

ABSTRACT
The establishment of human cytomegalovirus (HCMV) latency and persistence relies on the successful infection of hematopoietic cells, which serve as sites of viral persistence and contribute to viral spread. Here, using blocking antibodies and pharmacological inhibitors, we document that HCMV activation of the epidermal growth factor receptor (EGFR) and downstream phosphatidylinositol 3-kinase (PI3K) mediates viral entry into CD34+ human progenitor cells (HPCs), resulting in distinct cellular trafficking and nuclear translocation of the virus compared to that in other immune cells, such as we have documented in monocytes. We argue that the EGFR allows HCMV to regulate the cellular functions of these replication-restricted cells via its signaling activity following viral binding. In addition to regulating HCMV entry/trafficking, EGFR signaling may also shape the early steps required for the successful establishment of viral latency in CD34+ cells, as pharmacological inhibition of EGFR increases the transcription of lytic IE1/IE2 mRNA while curbing the expression of latency-associated UL138 mRNA. EGFR signaling following infection of CD34+ HPCs may also contribute to changes in hematopoietic potential, as treatment with the EGFR kinase (EGFRK) inhibitor AG1478 alters the expression of the cellular hematopoietic cytokine interleukin 12 (IL-12) in HCMV-infected cells but not in mock-infected cells. These findings, along with our previous work with monocytes, suggest that EGFR likely serves as an important determinant of HCMV tropism for select subsets of hematopoietic cells. Moreover, our new data suggest that EGFR is a key receptor for efficient viral entry and that the ensuing signaling regulates important early events required for successful infection of CD34+ HPCs by HCMV.
IMPORTANCE HCMV establishes lifelong persistence within the majority of the human population without causing overt pathogenesis in healthy individuals. Despite this, reactivation of HCMV from its latent reservoir in the bone marrow causes significant morbidity and mortality in immunologically compromised individuals, such as bone marrow and solid organ transplant patients. Lifelong persistent infection has also been linked with the development of various cardiovascular diseases in otherwise healthy individuals. Current HCMV therapeutics target lytic replication, but not the latent viral reservoir; thus, an understanding of the molecular basis for viral latency and persistence is paramount to controlling or eliminating HCMV infection. Here, we show that the viral signalosome activated by HCMV binding to its entry receptor, EGFR, in CD34+ HPCs initiates early events necessary for successful latent infection of this cell type. EGFR and associated signaling players may therefore represent promising targets for mitigating HCMV persistence.
KEYWORDS: CD34+ HPC, EGFR, HCMV, latency, virus entry
INTRODUCTION
Human cytomegalovirus (HCMV) establishes a persistent infection that remains for the lifetime of its human host and is marked by periods of latency and reactivation (1, 2). HCMV infection causes severe disease in immunocompromised patients, including allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant (SOT) patients, and in congenitally infected neonates (1, 3–5). In the immunocompetent, HCMV is associated with chronic inflammatory diseases and some tumors (4, 6–10). HCMV pathogenesis is due to infection of most host organs and the resulting overt organ disease (1, 3, 11–13). During primary infection, HCMV spreads to peripheral organ systems via a hematogenous route (13, 14). Following tissue dissemination, multiple organ systems are infected, allowing spread and the establishment of persistence in the bone marrow, where HCMV remains latent in cells of the myeloid lineage (15), although the regulatory mechanisms involved in latency remain largely undefined.
Hematopoietic cells play an indispensable role in the persistence strategy of HCMV, functioning as carrier cells for viral dissemination and serving as sources of viral latency. Circulating blood monocytes play a key role in HCMV hematogenous dissemination (16), and monocytes, as well as their predecessors, CD34+ human progenitor cells (HPCs) in the bone marrow, are crucial reservoirs for HCMV latency (17–19) that contribute to viral pathogenesis. Reactivation of latent virus from these cells is a major cause of morbidity and mortality in immunosuppressed individuals, such as HSCT and SOT recipients and cancer patients undergoing immunosuppressive therapies (4). Indeed, some 35% of SOT patients and 80% of allogeneic HSCT recipients develop HCMV infection requiring antiviral therapy (5, 20). Common clinical manifestations of HCMV infection in the transplant patient, termed CMV syndrome, are characterized by evidence of HCMV viremia accompanied by either fever, myelosuppression (leukopenia or thrombocytopenia), or hepatitis (5, 21, 22). In addition, HCMV has been implicated in graft failure after HSCT, as determined by the presence of severe pancytopenia, bone marrow hypoplasia, and detection of HCMV in the bone marrow (21, 23–25). HCMV-associated leukopenia also puts patients at risk for life-threatening secondary bacterial and fungal infections (26, 27).
Interestingly, although infection of monocytes and CD34+ HPCs is central to the viral persistence strategy, these cell types are highly restrictive of viral replication (16, 17, 28–34). This creates a considerable obstacle for HCMV to overcome in gaining control of infected cells without the benefit of the de novo expressed viral gene products that are known to regulate a variety of cellular functions in replication-permissive cell types. A major focus of our laboratory has been defining the complex mechanisms that HCMV has evolved to reprogram infected monocytes to serve as viral carriers in the absence of viral gene expression (16, 30–32, 35–45). We have shown that viral binding to and activation of the epidermal growth factor receptor (EGFR) (39) and cellular integrins (42, 43) on the surfaces of monocytes induce a distinct cellular signaling cascade resulting in functional and molecular changes that prime infected monocytes for their role in viral dissemination (31, 37–39, 42, 43). Among these functional changes is enhanced motility of HCMV-infected monocytes compared to mock-infected monocytes or to those stimulated with alternative activating agents (lipopolysaccharide [LPS] or phorbol 12-myristate 13-acetate [PMA]), leading to increased transendothelial migration of infected cells into the surrounding organ tissues (16, 30, 32, 44, 45). In addition, HCMV drives monocyte-to-macrophage differentiation in infected cells (16, 38) to create a cell type capable of promoting viral gene expression and replication. This differentiation process also results in distinct macrophage polarization, likely serving to balance viral gene expression and replication with immune evasion (31, 35). The HCMV signalosome created by activation of EGFR and integrins also promotes extended survival of infected monocytes, allowing HCMV to overcome the biologically limited life span of monocytes (37) and to combat the antiviral proapoptotic response to HCMV infection (46).
EGFR also modulates both viral replication and latency in CD34+ HPCs by functioning as a molecular switch that controls the replicative state of HCMV (47). EGFR activity favors the long-term maintenance of latency in CD34+ HPCs, whereas inhibition of EGFR signaling promotes reactivation and replication (47). Two opposing viral determinants (UL138 and UL135, important for regulating latency and reactivation, respectively) target EGFR with opposite effects on its endocytic trafficking and activity (47). UL138, which promotes latent infection (48, 49), sequesters EGFR at the cell surface and sustains its signaling activity (47). In contrast, UL135, which promotes reactivation and replication, in part by overcoming UL138-mediated suppression (50), downregulates EGFR cell surface levels and activity (47).
With this important role for continued EGFR signaling in the maintenance of latency later during infection defined, we hypothesized that EGFR signaling is a critical determinant of the early events of HCMV infection of CD34+ HPCs and that it likely sets the stage for a successful infection leading to viral persistence in these cells. Because chronic EGFR signaling is required for the maintenance of latency in CD34+ HPCs (47) and because EGFR signaling is also required for early events, such as viral entry during infection of monocytes (39), we next wanted to explore the role(s) EGFR plays in the early steps of HCMV infection of CD34+ HPCs, such as entry, viral trafficking, and the nuclear translocation of the viral DNA. We hypothesized that EGFR may also dictate HCMV tropism for CD34+ HPCs and allow HCMV to control the critical cellular processes in the cell type that promote infection and the establishment of latency.
Here, we show that EGFR on the surfaces of CD34+ HPCs functions as an entry receptor for HCMV infection of the cells. EGFR, as well as downstream phosphatidylinositol 3-kinase (PI3K) signaling, is required for efficient HCMV entry in CD34+ cells. Following entry, HCMV DNA reaches the nuclei of CD34+ HPCs by 8 h postinfection (hpi), and this process requires EGFR kinase (EGFRK) activity, suggesting a role for continued signaling in viral trafficking following entry. In addition, EGFR signaling plays a role in the early steps in the establishment of latency in CD34+ cells, as inhibition of EGFR signaling leads to increased expression of the lytic IE1 and IE2 transcripts and decreased expression of the latency-associated UL138 transcript. HCMV-induced EGFR signaling also curbs the expression of the cellular interleukin 12 (IL-12) cytokine transcript. IL-12 is an important regulator of hematopoiesis, and thus, EGFR-dependent virus-induced regulation of this cellular factor may reflect a mechanism by which HCMV infection also shapes the differentiation outcome of infected bone marrow and blood cells to promote viral persistence. Overall, this study suggests that EGFR signaling induced by viral binding and the ensuing activation shape key early events required for successful infection of CD34+ HPCs.
RESULTS
EGFR/PI3K signaling is required for HCMV entry into CD34+ HPCs.
To define EGFR expression on the surfaces of isolated CD34+ HPCs, we used immunofluorescent antibodies to stain uninfected cells for EGFR. We observed clusters of EGFR on the cell surface (Fig. 1A). To investigate a role for surface EGFR in entry, we next performed HCMV entry assays (39, 42, 43) to test whether EGFR activation and downstream signaling are required for viral attachment and/or entry into CD34+ HPCs. We pretreated cells with control dimethyl sulfoxide (DMSO), AG1478 (an EGFRK inhibitor), LY294002 (a PI3K inhibitor), blocking antibodies against EGFR, or control IgG antibodies under conditions we previously defined in monocytes/macrophages (39, 42, 43), endothelial cells (51), and fibroblasts (43). The cells were then infected with the recombinant virus TB40-UL32-HCMV/E, attached to slides using a cytospin, and visualized by confocal microscopy to monitor the internalization of viral particles. Z stacks were acquired for individual cells in each experimental cohort to render complete images of the total cell area. The images shown in Fig. 1B represent a single focal plane captured either at the surface of the cell (Fig. 1B, top row) or within the intracellular space (Fig. 1B, bottom row). HCMV was detected by staining of the green fluorescent protein (GFP)-fused UL32 protein. Viral particles (GFP+) were present on the surfaces (Fig. 1B, top row) of CD34+ cells treated with AG1478, LY294002, or anti-EGFR (α-EGFR) antibodies, as well as on the 4°C binding control groups, while the DMSO-treated, TB40-UL32-HCMV/E-infected groups internalized the virus (Fig. 1B, bottom row). The percentage of viral particles detected either on the surfaces of or inside the cells was determined (an average of 5 cells per experimental arm) for two independent experiments with different donors using Leica software (Fig. 1C). We saw that 80 to 100% of viral particles detected in the HCMV-infected, DMSO-treated groups were internalized (Fig. 1C). This number was reduced to ∼10% and ∼20% internalized in cells pretreated with AG1478 or LY294002, respectively (Fig. 1C). Pretreatment with anti-EGFR antibodies also reduced the percentage of internalized virus to ∼15% (Fig. 1C). Taken together, these data confirm that the presence of EGFR and its downstream EGFRK and PI3K signaling are required for efficient HCMV entry into CD34+ HPCs.
