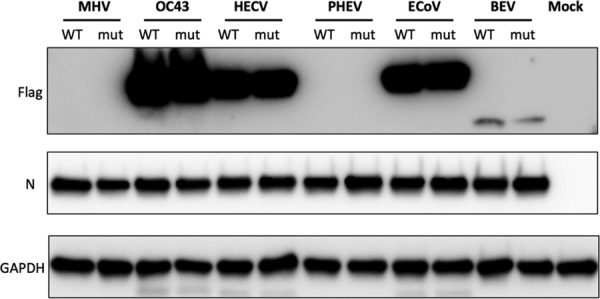
Expression of exogenous PDEs from chimeric viruses. L2 cells were infected with MHV or chimeric viruses, and proteins were harvested at 10 hpi and analyzed by Western immunoblotting. Blots were probed with antibody against Flag to detect PDEs, anti-nucleocapsid antibody to assess chimeric viral infection, and anti-GAPDH as a protein loading control. MHV NS2 (lanes 1 and 2) was not Flag tagged. Flag-tagged WT and mutant PDEs of OC43, HECoV, PHEV, ECoV, and BEV were detected as indicated. This blot was performed two times using proteins from independent infections with similar results.
Exogenous coronavirus and torovirus PDEs rescue replication of MHVMut in primary B6 BMMs through inhibition of RNase L activation.
To determine if the exogenous PDEs could antagonize RNase L in the context of infection, we infected BMMs from WT B6 mice and RNase L−/− mice with MHV, MHVMut, and the chimeric viruses and measured replication by plaque assay at 6, 9, 12, and 24 hpi. As expected, MHVMut was significantly attenuated in WT BMMs but replicated to titers equivalent to those of MHV in RNase L−/− BMMs (Fig. 6A). All of the chimeric viruses encoding WT exogenous PDEs from OC43, HECoV, ECoV, and BEV replicated to extents similar to that of WT A59 in B6 BMMs, indicating that these proteins effectively compensated for the inactive NS2H126R protein in MHV (Fig. 6A to E). In contrast, and similarly to MHVMut, the chimeras expressing catalytically inactive exogenous PDEs failed to replicate robustly in B6 BMMs but did replicate efficiently in RNase L−/− BMMs (Fig. 6A to E). The chimeric virus encoding PHEV NS2 was not assessed for replication in BMMs due to our inability to confirm PHEV NS2 expression (Fig. 5).
FIG 6.

Replication and activation of RNase L of chimeric viruses in bone marrow-derived macrophages (BMMs). (A to E) BMMs derived from WT B6 or RNase L−/− mice were infected with MHV (A) or chimeric MHVs expressing WT or mutant OC43 NS2 (B), HECoV NS2 (C), ECoV NS2 (D), or BEV pp1a-CTD (E). Viruses were titrated by plaque assay at each time point. Each time point is represented by three biological replicates titrated in duplicate, with variance expressed as SEM. Statistical significance was calculated by 2-way analysis of variance (ANOVA) in GraphPad Prism. **, P < 0.01; ***, P < 0.001. (F) Total RNA was isolated from WT B6 BMMs at 9 hpi, and rRNA integrity was assayed using an Agilent Bioanalyzer. These data are from one of at least two independent experiments with similar results.
To directly link antagonism of RNase L to the ability of the exogenous PDEs to rescue MHVMut replication, we assessed rRNA degradation in infected cells by use of a Bioanalyzer assay. We previously used this assay to demonstrate that MHV NS2, but not NS2H126R, inhibits RNase L-mediated RNA degradation and that a deficiency in RNase L obviates the requirement for NS2 in MHV replication (4). We infected WT B6 BMMs with MHV and the chimeric viruses and harvested total RNA at 9 hpi. We ran the total RNA on a Bioanalyzer to visualize the integrity of rRNA during infection with MHV and the chimeric viruses. MHV and the chimeric viruses encoding exogenous PDEs encoded by MHV, OC43, HECoV, ECoV, and BEV prevented rRNA degradation in WT B6 BMMs, while the viruses encoding the corresponding catalytically inactive PDEs failed to do so (Fig. 6F). This directly links the ability of the exogenous PDEs to rescue MHVMut replication to their antagonism of RNase L activation.
