

Abstract
Background
Mutational combinations of the cystic fibrosis transmembrane conductance regulator, CFTR, gene have different phenotypic manifestations at the molecular level with varying clinical consequences for individuals possessing such mutations. Reporting cystic fibrosis transmembrane conductance regulator mutations is important in understanding the genotype-phenotype correlations and associated clinical presentations in patients with cystic fibrosis. Understanding the effects of mutations is critical in developing appropriate treatments for individuals affected with cystic fibrosis, non-classic cystic fibrosis, or cystic fibrosis transmembrane conductance regulator-related disorders. This is the first report of related individuals possessing the R248G missense cystic fibrosis transmembrane conductance regulator mutation and we present their associated clinical histories.
Case presentation
All three patients are of Spanish descent. Deoxyribonucleic acid analysis revealed that all three siblings possessed a novel c.742A>G mutation, resulting in a p.Arg248Gly (R248G) amino acid change in exon 6 in trans with the known N1303K mutant allele. Case 1 patient is a 39-year-old infertile man presenting with congenital unilateral absence of the vas deferens and recurrent episodes of epigastric pain. Case 2 patient is a 32-year-old woman presenting with periods of infertility, two previous spontaneous abortions, recurrent epigastric pain, and recurrent pancreatitis. Case 3 patient is a 29-year-old woman presenting with recurrent pancreatitis and epigastric pain.
Conclusions
We report the genotype-phenotype correlations and clinical manifestations of a novel R248G cystic fibrosis transmembrane conductance regulator mutation: congenital unilateral absence of the vas deferens in males, reduced female fertility, and recurrent acute pancreatitis. In addition, we discuss the possible functional consequences of the mutations at the molecular level.
Electronic supplementary material
The online version of this article (doi:10.1186/s13256-016-1181-3) contains supplementary material, which is available to authorized users.
Keywords: CFTR, Missense mutation, Genotype-phenotype, Congenital absence of vas deferens
Background
Cystic fibrosis (CF), Online Mendelian Inheritance in Man (OMIM) #219700, is a life-threatening autosomal recessive monogenetic disease caused by mutations to the cystic fibrosis transmembrane conductance regulator (CFTR) gene (OMIM #602421). CFTR is an adenosine triphosphate (ATP)-binding cassette (ABC) transporter protein that functions as a chloride (Cl–) ion channel and is expressed in the apical membrane of epithelial cells lining the lungs, pancreas, gastrointestinal tract, and reproductive system [1]. Transport of Cl– ions is important in the movement of fluids required for the production of free flowing mucus in these epithelia [1]. Mutations to the CFTR gene phenotypically manifest themselves in variable clinical outcomes including: progressive decrease in lung function, recurrent colonization of opportunistic bacteria such as Pseudomonas aeruginosa and Staphylococcus aureus, pancreatic insufficiency (PI), and male infertility due to congenital bilateral absence of the vas deferens (CBAVD) or congenital unilateral absence of the vas deferens (CUAVD) [2, 3]. Accurate reporting of CFTR mutations is of clinical interest in improving genetic screening techniques, understanding CF genotype-phenotype correlations, and treatments of individuals affected with CF, non-classic CF, or CFTR-related disorders (CFTR-RD).
We present here the case of three related Spanish patients possessing a novel CFTR R248G missense mutation in trans with the N1303K mutant allele. The hospital network ethics committees and the institutional review boards approved the study. Informed consent was obtained from the patients. Molecular studies of genomic deoxyribonucleic acid (DNA) were performed from extracted peripheral blood samples of the patients. In the index case, the use of the “Cystic Fibrosis Genotyping assay” (Abbott) kit identified a c.3909C>G mutation responsible for a p.Asn1303Lys change. After clinical evaluation was carried out, the sequencing of exons and adjoining regions of the CFTR gene were conducted by direct sequencing (3130X ABI PRISM sequencer; Applied Biosystems, Foster City, CA, USA). The sequencing analysis confirmed the previous mutation and identified a c.742A>G mutation, resulting in a p.Arg248Gly amino acid change in exon 6 of the CFTR gene. This new mutation was passed paternally and is present in all three siblings.