FIG 1.
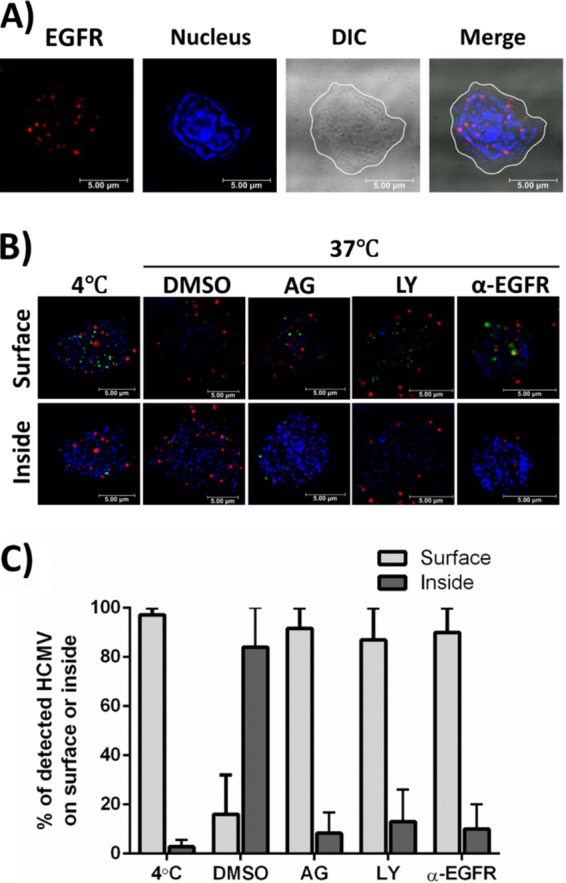
EGFR is expressed on the surfaces of CD34+ HPCs and is required for HCMV entry. (A) Uninfected CD34+ HPCs were stained with anti-EGFR antibodies and Alexa Fluor 594-conjugated secondary antibodies to detect EGFR expression on the cell surface (red). DAPI was used to counterstain the nucleus (blue). The contrast was adjusted in the differential interference contrast (DIC) image, and the cell membrane is outlined white in the DIC and merged images. Both adjustments were made so that the images could be more easily viewed. (B and C) Viral entry assays were performed in CD34+ HPCs. Cells were pretreated with AG1478 (AG) (EGFRK inhibitor), LY294002 (LY) (PI3K inhibitor), anti-EGFR blocking antibodies (α-EGFR), or DMSO/IgG antibody controls before HCMV infection. (B) Cells were infected with HCMV (TB40-UL32-HCMV/E) and stained with anti-EGFR antibodies and Alexa Fluor 594-conjugated secondary antibodies to detect EGFR (red). UL32-GFP was stained with an anti-GFP Alexa Flour 488-labeled antibody (green). DAPI was used to counterstain the nucleus (blue). Confocal microscopy was used to visualize HCMV bound to the cell surface or internalized into target cells. Representative slices from a Z stack are shown to compare the cell surface with intracellular space. (C) Quantification comparing the percentages of HCMV virions on the surfaces of or internalized into target cells. An average of 5 cells per experimental arm from two independent donors were used for counting. The means and standard errors of the mean (SEM) (error bars) of the results of two independent experiments performed.
To further evaluate the signaling required for HCMV entry into CD34+ HPCs, we infected cells with Towne/E, an additional clinically relevant strain of the virus. Following viral binding and entry, the cells were treated with proteinase K to remove bound but uninternalized virus, and total DNA was isolated. We assessed internalization of HCMV DNA by PCR using primers corresponding to the UL123 gene region of the HCMV genome. Consistent with our results following infection with TB40-UL32-HCMV/E (Fig. 1B and C), we found that Towne/E-infected cells treated with control DMSO had higher levels of internalized viral DNA than the 4°C binding-only controls for two independent donors (Fig. 2A). Cells treated with AG1478 or LY294002 contained levels of viral DNA similar to those seen in the 4°C controls (Fig. 2A). Similarly, cells treated with anti-EGFR antibodies had lower levels of internalized viral DNA than IgG antibody-treated controls (∼50% decrease for both donors) (Fig. 2B). Although these data show similar trends among the different donors, the reduction in viral entry was more striking in cells pretreated with the signaling inhibitors versus those treated with EGFR-blocking antibodies, highlighting the importance of downstream signaling to viral entry.
FIG 2.

EGFR/PI3K signaling is required for HCMV entry into CD34+ HPCs. (A and B) Viral entry assays were performed in CD34+ HPCs, as previously described in monocytes and fibroblasts (39, 42, 43). Cells were pretreated with AG1478 (EGFRK inhibitor), LY294002 (PI3K inhibitor), or DMSO solvent control (A) or anti-EGFR blocking antibodies (α-EGFR) or IgG control antibodies (B) before HCMV infection. The cells were infected with HCMV (low-passage-number Towne/E), and following proteinase K treatment, total DNA was isolated to detect viral entry by qPCR amplification of viral DNA corresponding to the UL123 gene region of the genome. The experiment was repeated with two different donors. For all the graphs, the levels of internalized HCMV DNA were normalized to the 4°C binding-only controls and to 18S rRNA as an internal control. Nontemplate controls (NTC) are also shown.
HCMV trafficking in CD34+ HPCs is enhanced by EGFR signaling activated during viral entry.
We have recently shown that cell-type-specific differences in signaling during the viral entry event give rise to distinct functional outcomes with respect to viral trafficking in infected cells (52). To investigate delivery of the viral genome to the nucleus in CD34+ HPCs, we infected cells with TB40-UL32-HCMV/E and stained them with anti-glycoprotein B (gB) and anti-GFP antibodies at 1 and 4 hpi, consistent with the time points used for our entry assays (Fig. 1B and C and 2). Virus particles (GFP+) and gB were visualized by confocal microscopy (Fig. 3A). Our data show that the viruses were internalized as early as 1 hpi. Furthermore, gB (a marker for the whole virus) colocalized with UL32 (a marker for the viral capsid) at 1 hpi, but not at 4 hpi (Fig. 3A), suggesting that de-envelopment occurs within this time frame. We wanted to test the possible involvement of EGFR signaling in virion trafficking in CD34+ HPCs. For this experiment, we treated cells with AG1478 at 30 min postinfection (mpi), as entry assays (Fig. 1B and C and 2) showed that pretreatment completely blocks viral entry. HCMV was able to enter the cells in the AG1478-treated groups by 1 hpi, similar to the DMSO-treated groups, as the viral particles detected were resistant to proteinase K treatment (Fig. 3A). However, at 1 hpi, we observed a distinct clustering of viral particles near the cell surface in the AG1478-treated cells compared to a more dispersed pattern in the DMSO-treated groups (Fig. 3A). Similar clustering of viral particles was seen during HCMV entry assays previously performed in monocytes following treatment with AG1478 (39). These results could therefore represent a slight temporal delay in the entry process resulting from inhibition of continuous EGFR signaling; virus particles in these images could be grouped together in an incompletely formed endocytic vesicle, for example. By 4 hpi, viral particles in both the DMSO-treated and the AG1478-treated cells were distributed similarly, but there appeared to be more colocalization between gB and UL32 in the AG1478-treated groups (Fig. 3A). These findings suggest that there is a delay in viral de-envelopment in the AG1478-treated groups when EGFR signaling is inhibited after initial activation following viral binding, consistent with the delay suggested by our results at 1 hpi (Fig. 3A). Taken together, these results suggest that EGFR signaling is involved in the endocytic trafficking of HCMV, possibly by directing the trafficking of the virus through various endocytic compartments and/or by simply enhancing the trafficking process so that it occurs in a timely manner. Future studies will investigate in more detail the specific involvement of EGFR signaling in the trafficking of HCMV to specific cellular compartments in CD34+ HPCs.
FIG 3.

HCMV trafficking in CD34+ HPCs is enhanced by EGFR signaling activated during viral entry. (A) CD34+ HPCs were infected with HCMV (TB40-UL32-HCMV/E) and treated with DMSO or AG1478 at 30 mpi. The cells were cytospun onto slides at 1 hpi and 4 hpi and then stained and visualized by confocal microscopy. The cells were stained with anti-gB antibody (Ab) and Alexa Fluor 594-conjugated secondary Ab to detect gB (red). UL32-GFP was stained with an anti-GFP Alexa Fluor 488-labeled antibody (green). DAPI was used to counterstain the nucleus (blue). (B) Cells were infected with HCMV (low-passage-number Towne/E) and stained with specific Alexa Fluor 488-labeled HCMV DNA probe (green) according to the FISH protocol. DAPI was used to counterstain the nucleus (blue). (C) Stained cells (an average of 50 for each experimental group) were analyzed and quantitated to calculate the percentage of total viral DNAs detected in the cytoplasm or nucleus. The means and SEM (error bars) of the results of two independent experiments performed are shown.
To further examine a potential role for EGFR in viral trafficking, we extended our time course to include an 8-hpi time point and monitored nuclear translocation of the viral DNA using fluorescence in situ hybridization (FISH) (Fig. 3B). These data show that the majority of HCMV input DNA remained in the cytoplasm at 1 hpi and then began to translocate into the nucleus at around 4 hpi. By 8 hpi, most of the viral DNA was present in the nucleus (Fig. 3B and C). Notably, when EGFRK signaling was blocked with AG1478 treatment beginning at 30 mpi to allow viral entry and entry-induced signaling, but to block continued EGFRK activity, nuclear translocation of the viral DNA was diminished and/or delayed (Fig. 3B and C). In the AG1478-treated experimental group, only ∼40% of the total viral genomes entered the nucleus by 8 hpi compared to ∼80% at 8 hpi in the DMSO-treated group (Fig. 3B and C). Taken together, these data suggest that HCMV trafficking and nuclear translocation in CD34+ HPCs is enhanced by EGFR signaling events initiated during viral entry that continue during the early course of infection.
HCMV-induced EGFR signaling favors latent over lytic viral gene expression in CD34+ HPCs.
Because signaling and entry ultimately set the stage for the successful infection of a target cell, we next determined how EGFR signaling during the entry process might contribute to the initial steps in the establishment of latency in CD34+ HPCs. Although HCMV latency is characterized by a limited viral transcription program (17, 28, 29), there is abundant evidence that the virus is able to manipulate a variety of host cellular processes during the establishment of latency to optimize the latency program or to prime cells for lytic reactivation (2, 53). For example, a limited number of latency-associated viral gene products, in association with a panel of cellular microRNAs (miRNAs), have been shown to alter viral replication and to activate cellular signaling pathways to control cell survival, immune surveillance, and transcription of cellular gene products (50, 54–63). Similarly, we have shown that HCMV usurps cellular signaling pathways via EGFR and integrin activation during quiescent infection of monocytes to promote viral dissemination and to prime infected cells for viral replication upon monocyte-to-macrophage differentiation (30–32, 37–39, 42, 43). We propose that unique viral-entry-induced cellular signaling events would also influence the establishment of latency in HPCs by directly controlling viral gene expression, in addition to altering the nature of the infected cell. To determine whether EGFR signaling affects viral gene expression programs in CD34+ HPCs, we treated cells from two independent donors with control DMSO or AG1478 at 4 hpi and examined the steady-state mRNA levels of the lytic gene products IE1 (Fig. 4A) and IE2 (Fig. 4B), as well as the latency gene product UL138 (Fig. 4C), at 24 hpi. Continued activation of EGFR appears to alter the cellular environment during HCMV infection so that inhibiting EGFR signaling after viral entry and nuclear translocation results in slightly higher levels of IE1 and IE2 steady-state mRNA (Fig. 4A and B) while lowering steady-state levels of the latency-associated UL138 mRNA transcript (Fig. 4C). These findings are functionally consistent with a defined role for EGFR in the maintenance of HCMV latency later during the course of HCMV infection (47) and further demonstrate a role for EGFR activity in modulating very early events following infection that impact the transcription program and may impact the establishment of latency. Increases in IE1 and IE2 expression alone in the presence of AG1478 are not sufficient to conclude that HCMV infection of CD34+ HPCs would become lytic in the absence of EGFR signaling. However, inhibition of EGFR signaling in combination with myeloid differentiation increases HCMV reactivation in CD34+ HPCs (47). Taking the data together, we conclude that HCMV-induced EGFR signaling creates an infection environment that favors latent over lytic transcription during early times following infection of CD34+ HPCs.
