

ABSTRACT
Autophagy functions as an intrinsic antiviral defense. However, some viruses can subvert or even enhance host autophagic machinery to increase viral replication and pathogenesis. The role of autophagy during avibirnavirus infection, especially late stage infection, remains unclear. In this study, infectious bursal disease virus (IBDV) was used to investigate the role of autophagy in avibirnavirus replication. We demonstrated IBDV induction of autophagy as a significant increase in puncta of LC3+ autophagosomes, endogenous levels of LC3-II, and ultrastructural characteristics typical of autophagosomes during the late stage of infection. Induction of autophagy enhances IBDV replication, whereas inhibition of autophagy impairs viral replication. We also demonstrated that IBDV infection induced autophagosome-lysosome fusion, but without active degradation of their contents. Moreover, inhibition of fusion or of lysosomal hydrolysis activity significantly reduced viral replication, indicating that virions utilized the low-pH environment of acidic organelles to facilitate viral maturation. Using immuno-transmission electron microscopy (TEM), we observed that a large number of intact IBDV virions were arranged in a lattice surrounded by p62 proteins, some of which lay between virions. Additionally, many virions were encapsulated within the vesicular membranes, with an obvious release stage observed by TEM. The autophagic endosomal pathway facilitates low-pH-mediated maturation of viral proteins and membrane-mediated release of progeny virions.
IMPORTANCE IBDV is the most extensively studied virus in terms of molecular characteristics and pathogenesis; however, mechanisms underlying the IBDV life cycle require further exploration. The present study demonstrated that autophagy enhances viral replication at the late stage of infection, and the autophagy pathway facilitates IBDV replication complex function and virus assembly, which is critical to completion of the virus life cycle. Moreover, the virus hijacks the autophagic vacuoles to mature in an acidic environment and release progeny virions in a membrane-mediated cell-to-cell manner. This autophagic endosomal pathway is proposed as a new mechanism that facilitates IBDV maturation, release, and reinternalization. This report presents a concordance in exit strategies among some RNA and DNA viruses, which exploit autophagy pathway for their release from cells.
KEYWORDS: IBDV, autophagy, maturation, release
INTRODUCTION
Infectious bursal disease is an immunosuppressive disease of young chickens that causes considerable economic loss to the poultry industry worldwide (1). The causative agent, infectious bursal disease virus (IBDV), belongs to the genus Avibirnavirus of the family Birnaviridae. IBDV is the most extensively studied virus in this family in terms of our collective understanding of its molecular (2–5) and cellular (6–9) biology, structure (10, 11), and pathogenesis (12); therefore, because the autophagic pathway and its function during double-stranded RNA (dsRNA) virus infection are poorly understood, IBDV is an important model system for dsRNA virus. Avibirnavirus VP2 binds to the pathogen receptor HSP90AA1 and induces autophagy by inactivating the AKT-mTOR pathway during the early stage of virus infection (13). The relationship between autophagic signaling and viral infection, the autophagic flux induced by IBDV during the late stage of virus infection, and the mechanism by which autophagy affects viral replication require further investigation.
Autophagy, a highly conserved cellular response to starvation, protein aggregation, organelle damage, and infectious agents, leads to sustained cellular homeostasis and to tissue development and acts as a defense mechanism against infection. There are at least three autophagy pathways: microautophagy, macroautophagy, and chaperone-mediated autophagy. Macroautophagy is a well-characterized pathway and has been implicated in both innate and adaptive immunity (14). In this report, autophagy specifically refers to macroautophagy. During autophagy, an isolation membrane forms around the substrate to generate an autophagic vesicle enclosed by two membranes. Bulk degradation of subcellular constituents occurs through formation of autolysosomes via fusion of autophagosomes with late endosomes and lysosomes (15, 16). Autophagy functions as an intrinsic antiviral defense mechanism, an activity known as xenophagy (17), by directly engulfing virions and/or viral components of herpes simplex virus 1 (18) or Sindbis virus (19) for lysosomal degradation.
However, some viruses subvert or enhance the host autophagic machinery for viral replication and pathogenicity. Several herpesviruses block autophagy at various steps, impairing autophagic vesicle induction, nucleation, or elongation (20–22). RNA viruses such as poliovirus (23), coxsackievirus B3 (24), human immunodeficiency virus type 1 (HIV-1) (25), and hepatitis C virus (26) induce the formation of vesicles resembling autophagosomes to enhance viral replication or nonlytic egress (27–29). Influenza A virus blocks the fusion of autophagosomes with lysosomes and, although this has no effect on virus replication, compromises the survival of infected cells (30). Avian reovirus nonstructural protein p17 induces autophagy, resulting in enhanced virus replication (31). Furthermore, autophagy is beneficial to the replication of Newcastle disease virus in both chicken cells and tissues (32). These data highlight the remarkable diversity in term of strategies by which viruses subvert the autophagy pathway and raise the possibility that autophagic degradation could itself promote virus production. Therefore, the role of autophagy in the defense of the host against viral infection and subversion of autophagy by the virus to facilitate viral replication are crucial factors in viral evolution and pathogenicity, which should be extensively studied.
The Birnaviridae family viruses integrate their genome into a complex structure known as the “viral factory,” which localizes to the cytoplasmic face of membrane of endocytic compartments. Additionally, the Golgi apparatus plays an important role in IBDV assembly (8). However, the mechanism of viral maturation and release needs further investigation. A recent study indicated that exocytosis of varicella-zoster virus virions involves a convergence of endosomal and autophagy pathways (33). The investigators demonstrated that some viral particles after secondary envelopment accumulated in a heterogeneous population of single-membraned vesicular compartments, which were decorated with components from both the endocytic pathway (Rab11) and the autophagy pathway (LC3B). Moreover, some RNA viruses exploit endosomal and autophagy pathways for their replication and release from cells (34). Poliovirus utilizes autophagosomes, or double-membraned vesicles, for nonlytic viral spread (35). A pathogenic picornavirus acquires an envelope by hijacking cellular membranes (36). Thus, in this study, the cellular aspects participating to the birnaviral replication process were investigated to gain insight into whether autophagy pathway is involved in viral maturation and release.
In this study, we demonstrated that IBDV induces autophagic signaling during the late stage of infection, which contributes to virus replication. Moreover, IBDV induces autophagosome fusion with acidic endosomes and lysosomes, leading to improved virus assembly and maturation. Additionally, many virions were encapsulated within the vesicular membranes with an obvious release stage. Our findings suggested a new mechanism for modulating autophagy pathway for avibirnavirus maturation, release, and reinternalization.
RESULTS
IBDV induces autophagic signaling during the late phase of infection.