Case presentation
Case 1 patient is a 39-year-old infertile man of Spanish descent who was referred to our CF Unit for non-classic CF evaluation after consulting with his primary care physician with regards to an inability to impregnate a woman in 2 years. A clinical evaluation confirmed the diagnosis of CUAVD on his left testicle and an in-depth past medical history was obtained. His history revealed that he had previously undergone surgical repair for right testicular hydrocele, recurrent episodes of acute pancreatitis with heartburn, and epigastric pain associated with increasing blood levels of amylase and lipase. Other potential causes of acute pancreatitis were ruled out (gallstones, endocrine problems and metabolic abnormalities, alcohol abuse, or others drugs). Pulmonary function tests suggested that case 1 patient does not have lung disease, although he has tested positive for S. aureus colonization. His sweat test averages of 47 mEq/l indicate borderline results for CF diagnosis. A review of his past clinical history was unremarkable for other CFTR-RDs. A non-classic CF clinical evaluation and evaluation of the past medical histories of his siblings were conducted as a means of examining particular genetic associations.
Case 2 patient is a 32-year-old Spanish woman whose past medical history was significant for periods of infertility with two previous spontaneous abortions. Her history also included recurrent episodes of epigastric pain and pancreatitis. The episodes of pancreatitis consisted of periodic acute periumbilical abdominal pain, vomiting, nausea, as well as a transient increase in enzymatic levels of amylase and pancreatic lipase. Similar to case 1, she had no prior medical or socio-behavioral histories that could account for the recurrent episodes of epigastric pain and pancreatitis. She exhibited normal lung function and has tested positive for S. aureus colonization. Her sweat test averages of 40 mEq/l indicate borderline results for CF diagnosis.
Case 3 patient is a 29-year-old Spanish woman with a past medical history of recurrent pancreatitis and epigastric pain. Her presentations of recurrent epigastric pain resembled that of the clinical history of case 1. Her episodes of recurrent pancreatitis were similar to the etiologies of case 2 with acute periumbilical abdominal pain, vomiting, nausea, as well as a transient increase in the enzymatic levels of amylase and pancreatic lipase. All other medical and socio-behavioral histories were also ruled out as potential causes of recurrent pancreatitis and epigastric pain. Her clinical history indicated previous periods of pneumonia and bronchitis. She exhibited normal lung function, has tested negative for S. aureus colonization, and has no prior history of reproductive problems. Her sweat test averages of 42 mEq/l indicate borderline results for CF diagnosis. Table 1 shows the summary of the clinical histories.
Table 1.
Clinical expression of three siblings with the N1303K/R248G cystic fibrosis transmembrane conductance regulator genotype
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Current age | 39-year-old man | 32-year-old woman | 29-year-old woman |
| Age of diagnosis | 32 years | 25 years | 22 years |
| Chloride sweat test (mEq/L) | 47 | 40 | 42 |
| FEV1 % | 82.2 | 86 | 84 |
| FVC % | 80.1 | 82 | 82.4 |
| Respiratory disease | No significant history | No significant history | 1 episode of bronchitis |
| 1 episode of pneumonia | |||
| Bacterial colonization | Staphylococcus aureus | Staphylococcus aureus | No significant history |
| Pancreatic and gastrointestinal manifestations | • Recurrent epigastric pain associated with pancreatitis | • 3 episodes of pancreatitis | • 4 episodes of pancreatitis |
| • Chronic heartburn | • Recurrent epigastric pain | ||
| Reproductive complications | • CUAVD | • 2 spontaneous abortions | No significant history |
| • Testicular hydrocele | • Periods of infertility |
CUAVD congenital unilateral absence of the vas deferens, FEV1 forced expiratory volume in 1 second, FVC forced vital capacity
Discussion
The N1303K allele is a product of a missense mutation in exon 21 of the CFTR gene [3], causing an asparagine to lysine amino acid change in the nucleotide-binding domain 2 (NBD2) of the CFTR protein (Cystic Fibrosis Mutation Database, http://www.genet.sickkids.on.ca/cftr). NBD2 is an ATPase domain of the CFTR protein that is involved in closing of the ion channel via the energy produced from ATP hydrolysis [4]. NBD2 mutations can result in destabilization of channel activity and significant reductions in channel permeability [4]. In addition, N1303K is a class II trafficking mutation that can cause folding or deficiency of protein maturation and consequently result in absence or reduced quantity of CFTR protein [1, 5]. This mutation has been heterogeneously associated with CBAVD and CUAVD in males [6, 7] and moderate to severe PI [8]. Figure 1a illustrates the two-dimensional model of the CFTR protein and major functional regions.
Fig. 1.
