

ABSTRACT
Clade 2.3.4.4 highly pathogenic avian influenza viruses (H5Nx) have spread from Asia to other parts of the world. Since 2014, human infections with clade 2.3.4.4 highly pathogenic avian influenza H5N6 viruses have been continuously reported in China. To investigate the genesis of the virus, we analyzed 123 H5 or N6 environmental viruses sampled from live-poultry markets or farms from 2012 to 2015 in Mainland China. Our results indicated that clade 2.3.4.4 H5N2/N6/N8 viruses shared the same hemagglutinin gene as originated in early 2009. From 2012 to 2015, the genesis of highly pathogenic avian influenza H5N6 viruses occurred via two independent pathways. Three major reassortant H5N6 viruses (reassortants A, B, and C) were generated. Internal genes of reassortant A and B viruses and reassortant C viruses derived from clade 2.3.2.1c H5N1 and H9N2 viruses, respectively. Many mammalian adaption mutations and antigenic variations were detected among the three reassortant viruses. Considering their wide circulation and dynamic reassortment in poultry, we highly recommend close monitoring of the viruses in poultry and humans.
IMPORTANCE Since 2014, clade 2.3.4.4 highly pathogenic avian influenza (H5Nx) viruses have caused many outbreaks in both wild and domestic birds globally. Severe human cases with novel H5N6 viruses in this group were also reported in China in 2014 and 2015. To investigate the genesis of the genetic diversity of these H5N6 viruses, we sequenced 123 H5 or N6 environmental viruses sampled from 2012 to 2015 in China. Sequence analysis indicated that three major reassortants of these H5N6 viruses had been generated by two independent evolutionary pathways. The H5N6 reassortant viruses had been detected in most provinces of southern China and neighboring countries. Considering the mammalian adaption mutations and antigenic variation detected, the spread of these viruses should be monitored carefully due to their pandemic potential.
KEYWORDS: H5N6, highly pathogenic avian influenza, reassortment
INTRODUCTION
Since the 1990s, outbreaks of highly pathogenic avian influenza (HPAI) have occurred frequently. The initial outbreak of HPAI H5N1 virus occurred in domestic geese in southern China in 1996 (1, 2). Following that, continued circulation of HPAI H5N1 virus in birds throughout many regions of the Eastern Hemisphere led to dozens of phylogenetic clades emerging through evolution (3). Human infections with serious clinical outcomes were sporadically reported in regions associated with epidemics.
The widely reported spread of HPAI H5 virus from Asia into the Middle East, Europe, and Africa was due to a clade 2.2 (Qinghai-like) H5N1 virus that infected wild birds (4). In 2014 and 2015, a new clade, 2.3.4.4, which included several subtypes (H5N1, H5N2, H5N3, H5N5, H5N6, and H5N8), raised great concern when it was detected in Asia, Europe, and North America. The clade 2.3.4.4 H5 virus, which had acquired the N8 neuraminidase (NA) gene and was represented by the A/Duck/Korea/Buan2/2014 (Buan2)-like H5N8 virus, had spread following the bird migration pathway to Europe and North America (5–8). During the same period in East Asia, where the 2.3.4.4 clade virus originated, wide outbreaks of HPAI H5N8, H5N6, and H5N2 were reported (http://www.oie.int/). In April 2014, the first fatal human case of HPAI H5N6 virus infection was identified in China (9). Up to 8 June 2016, a total of 14 H5N6 cases had been reported in China (http://www.who.int/en/), raising the threat of this viral infection to public health.
The HPAI H5 virus is a significant threat not only to animal health, in particular for the poultry industry, but also to human health due to its pandemic potential. As for clade 2.3.4.4 H5 viruses, HPAI H5N6, which has been detected in China, Laos (10), and Vietnam (11), is the only subtype to date that has been reported to infect humans. It is important to understand the origins and genesis of these subtype viruses and to determine their relationships with other subtype viruses in this clade. Previous studies revealed that the HPAI H5N6 virus contained the hemagglutinin (HA) gene of H5 clade 2.3.4.4, the internal genes of H5 clade 2.3.2.1, and the NA gene from the H6N6 avian virus (9, 12, 13). Based on the genetic evidence, HPAI H5N6 virus likely originated from migratory waterfowl (14). Further reassortment with other avian influenza viruses generated multiple genotypes of HPAI H5N6 viruses (15–19). The origins and mechanisms of dissemination of the HPAI H5N6 viruses remain to be fully elucidated, and this information is urgently needed for the development of effective disease control and prevention strategies.
RESULTS
Surveillance results.
A total of 123 H5 or N6 environmental viruses (1 H4N6, 44 H5N1, 7 H5N2, 56 H5N6, 1 H5N8, and 14 H6N6 viruses) sampled from 2012 to 2015 in Mainland China were sequenced (Fig. 1). Based on evolutionary analysis (Fig. 2A) and H5 clade nomenclature designated by the WHO/OIE/FAO H5N1 Evolution Working Group, the 108 H5 viruses from our study grouped into clades 2.3.4.4 (68/108), 2.3.2.1b (5/108), 2.3.2.1c (33/108), and 7.2 (2/108). These environmental isolates shared high similarity with duck or chicken isolate viruses from public databases, for example, A/Environment/Hubei/38005/2014 (H5N6) and A/Duck/Wuhan/WHYF03/2015 (H5N6) (see Fig. S1A in the supplemental material).
FIG 1.

Time distribution of H5 or N6 viruses isolated in this study. The number of isolates of each subtype was accumulated by month.
FIG 2.

Evolutionary analysis of H5 gene sequences. (A) Maximum likelihood phylogenetic analysis of HA genes of 108 HPAI H5 viruses. The viruses isolated in our study are highlighted in red. (B) Bayesian maximum clade credibility (MCC) phylogeny of clade 2.3.4.4 HPAI H5Nx viruses. The phylogenetic clades that included clade 2.3.4.4 HPAI H5N2/N6/N8 viruses were obtained from the dated phylogeny of HA gene segments constructed by molecular phylogenetic analysis and are further aligned onto the same time scale. The branches in purple, blue, red, gray, and black represent clade 2.3.4.4 HPAI H5N2, H5N8, H5N6, H5N5, and H5N1 viruses, respectively. The tMRCAs of the clade HPAI H5Nx viruses and the two H5N6 subgroups are indicated by red, blue, and yellow dots. The posterior probabilities of the main branches are shown as numbers. For details of the phylogeny, see Fig. S1 in the supplemental material.
Genetic divergence of clade 2.3.4.4 HPAI H5 genes.