In order to investigate whether the cellular autophagy pathway levels are altered in response to infection with IBDV, DF-1 cells were infected with IBDV strain Gt at a multiplicity of infection (MOI) of 1. A previous study indicated that the percentage of apoptotic cells increased with the time phase of IBDV infection (37). The release of cytochrome c from mitochondria to cytosol indicates the activation of intrinsic apoptotic pathway (38). Thus, we isolated the mitochondria from uninfected cells or cells infected with IBDV from 12 to 36 h postinfection (p.i.). The level of cytochrome c in mitochondria was used to evaluate the apoptosis in protein level with Western blotting (WB). Cytochrome c in mitochondria began to decrease at 24 h p.i. and cytochrome c decreased significantly at 36 h p.i. in IBDV-infected cells compared with uninfected cells, which indicated that apoptosis was induced significantly at 36 h p.i. (data not shown). Thus, we chose to analyze cells at 12 and 24 h p.i. The level of autophagosomes was analyzed by WB at the amount of endogenous lipidated LC3 (LC3-II) and p62. When lysates of infected and uninfected cells were analyzed by anti-LC3, -p62, or -(p)VP2 immunoblotting, the viral protein showed mainly as the precursor structural protein, pVP2, and the level of the autophagic protein LC3-II began to increase, while the p62 level was similar to that in uninfected cells at 12 h p.i. The precursor structural protein, pVP2, increased at 24 h p.i. and matured into VP2. And the level of the lipidated and therefore faster-migrating LC3-II form showed a significant increase in IBDV-infected cells, but the p62 level was not changed significantly (Fig. 1A and B). The increased level of autophagosomes was also observed by confocal microscopy; cells were infected with IBDV strain Gt at a multiplicity of infection (MOI) of 1 and observed at 12 and 24 h p.i. compared with mock-infected cells. IBDV-infected cells showed a significant increase in the number of puncta representing LC3+ autophagosomes (Fig. 1C and D). Transmission electron microscopy (TEM) was used to explore changes in the ultrastructure of DF-1 cells infected with IBDV versus that of uninfected cells. At 24 h p.i., a considerable increase in perinuclear and cytoplasmic double-membrane vesicles with morphological characteristics typical of autophagic vacuoles was observed in virus-infected cells. In contrast, similar vesicles were rarely detected in uninfected cells, of which morphologically normal organelles were in the cytoplasm (Fig. 1E). The number of autophagosomes per cell significantly increased in infected cells compared with that in mock-infected cells (Fig. 1F). Interestingly, the novel membrane crescent was observed in infected cells, which sequester cytoplasmic contents in double-membrane vesicles, expand, and self-fuse, together referred to as the induction of autophagosomes (data not shown). Taken together the results showed that autophagosomes accumulated in DF-1 cells infected with IBDV, suggesting that autophagic signaling is induced and regulated by IBDV during the late phase of infection.
FIG 1.

IBDV induces autophagic signaling during the late phase of infection. (A) DF-1 cells were infected with IBDV Gt at an MOI of 1; cells were harvested at 12 and 24 h p.i. Cell lysates were separated by SDS-PAGE and analyzed by WB with anti-LC3B, p62, or β-actin for host proteins and anti-(p)VP2 for viral protein. After incubation with IRDye 800CW secondary antibody, proteins were visualized and quantified using the Odyssey system. (B) Intensity band ratio of LC3-II/actin was normalized to the control. Means ± SDs were determined for three independent experiments. t test was used for comparison to the control. (C) DF-1 cells cultured in dishes with coverslip inserts were infected with IBDV Gt at an MOI of 1 for 12 and 24 h p.i. Cells were observed by confocal microscopy for fluorescence puncta. Viral protein is represented by red fluorescence, endogenous LC3 molecule appears green, and the nucleus was stained blue by DAPI. The scale bar is indicated at the bottom right. (D) Puncta per cell were quantified in mock- or IBDV-infected DF-1 cells. Data were analyzed by t test. (E) DF-1 cells were infected with IBDV Gt at an MOI of 1. Cells were fixed at 24 h p.i., and ultrastructure was observed by TEM. Arrows indicate vesicles with morphological characteristics typical of autophagic vacuoles of perinuclear double-membrane vacuoles measuring 0.3 to 2.0 μm. N, nucleus. The scale bar is indicated at the bottom right. (F) Autophagosomes per cell were quantified in mock- or IBDV-infected DF-1 cells. Data were analyzed by t test.
Induction of autophagy enhances viral replication, whereas inhibition of autophagy impairs viral replication.
In order to determine whether autophagy induction performs an antiviral function or facilitates replication of IBDV, DF-1 cells were treated with the pharmacological reagent rapamycin, which induces autophagy by inhibiting the mTOR pathway (39). At 24 h p.i., infectious progeny virus titer, both intracellular and extracellular, was titrated by 50% tissue culture infective dose (TCID50) assay, and intracellular viral protein yield was determined with WB. The intracellular loads of pVP2 were similar with and without rapamycin treatment, but the intracellular and extracellular virus titers increased significantly in the presence of rapamycin (data not shown). A previous study showed that rapamycin does not induce obvious autophagy in DF-1 cells as it does in mammalian cells (32). Cells were starved of amino acids, a condition also known to induce autophagy (40). As shown in Fig. 2A, the level of LC3-II was increased obviously under starvation conditions, which indicated that starvation was potent to autophagy induction in DF-1 cells. As shown in Fig. 2D, pVP2 levels significantly increased with starvation, and the infectious progeny virus titers, both intracellular and extracellular, significantly increased with autophagy induction by nutrient deprivation (Fig. 2E and F). These results indicated that induction of autophagy through pharmacological treatment or starvation significantly enhanced viral replication.
FIG 2.

Induced autophagy enhances IBDV replication, whereas inhibition of autophagy impairs IBDV replication. DF-1 cells were pretreated via starvation with DMEM without fetal bovine serum (A), complete medium containing 10 nM wortmannin (B), 5 mM 3-MA under starvation conditions (C), or corresponding solvent for 4 h and then subjected to a 1.5-h absorption with IBDV Gt (MOI = 0.1). Cells were cultured in fresh medium with starvation (A), 10 nM wortmannin (B), 5 mM 3-MA plus starvation (C) or corresponding solvent for 24 h p.i., and the intracellular and extracellular viral loads were analyzed by WB and TCID50 assay. Antibodies against LC3B, β-actin, and (p)VP2 were used for WB, and intensity band ratio of pVP2/actin (D) was normalized to the control. Progeny viral loads, both intracellular (E) and extracellular (F), were titrated in a TCID50 assay with secondary CEFs. Means ± SDs were determined for three independent experiments. t test was used for comparison to the control group.
To determine whether inhibition of autophagy can affect viral replication, DF-1 cells infected with IBDV were treated with wortmannin (WM), a phosphatidylinositol 3-kinase/PtdIns 3-kinase inhibitor known to inhibit autophagosome formation (41). As shown in Fig. 2B and D, WM treatment reduced the viral pVP2 load, but the difference was not significant compared with the value for untreated cells. However, it significantly reduced the intracellular and extracellular viral titers (Fig. 2E and F). To verify that autophagy inhibition impairs IBDV replication, cells infected with IBDV were treated with 3-methyladenine (3-MA) under nutrient-deprived conditions, which reduces autophagy signaling by inhibiting type III phosphatidylinositol 3-kinase activity (42, 43). As shown in Fig. 2C, autophagy was induced with increased LC3-II by starvation, and 3-MA inhibited the autophagy, with LC3-II decreasing under starvation conditions. Starvation induced autophagy and increased the level of viral protein pVP2, while 3-MA inhibited autophagy and reduced viral protein levels (Fig. 2C and D). Moreover, treatment with 3-MA significantly reduced both intracellular and extracellular viral titers (Fig. 2E and F). Thus, autophagy inhibition by WM or 3-MA significantly impaired IBDV replication.
Knockdown of autophagic signaling impairs IBDV replication.
As autophagic signaling is induced, increasing amounts of the cellular autophagic protein LC3 become conjugated to the lipid phosphatidylethanolamine. This conjugation confers membrane association and is required for autophagosome formation and membrane expansion (44). Avian LC3B (Av-LC3B) was knocked down with a specific short hairpin RNA (shRNA) in DF-1 cells. A competent shRNA against Av-LC3B was chosen for this assay based on small interfering RNA (siRNA) efficiency scanning (data not shown). Viral protein pVP2 levels exhibited a significant decrease in LC3 knockdown cells, and the infectious progeny viruses, both intracellularly and extracellularly, were significantly reduced by LC3B shRNA versus scrambled shRNA (Fig. 3A and C to E). These results indicated that the autophagic vacuole membrane structure is important for viral replication.
FIG 3.

knockdown of autophagic signaling impairs IBDV replication. DF-1 cells were transfected with competent shRNA targeted to Av-LC3B (A), siRNA targeted to Av-ATG5 (B), or scrambled shRNA or scrambled siRNA using Lipofectamine 2000. At 48 h posttransfection, cells were infected with IBDV Gt at an MOI of 0.1. The intracellular and extracellular viral loads were analyzed by WB and TCID50 assay at 24 h p.i. Antibodies against LC3B, β-actin, ATG5, and (p)VP2 were used for WB, and the intensity band ratio of pVP2/actin (C) was normalized to the control. Progeny viral loads both intracellular (D) and extracellular (E) were titrated in a TCID50 assay with secondary CEFs. Means ± SDs were analyzed for three independent experiments. t test was used for comparison to the controls.