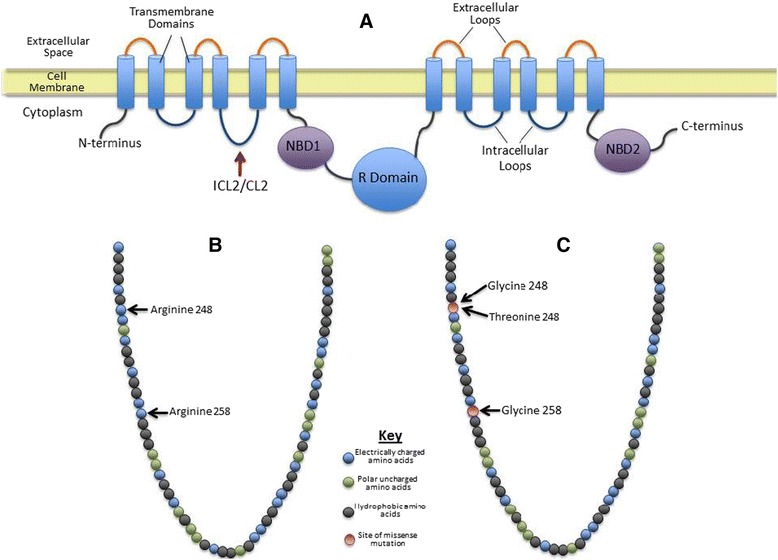
Cystic fibrosis transmembrane conductance regulator protein. a Two-dimensional model of the cystic fibrosis transmembrane conductance regulator protein with major functional components. b Wild-type intracellular loop 2/cellular loop 2 primary protein structure depicting amino acid properties. c Intracellular loop 2/cellular loop 2 regions clinically associated with congenital bilateral absence of the vas deferens or congenital unilateral absence of the vas deferens resulting from a reported missense mutation. ICL2/CL2 intracellular loop 2/cellular loop 2, NBD1 nucleotide-binding domain 1, NBD2 nucleotide-binding domain 2
The clinical phenotype of CUAVD in case 1 patient is consistent with previous studies of the N1303K mutation. Although correlations between CFTR mutations and reproduction have been thoroughly investigated in male patients, studies also suggest that CFTR mutations can result in reduced fertility in females due to the production of thick cervical mucus, and increase the likelihood of spontaneous abortions [9]. We propose that the N1303K mutation is responsible for the periods of infertility in case 2 patient and could also be related to her two spontaneous abortions. The recurrent pancreatic problems observed in all three patients may not be associated with the N1303K mutation despite its classification as a moderate to severe PI mutation [8]. Recurrent acute pancreatitis is observed in significantly higher frequencies among patients with CF carrying one or two mild PI mutations than patients carrying only one moderate to severe PI mutation [8].
All three patients in this case study possess a novel R248G missense mutation in exon 6 of the CFTR gene, causing an arginine to glycine amino acid change. Arginine-248 is located in intracellular loop 2/cellular loop 2 (ICL2/CL2) of the wild-type CFTR protein (http://www.uniprot.org). Figure 1b illustrates the wild-type primary protein structure based on the properties of the amino acids. ICL2/CL2 is both the largest and most hydrophilic loop of the CFTR protein that stabilizes the full conductance state of the channel [10]. Increased mean closed times are associated with ICL2/CL2 mutations of the CFTR protein [10].
In this study we conducted an extensive database and literature review, where the R248G mutation is not reported as an identified CFTR mutation. A similar missense mutation, R248T, has been previously reported as a mild CFTR-RD mutation that is associated with CBAVD and no other clinical phenotype (see Fig. 1c) [11]. The R248G mutation may alter the normal ICL2/CL2 function more than the R248T mutation based on the clinical phenotypes of the three patients. An arginine to threonine amino acid substitution alters residue charge from positive to neutral but does not change polarity. An arginine to glycine substitution changes the residue charge from positive to neutral as well as amino acid polarity. In addition, an R258G missense mutation located in the ICL2/CL2 domain was reported in a French male with CBAVD [12]. Based on these previous observations and three cases presented in this study, we suggest that amino acid substitution of arginine residues in the ICL2/CL2 domain of the CFTR protein with amino acids of contrasting molecular properties in charge and polarity could be associated with CBAVD and/or CUAVD (see Fig. 1c). The missense mutation on the ICL2/CL2 domain of the protein could result in increased mean closed times of normal CFTR function and contribute to increased thick epithelial viscosity of the reproductive tract of female patients. This could explain the clinical histories of spontaneous abortions and periods of infertility in case 2 patient. We also postulate that this mutation may have had an impact on the normal development of the male reproductive organs in case 1 patient that resulted in CUAVD. We have incorporated Additional files 1 and 2 to show the chromatogram and the amino acid sequence confirming the presence of the c.742A>G.