To elucidate the timing and pattern of divergence, we integrated the evolutionary analysis of the HA gene of HPAI H5Nx viruses onto the same time scale. As shown in Fig. 2B, HA genes of clade 2.3.4.4 HPAI H5Nx viruses can be divided into four subclades, I, II, III, and IV. The H5 viruses reassorted with different NA subtypes, including N2, N6, and N8. HPAI H5N6 mainly distributed into subclades I and II along with a few HPAI H5N2 and H5N1 viruses, respectively. The majority of viruses in subclade III were HPAI H5N2 and H5N8 viruses, and in subclade IV were mainly HPAI H5N8 viruses. The median time to the most recent common ancestor (tMRCA) of these four subclades was estimated to be early 2009 (95% highest posterior density [HPD], November 2008 to August 2009).
HPAI H5N8 viruses have evolved into two subgroups, the larger being subclade III and the smaller being subclade IV, which was in agreement with an early study on Korean H5N8 lineages (20). The smaller subclade contained the viruses isolated in Korea and neighboring regions, whereas the larger subclade included viruses from Asia, Europe, and North America. This result indicated that after the reassortment of clade HPAI H5 with N8 genes, some viruses circulated regionally, while others (Buan2-like viruses) dispersed following bird migration pathways and disseminated worldwide.
Similar to HPAI H5N8, two subgroups of HPAI H5N2 viruses have been observed. HPAI H5N2 viruses in subclade I were isolated in Mainland China from 2010 to 2014. This subclade can be distinguished from H5N8 viruses isolated in 2009. In subclade III, HPAI H5N2 viruses were isolated from 2014 to 2015 and grouped together with Buan2-like HPAI H5N8 viruses. In this subclade, the H5N2 viruses which have been responsible for outbreaks in North America were derived from the reassortment of H5N8 and local avian influenza viruses.
HPAI H5N6 viruses mainly circulated in Southern China, Laos, and Vietnam. The HA genes of subclade I viruses may have originated from HPAI H5N2 viruses, while the HA genes of subclade II may be derived from HPAI H5N8 viruses. The tMRCA of subclades I and II was estimated to be late 2011 (95% HPD, June 2011 to June 2012) and mid-2012 (95% HPD, December 2011 to December 2012), respectively. These results indicated that clade 2.3.4.4 H5N6 viruses might have been generated via different reassortment events. To examine this assumption, we next investigated the NA and internal genes.
Genetic divergence of NA genes of clade 2.3.4.4 HPAI H5N6 viruses.
Similar to the HA genes, a maximum clade credibility phylogenetic tree of the NA genes of HPAI H5N6 viruses divided into two subclades (Fig. 3). A significant difference between the two subclades was that subclade II consisted of H6N6 viruses with 59 to 69 deletions in the NA protein stalk. In late 2003, N6 genes evolved into two subclades based on the tMRCA estimation (June 2002 to November 2004). Thereafter, the N6 genes reassorted with two subclades of HPAI H5 viruses during the middle of 2011 (95% HPD, December 2010 to July 2012) and the middle of 2012 (95% HPD, October 2011 to December 2012), generating two subclades of H5N6 viruses circulating in ducks.
FIG 3.
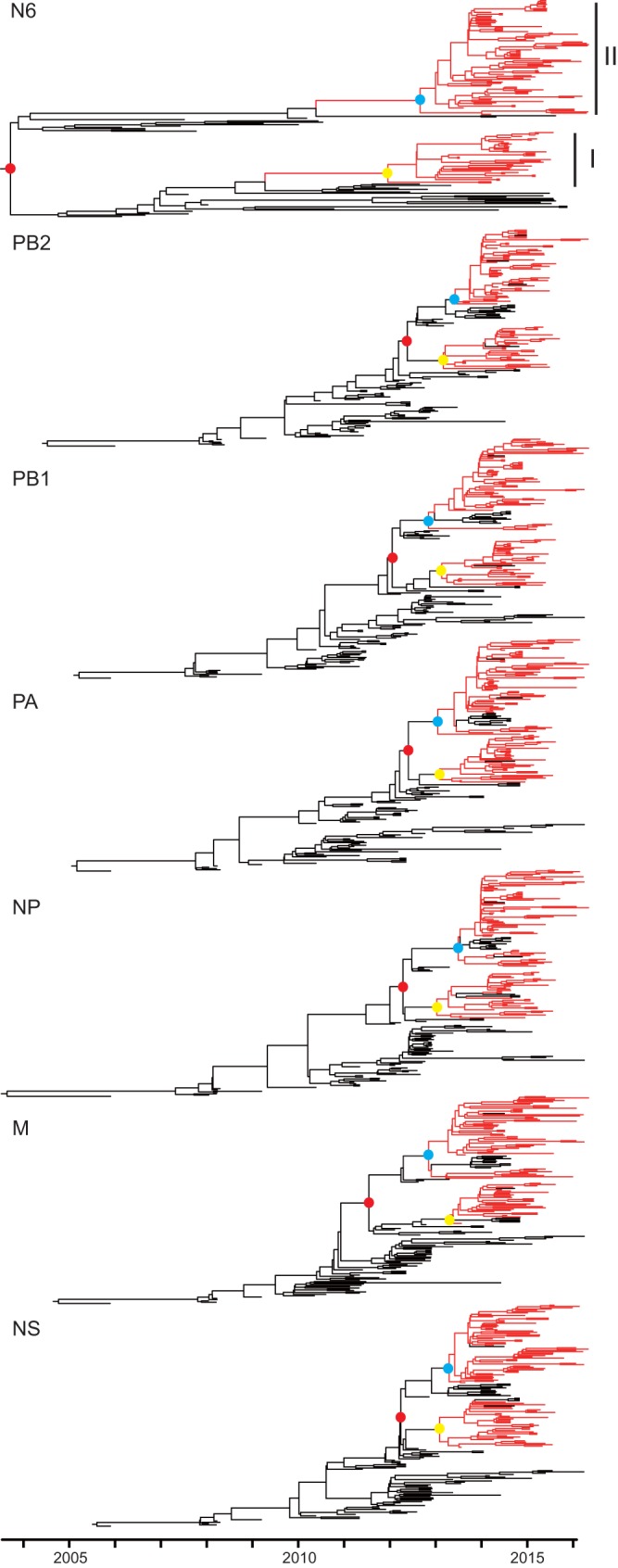
Genomic divergence of NA and six internal genes of clade 2.3.4.4 HPAI H5N6 viruses. The red and black branches represent the clade 2.3.4.4 HPAI H5N6 viruses and other related viruses, respectively. The tMRCAs of the two subgroups and each subgroup of H5N6 viruses are highlighted in solid red, blue, and yellow, respectively. For details of the phylogeny, see Fig. S2 to S8 in the supplemental material.
Thus, the two subclades of HPAI H5N6 viruses may have been generated by two independent reassortment events. Specifically, subclade I H5N2 viruses might have reassorted with H6N6 viruses without NA deletion in late 2011. Then subclade II H5N8 viruses might have reassorted with the H6N6 viruses with 59 to 69 deletions in mid-2012.
Dynamic reassortment of internal genes of clade 2.3.4.4 HPAI H5N6 viruses.