The autophagic protein ATG5 is essential for the activation of the conventional autophagy and autophagosome formation (45). In order to confirm knockdown of autophagic signaling impairing IBDV replication, the competent siRNA targeted to Av-ATG5 was scanned (data not shown). DF-1 cells were infected with IBDV after transfection with an siRNA targeting Av-ATG5 or scrambled siRNA and the infectious progeny viruses, both intracellular and extracellular, and viral protein were used to evaluate the effect of Av-ATG5 knockdown on viral replication. As shown in Fig. 3B, ATG5 was effectively knocked down and the level of LC3-II was also reduced obviously, which indicated that the autophagic signaling was effective knocked down based on ATG5 silencing. The results showed that the level of pVP2 was decreased significantly compared to that with the scrambled siRNA (Fig. 3C). The infectious progeny virus titers, both intracellular and extracellular, were reduced significantly in ATG5 knockdown cells (Fig. 3D and E). Taken together, these results suggest that knockdown of autophagic signaling targeting both Av-LC3B and Av-ATG5 impairs IBDV replication.
IBDV induces amphisome and/or autolysosome formation, but without active degradation.
In order to determine whether IBDV induces an autophagic flux for lysosomal hydrolysis, the tandem reporter construct monomeric red fluorescent protein (mRFP)-enhanced green fluorescent protein (EGFP)-LC3 (46) was used to observe amphisomal and/or lysosomal turnover of autophagosomes in IBDV-infected cells. The GFP moiety of this tandem reporter is sensitive to lysosomal proteolysis and quenching in acidic pH, while mRFP is not. Thus, the green fluorescent component of the composite yellow fluorescence for this mRFP-EGFP-LC3 reporter is lost upon autophagosome fusion with vesicles of the endocytic pathway to form amphisomes and/or lysosomes. This change from yellow to red fluorescence can be used to visualize acidic proteolysis and localization in acidified compartments of autophagy-targeted GFP (30, 46). After IBDV infection, we observed an increase in larger yellow autophagosomes, indicating their formation of amphisomes and/or lysosomes, followed by a gradual transition to red. The amphisomes and/or autolysosomes gradually increased in size with continuing fusion with other autophagic and endocytic vesicles. Thus, large red amphisomes and/or autolysosomes were observed. Finally, the cell was destroyed by the virus (Fig. 4). Autophagosomes are transient vesicles that deliver cargo for lysosomal hydrolysis (47); we used LC3 as an autophagosome marker and p62/sequestosome 1 (SQSTM1), an autophagy substrate and a marker of autophagic degradation (47, 48). The level of p62 did not alter significantly in IBDV-infected cells compared with that in mock-infected cells at 24 h p.i. (data not shown). Thus, IBDV infection induces amphisome and/or autolysosome formation but does not lead to active degradation.
FIG 4.

IBDV induces amphisome and/or autolysosome formation. DF-1 cells were transfected with ptfLC3/mRFP-EGFP-LC3 using Lipofectamine 2000. At 24 h posttransfection, cells were mock infected or infected with IBDV at an MOI of 1. After absorption for 1.5 h, cells were observed over time by live cell imaging with RFP and EGFP channels. Serial pictures obtained from the video at the indicated time points are shown.
IBDV proteins accumulated around acidic compartments.
LysoTracker can be used to trace acidic compartments, including late endosomes, amphisomes, lysosomes, and autolysosomes, in living cells (30). In order to observe the localization of IBDV in acidic organelles, the proteins were coexpressed with EGFP. EGFP-IBDV proteins were observed in LysoTracker red-traced acidic endosomes, amphisomes, lysosomes, and autolysosomes by live cell imaging. The serial pictures obtained from live cell imaging showed that the precursor of viral structure protein pVP2 was homogeneously distributed at the beginning throughout the whole cell, gradually accumulated to form spots, which increased in size, and partially concentrated around acidic organelles. The distribution of viral VP1 was similar to that of pVP2 but with a small spot at the beginning. Viral VP3 accumulated as small spots at the beginning and gradually concentrated. VP3 spots were also distributed around acidic organelles. Since the viral proteins pVP2, VP3, and VP4 are derived from the polyprotein (5), the distribution of viral polyprotein was assessed in live cell imaging. The results showed that viral polyprotein distributed throughout the cytoplasm with a few small spots at the beginning and then gradually concentrated to form spots, some of which accumulated around acidic organelles while others colocalized with acidic organelles without quenching in acidic pH, indicating that the active degradation of viral polyproteins was not induced because the EGFP reporter is sensitive to lysosomal proteolysis (Fig. 5). Taken together, these results indicate that viral assembly proteins distributed around acidic organelles.
FIG 5.

IBDV proteins accumulated around acidic compartments. DF-1 cells were transfected with pEGFP-N1-IBDV VP1, -IBDV VP3, -IBDV pVP2, and -IBDV PP (polyprotein) and pEGFP-N1 using Lipofectamine 2000 for 24 h. To stain the acidic compartments, cultures were incubated in medium containing 50 nM LysoTracker red (Molecular Probes) for 30 min; then nuclei were stained with Hoechst 33342 for 1 h. The distribution of proteins was observed by live cell imaging (green, viral proteins; red, acidic compartments; blue, nucleus). Serial pictures obtained from the video at the indicated time points are shown.
Viral assembly factors, pVP2, VP3, and dsRNA, colocalized with LAMP1.
IBDV assembly is mediated by three essential elements: pVP2, VP3, and dsRNA (4). In order to investigate whether endosomes, amphisomes, lysosomes, and autolysosomes have effects on viral assembly, we examined the distribution of pVP2, VP3, and dsRNA in IBDV-infected cells stained with endogenous lysosome-associated membrane protein 1 (LAMP1). As shown in Fig. 6, LAMP1 showed a distribution in IBDV-infected cells different from that in mock-infected cells. In mock-infected cells, LAMP1 was distributed homogeneously throughout the cells at a low level. However, in virus-infected cells, LAMP1 was much more concentrated and presented as spots around the viral components pVP2, VP3, and dsRNA, with partial colocalization. In addition, the expression of LAMP1 increased relative to that in mock-infected cells. To confirm this increase of LAMP1 in IBDV-infected cells, Western blotting was carried out to measure the load of LAMP1. The results showed that both the 120-kDa glycosylated LAMP1 and the 35-kDa polypeptide core increased in IBDV-infected cells relative to the proteins in mock-infected cells (data not shown). These results indicated that the essential viral assembly factors pVP2, VP3, and dsRNA accumulate with endosomes, amphisomes, lysosomes, and autolysosomes and may be associated with IBDV assembly.
FIG 6.
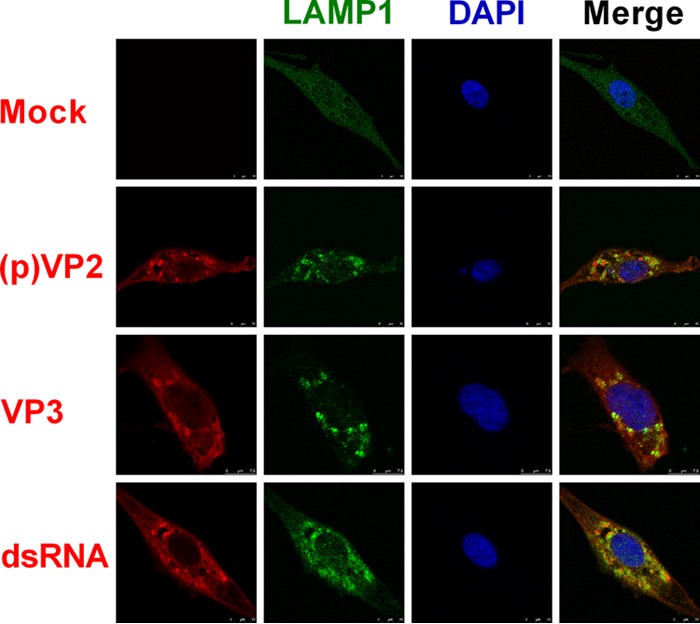
Viral assembly factors pVP2, VP3, and dsRNA colocalized with LAMP1. DF-1 cells were infected with IBDV Gt at an MOI of 1 for 24 h; then they were fixed and treated with antibodies against (p)VP2, VP3, or dsRNA or acidic endosomes, amphisomes, lysosomes, and autolysosome marker protein LAMP1 and stained with DAPI. The distribution of proteins was observed by confocal microscopy (red, viral proteins and dsRNA; green, LAMP1; blue, nucleus). The scale bar is indicated at the bottom right.