FIG 4.

HCMV-induced EGFR signaling favors latent over lytic viral gene expression in CD34+ HPCs. (A to C) CD34+ HPCs were mock infected or HCMV infected for 4 h and then treated with control DMSO or AG1478 (EGFRK inhibitor). RNA was isolated for RT-qPCR at 24 hpi, and expression of lytic IE1 mRNA (A) and lytic IE2 mRNA (B) versus that of latency-associated UL138 mRNA (C) was examined for two independent donors. The expression levels were normalized to 18S rRNA as an internal control. (A and C) Reverse transcriptase negative (RT−) and nontemplate (NTC) controls are also shown.
To ask whether EGFR-dependent altered transcription of viral genes occurs in a cell-type-specific manner among hematopoietic cells associated with HCMV latency, we performed a similar experiment using primary monocytes, which are further along the differentiation continuum. Monocytes undergo a period of quiescence during the first 2 weeks following HCMV infection (16). During this time, HCMV-induced signaling drives monocyte-to-macrophage differentiation of infected cells, resulting in a macrophage that is capable of supporting viral gene expression (16, 31). For this reason, monocytes/macrophages provide an ideal model with which to compare the role of EGFR signaling in viral transcription among cells representing a differentiation continuum of hematopoietic cells (pluripotent CD34+ HPCs versus more differentiated monocytes/macrophages). Monocytes were mock infected or HCMV infected and then treated with AG1478 beginning at either day 7, day 10, or day 14 (during the quiescent phase of HCMV infection, when no de novo viral gene expression is seen [16, 37]). At 3 weeks postinfection (when monocyte-to-macrophage differentiation has occurred and infected cells are permissive for viral gene expression [16]), total RNA was harvested and steady-state mRNA levels of lytic IE1 were quantified by reverse transcriptase quantitative PCR (RT-qPCR) (Fig. 5). Inhibition of EGFR reduced IE1 gene expression when applied at day 7 or 10 but had no effect at day 14 relative to the DMSO control. These findings suggest that continued EGFR signaling promotes IE1 gene expression in monocytes. In contrast, continued EGFR signaling in CD34+ HPCs promotes latent gene expression (Fig. 4) and latency (47), supporting the notion that HCMV exploits cell-type-specific differences in EGFR signaling to direct distinct cell-type-dependent infection outcomes.
FIG 5.
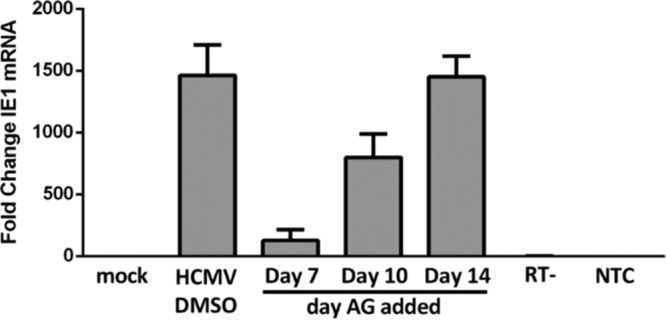
HCMV-induced EGFR signaling enhances IE1 expression in primary monocytes. Isolated monocytes were plated in 6-well dishes and mock infected or HCMV infected for 3 weeks. Beginning at 7, 10, or 14 dpi, control DMSO or AG1473 (EGFRK inhibitor) was added daily for the remainder of the infection period. No changes in cell survival were seen when they were added at these time points. After 3 weeks, RNA was isolated, and RT-qPCR was performed to determine expression of the IE1 viral transcript. The graph is representative of three replicates using different blood donors; the means and SEM (error bars) of the results of three independent experiments performed are shown. The expression levels of IE1 were normalized to that of 18S rRNA as an internal control; RT− and nontemplate controls are shown.
HCMV infection alters the transcription of cellular hematopoietic factors, in part via EGFR signaling.
The finding that lytic reactivation of HCMV occurs upon differentiation of an infected CD34+ HPC into a mature myeloid cell (33, 64, 65) has led to an intriguing hypothesis that HCMV may direct the differentiation of progenitor cells to promote the viral persistence strategy. In further support of this notion, HCMV infection of hematopoietic cells in the bone marrow has been shown to promote hematopoietic dysregulation, an effect that could also be a potential mechanism by which HCMV infection contributes to graft rejection in allogeneic HSCT patients (21, 66–68). Therefore, we next examined the effects of HCMV infection and of HCMV-induced EGFR signaling in CD34+ HPCs on the expression of cytokines known to play a role in hematopoiesis. HPCs were infected with Towne/E for 4 h to allow viral entry, appropriate trafficking, and partial nuclear translocation of the HCMV DNA and then treated with control DMSO or with AG1478 (an EGFRK inhibitor). RNA from two independent donors was isolated at 4 and 24 hpi, and RT-qPCR was performed. We initially focused our examination on the expression of IL-12 and transforming growth factor β (TGF-β) transcripts. IL-12, a T cell-stimulating factor involved in the differentiation of naive T cells into Th1 cells, has been shown to promote hematopoiesis and stem cell engraftment after ionizing radiation (69, 70). TGF-β is also involved in stem cell differentiation and T cell regulation/differentiation and suppresses general hematopoiesis (71). Our data indicate that both IL-12 and TGF-β are induced following HCMV infection of CD34+ HPCs (Fig. 6). IL-12 transcripts were increased in HCMV-infected cells at both 4 and 24 hpi (∼3-fold and ∼8-fold over mock infection in donors 1 and 2, respectively) (Fig. 6A, top row). Inhibition of EGFR signaling beginning at 4 hpi increased IL-12 expression by ∼15-fold (donor 1) and ∼18-fold (donor 2) over mock-infected cells at 24 h (Fig. 6A, top row). Because AG1478 treatment of mock-infected cells does not result in similar increases in IL-12 steady-state mRNA (Fig. 6A, bottom), these data indicate that EGFR signaling suppresses HCMV-induced IL-12 transcription. TGF-β was upregulated at later times in infection; its expression was higher at 24 hpi than at 4 hpi in both donors (Fig. 6B). Inhibition of EGFR signaling after entry at 4 hpi did not impact TGF-β levels in either donor (Fig. 6B), suggesting that the upregulation of TGF-β occurs independently of EGFR signaling. To determine whether increased expression of the IL-12 and TGF-β transcripts following infection results in higher levels of protein, we examined IL-12 (Fig. 6C) and TGF-β (Fig. 6D) proteins by enzyme-linked immunosorbent assay (ELISA) and a multiplexed bead-based cytokine immunoassay, respectively. IL-12 protein increased from ∼10 pg/ml in mock-infected cells to ∼15 pg/ml in HCMV-infected cells. While inhibition of EGFR in uninfected cells did not alter IL-12 protein levels, inhibition of EGFR in the context of infection resulted in a 2-fold increase in IL-12 protein compared to infection alone (DMSO control) (Fig. 6C), consistent with our IL-12 transcript data (Fig. 6A). Consistent with our TGF-β transcript data (Fig. 6B), TGF-β1 protein increased slightly (from ∼8 pg/ml in mock-infected cells to ∼11 pg/ml in HCMV-infected cells) following HCMV infection, and these levels were not affected by the inhibition of EGFR (Fig. 6D). No changes were observed for TGF-β2 and TGF-β3 during this time frame. Further studies will be needed to address the long-term secretion of the factor. To further expand our analysis, multiplexed bead-based immunoassays were used to measure the expression of 40 additional human cytokines/chemokines. Table 1 shows a summary of the findings from these experiments; we limited our analysis to only those chemokines that are known to play a role in hematopoiesis or in the regulation of hematopoietic cells and that had substantial differences in secretion following HCMV infection or treatment with AG1478 (Table 1). The data indicate that a number of hematopoietic chemokines are differentially expressed/secreted following HCMV infection (for example, CXCL1, CXCL5, IL-6, IL-8, MCP-1, migration inhibition factor [MIF], and thymus-expressed chemokine [TECK] all show enhanced secretion following infection [Table 1]) and that EGFR signaling plays a role in shaping the secretomes (e.g., IL-12 [Fig. 6C], CXCL1, CXCL2, CXCL5, IL-8, and TECK [Table 1]) of infected (and uninfected) CD34+ HPCs. Taken together, the results of these analyses demonstrate a global change in the expression of hematopoietic proteins following HCMV infection and show that these changes are, in part, the result of HCMV-induced EGFR signaling.
FIG 6.

HMCV infection alters the transcription of cellular hematopoietic factors, in part via EGFR signaling. (A and B) CD34+ HPCs were mock infected or HCMV infected for 4 h to allow viral entry and trafficking to the nucleus. (A, top row, and B) At 4 hpi, RNA was harvested from a single cohort of HCMV-infected cells to identify a baseline level of cellular transcripts in infected cells (4 h). DMSO or AG1478 (EGFRK inhibitor) was added to the other cohorts at 4 hpi, and RNA was isolated from the groups at 24 hpi (24 h and AG). (A, bottom) DMSO or AG1478 was added at 4 hpi, and RNA was isolated at 24 hpi. RT-qPCR was performed on all the samples to quantitate IL-12 (A) and TGF-β (B) transcripts. Expression levels for both IL-12 and TGF-β were normalized to 18S rRNA as an internal control; RT− and nontemplate controls are shown. (A, top row, and B) Fold changes in cellular transcripts examined in two different donors. (A, bottom) Averages from three independent experiments with different donors (the means and SEM [error bars] of the results of three independent experiments performed) are shown. (C and D) CD34+ HPCs were mock infected for 4 h before treatment with DMSO or AG1478. At 24 hpi, the supernatants were collected for quantification of IL-12 by ELISA (C) and TGF-β by multiplexed bead-based immunoassay (D). The graphs represent the average expression levels of IL-12 and TGF-β from three independent experiments with different donors. The means and SEM (error bars) of the results of three independent experiments performed are shown.
TABLE 1.