Conclusions
We suggest that the R248G mutation may be associated with the recurrent acute pancreatitis exhibited by case 2 and case 3 patients because it is unlikely that the N1303K moderate to severe PI mutation is the underlying molecular cause when taking into consideration that each patient has only one copy of this mutant allele [8]. The recurrent episodes of epigastric pain and heartburn observed in case 1 and case 2 patients could be symptomatically masking the clinical signs of recurrent acute pancreatitis [13, 14].
In this case series we report the potential genotype-phenotype correlations with the novel R248G CFTR mutation: CUAVD in males, reduced female fertility, and recurrent acute pancreatitis. Future structure-functional studies on the CFTR protein can provide further insight on the impact of the R248G mutation at the molecular level.
Acknowledgements
Not applicable.
Funding
This work was supported in part by grant MD001452 from the National Center on Minority Health and Health Disparities of the National Institutes of Health, Dr. Luz Claudio, Principal Investigator.
Availability of data and materials
The datasets supporting the conclusions of this article are available in: the Cystic Fibrosis Mutation Database, http://www.genet.sickkids.on.ca/cftr; the Universal Protein Resource (UniProt) repository, UniProtKB - P13569 (CFTR_Human), http://www.uniprot.org/uniprot/P13569; and the e!Ensembl repository, CFTR-001 ENST00000003084, http://useast.ensembl.org/Homo_sapiens/Transcript/Sequence_cDNA?db=core;g=ENSG00000001626;r=7:117479963-117668665;t=ENST00000003084.
Authors’ contributions
SV, GG, and JAO analyzed and interpreted the patients’ clinical histories after confirming R248G mutation in all three patients. RM and MM interpreted probable associations between mutations and clinical manifestations of the patients. PM, MR, and MP conducted the initial clinical evaluation of the patients and their clinical histories. All authors approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patients for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Ethics approval and consent to participate
The study was approved by the Ethics and Research Committee of the Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain.
Abbreviations
- ATP
Adenosine triphosphate
- CBAVD
Congenital bilateral absence of the vas deferens
- CF
Cystic fibrosis
- CFTR
Cystic fibrosis transmembrane conductance regulator
- CFTR-RD
CFTR-related disorders
- Cl– ion
Chloride ion
- CUAVD
Congenital unilateral absence of the vas deferens
- ICL2/CL2
Intracellular loop 2/cellular loop 2
- NBD2
Nucleotide-binding domain 2
- OMIM
Online Mendelian Inheritance in Man
- PI
Pancreatic insufficiency
Additional files
Direct sequencing of exon 6 of cystic fibrosis transmembrane conductance regulator gene by the standard Sanger method confirming the presence of the c.742A>G change in all three patients. A.) Representative chromatogram for the wild-type forward strand 5′ → 3′ sequence. B.) Representative chromatogram of the mutated forward strand 5′ → 3′ sequence. (DOCX 106 kb)
Human cystic fibrosis transmembrane conductance regulator nucleotide and amino acid sequence. Unique nucleotide sequence GATGATGAAGTACA is present in wild-type cystic fibrosis transmembrane conductance regulator protein. GATGATGAAGTAACG sequence observed in all three patient cases is due to a c.742A>G mutation, resulting in codon change from AGA to GGA and consequent arginine to glycine change. (RTF 600 kb)
Contributor Information
Seiichi Villalona, Email: svillalona@mail.usf.edu.
Guillermo Glover-López, Email: guillermo.glover@carm.es.
Juan Antonio Ortega-García, Email: ortega@pehsu.org.
Rosa Moya-Quiles, Email: rosa.moya2@carm.es.
Pedro Mondejar-López, Email: pmondejarlopez@yahoo.es.
Maria C. Martínez-Romero, Email: training@pehsu.org
Mariano Rigabert-Montiel, Email: mrigabert@ono.com.
María D. Pastor-Vivero, Email: mdpastor@gmail.com
Manuel Sánchez-Solís, Email: msolis@um.es.