Acidic amphisomes and/or autolysosomes enhance IBDV replication.
To characterize the influence of different steps of autophagy pathway on IBDV replication, we employed pharmacological reagents that target autophagy at various stages. We analyzed intracellular viral protein yields and infectious progeny virus both in intracellular and extracellular parts treated with bafilomycin A1 (BAF A1), which blocks the fusion of autophagosomes with lysosomes (47). As shown in Fig. 7A, BAF A1 treatment resulted in inhibition autophagosomes fusion with lysosomes, with increased levels of LC3-II and p62. Starvation conditions were used as the positive control of autophagy induction with an increase in the LC3-II level. Cells both starved and treated with BAF A1 showed that autophagic signaling was induced but that fusion of autophagosomes with lysosomes was inhibited with an increase in both LC3-II and p62. As shown in Fig. 7A, C, and D, viral proteins pVP2 and VP2 showed a significant decrease in the presence of BAF A1. Inhibition of autophagosome fusion significantly reduced the infectious progeny virus titers both intracellularly and extracellularly (Fig. 7E and F). Thus, inhibition of autophagosome fusion with lysosomes reduced IBDV replication.
FIG 7.

Acidic amphisomes and/or autolysosomes enhance IBDV replication. DF-1 cells were pretreated with 20 nM BAF A1 in complete medium or under starvation conditions (A), 50 μM CQ in complete medium or under starvation conditions (B), or corresponding solvent for 4 h; then they were subjected to 1.5 h of absorption of IBDV Gt (MOI = 1). Cells were further cultured in fresh medium with 20 nM BAF A1 (A), 50 μM CQ (B), or corresponding solvent for 24 h p.i., and intracellular and extracellular viral loads were analyzed by WB and TCID50 assay. Antibodies against LC3B, p62, β-actin, and (p)VP2 were used for WB, and intensity band ratio of pVP2/actin (C) or VP2/actin (D) was normalized to the control. Progeny viral loads, both intracellular (E) and extracellular (F), were titrated in a TCID50 assay with secondary CEFs. (G) DF-1 cells seeded in ViewPlate-96 F TC plates were infected with IBDV at an MOI of 0.1. After a 1.5 h absorption, cells were further cultured in fresh medium with 50 μM CQ or corresponding solvent. At 24 h p.i., cells were fixed, permeabilized, inoculated with anti-(p)VP2 primary antibody, incubated with FITC-conjugated secondary antibody, and stained for nuclei with DAPI. Fluorescence was detected with the Operetta high-content screening platform, and the HarmonyTM 2.1.1 system was used for image acquisition; original image data were analyzed with the Columbus 2.3.2 database. One representative image from one field view is shown. (H) Percentage of FITC cells per DAPI cells infected with IBDV with or without CQ treatment. Means ± SDs were analyzed for three independent experiments. t test was used to analyze differences from the control group.
Cargo is hydrolyzed in the acidic lysosomes. Chloroquine (CQ) inhibits lysosome acidification, thereby inhibiting the hydrolysis activity of lysosomes (30, 49). In order to elucidate whether IBDV replication can be influenced by the loss of lysosomal hydrolysis activity in the presence of CQ, IBDV-infected DF-1 cells were treated with CQ. After IBDV infection, intracellular and extracellular infectious progeny viruses and intracellular viral protein yields were determined with or without CQ treatment by TCID50 assay and WB, respectively. We also employed starvation as the positive control for autophagy induction in this experiment. As shown in Fig. 7B, CQ treatment resulted in an increase in LC3-II and p62 levels. Cells both starved and treated with CQ showed an increase in both LC3-II and p62, indicating that autophagic signaling was induced and hydrolysis by lysosomes was inhibited. As shown in Fig. 7B to D, the amount of pVP2 significantly increased with CQ treatment, whereas the amount of VP2 significantly decreased in CQ-treated cells, suggesting that maturation of VP2 derived from pVP2 decreased because of the loss of lysosomal hydrolysis activity. The infectious progeny virus titers both in the supernatant and in cell cultures were significantly reduced in CQ-treated versus untreated cells (Fig. 7E and F). To directly observe IBDV replication with or without CQ treatment, the infection rate was evaluated by high-throughput analysis. The results showed that the infection rate was significantly reduced by CQ treatment (Fig. 7G and H). Thus, inhibition of lysosomal hydrolysis activity by CQ significantly reduced viral infection. We also investigated IBDV replication in cells after inhibition of lysosomal protease activity with E-64d. The results showed that the amount of pVP2 significantly increased but VP2 significantly decreased in E-64d-treated cells. The progeny virus titer in the supernatant was also significantly reduced in E-64d-treated cells (data not shown). Thus, IBDV replication was impaired by inhibition of lysosomal protease activity with E-64d. Taken together, the results show that acidic amphisomes and/or autolysosomes enhance IBDV replication.
Autophagic vacuoles facilitate IBDV maturation, release, and reinternalization.
To determine whether intact IBDV virions are present in endosomes, autophagosomes, amphisomes, lysosomes, or autolysosomes, the ultrastructure of infected DF-1 cells was examined by TEM. As shown in Fig. 8A, a small number of IBDV virions were encapsulated within the vacuole similar to the cell entry process, since cells presented no obvious cytopathic effect (CPE). Mitochondria presented a clear structure, and a few vacuoles appeared in the cytoplasm. Figure 8B shows that large numbers of IBDV virions were encapsulated within the cytoplasmic vacuole and cells presented obvious CPE, with some vacuoles and leakage of the cytoplasm content. Moreover, large numbers of mature membrane-encircled IBDV virions in lytic cells infected adjacent cells in a cell-to-cell manner (Fig. 8C). These results indicated that autophagic vacuoles are hijacked to facilitate virus release and reinternalization, consistent with the findings of a previous study (50). To investigate if IBDV virions can exist in amphisomes and/or autolysosomes, the cells were stained with p62 for immuno-transmission electron microscopy. We determined that a large number of intact IBDV virions were arranged in a lattice surrounded by p62 proteins, some of which lay between virions (Fig. 8D). The uninfected cells and an isotype matched antibody were employed as controls in immuno-TEM. As shown in Fig. 8E, the mock-infected cells can be labeled with p62 proteins. We applied the rabbit IgG polyclonal primary antibody as the isotype control. As Fig. 8F shows, there was no nonspecific reactivity to either cellular components or virions. These results suggest that intact IBDV virions can exist in autolysosomes and avoid hydrolysis. Thus, autophagic vacuoles facilitate IBDV maturation, release, and reinternalization.
FIG 8.

Autophagic vacuoles facilitate IBDV maturation, release, and reinternalization. (A to C) DF-1 cells were infected with IBDV strain Gt at an MOI of 1 for 24 h. Virions and endosomes, autophagosomes, amphisomes, lysosomes, and autolysosomes were observed by TEM. The square-enclosed region provides a higher-magnification view. The arrow indicates vacuole membrane and the asterisk indicates virions. Scale bar: 500 nm. (D) DF-1 cells were infected with IBDV Gt at an MOI of 1 for 24 h. Autolysosomes labeled with p62 antibodies and virions were observed by immuno-TEM. The arrow indicates colloidal gold dots representing p62. Scale bar: 200 nm. (E) Uninfected cells labeled with p62 were used as a control. Scale bar: 200 nm. (F) Rabbit IgG polyclonal antibody was used as the isotype control in an immuno-TEM assay. Scale bar: 500 nm.