OC43 NS2 and BEV pp1a-CTD restore MHVMut replication and pathogenesis in vivo.
MHV causes profound hepatitis and associated liver pathology in B6 mice, with its replication and pathogenesis in the liver being dependent on NS2-mediated antagonism of RNase L (Fig. 7) (4). To determine whether exogenous viral PDEs can rescue replication of and restore pathogenesis to MHVMut, we infected B6 and RNase L−/− mice with MHV, MHVMut, and chimeric viruses expressing either the WT or catalytic mutant PDE from OC43 (NS2) or BEV (pp1a-CTD). At 5 days postinfection, at the time of peak titer, the mice were sacrificed and their livers harvested for virus titration by plaque assay. In WT B6 mice, chimeric viruses expressing either WT OC43 NS2 or BEV pp1-CTD replicated robustly in the liver, similarly to MHV. In contrast, and like MHVMut, the chimeric viruses expressing mutant OC43 NS2 (Fig. 7B) or BEV pp1a-CTD (Fig. 7C) were dramatically restricted, replicating only to titers below or just above the limit of detection, whereas all of the chimeric viruses replicated robustly in the livers of RNase L−/− mice (Fig. 7A to C).
FIG 7.
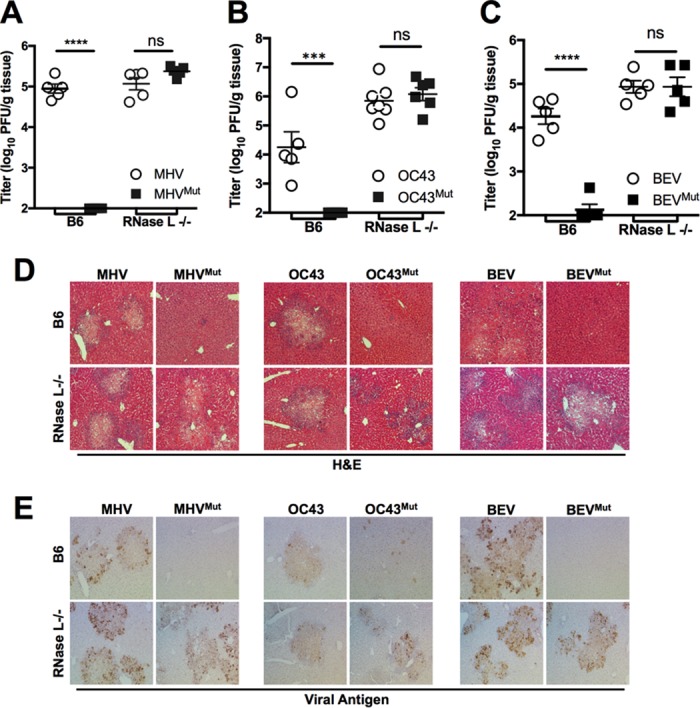
Replication and pathogenesis of chimeric viruses in vivo. (A to C) WT B6 or RNase L−/− mice (n = 5 to 7) were infected intrahepatically with MHV and MHVMut (A) or with chimeric viruses encoding WT or mutant OC43 NS2 (B) or BEV pp1a-CTD (C). At 5 days postinfection, livers were harvested and viruses titrated by plaque assay. Each data point represents a single mouse liver titrated in duplicate, with variance expressed as SEM. Statistical significance was determined by 1-way ANOVA in GraphPad Prism. ***, P < 0.001; ns, not significant. Liver sections from infected mice were stained with H&E to identify hepatic pathology (D) or with antibody to detect the MHV nucleocapsid protein (E).
To assess hepatitis in these infected mice, liver sections were assessed for viral antigen and pathological changes. Like A59, chimeric viruses expressing WT OC43 NS2 or BEV pp1a-CTD caused hepatitis in B6 mice, as indicated by pathological foci in hematoxylin-and-eosin (H&E)-stained livers, with widely observed viral antigen staining (Fig. 7D and E). Chimeric viruses expressing catalytically inactive OC43 NS2 or BEV pp1a-CTD did not cause liver pathology in B6 mice, and viral antigen was absent, consistent with the lack of replication (Fig. 7D and E). In RNase L−/− mice, all of the chimeric viruses replicated robustly and caused pathology similar to that with MHV A59 (Fig. 7D and E), further demonstrating that the restriction of the viruses expressing mutant PDEs in B6 mice was mediated by RNase L and that the exogenous PDEs functioned equivalently to MHV NS2.