Chemokines known to be involved in hematopoiesis and showing meaningful differences in secretion following HCMV infection or treatment with AG1478
| Chemokine | Hematopoietic function | Expression level (pg/ml) ± SEMa |
Fold changeb |
||||
|---|---|---|---|---|---|---|---|
| Mock | Mock + AG | HCMV | HCMV + AG | Mock + DMSO vs. HCMV + DMSO | HCMV + DMSO vs. HCMV + AG | ||
| CXCL1/GROα | Neutrophil chemoattractant | <LOD | <LOD | 8.7 ± 5.0 | 17.2 ± 1.5 | − | +2.0 |
| CXCL2/GROβ | Neutrophil chemoattractant | 7.2 ± 1.0 | 8.8 ± 0.6 | 3.9 ± 2.2 | 3.5 ± 2.0 | −0.5 | −1.1 |
| CXCL5 | Granulocyte production | 45.0 ± 26.0 | 32.8 ± 17.6 | 2.4E2 ± 98.2 | <LOD | +5.4 | − |
| IL-6 | Neutrophil and B cell production Antagonistic to T cells | <LOD | 0.03 ± 0.0 | 58.0 ± 3.9 | 54.2 ± 3.0 | − | −0.9 |
| IL-8 | Leukocyte trafficking | 10.8 ± 1.8 | 3.0 ± 0.9 | 6.7E2 ± 1.6E2 | 2.6E2 ± 34.3 | +62.2 | −2.6 |
| MCP-1 | Leukocyte trafficking | <LOD | <LOD | 7.8 ± 0.6 | 10.4 ± 2.0 | − | +1.3 |
| MIF | Macrophage migration | 1.1E3 ± 2.2E2 | 9.5E2 ± 2.2E2 | 5.2E3 ± 3.7E2 | 4.5E3 ± 3.2E2 | +4.8 | −1.2 |
| TECK | T cell development | 4.4 ± 1.3 | 6.2 ± 1.5 | 11.5 ± 0.9 | 19.8 ± 1.6 | +2.6 | +1.7 |
HCMV infection results in global expression changes of hematopoietic proteins, in part due to EGFR signaling. CD34+ HPCs were mock infected or HCMV infected for 4 h and then treated with AG1478 (AG) or control DMSO. At 24 hpi, the supernatants were collected for analysis by multiplexed bead-based immunoassay. A select group of chemokines known to be involved in hematopoiesis and showing meaningful differences in secretion following HCMV infection or treatment with AG1478 are shown. The expression levels are averages of the results of three different experiments ± SEM. <LOD (limit of detection), sample was below the limit of detection for the assay.
Fold changes in mean expression are shown as follows: mock infected plus DMSO versus HCMV infected plus DMSO and HCMV infected plus DMSO versus HCMV infected plus AG1478. −, fold change was below the LOD. The three independent experiments were run as a technical replicate on a single array.
DISCUSSION
Hematopoietic cells play a crucial role in the HCMV infection cycle. Infection of circulating monocytes and their CD34+ progenitors leads to the establishment of viral persistence both within an individual host and within the general human population. Although infection of both cell types contributes to HCMV persistence, the functional outcomes of infection differ between the two. We have previously shown that HCMV-induced signaling through EGFR and cellular integrins results in functional and molecular changes in the cellular environment that optimize HCMV infection of circulating monocytes. These HCMV-induced changes promote hematogenous dissemination of the virus to multiple host organ tissues by enhancing infected monocyte motility and increasing transendothelial migration of infected cells (16, 30, 39, 42, 44). Upon reaching peripheral organ sites, infected monocytes are induced to undergo monocyte-to-macrophage differentiation to promote viral replication and spread within the host, as well as to additional hosts (31, 38). In contrast, successful infection of CD34+ HPCs entails the establishment of latency so that viral replication is restricted and HCMV can persist within the host while minimizing the antiviral immune response (15, 17, 68, 72, 73). Latently infected CD34+ cells also serve as a reservoir for infectious virus that can become reactivated upon differentiation into mature myeloid cell types (14, 19, 53, 72, 74). Recently, Buehler et al. defined a key role for chronic EGFR signaling in the maintenance of HCMV latency in CD34+ HPCs (47). In this study, we confirm and expand on these findings to demonstrate a role for EGFR signaling during the early steps of HCMV infection of CD34+ cells. Modulation of EGFR upon virion entry tailors many of the early events required for successful infection of this cell type.
The findings reported here are the first describing the early steps during HCMV infection of CD34+ HPCs. We have previously shown that the majority of transcriptional changes induced by the HCMV signalosome in infected monocytes occur by 24 hpi, although the altered transcriptome continues to produce functional changes for up to 2 weeks following infection (31). These findings highlight the importance of early signaling in optimizing the cellular environment for HCMV infection. From the data generated in this study, we constructed a model for the role of EGFR signaling following viral binding to CD34+ HPCs (Fig. 7). HCMV activation of EGFR mediates viral entry via PI3K-dependent signaling. The entry process occurs within the first hour following infection, with HCMV de-envelopment occurring between 1 and 4 hpi. HCMV DNA begins its translocation into the nucleus at around 4 hpi (the process is completed by 8 hpi), where a latency-associated viral gene expression program is favored over lytic transcription (IE1 and IE2 expression is decreased and UL138 expression is increased) (48). HCMV infection also alters host cellular gene expression; IL-12 and TGF-β, two regulators of hematopoiesis, are differentially expressed following HCMV infection.
FIG 7.

Model for early HCMV infection of CD34+ HPCs. (Left) Various treatments used in this study to investigate HCMV-induced EGFR signaling in CD34+ HPCs. Pretreatment with anti-EGFR blocking antibodies was used to block HCMV-EGFR engagement at the cell surface and any direct signaling downstream of that specific engagement. Pretreatment with the signaling inhibitors AG1478 (EGFRK inhibitor) and LY294002 (PI3K inhibitor) was used to attenuate HCMV-induced molecular signaling associated with the viral binding and entry events. AG1478 was also used at 30 mpi or at 4 hpi to attenuate EGFR signaling after viral entry and trafficking commenced. (Right) Summary of our findings from this investigation. HCMV enters CD34+ HPCs via engagement of EGFR on the cell surface. The entry event is dependent upon both EGFRK and downstream PI3K signaling. HCMV de-envelopment occurs within 1 and 4 hpi, with nuclear translocation of the viral DNA occurring between 4 and 8 hpi. Both de-envelopment and delivery of HCMV DNA to the nucleus are enhanced by EGFR signaling. Once in the nucleus, HCMV initiates the appropriate cell-type-specific viral transcription program and also alters the transcription of the cellular genes that favor infection of CD34+ HPCs. EGFR signaling plays a role in shaping the induced viral and cellular transcriptional profiles during the initial stages of infection.
In examining these early events that occur during HCMV infection of CD34+ HPCs, we have uncovered a number of exciting findings that point to an important role for HCMV-induced EGFR signaling in optimizing HCMV infection of CD34+ cells: (i) we identified a role for EGFR/PI3K signaling in the HCMV entry process in CD34+ cells, (ii) we uncovered differences in HCMV-induced signaling downstream of EGFR that likely result in distinct viral trafficking when infections of multiple cell types were compared, (iii) we defined a role for HCMV-induced EGFR signaling in the early steps of the establishment of latency in CD34+ cells, and (iv) we showed that HCMV infection alters the expression of numerous cellular cytokines important to hematopoiesis, in part via EGFR signaling.
Buehler et al. demonstrated EGFR surface expression on CD34+ cells using a fluorescently conjugated EGFR ligand, EGF-647 (47). Epidermal growth factor (EGF) is widely accepted as a ligand that binds exclusively to EGFR and can therefore serve as a reliable indicator for EGFR surface expression (47). In this study, we confirmed and expanded on these findings by demonstrating bona fide EGFR expression on the surfaces of CD34+ HPCs via direct immunofluorescent staining with an EGFR-specific antibody (Fig. 1A). We have previously shown that EGFR is expressed on the surfaces of circulating blood monocytes, but not on the surfaces of B cells or T cells (39). Because circulating monocytes have been shown to carry HCMV DNA, whereas B cells and T cells do not (75, 76), we suggest that EGFR may function as a determinant of HCMV tropism that restricts infection of hematopoietic cells to those of the myeloid lineage. The expression of EGFR on CD34+ HPCs lends further support for this idea, and we propose that EGFR could serve as a tropism determinant across the hematopoietic lineage from the most pluripotent CD34+ cells to fully committed mature myeloid cells (e.g., monocytes/macrophages and dendritic cells). It is important to note that EGFR is not expressed on every cell type that HCMV infects. Other cellular receptors and/or coreceptors with signaling capabilities have also been shown to play a role in the HCMV entry process (e.g., platelet-derived growth factor receptor alpha [PDGFRα] [77] and cellular integrins [78–80]). Nonetheless, we believe that this and other studies demonstrate an important role for EGFR in HCMV infection of a variety of cell types.
Although EGFR functions as an entry receptor for HCMV in multiple cell types, including fibroblasts (81), monocytes (39), and CD34+ HPCs (Fig. 1B and C and 2), there appear to be differences in the signaling events that occur downstream following viral binding and activation of EGFR. For example, we saw that HCMV entry into monocytes occurs in an EGFR-dependent, PI3K-independent manner (39), whereas entry into CD34+ HPCs is the result of signaling through both EGFR and PI3K (Fig. 1B and C and 2). These findings are consistent with the idea that cell-type-specific differences in virus-induced cellular signaling result in differential infection outcomes that vary according to cell type. Because cellular signaling has been shown to play a role in the entry processes of several viruses (82), it stands to reason that cell-type-specific differences begin during the entry process itself. Although the specific mode of entry has not yet been examined in CD34+ cells, this certainly appears to be the case among other, more commonly studied cell types. For example, HCMV entry is thought to occur via pH-independent fusion in fibroblasts (83) or at the plasma membrane following macropinocytosis (84) and via endocytosis/macropinocytosis in monocytes, endothelial cells, and epithelial cells (42, 85, 86). These differences could be the result of differential signaling in these cell types, as HCMV induces a bidirectional cross talk between the EGFR and integrin signaling axes in fibroblasts (87) versus unidirectional signaling from integrins to EGFR in monocytes (30). Consistent with this idea, our data showed that the reduction in HCMV entry in CD34+ HPCs resulting from inhibition of EGFR is more striking in cells pretreated with the EGFR and PI3K signaling inhibitors (Fig. 2A) than in those treated with EGFR-blocking antibodies (Fig. 2B). These data highlight the importance of signaling downstream of EGFR in the viral entry process, as the inhibitors effectively abolish signaling, whereas the antibodies block only viral engagement of the receptor and not the kinase activity of EGFR. As such, partial activation of signaling players downstream of EGFR via cross talk with a cellular coreceptor, such as an integrin (30, 78), may result in some level of viral internalization in cells treated with the EGFR-blocking antibodies compared to those treated with signaling inhibitors. It is possible that the differences seen between AG1478-treated groups and those treated with EGFR-blocking antibodies are due to an off-target effect of AG1478 (88–91); however, our previous studies using both AG1478 and gefitinib, which target EGFRK differently, showed similar results for each inhibitor with respect to the maintenance of HCMV latency later during infection (47), providing supporting evidence that EGFR signaling, specifically, plays an important role in HCMV infection of CD34+ HPCs.