DISCUSSION
IBDV is the most extensively studied virus in terms of molecular characteristics and pathogenesis; however, mechanisms underlying the IBDV life cycle require further exploration. In this study, we found that autophagic vacuoles were subverted to promote viral maturation and release. We boldly propose a new mechanism for IBDV maturation, release, and reinternalization, as illustrated in Fig. 9. Hsp90 and α4β1 integrin are shown to be components of the cellular receptor complex that mediates IBDV infection (6, 7). Additionally, IBDV uptake involves macropinocytosis and trafficking to early endosomes in a Rab5-dependent manner; therefore, a functional endocytic pathway is critical for viral infection (9). Uncoating of the virus occurs within the endosome in response to a low-pH environment (3). One viral peptide, pep46, generated during the processing of pVP2 maturation is suggested to deform biological membranes, leading to the formation of pores. The pores promote the exchange of small molecules between endosomal ghosts and the cytoplasm, allowing initial transcription from the genome (3, 8). In addition, the virus establishes replication factories associated with the endosomal membranes (8). We revealed that IBDV induces typical double-membrane vesicles, termed autophagosomes, which can fuse with vesicles of the endocytic pathway to form amphisomes and/or autolysosomes. Viral maturation is enhanced by the low-pH environment of amphisomes and autolysosomes. Virions are packaged within the cellular membrane, which facilitates fusion with the membranes of adjacent cells and delivers a large number of virions directly and rapidly into uninfected cells in cell-to-cell way.
FIG 9.

Proposed model for IBDV replication cycle. Hsp90 and α4β1 integrin are components of the cellular receptor complex mediating IBDV infection (6, 7). Additionally, IBDV uptake involves macropinocytosis and trafficking to early endosomes in a Rab5-dependent manner, and a functional endocytic pathway is critical for viral infection (9). Uncoating of the virus occurs within the endosome in response to a low-pH environment (3). One viral peptide, pep46, generated during the processing of pVP2 maturation is suggested to deform biological membranes, leading to the formation of pores. The pores promote an exchange of small molecules between endosomal ghosts and the cytoplasm, allowing the initial transcription of the genome (3, 8). In addition, virus builds up its replication factory associated with the endosomal membrane (8). We revealed that IBDV induces typical double-membrane vesicles termed autophagosomes, which can fuse with vesicles of the endocytic pathway to form amphisomes and/or autolysosomes. Viral maturation is enhanced by the low-pH environment of amphisomes and/or autolysosomes. Virions are packaged within the cellular membrane, which facilitates fusion with the membranes of adjacent cells and delivers a large number of virions directly and rapidly into uninfected cells in cell-to-cell way. We propose that this may be a new mechanism for IBDV maturation, release, and reinternalization.
In this study, we revealed that IBDV induces autophagy during the late phase of infection, fusing autophagosomes with lysosomal systems. Although autophagosomes typically fuse with lysosomes to hydrolyze cargo, IBDV is not degraded. Interestingly, viral replication is promoted by the induction of autophagy with rapamycin or starvation, whereas inhibition of autophagy by 3-MA or wortmannin reduced viral replication, indicating that autophagosomes supplied more vesicle membranes as the viral replication sites for IBDV (8). Several viruses, in particular, members of the family Picornaviridae, have been found not only to subvert autophagy but also to actively induce the pathway and hijack the autophagy machinery to increase virus replication. Both poliovirus- and coxsackievirus B3-induced double-membrane-bound vesicles strongly resemble cellular autophagosomes (17, 24, 27). Autophagic protein LC3 conjugated to the lipid phosphatidylethanolamine confers membrane association and is required for autophagosome formation and membrane expansion. In our study, LC3B silencing decreased virus yields. In addition, the decrease in progeny virus released into the extracellular medium was greater than the decrease in intracellular viral proteins. This is similar to results with poliovirus, with which siRNA knockdown of LC3 or ATG12 significantly reduced progeny virus released, suggesting that hijacked autophagosomes provide a site for viral replication and allow virions within the cytoplasmic lumen of double-membrane vesicles to be released into the extracellular milieu. As a part of the host defense system, autophagy contributes to the elimination of invading microorganisms by degrading them in the lysosomal compartment (17, 51). We demonstrated the formation of amphisomes and/or autolysosomes via fusion of autophagosomes with endocytic vacuoles, consistent with the observation of changes in fluorescence with expression of mRFP-EGFP-LC3. Surprisingly, endogenous p62 levels were not significantly altered, indicating that autolysosomes formed without active degradation of contents. In rubella virus- and Semliki Forest virus-infected cells, modified endosomes and lysosomes constitute special vesicular structures that serve as RNA replication sites and replicative intermediates (52). In rubella virus infection, replication complexes are membrane-bound cytoplasmic vacuoles. These replication complexes are virus-modified lysosomes (53). Studies on poliovirus have indicated that intracellular vesicle acidification, a hallmark of autophagosome maturation, promotes maturation of infectious poliovirus particles (54–57). In IBDV-infected cells, pVP2, VP3, and dsRNA, which are essential components for IBDV assembly (4), colocalized with LAMP1 in autophagic endosomal pathway. HIV initiates assembly in the late endosome (58). Thus, the formation of amphisomes and/or autolysosomes and the activity of amphisomes and autolysosomes facilitate IBDV maturation and assembly. Live cell imaging showed that viral proteins accumulated around acidic organelles labeled with LysoTracker red and exhibited partial colocalization with them. The proteins were not degraded by the acidic environment; rather, the amount increased with concentrated fluorescent signal. The fusion of autophagosomes with lysosomes was inhibited by BAF A1, which reduced intracellular and extracellular virus yields. Lysosomal hydrolysis activity was inhibited by chloroquine, significantly reducing viral titers and resulting in the loss of mature VP2 derived from pVP2 due to the loss of lysosomal hydrolysis activity. Inhibiting the activity of lysosomal proteases by E-64d also impaired virus replication (data not shown). Endosomes, autophagosomes, amphisomes, lysosomes, and autolysosomes facilitate low-pH-mediated maturation of viral proteins and membrane-mediated progeny virus release. Enteroviruses such as poliovirus are relatively resistant to low pH and proteases and may survive in autophagosomes and lysosomes (59). IBDV, similar to other viruses, likely completes the morphogenetic process associated with the autophagic endosomal pathway and Golgi stacks (60–62). IBDV virions were not degraded in the lysosomes or autolysosomes. Indeed, IBDV utilized their low-pH environment to facilitate viral protein maturation and replication. A large number of IBDV virions were encapsulated in vacuoles, and well-organized virions were surrounded and permeated with p62, a marker of autolysosomes. Taken together, the results show that the autophagic endosomal pathway is one mechanism that facilitates IBDV maturation, release, and reinternalization. A recent study claims that a subpopulation varicella-zoster virus particles are sorted into a vesicle resembling an amphisome for their final trafficking to the outer cell membrane (33). Overall, these researches suggest that highly divergent DNA and RNA viruses may share similar exit pathways.
MATERIALS AND METHODS
Cells, viruses, plasmids, and reagents.