DISCUSSION
We previously demonstrated 2-5A cleavage and RNase L antagonism by 2′,5′-PDEs encoded by a lineage A and a lineage C betacoronavirus (MHV and MERS-CoV, respectively) and group A rotaviruses as well as by the cellular AKAP7 CD (4, 6–8). Here we extend these findings to show that additional lineage A betacoronaviruses as well as a related torovirus family member encode 2′,5′-PDEs capable of antagonizing RNase L by cleaving 2-5A. The presence of genes encoding these proteins in multiple lineage A betacoronaviruses suggests that the gene was acquired by an ancient common ancestor of this lineage. Whether this virus was also ancestral to toroviruses and lineage C betacoronaviruses or whether 2′,5′-PDEs were acquired by viruses in multiple independent events is unclear. The maintenance of this highly conserved protein throughout lineage A betacoronaviruses supports the idea that this protein mediates an essential function in the diverse natural hosts of these viruses, spanning multiple mammalian families. Our finding of homologous PDEs in some groups of rotaviruses (8), a virus family unrelated to coronaviruses, is intriguing. A coronavirus recently isolated from bats was found to encode a protein likely to have originated from a bat orthoreovirus, which, like rotaviruses, has a dsRNA genome, suggesting the possibility of recombination between a coronavirus and a dsRNA virus (13). Further support for this idea comes from a recent report of isolation of a MERS-CoV-like coronavirus and a rotavirus from the feces of Korean bats (14). Additionally, the viruses encoding the PDEs we describe here infect different tissues within their hosts (1, 15, 16), indicating that RNase L antagonism may be required for robust replication in diverse cell types. For example, although MHV is hepatotropic, OC43 infects the upper airway, while other PDEs described here are encoded by enterotropic viruses (1, 15, 16).
The PDEs encoded by OC43, HECoV, ECoV, and BEV antagonized RNase L and rescued the replication of MHVMut in primary WT B6 BMMs, indicating not only that they are enzymatically active 2′,5′-PDEs but also that they functionally compensate for an inactive MHV NS2 protein (Fig. 3, 6, and 7). Interestingly, the BEV-encoded PDE was able to antagonize RNase L and to rescue MHVMut replication both in vitro and in vivo, despite the apparently low level of expression (Fig. 5 to 7). This is not surprising, as the MERS-CoV NS4b PDE can rescue MHVMut despite its very low expression level in the cytoplasm (6). PHEV NS2 is less enzymatically active than the other PDEs (Fig. 3), suggesting that it may be less able to antagonize RNase L. However, since we could not detect its expression by Western blotting of the PHEV PDE from a chimeric virus (Fig. 5), further work will be needed to determine if it has RNase L antagonist activity in the context of infection. Interestingly, both the BEV and PHEV PDEs are truncated at their carboxy termini, similarly to the rotavirus PDE (Fig. 3) (8); clearly, the carboxy-terminal sequences are not required for cleavage of 2-5A or for RNase L antagonism, as the rotavirus VP3-CTD and BEV PDEs have activities similar to that of MHV NS2 (Fig. 3) (8). Nevertheless, the diminished enzymatic activity of PHEV NS2 relative to that of the other PDEs suggests that while the PDE may have been essential in the PHEV ancestor, it may not be required in the cells targeted by PHEV in its porcine host. However, RNase L is likely actively antiviral in other porcine tissues or stages of development, as suggested by the presence of an RNase L antagonist in protein 7 of transmissible gastroenteritis virus (17).
Although the chimeric MHVs encoding OC43 NS2 and BEV pp1a-CTD did not replicate quite as well as MHV in vivo (Fig. 7), this is unlikely to be due to disruption of the ORF4 gene by insertion of the exogenous PDE genes, as ablation of ORF4 expression within the genome of MHV strain JHM had no effect on replication in vitro or on in vivo pathogenesis and the MHV strain A59 ORF4 is disrupted by a termination codon (18). Nevertheless, these chimeric viruses replicated robustly in vivo, causing hepatitis, and their respective mutants replicated to wild-type titers in the livers of RNase L−/− mice, indicating that restriction of the mutants in WT B6 mice is due to RNase L activity.