Consistent with the HA and NA genes, six internal genes of HPAI H5N6 viruses that originated from clade 2.3.2.1c H5N1 viruses divided into two subclades. The tMRCA of the two clades was dated to be between mid-2012 and mid-2013 (95% HPD, March 2012 to July 2013 [Fig. 3]), similar to the case with the HA and NA genes. On the basis of combining the results from all eight genes, the HPAI H5N6 viruses likely underwent two independent reassortment events.
As shown in Fig. 4, clade 2.3.2.1c HPAI H5N1 viruses provided six internal genes to the majority of subgroup I and II H5N6 viruses. However, subgroup II H5N6 viruses isolated in Yunnan and Guangdong provinces from late 2015 had acquired the six internal genes from H9N2 viruses. Another two viruses from subgroup I H5N6 acquired the PB2 or M genes from H9N2 viruses. Additionally, several subgroup II H5N6 viruses acquired the PB2 gene from H6N6 viruses. Reassortment among viruses from the two H5N6 subclades has occasionally been observed.
FIG 4.
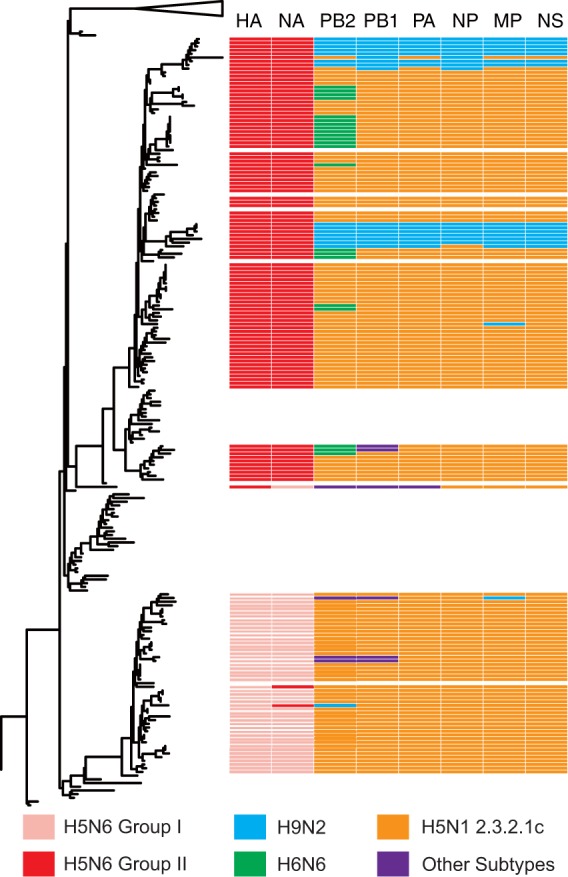
Reassortant patterns of the clade 2.3.4.4 HPAI H5N6 viruses and their potential donor-like viruses. A phylogenetic tree demonstrating the similar evolutionary pattern of clade 2.3.4.4 HPAI H5Nx HA gene in Fig. 2 is shown on the left. The unrooted tree was based on the full-length HA gene sequences. Reassortant patterns of the H5N6 viruses are listed on the right. Eight gene segments are indicated at the top of each bar. The colors of the bars represent the potential donor-like viruses listed at the bottom.
Possible evolutionary pathways of clade 2.3.4.4 HPAI H5N6 viruses.
The HA gene of four subclades of H5Nx viruses originated from clade 2.3.4 HPAI H5N1 viruses that had been introduced into and established among chickens in China since 2005. In 2007, the virus evolved into clade 2.3.4.4. From 2009 to 2012, the clade 2.3.4.4 H5 viruses reassorted with different NA subtype viruses from waterfowl, including the N2, N6, and N8 subtypes, generating the HPAI H5N2/N6/N8 viruses (Fig. 5).
FIG 5.

Possible evolutionary pathways toward the generation of clade 2.3.4.4 HPAI H5N2/N6/N8 viruses and the diverse genotypes of HPAI H5N6 viruses. Virus particles are represented by colored ovals containing horizontal bars that represent the eight gene segments (from top to bottom: PB2, PB1, PA, HA, NP, NA, M, and NS). Segments in descendant viruses are colored according to their corresponding source viruses (top) to illustrate gene ancestry through reassortment events. Source viruses for a reassortment are adjacent to arrow tails; arrowheads point to the resulting reassortants. A broken bar in segment 6 (NA) indicates a stalk region deletion. The timeline on the left indicates the possible time of virus emergence or reassortment events.
Phylogenetic analysis and tMRCA results suggested that two independent reassortment pathways generated two subclades of H5N6 viruses from 2011 to 2013. In the first pathway, H5N2 viruses with the clade 2.3.4.4 HA gene may have reassorted H6N6 with the full-length NA gene between mid-2011 and mid-2012; then a further reassortment might have occurred with six internal genes from chicken clade 2.3.2.1c H5N1 viruses, generating the reassortant A viruses. In the second pathway, H5N8 viruses with the clade 2.3.4.4 HA gene, and H6N6 viruses with the NA gene containing the deletion from positions 59 to 69 in the stalk region, may have reassorted with six internal genes from poultry clade 2.3.2.1c H5N1 viruses, generating reassortant B H5N6 viruses. Since 2015, consecutive reassortment of reassortant B H5N6 viruses with six internal genes from chicken H9N2 viruses generated reassortant C H5N6 viruses (Fig. 5). However, alternative reassortant pathways and hosts cannot be definitively excluded.
Geographic distribution of three reassortant A, B, and C viruses.
As shown in Fig. 6, almost all of the H5N6 viruses were distributed in southern China. Only reassortant A viruses have been detected in the Xinjiang and Jilin provinces in northern China. Both reassortant A and B viruses have been detected in neighboring countries, including Laos and Vietnam. Reassortant C viruses have been reported in the Yunnan and Guangdong provinces.
FIG 6.

Geographic distributions of three reassortant HPAI H5N6 viruses in Mainland China and neighboring Laos and Vietnam. Provinces in which clade 2.3.4.4 HPAI H5N6 viruses were isolated in Mainland China, Laos, and Vietnam are in dark gray. Solid red squares, blue triangles, and cyan circles indicate the reassortant A, B, and C H5N6 viruses, respectively. This map was drawn using ArcGIS (ESRI) software version 9.
All three reassortant viruses have been reported to cause human infections. The virus from the first reported H5N6 human case in Sichuan Province in 2014 belonged to the reassortant A viruses. Viruses isolated in Guangdong Province in late 2014 possessed the gene cassette from reassortant B viruses, and human isolates from the Yunnan and Guangdong provinces since 2015 belonged to the reassortant C viruses.
Molecular characteristics and antigenic analysis of three reassortant A, B, and C isolates.