DF-1 cells were cultured in Dulbecco's modified Eagle medium (DMEM; HyClone; SH30022.01B) supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified 5% CO2 incubator. Primary chicken embryo fibroblasts (CEFs) were prepared from 9-day-old specific-pathogen-free chicken embryos purchased from the Experimental Animal Center of Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences. The cell-adapted IBDV Gt strain was saved in our laboratory. The plasmid EGFP-Av-LC3B was described previously (41). The plasmids pcaggs, pGPU6/RFP/Neo, ptfLC3, and pEGFP-N1 (Clontech; 6085-1) were saved in our laboratory. Mouse monoclonal anti-IBDV (p)VP2 and anti-VP3 were saved in our laboratory. The following were purchased from Abcam: rabbit monoclonal anti-ATG5 (ab108327), rabbit polyclonal anti-LAMP1 (ab24170), and rabbit IgG isotype polyclonal antibody (ab27478). The following were purchased from Sigma-Aldrich: rabbit polyclonal anti-LC3B (L7543) and anti-p62/SQSTM1 (P0067); mouse monoclonal anti-β-actin (A1978) and anti-c-Myc (M4439); anti-mouse IgG (whole molecule)-tetramethyl rhodamine isothiocyanate (TRITC) antibody produced in goat (T5393); anti-mouse IgG (whole molecule)-fluorescein isothiocyanate (FITC) antibody produced in goat (F9006); and anti-rabbit IgG (whole molecule)-FITC antibody produced in goat (F9887). Mouse monoclonal anti-double-stranded RNA (J2-1303) was purchased from English and Scientific Consulting, Hungary. Rapamycin (R0395), 3-methyladenine (M9281), bafilomycin A1 (B1793), chloroquine diphosphate salt (C6628), E-64d (E3132), and a mitochondria isolation kit (MITOISO2) were purchased from Sigma-Aldrich. Wortmannin (12-338) was purchased from Millipore. IRDye 800CW goat anti-rabbit IgG (H+L) (926-32211) and IRDye 800CW goat anti-mouse IgG (H+L) (926-32210) were purchased from Li-Cor Biosciences.
Virus infection and IBDV titration.
DF-1 cells were seeded on six-well plates, cultured for about 24 h, and then infected with IBDV Gt at a multiplicity of infection (MOI) of 0.1 or 1 with a 1.5-h absorption period at 37°C. Unattached viruses were removed by aspiration, and the cells were cultured in fresh complete medium containing 10% FBS. The secondary CEF cultures were used to titrate infectious progeny virus after various treatments. The supernatants were collected at various time points to determine the titer of infectious viral progeny in terms of TCID50/100 μl using the Reed-Muench formula. All experiments were repeated three times, and the means and standard deviations (SDs) were calculated.
Lysate preparation, SDS-PAGE, and immunoblotting.
Cells were washed with phosphate-buffered saline (PBS) and were lysed with lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, sodium pyrophosphate, β-glycerophosphate, and 1× complete cocktail protease inhibitor. Whole lysates were boiled for 10 min in the presence of 5× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. After centrifugation at 12,000 × g for 2 min, equivalent sample amounts were separated by 12% SDS-PAGE and transferred to pure nitrocellulose blotting membranes (PALL; 66485). After blocking with 5% skim milk, the membranes were incubated with primary antibody at 37°C for 1.5 h, followed by IRDye 800CW secondary antibody for 1 h at 37°C. Proteins were visualized and quantified using the Odyssey system (Li-Cor).
Confocal microscopy.
DF-1 cells were cultured to ∼70% confluence in 35-mm culture dishes with 20-mm coverslip inserts (NEST; 801001) overnight and were infected with IBDV at an MOI of 1. The cells were further cultured in fresh complete medium for 12 and 24 h, then fixed with 4% paraformaldehyde for 30 min at room temperature (RT), and permeabilized with 0.1% Triton X-100 for 15 min at RT. After three washes with 0.05% Tween 20–PBS (PBST), cells were incubated with rabbit polyclonal anti-LC3B and mouse monoclonal anti-(p)VP2 primary antibodies for 1.5 h at RT. After three washes with PBST, cells were incubated for 1 h with 1:200 dilutions of goat anti-rabbit secondary antibodies conjugated to fluorescein isothiocyanate and goat anti-mouse secondary antibodies conjugated to tetramethyl rhodamine isothiocyanate simultaneously in the dark. After three washes with PBST, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min at RT. After three washes with PBST, the distribution of viral proteins (p)VP2 and endogenous LC3 was observed by confocal microscopy.
DF-1 cells were cultured to ∼70% confluence in 35-mm culture dishes with 20-mm coverslip inserts; then they were infected with IBDV Gt at an MOI of 1. At 24 h p.i., cells were fixed and permeabilized; then they were incubated with mouse monoclonal anti-(p)VP2, -VP3, or -dsRNA and rabbit polyclonal anti-lysosome-associated membrane protein 1 (anti-LAMP1) antibody for 1.5 h at RT. After three washes with PBST, cells were simultaneously incubated for 1 h with 1:200 dilutions of TRITC-conjugated goat anti-mouse and FITC-conjugated goat anti-rabbit secondary antibodies. Nuclei were stained with DAPI and observed by confocal microscopy after three washes with PBST.
Live cell imaging.
DF-1 cells were cultured to ∼70% confluence in 35-mm culture dishes with 20-mm coverslip inserts; then they were transfected with ptfLC3, a tandem reporter construct of mRFP-EGFP-LC3, using Lipofectamine 2000 (Invitrogen; 11668-019). At 24 h posttransfection, cells were mock infected or infected with IBDV at an MOI of 1. After 1.5 h of absorption, cells were observed by long-term live cell imaging with the RFP and EGFP channels to produce time-lapse movies. All movies were edited using Volocity software (PerkinElmer, Waltham, MA).
DF-1 cells were cultured to ∼70% confluence in 35-mm culture dishes with 20-mm coverslip inserts and transfected with pEGFP-N1-IBDV VP1, pEGFP-N1-IBDV VP3, pEGFP-N1-IBDV pVP2, pEGFP-N1-IBDV PP, or pEGFP-N1 using Lipofectamine 2000. At 24 h posttransfection, cells were incubated in medium containing 50 nM LysoTracker red (Molecular Probes, Life Technologies; L7528) for 30 min and then incubated in medium containing Hoechst 33342 for 1 h. Cells were observed by long-term live cell imaging using three channels (green, red, and blue) to produce time-lapse movies. All movies were edited using Volocity software.
High-throughput image analysis.
DF-1 cells were seeded in ViewPlate-96 F TC (PerkinElmer; 6005182) plates overnight and infected with IBDV at an MOI of 0.1. After 1.5 h of absorption, cells were cultured in fresh medium in the presence or absence of 50 μM CQ. At 24 h p.i., cells were fixed and permeabilized. After three washes with PBST, cells were inoculated with primary antibodies of mouse monoclonal anti-(p)VP2 for 1.5 h at RT. After three washes with PBST, cells were incubated for 1 h with 1:200 dilutions of FITC-conjugated goat anti-mouse secondary antibodies in the dark. Cells were stained with DAPI, and fluorescence was detected with the Operetta high-content screening platform (PerkinElmer). The Harmony 2.1.1 system (PerkinElmer) was used for high-throughput image acquisition. Thirty random individual imaging fields per well were analyzed with blue for nucleus (DAPI) and green for viral protein (FITC) channels on a single Z-plane. The original image data were imported into and analyzed with Columbus 2.3.2 (PerkinElmer). The number of FITC cells/DAPI cells was used to quantify virus infection rate. Cell nuclei were first identified using the DAPI channel image, and green fluorescence was detected using the FITC channel image. The quantified nuclei was generated a first population of objects (Nuclei), and the second population of objects (FITC-positive cells) was generated based on the Calculate Intensity Properties function for green fluorescence. The FITC-positive cell/DAPI cell ratio was calculated and analyzed with or without CQ treatment.
Transmission electron microscopy.
For ultrastructural analysis, DF-1 cells were mock infected or infected with IBDV Gt at an MOI of 1 for 24 h. Cells were collected at 900 × g for 15 min and fixed in 2.5% glutaraldehyde in phosphate buffer, pH 7.2, for 2 h at 4°C. Following three 15-min washes in phosphate buffer, cells were postfixed in 1% osmium tetroxide for 1 h at 4°C. The samples were then rinsed extensively in three 15-min washes with distilled H2O. Cells were dehydrated in a graded series of acetone and embedded in LR-White resin. Sections of 50 to 70 nm were prepared after polymerization for 2 days at 80°C. Sections were doubly stained with uranyl acetate for 15 min and lead citrate for 10 min at RT. Autophagosomes were defined as double-membrane vacuoles measuring 0.3 to 2.0 μm with clearly recognizable cytoplasmic contents.