Overall, we have demonstrated that active 2′,5′-PDEs are a conserved feature of lineage A betacoronavirus genomes and that a homologous domain is encoded in the first open reading frame of a related nidovirus, BEV. This suggests that RNase L is a potent antiviral effector in diverse species and tissues due to the wide host range represented by the viruses encoding these newly characterized PDEs. Thus far, the group of PDE expressing viruses includes the lineage A and lineage C betacoronaviruses as well as the related toroviruses and the unrelated group A rotaviruses (4, 6, 8). Finally, since 2′,5′-PDEs are potent antagonists of host antiviral defenses encoded by multiple and diverse viruses within the Coronaviridae, this class of protein may have the potential to be a broad-spectrum therapeutic target for human viruses, including OC43, a ubiquitous agent of the common cold, and MERS-CoV.
MATERIALS AND METHODS
Cell lines and mice.
Murine fibroblast L2 (L2) cells, murine 17 clone 1 (17CL1) cells, and baby hamster kidney cells expressing the MHV receptor (BHK-R cells) were cultured as previously described (19, 20). C57BL/6 (B6) mice were originally procured from the National Cancer Institute's mouse repository, and RNase L−/− mice on a B6 genetic background were derived by Robert Silverman (21) and subsequently bred in the University of Pennsylvania animal facility. All experiments involving mice were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. Primary bone marrow-derived macrophages (BMMs) were derived from marrow harvested from the hind limbs (tibias and femurs) of 4- to 6-week-old B6 or RNase L−/− mice as described previously (4, 22). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (HyClone) and 20% L929 cell-conditioned medium for 6 days before infection.
Plasmids.
NS2 genes from the lineage A betacoronaviruses OC43, HECoV, ECoV, and PHEV and the pp1a carboxy-terminal domain (CTD) from torovirus Berne were synthesized and cloned into pUC57 by BioBasic, yielding pUC-OC43NS2, pUC-HECoVNS2, pUC-ECoVNS2, pUC-PHEVNS2, and pUC-pp1a. The second catalytic His-to-Arg substitutions were made by site-directed mutagenesis in all plasmids, resulting in pUC-OC43NS2H129R, pUC-HECVNS2H129R, pUC-NC99NS2H129R, pUC-PHEVNS2H129R, and pUC-pp1aH4516R. Select genes were subsequently subcloned into the pMal parallel-2 expression vector, resulting in pMAL-OC43NS2, pMAL-OC43NS2H129R, pMAL-PHEVNS2, pMAL-PHEVNS2H129R, pMAL-pp1a, and pMAL-pp1aH4516R. MHV NS2 and NS2H129R had previously been cloned into pMAL-c2 (4).
Purification of recombinant PDEs from E. coli and FRET assay.
MBP-PDE fusion proteins were expressed from pMAL plasmids in E. coli BL21 T7 expression competent cells (NEB, Inc., Ipswich, MA) and purified by affinity chromatography followed by ion-exchange chromatography on a MonoQ GL10/100 column, using a NaCl gradient from 0 to 1 M in 20 mM NaCl as previously described (4, 23). The integrity and purity of the purified MBP fusion proteins were determined by SDS-PAGE and Coomassie blue R250 staining. The extents of purity were similar for all of the enzymes as assessed by SDS-PAGE analysis. To assess enzymatic activity, purified protein (10 μM MBP [420 μg/ml] as a control or 1 μM OC43 [75 μg/ml], BEV [60 μg/ml], PHEV [65 μg/ml], or MHV [70 μg/ml] MBP-PDE fusion protein) in 150 μl of assay buffer (20 μM HEPES [pH 7.2], 10 mM MgCl2, 1 mM dithiothreitol) was incubated at 30°C with (2′-5′)p3A3 (2-5A). After 1 h, reactions were stopped by heat inactivation at 95°C for 3 min followed by 30 min of centrifugation at 20,000 × g (4°C), and supernatants were carefully removed. A fluorescence resonance energy transfer (FRET) assay was used to assess enzymatic activity by measuring the amount of uncleaved, intact 2-5A left in the reaction mixture, as previously described (11). The ability of recombinant enzyme to degrade 2-5A was determined by a FRET-based RNase L activation assay using an authentic 2-5A trimer as described earlier (4, 6, 8, 11). Assays were performed three times in triplicate, using two separate enzyme preparations.
