

Abstract
NOD mice, a model strain for human type 1 diabetes, express proinsulin (PI) in the thymus. However, insulin-reactive T cells escape negative selection, and subsequent activation of the CD8+ T-cell clonotype G9C8, which recognizes insulin B15-23 via an αβ T-cell receptor (TCR) incorporating TRAV8-1/TRAJ9 and TRBV19/TRBJ2-3 gene rearrangements, contributes to the development of diabetes. In this study, we used fixed TRAV8-1/TRAJ9 TCRα-chain transgenic mice to assess the impact of PI isoform expression on the insulin-reactive CD8+ T-cell repertoire. The key findings were: 1) PI2 deficiency increases the frequency of insulin B15-23–reactive TRBV19+CD8+ T cells and causes diabetes; 2) insulin B15-23–reactive TRBV19+CD8+ T cells are more abundant in the pancreatic lymph nodes of mice lacking PI1 and/or PI2; 3) overexpression of PI2 decreases TRBV19 usage in the global CD8+ T-cell compartment; 4) a biased repertoire of insulin-reactive CD8+ T cells emerges in the periphery regardless of antigen exposure; and 5) low-avidity insulin-reactive CD8+ T cells are less affected by antigen exposure in the thymus than in the periphery. These findings inform our understanding of the diabetogenic process and reveal new avenues for therapeutic exploitation in type 1 diabetes.
Introduction
Autoreactive T cells are key players in the process of immune-mediated β-cell destruction, which culminates in the development of type 1 diabetes. Immature CD4+CD8+ thymocytes that recognize self-derived peptide-loaded major histocompatibility complex (MHC) molecules via high-affinity interactions with a clonotypically expressed T-cell receptor (TCR) are typically removed by negative selection to prevent the egress of such autoreactive T cells. Similar deletional events may also occur in the periphery (1). However, these tolerogenic mechanisms are not infallible, and self-derived antigen-specific T-cell escape likely underpins a number of autoimmune diseases.
Proinsulin (PI) is a major diabetogenic autoantigen in humans (2–5) and mice (6–10). PI is generated from a larger preprohormone by cleavage of the signal peptide and then further processed in the pancreatic β-cells to insulin, which is the metabolically active hormone. Self-derived antigens, including PI, are expressed in the thymus (11,12) under the control of the autoimmune regulator (AIRE) protein, a transcription factor found in medullary thymic epithelial cells (13). Central exposure to these antigens limits the development of autoreactive T cells. Although the MHC locus, encompassing both class I and II genes, is the most important genetic susceptibility factor for type 1 diabetes (14), the insulin 5′-variable number tandem repeat region also plays a significant role by regulating PI expression in the thymus and pancreas (15). There are two types of PI in mice, known as PI1 and PI2, which are separately encoded in the genome. PI2 is expressed in the thymus and pancreas, whereas PI1 is thought to occur primarily in the pancreas (11,16), with only low-level expression in the thymus (17). For this reason, it has been suggested that PI2 may be more pivotal in the development of T-cell tolerance. Investigation of individual PI1 knockout (PI1−/−) mice backcrossed to the NOD background has shown that loss of PI1 reduces the incidence of diabetes (18). In contrast, loss of PI2 in the corresponding PI2−/− model accelerates the development of diabetes in 100% of mice (19). Conversely, thymic overexpression of PI2 on the MHC class II promoter (NODPI2tg) leads to a decreased incidence of diabetes (20,21). These findings suggest that PI2 is important for both central and peripheral tolerance.
Insulin-reactive CD4+ and CD8+ T cells have been shown to be important for the development of diabetes in humans and mice (6–8,10,22). The dominant CD8+ T-cell epitope in NOD mice, insulin B15-23 (8,22), overlaps with a longer peptide recognized by diabetogenic CD4+ T cells (23). In a study where both PI1 and PI2 were eliminated and an altered insulin expressing alanine at position B16 instead of tyrosine was substituted for native insulin, the resulting PI1−/−PI2−/−Y16Atg mice were protected from diabetes due to removal of the cognate CD4+ and CD8+ autoreactive T-cell epitopes (9). However, if both PI1 and PI2 are deleted without insulin substitution, mice die rapidly from metabolic problems related to insulin deficiency. It is also notable that insulin autoimmunity is required for the development of islet-specific glucose-6-phosphatase catalytic subunit–related protein (IGRP) reactivity (24,25). Modifying the development of insulin autoimmunity may therefore prove to be a key therapeutic intervention.