A list of amino acid substitutions of biological relevance for mammalian adaptations or reducing drug susceptibility was investigated for the three reassortant isolates (Table 1). Although the most well-known receptor binding sites (222 to 224, H5 numbering) were still avian-like (QSG) in the three reassortant isolates (21), other mutations that may alter receptor specificity were detected, such as S123P, I151T, and T156A (22–24). The reassortant C isolates carried a deletion at position 126 in the HA, adding a potential N-glycosylation at site 124. The R152K mutation in the NA in Yunnan human isolates may affect susceptibility to inhibitors (25). Mutations E627K and D701N, which that enhanced virulence in mammals, were found in the PB2 protein of human isolates (26). Some mutations in PB1, PA, M1, and M2 associated with viral transmissibility, species specificity, virulence, or adamantine resistance were detected in reassortant C viruses due to their internal gene from H9N2 virus (27–30). The reassortant A and B viruses with NS1 genes from H5N1 carried mutations that increased virulence in mice (31, 32).
TABLE 1.
Characterization of selected molecular markers of H5N6 isolatesa
| Gene | Phenotypic characteristic(s) | Mutation | Reassortant A |
Reassortant B |
Reassortant C |
|||
|---|---|---|---|---|---|---|---|---|
| Birds or Ev | SC/26221 | Birds or Ev | GD/99710 | Birds or Ev | YN/44625 | |||
| HAb | Altered receptor specificity | S123P | T(41) | T | P(58) | P | P (7) | P |
| Add N-glycosylation site at position 124 | Deletion at position 126 | E (41) | E | E (57), deletion (1) | E | Deletion (7) | Deletion | |
| Altered receptor specificity | I151T | I (36), V (5) | I | I (8), T (50) | T | T (7) | T | |
| Altered receptor specificity, reduced transmission | T156A | A (41) | A | A (58) | T | A (7) | A | |
| Highly pathogenic cleavage peptides | R/KEKRRKR↓G | REKRRKR↓G | R/KERRRKR↓G | RERRRKR↓G | RERRRKR↓G | RERRRKR↓G | ||
| NAc | Deletion at positions 59–69 | No (41) | No | Yes (58) | Yes | Yes (7) | Yes | |
| Reduced susceptibility to NA inhibitors | R152K | R (41) | R | R (58) | R | R (7) | K | |
| PB2 | Altered virulence in mice | E627K | E (41) | E | E (58) | K | E (7) | K |
| D701N | D (41) | N | D (58) | D | D (7) | D | ||
| PB1 | Increased transmission in ferrets | I368V | I (41) | I | I (57), V (1) | I | V (7) | V |
| PB1-F2 | Increased pathogenicity in mice | 87–90 aa in length | 57 aa (40) | 57 aa | 11 aa (36) | 11 aa | 90 aa (7) | 90 aa |
| 90 aa (1) | 57 aa (21) | |||||||
| 90 aa (1) | ||||||||
| PA | Species-associated signature positions | V100A | V (41) | V | V (58) | V | V (3), A (4) | A |
| K356R | K (41) | K | K (57), R (1) | K | R (7) | R | ||
| S409N | S (41) | S | S (56), N (1), G (1) | S | N (7) | N | ||
| M1 | Altered virulence in mice | T139A | T (41) | T | T (58) | T | T (3), A (4) | A |
| M2 | Resistance to adamantine derivatives | S31N | S (41) | S | S (58) | S | N (7) | N |
| NS1 | Altered virulence in mice | Deletion at positions 80–84 | Yes (40), no (1) | Yes | Yes (58) | Yes | No (7) | No |
| D92E | E (41) | E | E (58) | E | D (7) | D | ||
| L103F | F (41) | F | F (52), S (6) | F | L (7) | L | ||
| I106 M | M (41) | M | M (54), K (4) | M | I (106) | I | ||
| Altered virulence in mice, PDZ motif | 227–230 | ESEV (41) | ESEV | ESEV (55) | ESEV | Truncated (7) | Truncated | |
| ESKV (1) | ||||||||
| ESEI (1) | ||||||||
Numbers in parentheses correspond to the numbers of isolates. Abbreviations: Ev, environment; aa, amino acid; SC/26221, A/Sichuan/26221/2014; GD/99710, A/Guangdong/99710/2014; YN/44625, A/Yunnan/44625/2015.
The H5 numbering system is used.
The N6 numbering system is used.
Hemagglutination inhibition assays were conducted, using available H5N1, H5N6, and H5N8 viruses from distinct clades and subclades, according to standard protocols using 0.5% turkey red blood cells in a biosafety level 3 (BSL3) laboratory. The results showed that viruses from clade 2.3.4.4 were antigenically different from other clades, and antigenicity varied within clades (Table 2). The Sichuan and Guangdong human isolates (representing the reassortant A and B viruses, respectively) were similar to each other but different from subclade III H5N8 virus. The Yunnan human isolate, representing the reassortant C viruses, showed very low activity with all antisera of reference viruses.
TABLE 2.
Hemagglutination inhibition reactions of influenza H5Nx viruses in experiments conducted on 10 August 2016
| Reference virus | Type of virus or reassortant | Clade | Titer obtained with viruse |
Passage | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2.2.1,H5N1,Turkey/1 | 2.3.4,H5N1,Anhui/1 | 2.3.4,H5N1,HK/AP156 | 2.3.4.2,H5N1,GZ/1 | 2.3.4.4 I,H5N6,SC/26221 | 2.3.4.4, II,H5N6,GD/99170 | 2.3.4.4, III,H5N8,GYR/41088-6 | ||||
| A/turkey/Turkey/1/2005 (H5N1) NIBRG-23a | Reverse genetics | 2.2.1 | 80 | <20 | 160 | 20 | <20 | 20 | <20 | C1E3 |
| A/Anhui/1/2005 (H5N1) IBCDC-RG6b | Reverse genetics | 2.3.4 | 20 | 320 | 1,280 | 320 | <20 | 80 | <20 | E2 |
| A/chicken/Hong Kong/AP156/2008 (H5N1) SJ002c | Reverse genetics | 2.3.4 | <20 | <20 | 640 | 40 | <20 | 20 | <20 | E2 |
| A/Guizhou/1/2013 (H5N1) IDCDC-RG35b | Reverse genetics | 2.3.4.2 | <20 | <20 | 320 | 320 | <20 | <20 | <20 | E2 |
| A/Sichuan/26221/2014 (H5N6) | Wild type | 2.3.4.4 I | <20 | <20 | 20 | <20 | 80 | 320 | <20 | E2 |
| A/Sichuan/26221/2014 (H5N6) IDCDC-RG42Ab | Reverse genetics | 2.3.4.4 I | <20 | <20 | 80 | 20 | 160 | 1280 | 40 | C1E3 |
| A/Guangdong/99710/2014 (H5N6) | Wild type | 2.3.4.4 II | <20 | <20 | 80 | 20 | 320 | 1,280 | <20 | E2 |
| A/Guangdong/99170/2014 (H5N6) RG NICd | Reverse genetics | 2.3.4.4 II | <20 | <20 | 160 | 20 | 320 | 1,280 | <20 | C1E3 |
| A/gyrfalcon/Washington/41088-6/2014 (H5N8) IDCDC-RG43Ab | Reverse genetics | 2.3.4.4 III | <20 | <20 | <20 | <20 | 160 | 160 | 80 | C1E3 |
| Test virus | ||||||||||
| A/Yunnan/44625/2015 (H5N6) | Wild type | 2.3.4.4 II | <20 | <20 | 20 | <20 | <20 | 20 | <20 | E1 |
Virus was generated by the National Institute for Biological Standards and Control, UK.