Differences in the viral entry process appear to shape many of the downstream events in the infectious cycle, a prime example being the intracellular trafficking and nuclear translocation of an infecting virus. Consistent with this theme, we saw differences in the cellular trafficking of HCMV when various cell types were compared, most notably in the temporal kinetics of viral de-envelopment and nuclear translocation of the viral DNA. Viral de-envelopment is an essential step required in all cell types for the nuclear translocation of viral DNA to occur; however, it appears to proceed differently in CD34+ HPCs than in other cell types. We show in this study that HCMV de-envelopment occurs between 1 and 4 hpi in CD34+ HPCs (Fig. 3A). In contrast, de-envelopment occurs rapidly in fibroblasts during the initial fusion of the viral envelope with the cellular membrane (83) or during a rapid macropinocytosis event (84). De-envelopment occurs with similarly rapid kinetics in epithelial and endothelial cells following fusion with an endocytic vesicle (86). In monocytes, a CD34+ HPC derivative, de-envelopment is delayed until ∼2 days postinfection (dpi) and occurs in recycling endosomes (52). In addition, we saw that nuclear translocation of HCMV DNA occurs within 30 min of infection in permissive fibroblasts and endothelial cells (52). In monocytes, which are also part of the hematopoietic lineage and similarly nonpermissive for viral replication, nuclear translocation is delayed until 3 days postinfection (52). This is strikingly different from our findings in this study that nuclear translocation occurs between 4 and 8 hpi in CD34+ HPCs (Fig. 3B and C). We proposed in previous studies that differences in nuclear translocation of the viral DNA were due to differences in virus-induced signaling and the requirements for cell-type-specific downstream signaling components among the various cell types (52). This claim is supported by our recent finding that infection of monocytes with a strain of HCMV lacking the pentameric gH–gL–UL128-131 complex fails to traffic properly and that the viral DNA fails to translocate to the nucleus (52). We believe this change is due to defective signaling induced by the binding of β1 and β3 integrins to the gH-gL-gO trimeric complex rather than to the gH–gL–UL122-131 pentameric complex (43, 52). Because the integrin signaling axis amplifies EGFR signaling during HCMV infection of monocytes (30), these results could also suggest some level of involvement for EGFR signaling in HCMV trafficking in monocytes, as well. In this study, we show that EGFR does, in fact, play a role in the cellular trafficking and nuclear translocation of the viral DNA following HCMV infection of CD34+ HPCs, as inhibition of EGFR signaling resulted in diminished and/or delayed nuclear translocation of the infectious DNA (Fig. 3). Taken together, these data suggest that HCMV utilizes an entry and/or nuclear translocation pathway in CD34+ HPCs different from those in other cell types, due in part to EGFR signaling events initiated during viral entry.
In addition to effects directly downstream of entry, such as trafficking and nuclear translocation of the viral DNA, we also examined some of the early changes in the cellular environment that may be controlled by HCMV-induced EGFR signaling. HCMV is a master of controlling the cellular environment during infection to promote proviral outcomes (32, 92, 93). We suggest that different environmental conditions lead to different inflection outcomes and that EGFR, which casts a wide net of regulation over cellular processes, is a central candidate for HCMV to exploit for this purpose. When HCMV infection of CD34+ HPCs was compared to infection of monocytes, the ideal functional outcome of infection varied for each cell type. For example, the most favorable infection outcome for the virus in CD34+ cells appears to be the establishment of latency with limited viral gene expression so that the virus can maintain a persistent infection while successfully evading the host immune system (15, 17, 68, 72, 73). In contrast, successful infection of monocytes entails viral dissemination to peripheral organs and the transition of the infected cell into a replication-permissive macrophage that can serve as a platform for viral replication and spread in the host organ tissues (16, 30, 31, 65, 94–97). A role for EGFR signaling in the maintenance of HCMV latency (47) led us to examine the role of EGFR signaling in the early steps during establishment of latency. In this study, we saw that EGFR signaling promotes early required steps in the establishment of latency in CD34+ HPCs by curbing the expression of the lytic transcripts IE1 (Fig. 4A) and IE2 (Fig. 4B) while increasing the expression of the latency-associated UL138 transcript (Fig. 4C). In monocytes, EGFR signaling enhances IE1 lytic viral transcription (Fig. 5), perhaps to prime these cells for viral replication once monocyte-to-macrophage differentiation has occurred. Furthermore, these data indicate that lytic viral gene expression increases as cells mature along the myeloid continuum, at least in part due to continued EGFR signaling following viral binding (Fig. 4 and 5). This finding is consistent with previous reports that lytic reactivation of HCMV is synchronous with differentiation into mature myeloid cells (dendritic cells and macrophages) (33, 64, 65). In fact, we have shown that HCMV infection of blood monocytes drives monocyte-to-macrophage differentiation and that these infected macrophages ultimately become productive for the original input virus (16, 31, 35, 36, 38). This process is likely manipulated by the virus in order to optimize the environment of infected monocytes/macrophages to support viral gene expression and replication. Taken together, these data suggest an important role for EGFR signaling in determining the cell-type-specific outcomes of HCMV infection and in regulating states of latency and replication in the infected cell.
In addition to examining the effects of EGFR signaling on viral gene expression, we also explored how EGFR affects the expression of cellular genes that shape the infection environment. In particular, we looked at the transcription of cellular factors associated with hematopoiesis, as they may play a role in a potential viral strategy designed to control the differentiation outcome of infected CD34+ HPCs. We report here that although HCMV infection induces transcriptional upregulation of both IL-12 (Fig. 6A) and TGF-β (Fig. 6B), HCMV-induced EGFR signaling quells this effect in regard to IL-12 (Fig. 6A). The idea that HCMV-induced EGFR signaling dampens the upregulation of IL-12 that occurs following HCMV infection may initially seem counterintuitive. It could be, however, that lower levels of IL-12 exert a proviral effect, as other proinflammatory players have been shown to do (31), and that HCMV-induced signaling functions to optimize its expression level for the benefit of the virus. Interestingly, because IL-12 is known to promote hematopoiesis and stem cell engraftment after ionizing radiation, the EGFR-dependent control of its expression could be among the functional mechanisms that lead to graft rejection in HCMV-infected transplant patients. Taken together, our data indicate that HCMV infection results in the EGFR-independent upregulation of the hematopoietic suppressor TGF-β, while virus-induced signaling through EGFR limits the expression of proinflammatory IL-12 (Fig. 6). Furthermore, we showed that HCMV infection alters the expression of a number of cytokines and chemokines (CXCL1, CXCL2, CXCL5, IL-6, IL-8, MCP-1, MIF, and TECK) known to shape the hematopoietic process (Table 1). This altered pattern of cytokine expression could reflect a viral objective for infection of HPCs that exists in two parts: (i) to bias differentiation toward a cell type that promotes viral dissemination and persistence and (ii) to attenuate the antiviral immune response by reducing the production or altering the function of lymphoid cells. Some of the secretory changes we observed are consistent with a viral strategy to bias hematopoietic differentiation (e.g., increases in CXCL5, which is involved in granulocyte production, and IL-6, which enhances the production of neutrophils and B cells). Others seem to support the notion that HCMV changes the chemokine expression profile to alter the immune response. Among them, we noted that EGFR signaling seems to attenuate the HCMV-induced expression of CXCL2/GRO-β, a neutrophil chemoattractant. We also observed increases in secretion of MCP-1 and IL-8 (two cytokines that function in leukocyte trafficking), as well as HCMV-induced increases in TECK and IL-6 (cytokines that affect T cell development and function). The expression profiles of these cytokines following HCMV infection could reflect a strategy by which HCMV alters both the innate and adaptive immune responses in the infected host. In further support, secretome analysis of latently infected CD34+ cells identified changes in multiple proteins known to be involved in regulating the immune response (62). In the study by Mason et al., CCL8 expression was increased, resulting in recruitment of CD4+ T cells; however, other latency-associated changes inhibited the cytotoxicity of HCMV-specific T cells and their secretion of cytokines, such as gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), and TNF-β (62). Consistent with our data, this study also identified an increase in TGF-β secretion from both infected and bystander CD34+ cells, as well as an increase in the secretion of IL-10, a cytokine that suppresses T cell proliferation and effector function (62). Mason et al. reported that neutralization of IL-10 and TGF-β resulted in complete recovery of cytotoxic effector function (62), further supporting the idea that HCMV alters the expression of hematopoietic factors, at least in part, to attenuate the antiviral immune response.
This study, compared to previously published studies using our primary monocyte system, suggests that EGFR signaling is required for viral entry and contributes to a variety of infectious processes in multiple cell types, most notably controlling the establishment of persistence, a hallmark event that occurs from bone marrow to blood in a range of hematopoietic cell types. Importantly, there appear to be differences in the various cell types infected, both in downstream signaling events and in functional outcomes, a fact that may reflect the requirement for different persistence goals (CD34+ HPCs for long-term persistence within the host versus monocytes/tissue macrophages for viral dissemination, replication, and shedding). Taken together, these data reveal that although there are some similarities in the requirement for EGFR signaling in different cell types, there are also dramatic differences downstream in the outcomes of infection. Defining these cell-type-specific differences may therefore be a key aspect of understanding the viral persistence strategy and developing therapeutic interventions to combat viral reactivation in immunocompromised patients.
MATERIALS AND METHODS
Virus preparation.
HCMV (strains Towne/E [passages 38 to 41] [98] and TB40-UL32-HCMV/E [99]) was cultured in human embryonic lung fibroblasts in Dulbecco's modified Eagle medium (DMEM) with 4% fetal bovine serum (FBS) (Mediatech, Inc., Manassas, VA) (16, 31, 35–46, 51, 100–102). The supernatants were collected, and virus was purified through a 0.5 M sucrose gradient and then resuspended in Roswell Park Memorial Institute (RPMI) 1640 medium (Mediatech, Inc.) (16, 31, 35–46, 51, 100–102). Cells were infected at a multiplicity of infection (MOI) of 10 unless otherwise specified. Mock infection of cells was achieved using equivalent volumes of virus-free RPMI 1640. TB40-UL32-HCMV/E is a recombinant virus expressing enhanced green fluorescent protein (EGFP) fused to the C terminus of the capsid-associated tegument protein pUL32 (pp150) (43, 52, 99, 100). Because GFP is incorporated into a structural protein, viral particles can be tracked within the cell with no requirement for viral gene expression.
CD34+ HPC isolation.
CD34+ HPCs were isolated from human cord blood using a CD34 MicroBead kit (magnetically activated cell sorting [MACS] kit; Miltenyi Biotec, San Diego, CA), yielding 80 to 85% pure CD34+ cells as determined by flow cytometry with phycoerythrin (PE)-conjugated mouse anti-CD34+ antibody (BD Biosciences, Franklin Lakes, NJ). The isolated cells were resuspended in DMSO and stored in liquid nitrogen prior to infection. Infected cells were cultured at 37°C and 5% CO2 in hematopoietic infection medium (Iscove's modified Dulbecco's medium [IMDM] (Mediatech, Inc.) supplemented with 10% BIT 9500 serum substitute (StemCell Technologies, Vancouver, BC, Canada), 50 μM 2-mercaptoethanol, 2 nM l-glutamine, 20 ng/ml human low-density lipoprotein (Sigma-Aldrich Co., St. Louis, MO), and penicillin-streptomycin (pen-strep). The procedure was approved by the Institutional Review Board for the Protection of Human Research Subjects at the University of Arizona Medical Center. All specimens were deidentified to maintain donor anonymity.