We previously generated a highly diabetogenic murine CD8+ T-cell clone (G9C8) that expresses an αβ TCR encoded by TRAV8-1/TRAJ9 and TRBV19/TRBJ2-3 gene rearrangements (22). In vitro, this clone displayed potent cytotoxic and proliferative activity in response to islet cells. In vivo, G9C8 caused diabetes within 5–10 days in young prediabetic NOD and NOD.scid mice (22). Moreover, T cells that recognize the H-2Kd–restricted insulin B15-23 epitope targeted by G9C8 infiltrate the islets of NOD mice at 4 weeks of age, a time when very few T cells with other specificities are present (8,26). These observations further suggest that CD8+ T cells are important in the pathogenesis of autoimmune diabetes in the NOD mouse model. The insulin B15-23 peptide (LYLVCGERG) binds poorly to H2-Kd (27,28). Consequently, relatively high peptide concentrations are required for exogenous recognition of this antigenic complex, which typically elicits low-avidity T cells. Of note, the native B15-23 sequence is conserved in humans and common to murine PI1 and PI2.
It is established that CD8+ T cells in humans can recognize antigenic peptides derived from preproinsulin and destroy β-cells (4,5). However, the underlying mechanisms that allow the development and expansion of such disease-relevant CD8+ T-cell populations remain obscure. In this study, we generated fixed TRAV8-1/TRAJ9 TCRα-chain NOD mice (designated A22 for simplicity to reflect usage of mouse line 22) (29) with either normal PI1 and PI2 levels (A22Cα−/− mice), PI2 overexpression (A22Cα−/−PI2tg mice), PI2 deficiency (A22Cα−/−PI2−/− mice), PI1 deficiency (A22Cα−/−PI1−/− mice), or both PI1 and PI2 deficiency with a mutant transgene preventing recognition of the insulin B15-23 peptide (A22Cα−/−PI1−/−PI2−/−Y16Atg mice) (Table 1). These unique models were then used to investigate the role of PI expression on the insulin B15-23–reactive CD8+ T-cell repertoire.
Table 1.
Transgenic and knockout mice used in this study
| Genotype | TCRα-chain | TCRβ chain | PI1 expression | PI2 expression | Diabetes |
|---|---|---|---|---|---|
| A22Cα−/− | G9 TCRα transgene (insulin B15-23–specific) | Endogenous TCRβ chains | Normal | Normal | No diabetes |
| A22Cα−/−PI2tg | G9 TCRα transgene (insulin B15-23–specific) | Endogenous TCRβ chains | Normal | Overexpression (under MHC class II promoter) | No diabetes |
| A22Cα−/−PI1−/− | G9 TCRα transgene (insulin B15-23–specific) | Endogenous TCRβ chains | Deficient | Normal | No diabetes |
| A22Cα−/−PI2−/− | G9 TCRα transgene (insulin B15-23–specific) | Endogenous TCRβ chains | Normal | Deficient | Accelerated diabetes in males |
| A22Cα−/−PI1−/−PI2−/−Y16Atg | G9 TCRα transgene (insulin B15-23–specific) | Endogenous TCRβ chains | Lack both native PI genes but express a mutated insulin transgene | No diabetes | |
Research Design and Methods
Mice
Insulin B15-23–reactive TRAV8-1/TRAJ9 TCRα-chain transgenic mice were generated as described previously (29). Line 22 was selected for further characterization, hence the designation A22. TCRα-chain transgenic mice were crossed with NODCα−/− mice to generate A22Cα−/− mice exclusively expressing the TRAV8-1/TRAJ9 transgene. These A22Cα−/− mice were then crossed with NOD mice overexpressing PI2 under the MHC class II promoter (NODPI2tg) to generate A22Cα−/−PI2tg mice. In addition, A22Cα−/− mice were crossed with NOD mice lacking PI2, NOD mice lacking PI1, and NOD mice lacking PI1 and PI2 but reconstituted with a mutant transgene encoding a tyrosine-to-alanine mutation at position 16 of the insulin B chain (9) to generate A22Cα−/−PI2−/− mice, A22Cα−/−PI1−/− mice, and A22Cα−/−PI1−/−PI2−/−Y16Atg mice, respectively (Table 1). Mice were housed in microisolators or scantainers in the specific pathogen–free facility at Cardiff University. All procedures were performed in accordance with protocols approved by the U.K. Home Office.