Viruses were generated by the Centers for Disease Control and Prevention, USA.
Virus was generated by the St. Jude Children's Research Hospital, USA.
Virus was generated by the Chinese National Influenza Center, China.
Bold, underlined numbers indicate the homologous titers of the reference viruses.
DISCUSSION
There are several possible evolutionary pathways for the generation of diverse genotypes of influenza virus: (i) one-time reassortment, (ii) multiple independent reassortment events, or (iii) sequential multiple-step reassortment events (33). In this study, we found that two independent reassortment events underlie the generation of reassortant A and B H5N6 viruses. Reassortant B H5N6 viruses further reassorted with H9N2 viruses to generate reassortant C H5N6 viruses. It is probable that the reassortment pathway of the H5N6 virus was due to the epidemic of clade 2.3.4.4 H5 virus in both migratory and domestic birds. Unlike clade 2.2 (Qinghai-like) HPAI H5N1 virus, clade 2.3.4.4 H5 viruses have undergone reassortment activities with other NA subtypes (34). This novel combination of NA might facilitate the virus to circulate in migratory birds. The wide prevalence of clade 2.3.4.4 H5 viruses in wild birds could increase the probability of reassortment among H5 and other avian influenza viruses, generating new reassortants. It also may raise the reassortment frequency with viruses prevalent in poultry and generate poultry-adapted viruses, such as the reassortant A and B H5N6 viruses, through interaction between migratory birds and local poultry. H5N6 viruses could further exchange gene segments with viruses circulating in poultry, especially in live-poultry markets (16, 35, 36).
According to our surveillance data, clade 2.3.4.4 HPAI H5N6 viruses have been circulating in poultry in China. All reassortants may have originated in southern China based on the geographic distribution of the viruses (Fig. 6). Previously we reported that “genetic tuning” mediated the genesis and interspecies transmission of novel H7N9 viruses (37, 38). As HPAI H5N6 viruses circulate in environments similar to those of H7N9 viruses, the same forces of evolution may affect H5N6 viruses. Similar to H7N9 viruses, reassortant C H5N6 viruses contain an NA stalk deletion and internal genes derived from H9N2 viruses. Although the isolates in this study were from environmental samples, we speculated that reassortant C H5N6 viruses may have potential advantages for pathogenicity in chickens based on their genetic characteristics.
Our surveillance results revealed that multiple clades of HPAI H5 virus had been cocirculating in Mainland China. The evolution of distinct clades could lead to antigenic alterations. The hemagglutination inhibition assay results provided evidence that the three reassortant H5N6 viruses had undergone antigenic variation. Therefore, continued surveillance of these H5N6 reassortants, along with antigenic characterization, should be implemented to avoid the development of a mismatched vaccine when preparing for a pandemic. In addition, some studies have reported that clade 2.3.4.4 HPAI H5N6 viruses show highest affinity for the avian-like receptor (α-2,3 sialic acid), with lower but increased affinity for the human-like receptor (α-2,6 sialic acid) (14, 39, 40). The mutations identified in human isolates (S123P, I151T, T156A, and the deletion at position 126 in HA) should be further investigated to identify their effects on the receptor binding preferences of H5N6 viruses.
Previous studies showed that while avian influenza virus lacks the capacity for sustained human-to-human transmission, human infection with avian influenza virus resulted from direct contact with infected birds or exposure to contaminated environments, such as live-poultry markets (41–43). Over the past decade, viruses from both clades 2.3.2 and 2.3.4 have been circulating in poultry (44, 45) and have been responsible for most of the confirmed human cases in China (46). In this study, we detected clade 2.3.4.4 HPAI H5N6 virus in environmental samples from most provinces of southern China. In addition, over 30 outbreaks in poultry caused by H5N6 viruses have been reported by the World Organization for Animal Health (OIE). This revealed the risk of human infection posed by these reassortant viruses. Recently, we reported that the increased H5N6 virus positivity rate in the live-poultry markets coincided with subsequent human infections in Shenzhen in China (47). Human infections with HPAI H5N6 viruses resulted in high mortality rates (10/14) and clinical symptoms similar to those seen in HPAI H5N1 patients (9, 48). Therefore, H5N6 viruses pose a significant public health threat, which requires close monitoring similar to that for HPAI H5N1 viruses.
MATERIALS AND METHODS
Surveillance of avian influenza viruses from avian-linked environment in Mainland China from 2012 to 2015.
From January 2012 to December 2015, routine avian influenza virus surveillance was carried out in Mainland China. Samples were randomly collected from avian-linked environments (live-poultry markets or farms) once per month across 31 provinces. In total, 170,072 samples were collected, and 34,492 of these samples tested positive for influenza A virus at local centers for disease control and prevention using a real-time PCR assay. Of these, 16,341 positive samples were received by our laboratory for virus isolation. A preliminary test for HPAI H5 by real-time PCR was conducted. The positive samples were isolated in a BSL3 laboratory. Swab samples were treated with antibiotics for 30 min, and then 0.2 ml of the supernatant was inoculated into the allantoic cavities of 9-day-old specific-pathogen-free embryonated chicken eggs. After incubation at 35°C for 48 to 72 h, the presence of virus in the allantoic fluid of the embryos was determined by a hemagglutination test using 0.5% turkey red blood cells. The HA subtypes of all of the viruses were identified by quantitative reverse transcription-PCR (RT-PCR).
Virus sequencing.
RNA was extracted from viruses using the QIAamp viral RNA minikit (Qiagen, Hilden, Germany). Extracted RNA was subjected to reverse transcription and amplification using the SuperScript III One-Step RT-PCR system (Thermo Fisher, Waltham, MA) according to a previously described method (49). Whole-genome sequencing of FluA was implemented on the Ion Torrent PGM platform (Thermo Fisher) with a read length of 200 bp. Data analysis was mainly conducted using CLC Genomics Workbench 7.5.1 software. Low-quality reads were trimmed out using a CLC trimmer with the quality limit set at 0.05. The filtered reads were de novo assembled in CLC using the default parameters. Contigs with a coverage over 10 were extracted and used in a BLAST search against a database containing all of the FluA sequences collected from the NCBI and Global Initiative on Sharing All Influenza Data (GISAID) databases. The sequences with the highest levels of similarity were selected as references for reads mapping under the parameters “Length fraction = 0.8, Similarity fraction = 0.8.” FluA genome sequences were obtained by extracting the consensus sequences from the mapping results, with at least 100× coverage depth at each site for the eight segments.
Evolutionary analysis.