Human peripheral blood monocyte isolation.
Primary human monocytes were collected from healthy donors by venipuncture, as previously described. Briefly, 240 ml of whole blood was centrifuged through a Ficoll Histopaque 1077 gradient (Sigma-Aldrich Co.) at 200 × g for 30 min; then, the leukocyte fraction was collected and washed in saline to remove platelets. Cells were layered over a 45% and 52.5% isosmotic Percoll (GE Healthcare Life Sciences, Pittsburgh, PA) gradient and centrifuged at 400 × g, yielding >95% pure, unactivated monocytes with >95% survival by trypan blue exclusion. Isolated monocytes were resuspended in RPMI 1640 medium (Mediatech, Inc.) supplemented with 10% human serum (Sigma-Aldrich Co.) and cultured on fibronectin-coated 6-well plates at 37°C and 5% CO2. The procedure was approved by the Institutional Review Board for the Protection of Human Research Subjects at Louisiana State University Health Sciences Center—Shreveport and followed all the guidelines outlined by the Health Insurance Portability and Accountability Act.
Immunofluorescence.
HPCs were cytospun onto slides and fixed with 4% paraformaldehyde (Thermo Fisher Scientific, Waltham, MA). The cells were permeabilized with 0.1% Triton X-100 and blocked with 5% goat serum (Zymed Laboratories, San Francisco, CA). To detect EGFR on the surfaces of HPCs and the viral gB, the fixed cells were stained with primary anti-EGFR (Calbiochem, San Diego, CA) and anti-gB (Santa Cruz Biotechnology, Dallas, TX) antibodies, respectively, followed by incubation with a secondary Alexa Fluor 594-conjugated antibody (Life Technologies, Carlsbad, CA). An anti-GFP-Alexa Fluor 488 antibody (Life Technologies) was added to enhance detection of TB40-UL32-HCMV/E. The slides were mounted in Slow Fade Gold with DAPI (4′,6-diamidino-2-phenylindole) (Life Technologies), and images were captured using a Leica TCS SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany). Two methods were used to analyze the images via confocal microscopy. First, Z stacks were generated by the confocal microscope to quantitate the number of viral particles on the surfaces of or inside the cells (examples are shown in Fig. 1B and C). Second, line profiling was used with the Leica Application Suite Advanced Fluorescent (LAS AF) software (version 2.6.3) to confirm colocalization between gB and the viral capsid/UL32 (an example is shown in Fig. 2A).
Entry assays.
HCMV entry assays were performed as previously described in monocytes and fibroblasts (39, 42, 43). Briefly, CD34+ HPCs were treated with 20 μM AG1478 (EGFRK inhibitor), 20 μM LY294002 (PI3K inhibitor), control DMSO, 10 μg/ml anti-EGFR blocking antibodies, or 10 μg/ml IgG control antibodies at 37°C and 5% CO2 for 1 h prior to infection. The cells were cooled to 4°C for 1 h and then HCMV infected (MOI = 10) for 1 h to allow viral binding but not entry. The cells were then temperature shifted to 37°C (except for 4°C binding controls) for 4 h to allow viral entry. The cells were washed in phosphate-buffered saline (PBS) to remove unbound virus and then prepared for analysis. TB40-UL32-HCMV/E-infected cells were mounted on slides for microscopy. Towne/E-infected cells were treated with 2 mg/ml proteinase K (Promega, Madison, WI) in hematopoietic infection medium for 1 h at 4°C to remove virus bound to the cell surface before isolating DNA.
qPCR and RT-qPCR.
Total DNA (qPCR) or RNA (RT-qPCR) was isolated from CD34+ HPCs or monocytes/macrophages following treatment, infection, and/or the experimental procedures described. DNA was harvested with DNA STAT-60 (Tel-Test, Inc., Friendswood, TX), and qPCR was performed to detect viral entry by amplification of viral DNA corresponding to the UL123 gene region of the genome (primers: sense, 5′-CATGCAGGTGAACAACAAGG-3′; antisense, 5′-TGACGCCTTCGTTACAAGC-3′). RNA was harvested with RNA STAT-60 (Tel-Test, Inc.) and made into cDNA. RT-qPCR was performed using primers specific for IE1 (sense, 5′-AGTGACCGAGGATTGCAACG-3′; antisense, 5′-CCTTGATTCTATGCCGCACC-3′), IE2 (sense, 5′-TCCTCCTGCAGTTCGGCTTC-3′; antisense, 5′-TTTCATGATATTGCGCACCT-3′), UL138 (sense, 5′-TGCGCATGTTTCTGAGCTAC-3′; antisense, 5′-ACGGGTTTCAACAGATCGAC-3′, IL-12 (sense, 5′-ATGATGGCCCTGTGCCTTAGTAGT-3′; antisense, 5′-AGGGCCTGCATCAGCTCATCAATA-3′), and TGF-β (sense, 5′-ACACACTGCAAGTGGACATCAACG-3′; antisense, 5′-TCTTCTCCGTGGAGCTGAAGCAAT-3′) and 18S rRNA (sense, 5′-CGAGCCGCCTGGATACC-3′; antisense, 5′-CAGTTCCGAAAACCAACAAAATAG-3′) as an internal control.
FISH.
Alexa Fluor 488-labeled DNA probes were synthesized using a FISH Tag DNA Green kit (Invitrogen, Carlsbad, CA), according to the manufacturer's protocol. The whole HCMV Towne/E genome was used to synthesize the fluorescent probes (43). HPCs were infected with Towne/E (MOI = 10) and incubated at 37°C until each time point (1 hpi, 4 hpi, and 8 hpi). The cells were cytospun onto slides and fixed with 4% paraformaldehyde (Thermo Fisher Scientific). The fixed cells were stained with the Alexa Fluor 488-labeled DNA probes, as described previously (43). In brief, samples were dried and fixed with methanol (VWR International, Radnor, PA)-acetic acid (VWR International) (3:1) and then dehydrated with 70%, 85%, and 100% ethanol. Next, DNA in the cells was denatured with 70% formamide (Thermo Fisher Scientific) at 65°C, and the cells were dehydrated with 70%, 80%, and 95% ethanol. DNA probe was added, and the temperature was gradually decreased to 37°C to allow hybridization. The cells were washed with 50% formamide and mounted using Slow Fade Gold with DAPI (Life Technologies). Images were taken using a Leica TCS SP5 confocal microscope. Images were taken in the centers of cells along the z axis to identify viral particles in the cytoplasm or in the nucleus. For quantification, viral particles in the cytoplasm or in the nucleus were counted at an average of 50 cells per experimental group. The data are shown as percentages of the total number of detected virions in either the cytoplasm or the nucleus.
IL-12 ELISA.
CD34+ HPCs were mock infected or HCMV infected for 4 h before adding AG1478 (EGFRK inhibitor) or DMSO solvent control. The cells were incubated at 37°C and 5% CO2, and the supernatants were collected at 24 hpi for analysis by ELISA using a human IL-12 p70 Quantikine ELISA kit (R&D Systems, Minneapolis, MN) according to the manufacturer's protocol.
Multiplexed bead-based cytokine immunoassays.
CD34+ HPCs were mock infected or HCMV infected for 4 h before treatment with AG1478 (EGFRK inhibitor) or DMSO. After 24 h at 37°C and 5% CO2, the supernatants were collected and frozen at −20°C prior to analysis. Undiluted supernatants were analyzed to quantify the protein levels of TGF-β using a Bio-Plex Pro TGF-β immunoassay (Bio-Rad Laboratories, Hercules, CA) and a panel of 40 human chemokines using a Bio-Plex Pro human chemokine immunoassay (Bio-Rad) according to the manufacturer's protocol.
ACKNOWLEDGMENTS
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
This work was funded by a fellowship from the Center for Cardiovascular Diseases and Sciences at LSU Health Sciences Center—Shreveport and an American Heart Association Pre-Doctoral Fellowship (14PRE20310027). Research reported in this publication was supported by the National Institutes of Allergy and Infectious Diseases (NIAID) and by the National Institutes of General Medical Sciences (NIGMS), National Institutes of Health, under award numbers AI056077, AI079059, AI105062, and P30GM110703. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. A Malcolm Feist Cardiovascular Fellowship provided funding to Jung Heon Kim under grant number MFCF2015. The American Heart Association (AHA) provided funding to Donna Collins-McMillen under grant number 14PRE20310027. Jason C. Buehler is supported by National Cancer Institute Training Grant T32 CA009213. NIAID provided funding to Andrew D. Yurochko under grant number AI056077. NIGMS provided funding to Andrew D. Yurochko under grant number P30GM110703. NIAID provided funding to Felicia D. Goodrum under grant numbers AI079059 and AI105062.
We acknowledge Gloria McClure and the laboratory of John Vanchiere at LSU Health Sciences Center—Shreveport for technical assistance with the multiplexed bead-based immunoassays.