For immunoelectron microscopy, cells were collected and fixed in 4% paraformaldehyde in 0.1 M HEPES-NaOH buffer, pH 7.4, overnight at RT. Cells were washed three times in distilled H2O for 15 min; then they were dehydrated in a graded series of N,N-dimethylformamide, embedded in LR-white resin overnight at 4°C, and polymerized for 10 days at −20°C under UV light. Sections of 80 to 100 nm were prepared and blocked in 3% bovine serum albumin for 30 min at RT. Primary antibodies were incubated for 40 min at RT and washed three times in distilled H2O for 5 min. The secondary antibodies were conjugated to colloidal gold (10-nm diameter), incubated for 40 min at RT, and washed three times. The sections were stained with 3% uranyl acetate for 10 min at RT.
shRNA- or siRNA-mediated gene silencing.
The siRNA sequences were as follows: avian LC3B-1, 5′-GGUGAUCAUUGAAAGGUACTT-3′; LC3B-2, 5′-GCCAGAUCACGUCAACAUGTT-3′; LC3B-3, 5′-GCUUCCUGUACAUGGUGUATT-3′; avian ATG5-1, 5′-GGCAUUACCCAAUUGGUUUTT-3′; ATG5-2, 5′-GCAUCACAGUGCAUUUCAATT-3′; and ATG5-3, 5′-GGAUGUGAUUGAAGCUCAUTT-3′. DF-1 cells cultured to 60 to 70% confluence in 6-well plates were transfected with avian LC3B siRNA or ATG5 siRNA using Lipofectamine 2000. Briefly, 60 pmol of siRNA was diluted in 250 μl of serum-free Opti-MEM and 6 μl of Lipofectamine 2000 was also diluted in 250 μl of Opti-MEM, and both mixtures were incubated for 5 min at RT. The diluted RNA and Lipofectamine 2000 were combined and incubated again for 20 min at RT. The 500-μl mixture was then added dropwise to each culture well. The plates were incubated at 37°C in a CO2 incubator for 4 h; then the mixtures were replaced with fresh complete medium and incubated for an additional 44 h. Scrambled siRNAs were used as a negative control. The knockdown efficiency was evaluated based on protein expression level. Competent LC3B siRNAs were chosen to construct the shRNA expression vector with pGPU6/RFP/Neo, and shRNA or siRNA was used to investigate the influence on viral replication of target protein silencing. Sequences of primers used to clone avian ATG5 or IBDV proteins in vector pcaggs or pEGFP-N1 are shown in Table 1.
TABLE 1.
Sequences of primers used to clone avian ATG5 or IBDV proteins in vector pcaggs or pEGFP-N1
| Primer name | Sequence (5′–3′)a |
|---|---|
| EGFP-N1-VP1-F | CCGCTCGAGATGAGTGACATTTTCAACAGTC |
| EGFP-N1-VP1-R | CGGAATTCGGCGGCTCTCCTTTTGGCGT |
| EGFP-N1-pVP2-F | CGGAATTCATGACAAACCTGCAAGATCAAAC |
| EGFP-N1-pVP2-R | GGGGTACCGTGGCGAGGGTTAGCTGCCTTATG |
| EGFP-N1-VP3-F | CGGAATTCATGGCATCAGAGTTCAAAGAGAC |
| EGFP-N1-VP3-R | GGGGTACCGTCTCAAGGTCCTCATCAG |
| EGFP-N1-PP-F | CGGAATTCATGACAAACCTGCAAGATCAAAC |
| EGFP-N1-PP-R | GGGGTACCGTCTCAAGGTCCTCATCAG |
| pc-mycAtg5-F | GGGGTACCATGGCATCAATGCAGAAGCTGATCTCAGAGGAGGACCTG ACAGATGACAAAGATG |
| pc-Atg5-R | CCGCTCGAGTCAATCAGTAGGTCGGGGAATG |
Inserted restriction sites are underlined, and the c-Myc tag is italicized.
Statistical analysis.
All data are presented as means ± SDs based on three independent experiments. The significance of the variability between groups was determined by t test (shown in figures as follows: #, P > 0.05; *, P < 0.05; and **, P < 0.01).
ACKNOWLEDGMENTS
We thank Hualan Chen for providing valuable suggestions and Changjun Liu, Yanping Zhang, and Kai Li for their help.
This work was supported by a major project of the National Natural Science Foundation of China (no. 31430087) and the earmarked fund for the Modern Agro-industry Technology Research System (no. CARS-42-G07).
REFERENCES
- 1.Cosgrove AS. 1962. An apparently new disease of chickens: avian nephrosis. Avian Dis 6:385–389. doi: 10.2307/1587909. [DOI] [Google Scholar]
- 2.Luque D, Rivas G, Alfonso C, Carrascosa JL, Rodriguez JF, Caston JR. 2009. Infectious bursal disease virus is an icosahedral polyploid dsRNA virus. Proc Natl Acad Sci U S A 106:2148–2152. doi: 10.1073/pnas.0808498106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galloux M, Libersou S, Morellet N, Bouaziz S, Da Costa B, Ouldali M, Lepault J, Delmas B. 2007. Infectious bursal disease virus, a non-enveloped virus, possesses a capsid-associated peptide that deforms and perforates biological membranes. J Biol Chem 282:20774–20784. doi: 10.1074/jbc.M701048200. [DOI] [PubMed] [Google Scholar]
- 4.Maraver A, Oña A, Abaitua F, González D, Clemente R, Ruiz-Díaz JA, Castón JR, Pazos F, Rodriguez JF. 2003. The oligomerization domain of VP3, the scaffolding protein of infectious bursal disease virus, plays a critical role in capsid assembly. J Virol 77:6438–6449. doi: 10.1128/JVI.77.11.6438-6449.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birghan C, Mundt E, Gorbalenya AE. 2000. A non-canonical lon proteinase lacking the ATPase domain employs the ser-Lys catalytic dyad to exercise broad control over the life cycle of a double-stranded RNA virus. EMBO J 19:114–123. doi: 10.1093/emboj/19.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin TW, Lo CW, Lai SY, Fan RJ, Lo CJ, Chou YM, Thiruvengadam R, Wang AH, Wang MY. 2007. Chicken heat shock protein 90 is a component of the putative cellular receptor complex of infectious bursal disease virus. J Virol 81:8730–8741. doi: 10.1128/JVI.00332-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delgui L, Oña A, Gutiérrez S, Luque D, Navarro A, Castón JR, Rodríguez JF. 2009. The capsid protein of infectious bursal disease virus contains a functional α4β1 integrin ligand motif. Virology 386:360–372. doi: 10.1016/j.virol.2008.12.036. [DOI] [PubMed] [Google Scholar]
- 8.Delgui LR, Rodríguez JF, Colombo MI. 2013. The endosomal pathway and the Golgi complex are involved in the infectious bursal disease virus life cycle. J Virol 87:8993–9007. doi: 10.1128/JVI.03152-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gimenez MC, Aguirre JFR, Colombo MI, Delgui LR. 2015. Infectious bursal disease virus uptake involves macropinocytosis and trafficking to early endosomes in a Rab5-dependent manner. Cell Microbiol 17:988–1007. doi: 10.1111/cmi.12415. [DOI] [PubMed] [Google Scholar]
- 10.Coulibaly F, Chevalier C, Gutsche I, Pous J, Navaza J, Bressanelli S, Delmas B, Rey FA. 2005. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 120:761–772. doi: 10.1016/j.cell.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Saugar I, Luque D, Oña A, Rodríguez JF, Carrascosa JL, Trus BL, Castón JR. 2005. Structural polymorphism of the major capsid protein of a double-stranded RNA virus: an amphipathic α helix as a molecular switch. Structure 13:1007–1017. doi: 10.1016/j.str.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 12.Escaffre O, Le Nouën C, Amelot M, Ambroggio X, Ogden KM, Guionie O, Toquin D, Müller H, Islam MR, Eterradossi N. 2013. Both genome segments contribute to the pathogenicity of very virulent infectious bursal disease virus. J Virol 87:2767–2780. doi: 10.1128/JVI.02360-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu B, Zhang Y, Jia L, Wu H, Fan C, Sun Y, Ye C, Liao M, Zhou J. 2015. Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy 11:503–515. doi: 10.1080/15548627.2015.1017184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmid D, Munz C. 2007. Innate and adaptive immunity through autophagy. Immunity 27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. 2008. Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie Z, Klionsky DJ. 2007. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 17.Levine B. 2005. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 18.Tallóczy Z, Levine B. 2006. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2:24–29. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- 19.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. 1998. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 72:8586–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulvey M, Poppers J, Sternberg D, Mohr I. 2003. Regulation of eIF2alpha phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J Virol 77:10917–10928. doi: 10.1128/JVI.77.20.10917-10928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaumorcel M, Souquère S, Pierron G, Codogno P, Esclatine A. 2008. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 4:46–53. doi: 10.4161/auto.5184. [DOI] [PubMed] [Google Scholar]