The whole-genome sequences of 123 environmental H5/N6 isolates were obtained in this study. Phylogenetic analyses were conducted together with the available sequences from the GISAID. Approximate maximum likelihood (ML) phylogenetic trees for each of the genes were constructed using the FastTree 2.1 software with the general reversible GTR+Γ model and 1,000 bootstraps.
To estimate the time to the most recent common ancestor (tMRCA) of each of the segments of HPAI H5N6 virus, nonredundant sub-data sets were selected to run time-measured Bayesian Markov chain Monte Carlo analysis via BEAST v1.82 software (50). A root-to-tip regression plot was performed using TempEst software, and no outliers were found in the sub-data sets. The SRD06 substitution model (51) and the uncorrelated relaxed molecular clock model were used. The Bayesian skygrid coalescent was set as tree prior. The uniform distribution was used for prior CP1 + 2, CP3, and ucld.mean. The Bayesian MCMC was run for up to 5 × 107 steps, with sampling at each 5,000 steps to achieve convergence. Tracer v1.6 software was used to examine the effective sample sizes of the parameters in the log file, and these were all ≥200, implying reasonable convergence and posterior estimates. A maximum clade credibility tree with median node height was conducted after excluding the beginning 10 to 15% states.
Accession number(s).
The full genome sequences of the H5 and N6 viruses determined in our study have been deposited in the GISAID database under the accession numbers listed in Table S1 in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by National Mega-projects for Infectious Diseases (2014ZX10004002 to Yuelong Shu) and the National Key Research and Development Program (2016YFC1200200 to Yuelong Shu and 2016YFD0500208 to Dayan Wang).
We acknowledge the authors and laboratories who shared H5 sequences in GISAID's EpiFlu Database.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the views of the China CDC and other organizations.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.02199-16.
REFERENCES
- 1.Duan L, Campitelli L, Fan XH, Leung YH, Vijaykrishna D, Zhang JX, Donatelli I, Delogu M, Li KS, Foni E, Chiapponi C, Wu WL, Kai H, Webster RG, Shortridge KF, Peiris JS, Smith GJ, Chen H, Guan Y. 2007. Characterization of low-pathogenic H5 subtype influenza viruses from Eurasia: implications for the origin of highly pathogenic H5N1 viruses. J Virol 81:7529–7539. doi: 10.1128/JVI.00327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duan L, Bahl J, Smith GJ, Wang J, Vijaykrishna D, Zhang LJ, Zhang JX, Li KS, Fan XH, Cheung CL, Huang K, Poon LL, Shortridge KF, Webster RG, Peiris JS, Chen H, Guan Y. 2008. The development and genetic diversity of H5N1 influenza virus in China, 1996–2006. Virology 380:243–254. doi: 10.1016/j.virol.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO/OIE/FAO H5N1 Evolution Working Group. 29 October 2011. Continued evolution of highly pathogenic avian influenza A (H5N1): updated nomenclature. Influenza Other Respir Viruses doi: 10.1111/j.1750-2659.2011.00298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J, Xiao H, Lei F, Zhu Q, Qin K, Zhang XW, Zhang XL, Zhao D, Wang G, Feng Y, Ma J, Liu W, Wang J, Gao GF. 2005. Highly pathogenic H5N1 influenza virus infection in migratory birds. Science 309:1206. doi: 10.1126/science.1115273. [DOI] [PubMed] [Google Scholar]
- 5.Hill SC, Lee YJ, Song BM, Kang HM, Lee EK, Hanna A, Gilbert M, Brown IH, Pybus OG. 2015. Wild waterfowl migration and domestic duck density shape the epidemiology of highly pathogenic H5N8 influenza in the Republic of Korea. Infect Genet Evol 34:267–277. doi: 10.1016/j.meegid.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee DH, Torchetti MK, Winker K, Ip HS, Song CS, Swayne DE. 2015. Intercontinental spread of Asian-origin H5N8 to North America through Beringia by migratory birds. J Virol 89:6521–6524. doi: 10.1128/JVI.00728-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhagen JH, van der Jeugd HP, Nolet BA, Slaterus R, Kharitonov SP, de Vries PP, Vuong O, Majoor F, Kuiken T, Fouchier RA. 2015. Wild bird surveillance around outbreaks of highly pathogenic avian influenza A(H5N8) virus in the Netherlands, 2014, within the context of global flyways. Euro Surveill 20(12):pii=21069. [DOI] [PubMed] [Google Scholar]
- 8.Global Consortium for H5N8 and Related Influenza Viruses. 2016. Role for migratory wild birds in the global spread of avian influenza H5N8. Science 354:213–217. doi: 10.1126/science.aaf8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan M, Gao R, Lv Q, Huang S, Zhou Z, Yang L, Li X, Zhao X, Zou X, Tong W, Mao S, Zou S, Bo H, Zhu X, Liu L, Yuan H, Zhang M, Wang D, Li Z, Zhao W, Ma M, Li Y, Li T, Yang H, Xu J, Zhou L, Zhou X, Tang W, Song Y, Chen T, Bai T, Zhou J, Wang D, Wu G, Li D, Feng Z, Gao GF, Wang Y, He S, Shu Y. 2016. Human infection with a novel, highly pathogenic avian influenza A (H5N6) virus: virological and clinical findings. J Infect 72:52–59. doi: 10.1016/j.jinf.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 10.Butler J, Stewart CR, Layton DS, Phommachanh P, Harper J, Payne J, Evans RM, Valdeter S, Walker S, Harvey G, Shan S, Bruce MP, Rootes CL, Gough TJ, Rohringer A, Peck GR, Fardy SJ, Karpala AJ, Johnson D, Wang J, Douangngeun B, Morrissy C, Wong FY, Bean AG, Bingham J, Williams DT. 2016. Novel reassortant H5N6 influenza A virus from the Lao People's Democratic Republic is highly pathogenic in chickens. PLoS One 11:e0162375. doi: 10.1371/journal.pone.0162375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu DH, Okamatsu M, Matsuno K, Hiono T, Ogasawara K, Nguyen LT, Van Nguyen L, Nguyen TN, Nguyen TT, Van Pham D, Nguyen DH, Nguyen TD, To TL, Van Nguyen H, Kida H, Sakoda Y. 2016. Genetic and antigenic characterization of H5, H6 and H9 avian influenza viruses circulating in live bird markets with intervention in the center part of Vietnam. Vet Microbiol 192:194–203. doi: 10.1016/j.vetmic.2016.07.016. [DOI] [PubMed] [Google Scholar]