REFERENCES
- 1.Mocarski E Jr, Shenk T, Griffiths PD, Pass R. 2013. Cytomegaloviruses, p 1960–2014. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA, USA. [Google Scholar]
- 2.Goodrum F. 2016. Human cytomegalovirus latency: approaching the Gordian knot. Annu Rev Virol 3:333–357. doi: 10.1146/annurev-virology-110615-042422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. [DOI] [PubMed] [Google Scholar]
- 4.Nogalski MT, Collins-McMillen D, Yurochko AD. 2014. Overview of human cytomegalovirus pathogenesis, p 15–28. In Yurochko AD, Miller WE (ed), Human cytomegaloviruses: methods and protocols. Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 5.Ramanan P, Razonable RR. 2013. Cytomegalovirus infections in solid organ transplantation: a review. Infect Chemother 45:260–271. doi: 10.3947/ic.2013.45.3.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen CY, Ho MS, Chang SF, Yen MS, Ng HT, Huang ES, Wu CW. 1993. High rate of concurrent genital infections with human cytomegalovirus and human papillomaviruses in cervical cancer patients. J Infect Dis 168:449–452. doi: 10.1093/infdis/168.2.449. [DOI] [PubMed] [Google Scholar]
- 7.Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ. 2002. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 62:3347–3350. [PubMed] [Google Scholar]
- 8.Harkins L, Volk AL, Samanta M, Mikolaenko I, Britt WJ, Bland KI, Cobbs CS. 2002. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 360:1557–1563. doi: 10.1016/S0140-6736(02)11524-8. [DOI] [PubMed] [Google Scholar]
- 9.Harkins LE, Matlaf LA, Soroceanu L, Klemm K, Britt WJ, Wang W, Bland KI, Cobbs CS. 2010. Detection of human cytomegalovirus in normal and neoplastic breast epithelium. Herpesviridae 1:8. doi: 10.1186/2042-4280-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soderberg-Naucler C. 2006. Does cytomegalovirus play a causative role in the development of various inflammatory diseases and cancer? J Intern Med 259:219–246. doi: 10.1111/j.1365-2796.2006.01618.x. [DOI] [PubMed] [Google Scholar]
- 11.Toorkey CB, Carrigan DR. 1989. Immunohistochemical detection of an immediate early antigen of human cytomegalovirus in normal tissues. J Infect Dis 160:741–751. doi: 10.1093/infdis/160.5.741. [DOI] [PubMed] [Google Scholar]
- 12.Myerson D, Hackman RC, Nelson JA, Ward DC, McDougall JK. 1984. Widespread presence of histologically occult cytomegalovirus. Hum Pathol 15:430–439. doi: 10.1016/S0046-8177(84)80076-3. [DOI] [PubMed] [Google Scholar]
- 13.Sinzger C, Digel M, Jahn G. 2008. Cytomegalovirus cell tropism. Curr Top Microbiol Immunol 325:63–83. [DOI] [PubMed] [Google Scholar]
- 14.Reeves M, Sinclair J. 2008. Aspects of human cytomegalovirus latency and reactivation. Curr Top Microbiol Immunol 325:297–313. [DOI] [PubMed] [Google Scholar]
- 15.Goodrum F, Caviness K, Zagallo P. 2012. Human cytomegalovirus persistence. Cell Microbiol 14:644–655. doi: 10.1111/j.1462-5822.2012.01774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith MS, Bentz GL, Alexander JS, Yurochko AD. 2004. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol 78:4444–4453. doi: 10.1128/JVI.78.9.4444-4453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci U S A 99:16255–16260. doi: 10.1073/pnas.252630899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mendelson M, Monard S, Sissons P, Sinclair J. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol 77:3099–3102. doi: 10.1099/0022-1317-77-12-3099. [DOI] [PubMed] [Google Scholar]
- 19.Zhuravskaya T, Maciejewski JP, Netski DM, Bruening E, Mackintosh FR, St Jeor S. 1997. Spread of human cytomegalovirus (HCMV) after infection of human hematopoietic progenitor cells: a model of HCMV latency. Blood 90:2482–2491. [PubMed] [Google Scholar]
- 20.Ljungman P, Hakki M, Boeckh M. 2011. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol Oncol Clin North Am 25:151–169. doi: 10.1016/j.hoc.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Randolph-Habecker J, Iwata M, Torok-Storb B. 2002. Cytomegalovirus mediated myelosuppression. J Clin Virol 25(Suppl 2):S51–S56. [DOI] [PubMed] [Google Scholar]
- 22.Humar A, Lebranchu Y, Vincenti F, Blumberg EA, Punch JD, Limaye AP, Abramowicz D, Jardine AG, Voulgari AT, Ives J, Hauser IA, Peeters P. 2010. The efficacy and safety of 200 days valganciclovir cytomegalovirus prophylaxis in high-risk kidney transplant recipients. Am J Transplant 10:1228–1237. doi: 10.1111/j.1600-6143.2010.03074.x. [DOI] [PubMed] [Google Scholar]
- 23.Ljungman P, Griffiths P, Paya C. 2002. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis 34:1094–1097. doi: 10.1086/339329. [DOI] [PubMed] [Google Scholar]
- 24.Boeckh M, Leisenring W, Riddell SR, Bowden RA, Huang ML, Myerson D, Stevens-Ayers T, Flowers ME, Cunningham T, Corey L. 2003. Late cytomegalovirus disease and mortality in recipients of allogeneic hematopoietic stem cell transplants: importance of viral load and T-cell immunity. Blood 101:407–414. doi: 10.1182/blood-2002-03-0993. [DOI] [PubMed] [Google Scholar]
- 25.Boeckh M, Nichols WG, Papanicolaou G, Rubin R, Wingard JR, Zaia J. 2003. Cytomegalovirus in hematopoietic stem cell transplant recipients: Current status, known challenges, and future strategies. Biol Blood Marrow Transplant 9:543–558. doi: 10.1016/S1083-8791(03)00287-8. [DOI] [PubMed] [Google Scholar]
- 26.Salzberger B, Bowden RA, Hackman RC, Davis C, Boeckh M. 1997. Neutropenia in allogeneic marrow transplant recipients receiving ganciclovir for prevention of cytomegalovirus disease: risk factors and outcome. Blood 90:2502–2508. [PubMed] [Google Scholar]
- 27.Nichols WG, Corey L, Gooley T, Davis C, Boeckh M. 2002. High risk of death due to bacterial and fungal infection among cytomegalovirus (CMV)-seronegative recipients of stem cell transplants from seropositive donors: evidence for indirect effects of primary CMV infection. J Infect Dis 185:273–282. doi: 10.1086/338624. [DOI] [PubMed] [Google Scholar]
- 28.Hargett D, Shenk T. 2010. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc Natl Acad Sci U S A 107:20039–20044. doi: 10.1073/pnas.1014509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossetto CC, Tarrant-Elorza M, Pari GS. 2013. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog 9:e1003366. doi: 10.1371/journal.ppat.1003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan G, Nogalski MT, Stevenson EV, Yurochko AD. 2012. Human cytomegalovirus induction of a unique signalsome during viral entry into monocytes mediates distinct functional changes: a strategy for viral dissemination. J Leukoc Biol 92:743–752. doi: 10.1189/jlb.0112040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevenson EV, Collins-McMillen D, Kim JH, Cieply SJ, Bentz GL, Yurochko AD. 2014. HCMV reprogramming of infected monocyte survival and differentiation: a Goldilocks phenomenon. Viruses 6:782–807. doi: 10.3390/v6020782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yurochko AD. 2008. Human cytomegalovirus modulation of signal transduction. Curr Top Microbiol Immunol 325:205–220. [DOI] [PubMed] [Google Scholar]
- 33.Taylor-Wiedeman J, Sissons P, Sinclair J. 1994. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J Virol 68:1597–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinclair JH, Baillie J, Bryant LA, Taylor-Wiedeman JA, Sissons JG. 1992. Repression of human cytomegalovirus major immediate early gene expression in a monocytic cell line. J Gen Virol 73:433–435. doi: 10.1099/0022-1317-73-2-433. [DOI] [PubMed] [Google Scholar]
- 35.Chan GCT, Bivens-Smith ER, Smith MS, Smith PM, Yurochko AD. 2008. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an M1 macrophage. J Immunol 181:698–711. doi: 10.4049/jimmunol.181.1.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan GCT, Bivens-Smith ER, Smith MS, Yurochko AD. 2009. NF-kappaB and phosphatidylinositol 3-kinase activity mediates the HCMV-induced atypical M1/M2 polarization of monocytes. Virus Res 144:329–333. doi: 10.1016/j.virusres.2009.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan GCT, Nogalski MT, Bentz GL, Smith MS, Parmater A, Yurochko AD. 2010. PI3K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J Immunol 184:3213–3222. doi: 10.4049/jimmunol.0903025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan GCT, Nogalski MT, Yurochko AD. 2012. Human cytomegalovirus stimulates monocyte-to-macrophage differentiation via the temporal regulation of caspase 3. J Virol 86:10714–10723. doi: 10.1128/JVI.07129-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan GCT, Nogalski MT, Yurochko AD. 2009. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc Natl Acad Sci U S A 106:22369–22374. doi: 10.1073/pnas.0908787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeMeritt IB, Milford LE, Yurochko AD. 2004. Activation of the NF-kappaB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J Virol 78:4498–4507. doi: 10.1128/JVI.78.9.4498-4507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeMeritt IB, Podduturi JP, Tilley AM, Nogalski MT, Yurochko AD. 2006. Prolonged activation of NF-kappaB by human cytomegalovirus promotes efficient viral replication and late gene expression. Virology 346:15–31. doi: 10.1016/j.virol.2005.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nogalski MT, Chan G, Stevenson EV, Gray S, Yurochko AD. 2011. Human cytomegalovirus-regulated paxillin in monocytes links cellular pathogenic motility to the process of viral entry. J Virol 85:1360–1369. doi: 10.1128/JVI.02090-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nogalski MT, Chan GCT, Stevenson EV, Collins-McMillen D, Yurochko AD. 2013. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog 9:e1003463. doi: 10.1371/journal.ppat.1003463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith MS, Bentz GL, Smith PM, Bivens ER, Yurochko AD. 2004. HCMV activates PI(3)K in monocytes and promotes monocyte motility and transendothelial migration in a PI(3)K-dependent manner. J Leukoc Biol 76:65–76. doi: 10.1189/jlb.1203621. [DOI] [PubMed] [Google Scholar]
- 45.Smith MS, Bivens-Smith ER, Tilley AM, Bentz GL, Chan G, Minard J, Yurochko AD. 2007. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: strategy for viral persistence. J Virol 81:7683–7694. doi: 10.1128/JVI.02839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins-McMillen D, Kim JH, Nogalski MT, Stevenson EV, Chan GC, Caskey JR, Cieply SJ, Yurochko AD. 2015. Human cytomegalovirus promotes survival of infected monocytes via a distinct temporal regulation of cellular Bcl-2 family proteins. J Virol 90:2356–2371. doi: 10.1128/JVI.01994-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buehler J, Zeltzer S, Reitsma J, Petrucelli A, Umashankar M, Rak M, Zagallo P, Schroeder J, Terhune S, Goodrum F. 2016. Opposing regulation of the EGF receptor: a molecular switch controlling cytomegalovirus latency and replication. PLoS Pathog 12:e1005655. doi: 10.1371/journal.ppat.1005655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945. doi: 10.1182/blood-2007-01-070078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petrucelli A, Rak M, Grainger L, Goodrum F. 2009. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol 83:5615–5629. doi: 10.1128/JVI.01989-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Umashankar M, Rak M, Bughio F, Zagallo P, Caviness K, Goodrum FD. 2014. Antagonistic determinants controlling replicative and latent states of human cytomegalovirus infection. J Virol 88:5987–6002. doi: 10.1128/JVI.03506-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bentz GL, Yurochko AD. 2008. Human CMV infection of endothelial cells induces an angiogenic response through viral binding to EGF receptor and beta1 beta3 integrins. Proc Natl Acad Sci U S A 105:5531–5536. doi: 10.1073/pnas.0800037105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim JH, Collins-McMillen D, Caposio P, Yurochko AD. 2016. Viral binding-induced signaling drives a unique and extended intracellular trafficking pattern during infection of primary blood monocytes. Proc Natl Acad Sci U S A 113:8819–8824. doi: 10.1073/pnas.1604317113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poole E, Sinclair J. 2015. Sleepless latency of human cytomegalovirus. Med Microbiol Immunol 204:421–429. doi: 10.1007/s00430-015-0401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caviness K, Cicchini L, Rak M, Umashankar M, Goodrum FD. 2014. Complex expression of the UL136 gene of human cytomegalovirus results in multiple protein isoforms with unique roles in replication. J Virol 88:14412–14425. doi: 10.1128/JVI.02711-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petrucelli A, Umashankar M, Zagallo P, Rak M, Goodrum FD. 2012. Interactions between proteins encoded within the human cytomegalovirus UL133-UL138 locus. J Virol 86:8653–8662. doi: 10.1128/JVI.00465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, Goodrum FD. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog 7:e1002444. doi: 10.1371/journal.ppat.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grey F, Tirabassi R, Meyers H, Wu G, McWeeney S, Hook L, Nelson JA. 2010. A viral microRNA down-regulates multiple cell cycle genes through mRNA 5′UTRs. PLoS Pathog 6:e1000967. doi: 10.1371/journal.ppat.1000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Landais I, Pelton C, Streblow D, DeFilippis V, McWeeney S, Nelson JA. 2015. Human cytomegalovirus miR-UL112-3p targets TLR2 and modulates the TLR2/IRAK1/NFkappaB signaling pathway. PLoS Pathog 11:e1004881. doi: 10.1371/journal.ppat.1004881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T, Hancock M, Landais I, Jeng S, McWeeney S, Britt W, Nelson JA. 2014. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 15:363–373. doi: 10.1016/j.chom.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poole E, McGregor DSR, Colston J, Joseph RS, Sinclair J. 2011. Virally induced changes in cellular microRNAs maintain latency of human cytomegalovirus in CD34 progenitors. J Gen Virol 92:1539–1549. doi: 10.1099/vir.0.031377-0. [DOI] [PubMed] [Google Scholar]
- 61.Mason GM, Jackson S, Okecha G, Poole E, Sissons JG, Sinclair J, Wills MR. 2013. Human cytomegalovirus latency-associated proteins elicit immune-suppressive IL-10 producing CD4(+) T cells. PLoS Pathog 9:e1003635. doi: 10.1371/journal.ppat.1003635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mason GM, Poole E, Sissons JG, Wills MR, Sinclair J. 2012. Human cytomegalovirus latency alters the cellular secretome, inducing cluster of differentiation (CD)4+ T-cell migration and suppression of effector function. Proc Natl Acad Sci U S A 109:14538–14543. doi: 10.1073/pnas.1204836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poole E, Walther A, Raven K, Benedict CA, Mason GM, Sinclair J. 2013. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J Virol 87:4261–4271. doi: 10.1128/JVI.03497-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sinclair J, Reeves M. 2014. The intimate relationship between human cytomegalovirus and the dendritic cell lineage. Front Microbiol 5:389. doi: 10.3389/fmicb.2014.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soderberg-Naucler C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA. 2001. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J Virol 75:7543–7554. doi: 10.1128/JVI.75.16.7543-7554.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Torok-Storb B, Simmons P, Khaira D, Stachel D, Myerson D. 1992. Cytomegalovirus and marrow function. Ann Hematol. 64(Suppl):A128–A131. doi: 10.1007/BF01715365. [DOI] [PubMed] [Google Scholar]
- 67.Steffens HP, Podlech J, Kurz S, Angele P, Dreis D, Reddehase MJ. 1998. Cytomegalovirus inhibits the engraftment of donor bone marrow cells by downregulation of hemopoietin gene expression in recipient stroma. J Virol 72:5006–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sindre H, Tjoonnfjord GE, Rollag H, Ranneberg-Nilsen T, Veiby OP, Beck S, Degre M, Hestdal K. 1996. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 88:4526–4533. [PubMed] [Google Scholar]
- 69.Chen T, Burke KA, Zhan Y, Wang X, Shibata D, Zhao Y. 2007. IL-12 facilitates both the recovery of endogenous hematopoiesis and the engraftment of stem cells after ionizing radiation. Exp Hematol 35:203–213. doi: 10.1016/j.exphem.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 70.Gluzman-Poltorak Z, Mendonca SR, Vainstein V, Kha H, Basile LA. 2014. Randomized comparison of single dose of recombinant human IL-12 versus placebo for restoration of hematopoiesis and improved survival in rhesus monkeys exposed to lethal radiation. J Hematol Oncol 7:31. doi: 10.1186/1756-8722-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blank U, Karlsson S. 2015. TGF-beta signaling in the control of hematopoietic stem cells. Blood 125:3542–3550. doi: 10.1182/blood-2014-12-618090. [DOI] [PubMed] [Google Scholar]
- 72.Bego MG, St Jeor S. 2006. Human cytomegalovirus infection of cells of hematopoietic origin: HCMV-induced immunosuppression, immune evasion, and latency. Exp Hematol 34:555–570. doi: 10.1016/j.exphem.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 73.Maciejewski JP, St Jeor SC. 1999. Human cytomegalovirus infection of human hematopoietic progenitor cells. Leuk Lymphoma 33:1–13. doi: 10.3109/10428199909093720. [DOI] [PubMed] [Google Scholar]
- 74.Sinclair J. 2008. Human cytomegalovirus: latency and reactivation in the myeloid lineage. J Clin Virol 41:180–185. doi: 10.1016/j.jcv.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 75.Bevan IS, Daw RA, Day PJ, Ala FA, Walker MR. 1991. Polymerase chain reaction for detection of human cytomegalovirus infection in a blood donor population. Br J Haematol 78:94–99. doi: 10.1111/j.1365-2141.1991.tb04388.x. [DOI] [PubMed] [Google Scholar]
- 76.Taylor-Wiedeman J, Sissons JG, Borysiewicz LK, Sinclair J. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol 72:2059–2064. doi: 10.1099/0022-1317-72-9-2059. [DOI] [PubMed] [Google Scholar]
- 77.Soroceanu L, Akhavan A, Cobbs CS. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- 78.Campadelli-Fiume G, Collins-McMillen D, Gianni T, Yurochko AD. 2016. Integrins as herpesvirus receptors and mediators of the host signalosome. Annu Rev Virol 3:215–236. doi: 10.1146/annurev-virology-110615-035618. [DOI] [PubMed] [Google Scholar]
- 79.Feire AL, Koss H, Compton T. 2004. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc Natl Acad Sci U S A 101:15470–15475. doi: 10.1073/pnas.0406821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feire AL, Roy RM, Manley K, Compton T. 2010. The glycoprotein B disintegrin-like domain binds beta 1 integrin to mediate cytomegalovirus entry. J Virol 84:10026–10037. doi: 10.1128/JVI.00710-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 424:456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 82.Boulant S, Stanifer M, Lozach PY. 2015. Dynamics of virus-receptor interactions in virus binding, signaling, and endocytosis. Viruses 7:2794–2815. doi: 10.3390/v7062747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Compton T, Nepomuceno RR, Nowlin DM. 1992. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 191:387–395. doi: 10.1016/0042-6822(92)90200-9. [DOI] [PubMed] [Google Scholar]
- 84.Hetzenecker S, Helenius A, Krzyzaniak MA. 2016. HCMV induces macropinocytosis for host cell entry in fibroblasts. Traffic 17:351–368. doi: 10.1111/tra.12355. [DOI] [PubMed] [Google Scholar]
- 85.Bodaghi B, Slobbe-van Drunen ME, Topilko A, Perret E, Vossen RC, van Dam-Mieras MC, Zipeto D, Virelizier JL, LeHoang P, Bruggeman CA, Michelson S. 1999. Entry of human cytomegalovirus into retinal pigment epithelial and endothelial cells by endocytosis. Invest Ophthalmol Vis Sci 40:2598–2607. [PubMed] [Google Scholar]
- 86.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 and UL150 and occurs by endocytosis and low-pH fusion. J Virol 80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang X, Huang DY, Huong SM, Huang ES. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med 11:515–521. doi: 10.1038/nm1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Douglas MR, Morrison KC, Jacques SJ, Leadbeater WE, Gonzalez AM, Berry M, Logan A, Ahmed Z. 2009. Off-target effects of epidermal growth factor receptor antagonists mediate retinal ganglion cell disinhibited axon growth. Brain 132:3102–3121. doi: 10.1093/brain/awp240. [DOI] [PubMed] [Google Scholar]
- 89.Carrasco-Garcia E, Saceda M, Grasso S, Rocamora-Reverte L, Conde M, Gomez-Martinez A, Garcia-Morales P, Ferragut JA, Martinez-Lacaci I. 2011. Small tyrosine kinase inhibitors interrupt EGFR signaling by interacting with erbB3 and erbB4 in glioblastoma cell lines. Exp Cell Res 317:1476–1489. doi: 10.1016/j.yexcr.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 90.Pruvot B, Cure Y, Djiotsa J, Voncken A, Muller M. 2014. Developmental defects in zebrafish for classification of EGF pathway inhibitors. Toxicol Appl Pharmacol 274:339–349. doi: 10.1016/j.taap.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 91.Choi BH, Choi JS, Rhie DJ, Yoon SH, Min DS, Jo YH, Kim MS, Hahn SJ. 2002. Direct inhibition of the cloned Kv1.5 channel by AG-1478, a tyrosine kinase inhibitor. Am J Physiol Cell Physiol 282:C1461–C1468. doi: 10.1152/ajpcell.00398.2001. [DOI] [PubMed] [Google Scholar]
- 92.Jackson SE, Mason GM, Wills MR. 2011. Human cytomegalovirus immunity and immune evasion. Virus Res 157:151–160. doi: 10.1016/j.virusres.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 93.Spector DH. 2015. Human cytomegalovirus riding the cell cycle. Med Microbiol Immunol 204:409–419. doi: 10.1007/s00430-015-0396-z. [DOI] [PubMed] [Google Scholar]
- 94.Ibanez CE, Schrier R, Ghazal P, Wiley C, Nelson JA. 1991. Human cytomegalovirus productively infects primary differentiated macrophages. J Virol 65:6581–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maciejewski JP, Bruening EE, Donahue RE, Sellers SE, Carter C, Young NS, St Jeor S. 1993. Infection of mononucleated phagocytes with human cytomegalovirus. Virology 195:327–336. doi: 10.1006/viro.1993.1383. [DOI] [PubMed] [Google Scholar]
- 96.Plachter B, Sinzger C, Jahn G. 1996. Cell types involved in replication and distribution of human cytomegalovirus. Adv Virus Res 46:195–261. doi: 10.1016/S0065-3527(08)60073-1. [DOI] [PubMed] [Google Scholar]
- 97.Sinclair J, Sissons P. 1996. Latent and persistent infections of monocytes and macrophages. Intervirology 39:293–301. [DOI] [PubMed] [Google Scholar]
- 98.Plotkin SA, Furukawa T, Zygraich N, Huygelen C. 1975. Candidate cytomegalovirus strain for human vaccination. Infect Immun 12:521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sampaio KL, Cavignac Y, Stierhof YD, Sinzger C. 2005. Human cytomegalovirus labeled with green fluorescent protein for live analysis of intracellular particle movements. J Virol 79:2754–2767. doi: 10.1128/JVI.79.5.2754-2767.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bentz GL, Jarquin-Pardo M, Chan GCT, Smith MS, Sinzger C, Yurochko AD. 2006. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol 80:11539–11555. doi: 10.1128/JVI.01016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chan G, Bivins-Smith ER, Smith MS, Yurochko AD. 2008. Transcriptome analysis of NF-kappaB- and phosphatidylinositol 3-kinase-regulated genes in human cytomegalovirus-infected monocytes. J Virol 82:1040–1046. doi: 10.1128/JVI.00864-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nogalski MT, Podduturi JP, DeMeritt IB, Milford LE, Yurochko AD. 2007. The human cytomegalovirus virion possesses an activated casein kinase II that allows for the rapid phosphorylation of the inhibitor of NF-kappaB, IkappaBalpha. J Virol 81:5305–5314. doi: 10.1128/JVI.02382-06. [DOI] [PMC free article] [PubMed] [Google Scholar]