Viruses and chimeric recombinant virus construction.
Wild-type MHV strain A59 and the mutant virus NS2H126R (referred to as MHV and MHVMut, respectively, herein) were described previously (20). The chimeric viruses were constructed based on the infectious cDNA clone icMHV-A59 (19, 24). The wild-type and mutant PDE genes were PCR amplified from the pUC plasmids described above by use of primers bearing SalI and NotI restriction sites. After purification and digestion with SalI and NotI, the fragments were cloned into icMHV-A59 fragment G encoding an NS2H126R mutation, as previously described (8), and confirmed by DNA sequencing. The full-length A59 genomic cDNA was assembled, and the recombinant viruses were recovered in BHK-R cells as previously described (8, 19, 24). When virus cytopathology was observed, virus was plaque purified from the supernatant and amplified on 17CL1 cells for use. The pairs of chimeric viruses expressing WT and mutant PDEs were named by the source of the PDE, as follows: OC43 and OC43Mut, HECoV and HECoVMut, PHEV and PHEVMut, ECoV and ECoVMut, and BEV and BEVMut. The PDE gene and flanking regions were amplified by PCR from the cloned chimeric virus genomes and the sequences verified. The primers used for sequencing were Fns4 (5′-TTGTTGTGATGAGTATGGAG), which maps 136 nucleotides upstream of the ATG start codon of the PDE gene, and Rns4 (5′-GCGTAACCATGCATCACTCAC), which maps 139 nucleotides downstream of the PDE open reading frame. The sequenced region included the SalI and NotI restriction sites as well as the transcription regulatory sequence (TRS) for ORF4 and ORF5a.
Chimeric MHV infections of BMMs.
BMMs were mock infected or infected at a multiplicity of infection (MOI) of 1 PFU/cell (in triplicate) and allowed to adsorb for 1 h at 37°C. Cultures were washed with phosphate-buffered saline (PBS) 3 times and fed with medium. At the indicated times, cells were lysed and analyzed for degradation of RNA (described below), or supernatants were harvested for quantification of viral titers by plaque assay on L2 cells (8).
Immunoblotting.
L2 cells were infected with MHV or chimeric viruses (MOI = 1 PFU/cell). At 10 hpi, cells were lysed in Nonidet P-40 (NP-40) buffer (1% NP-40, 2 mM EDTA, 10% glycerol, 150 mM NaCl, and 50 mM Tris, pH 8.0) containing protease inhibitors (Roche). Protein concentrations were measured using a DC protein assay kit (Bio-Rad). Supernatants were mixed 3:1 with 4× SDS-PAGE sample buffer. Samples were boiled, separated by 4 to 15% SDS-PAGE, and transferred to polyvinylidene difluoride (PVDF) membranes. Blots were blocked with 5% nonfat milk and probed with the following antibodies: mouse anti-Flag monoclonal antibody M2 (1:1,000; Agilent), mouse anti-MHV nucleocapsid monoclonal antibody (1:400; a gift from Julian Leibowitz), and mouse anti-GAPDH monoclonal antibody (1:1,000; Thermo Scientific). Horseradish peroxidase (HRP)-conjugated anti-mouse (1:5,000; Santa Cruz) secondary antibodies were used to detect the primary antibodies. The blots were visualized using Super Signal West Dura Extended Duration substrate (Thermo Scientific). Blots were probed sequentially with antibodies, with the blots stripped between antibody treatments.
Analysis of RNase L-mediated rRNA degradation.
RNAs from WT B6 BMMs infected with MHV or with chimeric viruses encoding WT and catalytically inactive PDEs were harvested at the indicated time points by use of a Qiagen RNeasy kit. RNase was denatured at 70°C for 2 min and analyzed with an Agilent Bioanalyzer 2100 on a eukaryotic total RNA nanochip. The Bioanalyzer instrument converts the electropherogram generated for each sample into a pseudogel, as depicted in Fig. 6 (4).
Replication in mice.