References
- 1.Derichs N. Targeting a genetic defect: Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Eur Respir Rev. 2013;22:58–65. doi: 10.1183/09059180.00008412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.des Georges M, Guittard C, Altiéri JP, Templin C, Sarles J, Sarda P, Claustres M. High heterogeneity of CFTR mutations and unexpected low incidence of cystic fibrosis in the Mediterranean France. J Cyst Fibros. 2004;3:265–72. doi: 10.1016/j.jcf.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Bombieri C, Claustres M, De Boeck K, Derichs N, Dodge J, Girodon E, Sermet I, Schwarz M, Tzetis M, Wilschanski M, Bareil C, Bilton D, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10:86–102. doi: 10.1016/S1569-1993(11)60014-3. [DOI] [PubMed] [Google Scholar]
- 4.Tsai M, Li M, Hwang T. Stable ATP binding mediated by a partial NBD dimer of the CFTR chloride channel. J Gen Physiol. 2010;135:399–414. doi: 10.1085/jgp.201010399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogan MP, Stoltz DA, Hornick DB. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest. 2011;139:1480–90. doi: 10.1378/chest.10-2077. [DOI] [PubMed] [Google Scholar]
- 6.Mickle J, Milunsky A, Amos JA, Oates RD. Immunology: Congenital unilateral absence of the vas deferens: A heterogeneous disorder with two distinct subpopulations based upon aetiology and mutational status of the cystic fibrosis gene. Hum Reprod. 1995;10:1728–35. doi: 10.1093/oxfordjournals.humrep.a136164. [DOI] [PubMed] [Google Scholar]
- 7.Van Hoorenbeeck K, Storm K, van den Ende J, Biervliet M, Desager KN. N1303K and IVS8-5T, clinical presentation within a family with atypical cystic fibrosis. J Cyst Fibros. 2007;6:220–2. doi: 10.1016/j.jcf.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Ooi CY, Dorfman R, Cipolli M, Gonska T, Castellani C, Keenan K, Freedman SD, Zielenski J, Berthiaume Y, Corey M, Schibli S, Tullis E, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology. 2011;140:153–61. doi: 10.1053/j.gastro.2010.09.046. [DOI] [PubMed] [Google Scholar]
- 9.Chan HC, Ruan YC, He Q, Chen MH, Chen H, Xu WM, Chen WY, Xie C, Zhang XH, Zhou Z. The cystic fibrosis transmembrane conductance regulator in reproductive health and disease. J Physiol. 2009;587:2187–95. doi: 10.1113/jphysiol.2008.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seibert FS, Jia Y, Mathews CJ, Hanrahan JW, Riordan LTW, Clarke DM. Disease-associated mutations in cytoplasmic loops 1 and 2 of cystic fibrosis transmembrane conductance regulator impede processing or opening of the channel. Biochem. 1997;36(39):11966–74. doi: 10.1021/bi9712652. [DOI] [PubMed] [Google Scholar]
- 11.Dorfman R, Nalpathamkalam T, Taylor C, Gonska T, Keenan K, Yuan X, Keenan K, Yuan XW, Corey M, Tsui LC, Zielenski J, Durie P. Do common in silico tools predict the clinical consequences of amino-acid substitutions in the CFTR gene? Clin Genet. 2010;77:464–73. doi: 10.1111/j.1399-0004.2009.01351.x. [DOI] [PubMed] [Google Scholar]
- 12.Masvidal L, Giménez J, Ramos MD, Domingo C, Farré A, Bassas L, Casals T. The p.Arg258Gly mutation in intracellular loop 2 of CFTR is associated with CFTR-related disorders. Genet Test Mol Biomarkers. 2009;13:765–8. doi: 10.1089/gtmb.2009.0070. [DOI] [PubMed] [Google Scholar]
- 13.Sciume C, Geraci G, Pisello F, Li Volsi F, Facella T, Modica G. Aberrant pancreas: A rare cause of epigastralgia. Ann Ital Chir. 2004;75:483–6. [PubMed] [Google Scholar]
- 14.Fukumori K, Shakado S, Miyahara T, Fukuizumi K, Takemoto R, Nishi H, Sakai H, Muranaka T, Sata M. Atypical manifestations of pancreatitis with autoimmune phenomenon in an adolescent female. Intern Med. 2005;44:886–91. doi: 10.2169/internalmedicine.44.886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are available in: the Cystic Fibrosis Mutation Database, http://www.genet.sickkids.on.ca/cftr; the Universal Protein Resource (UniProt) repository, UniProtKB - P13569 (CFTR_Human), http://www.uniprot.org/uniprot/P13569; and the e!Ensembl repository, CFTR-001 ENST00000003084, http://useast.ensembl.org/Homo_sapiens/Transcript/Sequence_cDNA?db=core;g=ENSG00000001626;r=7:117479963-117668665;t=ENST00000003084.