- 12.Jiao P, Cui J, Song Y, Song H, Zhao Z, Wu S, Qu N, Wang N, Ouyang G, Liao M. 2016. New reassortant H5N6 highly pathogenic avian influenza viruses in Southern China, 2014. Front Microbiol 7:754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qi X, Cui L, Yu H, Ge Y, Tang F. 2014. Whole-genome sequence of a reassortant H5N6 avian influenza virus isolated from a live poultry market in China, 2013. Genome Announc 2(5):e00706-14. doi: 10.1128/genomeA.00706-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bi Y, Liu H, Xiong C, Di L, Shi W, Li M, Liu S, Chen J, Chen G, Li Y, Yang G, Lei Y, Xiong Y, Lei F, Wang H, Chen Q, Chen J, Gao GF. 2016. Novel avian influenza A (H5N6) viruses isolated in migratory waterfowl before the first human case reported in China, 2014. Sci Rep 6:29888. doi: 10.1038/srep29888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi Y, Mei K, Shi W, Liu D, Yu X, Gao Z, Zhao L, Gao GF, Chen J, Chen Q. 2015. Two novel reassortants of avian influenza A (H5N6) virus in China. J Gen Virol 96:975–981. doi: 10.1099/vir.0.000056. [DOI] [PubMed] [Google Scholar]
- 16.Yuan R, Wang Z, Kang Y, Wu J, Zou L, Liang L, Song Y, Zhang X, Ni H, Lin J, Ke C. 2016. Continuing reassortant of H5N6 subtype highly pathogenic avian influenza virus in Guangdong. Front Microbiol 7:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu W, Li X, Bai T, Zhao X, Zhao X, Zhang Y, Guo J, Li Z, Yang L, Wang D, Shu Y. 2016. A fatal case of infection with a further reassortant, highly pathogenic avian influenza (HPAI) H5N6 virus in Yunnan, China. Infect Genet Evol 40:63–66. doi: 10.1016/j.meegid.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 18.Hu T, Song J, Zhang W, Zhao H, Duan B, Liu Q, Zeng W, Qiu W, Chen G, Zhang Y, Fan Q, Zhang F. 2015. Emergence of novel clade 2.3.4 influenza A (H5N1) virus subgroups in Yunnan Province, China. Infect Genet Evol 33:95–100. doi: 10.1016/j.meegid.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Mok CK, Da Guan W, Liu XQ, Lamers MM, Li XB, Wang M, Zhang TJ, Zhang QL, Li ZT, Huang JC, Lin JY, Zhang YH, Zhao P, Lee HH, Chen L, Li YM, Peiris JS, Chen RC, Zhong NS, Yang ZF. 2015. Genetic characterization of highly pathogenic avian influenza A(H5N6) virus, Guangdong, China. Emerg Infect Dis 21:2268–2271. doi: 10.3201/eid2112.150809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong J, Kang HM, Lee EK, Song BM, Kwon YK, Kim HR, Choi KS, Kim JY, Lee HJ, Moon OK, Jeong W, Choi J, Baek JH, Joo YS, Park YH, Lee HS, Lee YJ. 2014. Highly pathogenic avian influenza virus (H5N8) in domestic poultry and its relationship with migratory birds in South Korea during 2014. Vet Microbiol 173:249–257. doi: 10.1016/j.vetmic.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Chutinimitkul S, van Riel D, Munster VJ, van den Brand JM, Rimmelzwaan GF, Kuiken T, Osterhaus AD, Fouchier RA, de Wit E. 2010. In vitro assessment of attachment pattern and replication efficiency of H5N1 influenza A viruses with altered receptor specificity. J Virol 84:6825–6833. doi: 10.1128/JVI.02737-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada S, Suzuki Y, Suzuki T, Le MQ, Nidom CA, Sakai-Tagawa Y, Muramoto Y, Ito M, Kiso M, Horimoto T, Shinya K, Sawada T, Kiso M, Usui T, Murata T, Lin Y, Hay A, Haire LF, Stevens DJ, Russell RJ, Gamblin SJ, Skehel JJ, Kawaoka Y. 2006. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature 444:378–382. doi: 10.1038/nature05264. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Lu B, Zhou H, Suguitan AL Jr, Cheng X, Subbarao K, Kemble G, Jin H. 2010. Glycosylation at 158N of the hemagglutinin protein and receptor binding specificity synergistically affect the antigenicity and immunogenicity of a live attenuated H5N1 A/Vietnam/1203/2004 vaccine virus in ferrets. J Virol 84:6570–6577. doi: 10.1128/JVI.00221-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe Y, Ibrahim MS, Ellakany HF, Kawashita N, Mizuike R, Hiramatsu H, Sriwilaijaroen N, Takagi T, Suzuki Y, Ikuta K. 2011. Acquisition of human-type receptor binding specificity by new H5N1 influenza virus sublineages during their emergence in birds in Egypt. PLoS Pathog 7:e1002068. doi: 10.1371/journal.ppat.1002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen HT, Fry AM, Gubareva LV. 2012. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir Ther 17:159–173. doi: 10.3851/IMP2067. [DOI] [PubMed] [Google Scholar]
- 26.Yamada S, Hatta M, Staker BL, Watanabe S, Imai M, Shinya K, Sakai-Tagawa Y, Ito M, Ozawa M, Watanabe T, Sakabe S, Li C, Kim JH, Myler PJ, Phan I, Raymond A, Smith E, Stacy R, Nidom CA, Lank SM, Wiseman RW, Bimber BN, O'Connor DH, Neumann G, Stewart LJ, Kawaoka Y. 2010. Biological and structural characterization of a host-adapting amino acid in influenza virus. PLoS Pathog 6:e1001034. doi: 10.1371/journal.ppat.1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2012. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336:1534–1541. doi: 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Q, Lu L, Sun Z, Chen GW, Wen Y, Jiang S. 2013. Genomic signature and protein sequence analysis of a novel influenza A (H7N9) virus that causes an outbreak in humans in China. Microbes Infect 15:432–439. doi: 10.1016/j.micinf.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Brown EG, Bailly JE. 1999. Genetic analysis of mouse-adapted influenza A virus identifies roles for the NA, PB1, and PB2 genes in virulence. Virus Res 61:63–76. doi: 10.1016/S0168-1702(99)00027-1. [DOI] [PubMed] [Google Scholar]
- 30.He G, Qiao J, Dong C, He C, Zhao L, Tian Y. 2008. Amantadine-resistance among H5N1 avian influenza viruses isolated in Northern China. Antiviral Res 77:72–76. doi: 10.1016/j.antiviral.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Long JX, Peng DX, Liu YL, Wu YT, Liu XF. 2008. Virulence of H5N1 avian influenza virus enhanced by a 15-nucleotide deletion in the viral nonstructural gene. Virus Genes 36:471–478. doi: 10.1007/s11262-007-0187-8. [DOI] [PubMed] [Google Scholar]