Four-week-old B6 or RNase L−/− mice (4, 6–8) were anesthetized with isoflurane (Abbott Laboratories, Chicago, IL) and inoculated intrahepatically with 2,000 PFU of virus in 50 μl of Dulbecco's PBS (DPBS) (Gibco) containing 0.75% bovine serum albumin (Sigma). Mice were euthanized with CO2 and perfused with DPBS (Gibco) and their livers harvested at day 5 postinoculation. Part of the liver was fixed for histology as described below, and the rest was homogenized and its viral titer determined by plaque assay of liver homogenates on L2 cells (8, 25). A piece of each liver was fixed overnight in 4% paraformaldehyde, embedded in paraffin, and sectioned. Sections were stained with hematoxylin and eosin (H&E) or blocked with 10% normal donkey serum and immunostained with a 1:20 dilution of a monoclonal antibody against the MHV nucleocapsid (N) protein (1:1,000 dilution). Staining was developed using avidin-biotin-immunoperoxidase (Vector Laboratories).
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) under awards F32AI114143 to J.M.T., R21AI114920 to S.R.W., and R01AI104887 to S.R.W. and R.H.S.
REFERENCES
- 1.Weiss SR, Leibowitz JL. 2011. Coronavirus pathogenesis. Adv Virus Res 81:85–164. doi: 10.1016/B978-0-12-385885-6.00009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Guan Y, Rozanov M, Spaan WJ, Gorbalenya AE. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol 331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazumder R, Iyer LM, Vasudevan S, Aravind L. 2002. Detection of novel members, structure-function analysis and evolutionary classification of the 2H phosphoesterase superfamily. Nucleic Acids Res 30:5229–5243. doi: 10.1093/nar/gkf645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. 2012. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverman RH. 2007. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol 81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thornbrough JM, Jha BK, Yount B, Goldstein SA, Li Y, Elliott R, Sims AC, Baric RS, Silverman RH, Weiss SR. 2016. Middle East respiratory syndrome coronavirus NS4b protein inhibits host RNase L activation. mBio 7:e00258. doi: 10.1128/mBio.00258-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gusho E, Zhang R, Jha BK, Thornbrough JM, Dong B, Gaughan C, Elliott R, Weiss SR, Silverman RH. 2014. Murine AKAP7 has a 2′,5′-phosphodiesterase domain that can complement an inactive murine coronavirus ns2 gene. mBio 5:e01312-14. doi: 10.1128/mBio.01312-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, Patton JT, Silverman RH, Weiss SR. 2013. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc Natl Acad Sci U S A 110:13114–13119. doi: 10.1073/pnas.1306917110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogden KM, Hu L, Jha BK, Sankaran B, Weiss SR, Silverman RH, Patton JT, Prasad BV. 2015. Structural basis for 2′-5′-oligoadenylate binding and enzyme activity of a viral RNase L antagonist. J Virol 89:6633–6645. doi: 10.1128/JVI.00701-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gold MG, Smith FD, Scott JD, Barford D. 2008. AKAP18 contains a phosphoesterase domain that binds AMP. J Mol Biol 375:1329–1343. doi: 10.1016/j.jmb.2007.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thakur CS, Xu Z, Wang Z, Novince Z, Silverman RH. 2005. A convenient and sensitive fluorescence resonance energy transfer assay for RNase L and 2′,5′ oligoadenylates. Methods Mol Med 116:103–113. [DOI] [PubMed] [Google Scholar]
- 12.Ontiveros E, Kuo L, Masters PS, Perlman S. 2001. Inactivation of expression of gene 4 of mouse hepatitis virus strain JHM does not affect virulence in the murine CNS. Virology 289:230–238. doi: 10.1006/viro.2001.1167. [DOI] [PubMed] [Google Scholar]
- 13.Huang C, Liu WJ, Xu W, Jin T, Zhao Y, Song J, Shi Y, Ji W, Jia H, Zhou Y, Wen H, Zhao H, Liu H, Li H, Wang Q, Wu Y, Wang L, Liu D, Liu G, Yu H, Holmes EC, Lu L, Gao GF. 2016. A bat-derived putative cross-family recombinant coronavirus with a reovirus gene. PLoS Pathog 12:e1005883. doi: 10.1371/journal.ppat.1005883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim HK, Yoon SW, Kim DJ, Koo BS, Noh JY, Kim JH, Choi YG, Na W, Chang KT, Song D, Jeong DG. 2016. Detection of severe acute respiratory syndrome-like, Middle East respiratory syndrome-like bat coronaviruses and group H rotavirus in faeces of Korean bats. Transbound Emerg Dis 63:365–372. doi: 10.1111/tbed.12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navas-Martin S, Weiss SR. 2003. SARS: lessons learned from other coronaviruses. Viral Immunol 16:461–474. doi: 10.1089/088282403771926292. [DOI] [PubMed] [Google Scholar]