- 22.Ku B, Woo JS, Liang C, Lee KH, Hong HS, E X, Kim KS, Jung JU, Oh BH. 2008. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog 4:239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor MP, Jackson WT. 2009. Viruses and arrested autophagosome development. Autophagy 5:870–871. doi: 10.4161/auto.9046. [DOI] [PubMed] [Google Scholar]
- 24.Wong J, Zhang JX, Gao G, Mao I, Mcmanus BM, Luo H. 2008. Autophagosome supports coxsackievirus B3 replication in host cells. J Virol 82:9143–9153. doi: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou D, Spector SA. 2008. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS 22:695–699. doi: 10.1097/QAD.0b013e3282f4a836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dreux M, Gastaminza P, Wieland SF, Chisari FV.. 2009. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A 106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson WT, Giddings TH Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. Plos Biol 3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richards AL, Jackson WT. 2013. That which does not degrade you makes you stronger: infectivity of poliovirus depends on vesicle acidification. Autophagy 9:806–807. doi: 10.4161/auto.23962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salonen A, Ahola T, Kääriäinen L. 2005. Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol 285:139–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer P, Lee M, Strowig T, Arrey F, Conenello G, Pypaert M, Andersen J, García-Sastre A, Münz C. 2009. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chi PI, Huang WR, Lai IH, Cheng CY, Liu HJ. 2013. The p17 nonstructural protein of avian reovirus triggers autophagy enhancing virus replication via activation of phosphatase and tensin deleted on chromosome 10 (PTEN) and AMP-activated protein kinase (AMPK), as well as dsRNA-dependent protein kinase (PKR). J Biol Chem 288:3571–3584. doi: 10.1074/jbc.M112.390245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y, Yu S, Ding N, Meng C, Meng S, Zhang S, Zhan Y, Qiu X, Tan L, Chen H. 2014. Autophagy benefits the replication of Newcastle disease virus in chicken cells and tissues. J Virol 88:525–537. doi: 10.1128/JVI.01849-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buckingham EM, Jarosinski KW, Jackson W, Carpenter JE, Grose C. 2016. Exocytosis of varicella-zoster virus virions involves a convergence of endosomal and autophagy pathways. J Virol 90:8673–8685. doi: 10.1128/JVI.00915-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson WT. 2015. Viruses and the autophagy pathway. Virology 479-480:450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bird SW, Kirkegaard K, Agbandje-Mckenna M, Freed EO. 2014. The ins and outs of viral infection: keystone meeting review. Viruses 6:3652–3662. doi: 10.3390/v6093652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng Z, Hensley L, Mcknight KL, Hu F, Madden V, Ping L, Jeong SH, Walker C, Lanford RE, Lemon SM. 2013. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496:367–371. doi: 10.1038/nature12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jungmann A, Nieper H, Müller H. 2001. Apoptosis is induced by infectious bursal disease virus replication in productively infected cells as well as in antigen-negative cells in their vicinity. J Gen Virol 82:1107–1115. doi: 10.1099/0022-1317-82-5-1107. [DOI] [PubMed] [Google Scholar]
- 38.Saraste A, Pulkki K. 2000. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res 45:528–537. doi: 10.1016/S0008-6363(99)00384-3. [DOI] [PubMed] [Google Scholar]
- 39.Blommaart EFC, Luiken JJFP, Meijer AJ. 1997. Autophagic proteolysis: control and specificity. Histochem J 29:365–385. doi: 10.1023/A:1026486801018. [DOI] [PubMed] [Google Scholar]
- 40.Codogno P, Meijer AJ. 2005. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 12(Suppl 2):S1509–S1518. [DOI] [PubMed] [Google Scholar]
- 41.Maier HJ, Cottam EM, Stevenson-Leggett P, Wilkinson JA, Harte CJ, Willeman T, Britton P. 2013. Visualizing the autophagy pathway in avian cells and its application to studying infectious bronchitis virus. Autophagy 9:496–509. doi: 10.4161/auto.23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seglen PO, Gordon PB. 1982. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. 2010. Dual Role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2003. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 22:4577–4577. doi: 10.1093/emboj/cdg454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. 2004. The role of autophagy during the early neonatal starvation period. Nature 432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 46.Kimura S, Noda T, Yoshimori T. 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 47.Klionsky DJ, Abeliovich H, Agostinis P. 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in highereukaryotes. Autophagy 4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 49.Joubert PE, Meiffren G, Grégoire IP, Pontini G, Richetta C, Flacher M, Azocar O, Vidalain PO, Vidal M, Lotteau V. 2009. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 6:354–366. doi: 10.1016/j.chom.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 50.Käufer I, Weiss E. 1976. Electron-microscope studies on the pathogenesis of infectious bursal disease after intrabursal application of the causal virus. Avian Dis 20:483–495. doi: 10.2307/1589381. [DOI] [PubMed] [Google Scholar]
- 51.Kim HJ, Lee S, Jung JU. 2010. When autophagy meets viruses: a double-edged sword with functions in defense and offense. Semin Immunopathol 32:323–341. doi: 10.1007/s00281-010-0226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JY, Marshall JA, Bowden DS. 1994. Characterization of rubella virus replication complexes using antibodies to double-stranded RNA. Virology 200:307–312. doi: 10.1006/viro.1994.1192. [DOI] [PubMed] [Google Scholar]
- 53.Magliano D, Marshall JA, Bowden DS, Vardaxis N, Meanger J, Lee JY. 1998. Rubella virus replication complexes are virus-modified lysosomes. Virology 240:57–63. doi: 10.1006/viro.1997.8906. [DOI] [PubMed] [Google Scholar]
- 54.Jackson WT. 2014. Poliovirus-induced changes in cellular membranes throughout infection. Curr Opin Virol 9:67–73. doi: 10.1016/j.coviro.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richards AL, Jackson WT. 2012. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog 8:1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richards AL, Jackson WT. 2013. How positive-strand RNA viruses benefit from autophagosome maturation. J Virol 87:9966–9972. doi: 10.1128/JVI.00460-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richards AL, Soares-Martins JA, Riddell GT, Jackson WT. 2014. Generation of unique poliovirus RNA replication organelles. mBio 5:e00833-13. doi: 10.1128/mBio.00833-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nydegger S, Foti M, Derdowski A, Spearman P, Thali M. 2003. HIV-1 egress is gated through late endosomal membranes. Traffic 4:902–910. doi: 10.1046/j.1600-0854.2003.00145.x. [DOI] [PubMed] [Google Scholar]
- 59.Wileman T. 2006. Aggresomes and autophagy generate sites for virus replication. Science 312:875–878. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- 60.Marsh M, Bron R. 1997. SFV infection in CHO cells: cell-type specific restrictions to productive virus entry at the cell surface. J Cell Sci 110(Part 1):95–103. [DOI] [PubMed] [Google Scholar]
- 61.Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu Rev Biochem 79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 62.Sodeik B. 2000. Mechanisms of viral transport in the cytoplasm. Trends Microbiol 8:465–472. doi: 10.1016/S0966-842X(00)01824-2. [DOI] [PubMed] [Google Scholar]