- 32.Spesock A, Malur M, Hossain MJ, Chen LM, Njaa BL, Davis CT, Lipatov AS, York IA, Krug RM, Donis RO. 2011. The virulence of 1997 H5N1 influenza viruses in the mouse model is increased by correcting a defect in their NS1 proteins. J Virol 85:7048–7058. doi: 10.1128/JVI.00417-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu A, Su C, Wang D, Peng Y, Liu M, Hua S, Li T, Gao GF, Tang H, Chen J, Liu X, Shu Y, Peng D, Jiang T. 2013. Sequential reassortments underlie diverse influenza H7N9 genotypes in China. Cell Host Microbe 14:446–452. doi: 10.1016/j.chom.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 34.de Vries E, Guo H, Dai M, Rottier PJ, van Kuppeveld FJ, de Haan CA. 2015. Rapid emergence of highly pathogenic avian influenza subtypes from a subtype H5N1 hemagglutinin variant. Emerg Infect Dis 21:842–846. doi: 10.3201/eid2105.141927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu H, Peng X, Peng X, Cheng L, Lu X, Jin C, Xie T, Yao H, Wu N. 2015. Genetic and molecular characterization of H9N2 and H5 avian influenza viruses from live poultry markets in Zhejiang Province, eastern China. Sci Rep 5:17508. doi: 10.1038/srep17508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma MJ, Chen SH, Wang GL, Zhao T, Qian YH, Wu MN, Liu Y, Gray GC, Lu B, Cao WC. 2016. Novel highly pathogenic avian H5 influenza A viruses in live poultry markets, Wuxi City, China, 2013-2014. Open Forum Infect Dis 3:ofw054. doi: 10.1093/ofid/ofw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu W, Shu Y. 2015. Genetic tuning of avian influenza A (H7N9) virus promotes viral fitness within different species. Microbes Infect 17:118–122. doi: 10.1016/j.micinf.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 38.Wang D, Yang L, Gao R, Zhang X, Tan Y, Wu A, Zhu W, Zhou J, Zou S, Li X, Sun Y, Zhang Y, Liu Y, Liu T, Xiong Y, Xu J, Chen L, Weng Y, Qi X, Guo J, Li X, Dong J, Huang W, Zhang Y, Dong L, Zhao X, Liu L, Lu J, Lan Y, Wei H, Xin L, Chen Y, Xu C, Chen T, Zhu Y, Jiang T, Feng Z, Yang W, Wang Y, Zhu H, Guan Y, Gao GF, Li D, Han J, Wang S, Wu G, Shu Y. 2014. Genetic tuning of the novel avian influenza A(H7N9) virus during interspecies transmission, China, 2013. Euro Surveill 19(25):pii=20836. [DOI] [PubMed] [Google Scholar]
- 39.Yang H, Carney PJ, Mishin VP, Guo Z, Chang JC, Wentworth DE, Gubareva LV, Stevens J. 2016. Molecular characterizations of surface proteins hemagglutinin and neuraminidase from recent H5Nx avian influenza viruses. J Virol 90:5770–5784. doi: 10.1128/JVI.00180-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun H, Pu J, Wei Y, Sun Y, Hu J, Liu L, Xu G, Gao W, Li C, Zhang X, Huang Y, Chang KC, Liu X, Liu J. 2016. Highly pathogenic avian influenza H5N6 viruses exhibit enhanced affinity for human type sialic acid receptor and in-contact transmission in model ferrets. J Virol 90:6235–6243. doi: 10.1128/JVI.00127-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Q, Zhou L, Zhou M, Chen Z, Li F, Wu H, Xiang N, Chen E, Tang F, Wang D, Meng L, Hong Z, Tu W, Cao Y, Li L, Ding F, Liu B, Wang M, Xie R, Gao R, Li X, Bai T, Zou S, He J, Hu J, Xu Y, Chai C, Wang S, Gao Y, Jin L, Zhang Y, Luo H, Yu H, He J, Li Q, Wang X, Gao L, Pang X, Liu G, Yan Y, Yuan H, Shu Y, Yang W, Wang Y, Wu F, Uyeki TM, Feng Z. 2014. Epidemiology of human infections with avian influenza A(H7N9) virus in China. N Engl J Med 370:520–532. doi: 10.1056/NEJMoa1304617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Q, Shi J, Deng G, Guo J, Zeng X, He X, Kong H, Gu C, Li X, Liu J, Wang G, Chen Y, Liu L, Liang L, Li Y, Fan J, Wang J, Li W, Guan L, Li Q, Yang H, Chen P, Jiang L, Guan Y, Xin X, Jiang Y, Tian G, Wang X, Qiao C, Li C, Bu Z, Chen H. 2013. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science 341:410–414. doi: 10.1126/science.1240532. [DOI] [PubMed] [Google Scholar]
- 43.Wan XF, Dong L, Lan Y, Long LP, Xu C, Zou S, Li Z, Wen L, Cai Z, Wang W, Li X, Yuan F, Sui H, Zhang Y, Dong J, Sun S, Gao Y, Wang M, Bai T, Yang L, Li D, Yang W, Yu H, Wang S, Feng Z, Wang Y, Guo Y, Webby RJ, Shu Y. 2011. Indications that live poultry markets are a major source of human H5N1 influenza virus infection in China. J Virol 85:13432–13438. doi: 10.1128/JVI.05266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang WM, Liu S, Chen J, Hou GY, Li JP, Cao YF, Zhuang QY, Li Y, Huang BX, Chen JM. 7 July 2010. Molecular epidemiological surveys of H5 subtype highly pathogenic avian influenza viruses in poultry in China in 2007–2009. J Gen Virol doi: 10.1099/vir.0.023168-0. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Shi J, Zhong G, Deng G, Tian G, Ge J, Zeng X, Song J, Zhao D, Liu L, Jiang Y, Guan Y, Bu Z, Chen H. 2010. Continued evolution of H5N1 influenza viruses in wild birds, domestic poultry and humans in China from 2004 to 2009. J Virol doi: 10.1128/JVI.00413-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu H, Gao Z, Feng Z, Shu Y, Xiang N, Zhou L, Huai Y, Feng L, Peng Z, Li Z, Xu C, Li J, Hu C, Li Q, Xu X, Liu X, Liu Z, Xu L, Chen Y, Luo H, Wei L, Zhang X, Xin J, Guo J, Wang Q, Yuan Z, Zhang K, Zhang W, Yang J, Zhong X, Xia S, Li L, Cheng J, Ma E, He P, Lee SS, Wang Y, Uyeki TM, Yang W. 2008. Clinical characteristics of 26 human cases of highly pathogenic avian influenza A (H5N1) virus infection in China. PLoS One 3:e2985. doi: 10.1371/journal.pone.0002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang S, Bai T, Yang L, Wang X, Peng B, Liu H, Geng Y, Zhang R, Ma H, Zhu W, Wang D, Cheng J, Shu Y. 2016. Sustained live poultry market surveillance contributes to early warnings for human infection with avian influenza viruses. Emerg Microbes Infect 5:e79. doi: 10.1038/emi.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang ZF, Mok CK, Peiris JS, Zhong NS. 2015. Human infection with a novel avian influenza A(H5N6) virus. N Engl J Med 373:487–489. doi: 10.1056/NEJMc1502983. [DOI] [PubMed] [Google Scholar]
- 49.Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza A viruses. J Virol 83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.