- 16.Navas-Martin SR, Weiss S. 2004. Coronavirus replication and pathogenesis: implications for the recent outbreak of severe acute respiratory syndrome (SARS), and the challenge for vaccine development. J Neurovirol 10:75–85. doi: 10.1080/13550280490280292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cruz JL, Sola I, Becares M, Alberca B, Plana J, Enjuanes L, Zuniga S. 2011. Coronavirus gene 7 counteracts host defenses and modulates virus virulence. PLoS Pathog 7:e1002090. doi: 10.1371/journal.ppat.1002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiss SR, Zoltick PW, Leibowitz JL. 1993. The ns4 gene of mouse hepatitis virus (MHV), strain A59 contains two ORFs and thus differs from ns4 of the JHM and S strains. Arch Virol 129:301–309. doi: 10.1007/BF01316905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yount B, Denison MR, Weiss SR, Baric RS. 2002. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J Virol 76:11065–11078. doi: 10.1128/JVI.76.21.11065-11078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth-Cross JK, Stokes H, Chang G, Chua MM, Thiel V, Weiss SR, Gorbalenya AE, Siddell SG. 2009. Organ-specific attenuation of murine hepatitis virus strain A59 by replacement of catalytic residues in the putative viral cyclic phosphodiesterase ns2. J Virol 83:3743–3753. doi: 10.1128/JVI.02203-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman RH. 1997. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J 16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caamano J, Alexander J, Craig L, Bravo R, Hunter CA. 1999. The NF-kappa B family member RelB is required for innate and adaptive immunity to Toxoplasma gondii. J Immunol 163:4453–4461. [PubMed] [Google Scholar]
- 23.Sheffield P, Garrard S, Derewenda Z. 1999. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr Purif 15:34–39. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- 24.Sperry SM, Kazi L, Graham RL, Baric RS, Weiss SR, Denison MR. 2005. Single-amino-acid substitutions in open reading frame (ORF) 1b-nsp14 and ORF 2a proteins of the coronavirus mouse hepatitis virus are attenuating in mice. J Virol 79:3391–3400. doi: 10.1128/JVI.79.6.3391-3400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gombold JL, Hingley ST, Weiss SR. 1993. Fusion-defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J Virol 67:4504–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luytjes W, Bredenbeek PJ, Noten AF, Horzinek MC, Spaan WJ. 1988. Sequence of mouse hepatitis virus A59 mRNA 2: indications for RNA recombination between coronaviruses and influenza C virus. Virology 166:415–422. doi: 10.1016/0042-6822(88)90512-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.St Jean JR, Jacomy H, Desforges M, Vabret A, Freymuth F, Talbot PJ. 2004. Human respiratory coronavirus OC43: genetic stability and neuroinvasion. J Virol 78:8824–8834. doi: 10.1128/JVI.78.16.8824-8834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Guy JS, Snijder EJ, Denniston DA, Timoney PJ, Balasuriya UB. 2007. Genomic characterization of equine coronavirus. Virology 369:92–104. doi: 10.1016/j.virol.2007.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vijgen L, Keyaerts E, Lemey P, Maes P, Van Reeth K, Nauwynck H, Pensaert M, Van Ranst M. 2006. Evolutionary history of the closely related group 2 coronaviruses: porcine hemagglutinating encephalomyelitis virus, bovine coronavirus, and human coronavirus OC43. J Virol 80:7270–7274. doi: 10.1128/JVI.02675-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snijder EJ, den Boon JA, Bredenbeek PJ, Horzinek MC, Rijnbrand R, Spaan WJ. 1990. The carboxyl-terminal part of the putative Berne virus polymerase is expressed by ribosomal frameshifting and contains sequence motifs which indicate that toro- and coronaviruses are evolutionarily related. Nucleic Acids Res 18:4535–4542. doi: 10.1093/nar/18.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]