Flow Cytometric Analysis of TCRVβ Expression
Thymus, spleen, pancreatic lymph nodes (PLNs), and mesenteric lymph nodes (MLNs) were harvested from mice aged 4–7, 8–10, and 12–16 weeks. Single-cell suspensions were generated and stained with monoclonal antibodies (mAbs) specific for CD4, CD8α, CD19, and 14 distinct TCRVβ chains. Live single CD8+ T cells were then gated to visualize TCRVβ expression patterns. Data were analyzed with FlowJo software version 7.6.5 (Tree Star Inc.).
Tetramer Analysis
Thymus, spleen, PLNs, and MLNs were harvested from TCRα transgenic mice and polyclonal NOD mice at 4–8 weeks of age. Single-cell suspensions were generated and stained serially with pretitrated concentrations of the H-2Kd-LYLVCGERG tetramer (National Institutes of Health Tetramer Core Facility), a viability dye, and the following mAbs: αCD4-PECy7, αCD8α-FITC, αCD11b-APC, and αCD19-PerCPCy5.5. Data were analyzed with FlowJo software version 7.6.5. G9C8 transgenic T cells (29) were used as a positive control. The minimal H-2Kd-AYAAAAAAV tetramer was used to determine nonspecific background.
TRBV19+CD8+ T-Cell Isolation and Expansion
Single-cell PLN suspensions from 6-week-old A22Cα−/−, A22Cα−/−PI2−/−, and A22Cα−/−PI1−/−PI2−/−Y16Atg mice were sorted by flow cytometry for live CD4−CD8+CD11b−CD19−TCRVβ6+ events. Cells were initially cultured in bulk with the addition of interleukin (IL)-2 (20 units/mL), IL-7 (2 ng/mL), and insulin B15-23 peptide (1 μg/mL) in RPMI complete medium (2 mmol/L l-glutamine, 100 units/mL penicillin, 0.1 mg/mL streptomycin, 5% FBS, and 0.05 mmol/L 2-mercaptoethanol in RPMI medium). Expanding cells were replated after limiting dilution and grown to sufficient numbers for functional analysis.
Cytotoxicity Assays
Expanded TRBV19+CD8+ T cells were washed twice in RPMI complete medium to remove cytokines from the culture. P815 cells (targets) were labeled with PKH-26 (Sigma) and incubated with TRBV19+CD8+ T cells (effectors) in the presence of insulin B15-23 peptide at an effector-to-target ratio of 10:1 for 16 h at 37°C. TO-PRO-3 iodide was added immediately before flow cytometric analysis. Single PKH-26+TO-PRO-3+ P815 cells were gated, and cytotoxicity was expressed as the percentage of dead cells/total targets (30). Specific target cell killing was corrected for spontaneous background death by subtracting the percentage of dead cells in the control sample comprising targets with T cells in the absence of peptide.
ELISAs
TRBV19+CD8+ T cells were cocultured with irradiated bone marrow–derived dendritic cells in the presence of insulin B15-23 peptide at an effector-to-target ratio of 10:1 for 48 h at 37°C. Supernatants were analyzed for macrophage inflammatory protein-1β (MIP1β) and interferon-γ (IFNγ) levels according to the manufacturer’s instructions (R&D Systems and eBioscience, respectively).
TCR Clonotyping
Clonotypic analysis of H-2Kd-LYLVCGERG tetramer-positive CD8+ T cells and oligoclonal lines was performed as described previously (31). Briefly, viable tetramer-positive CD8+ T cells (n = 43–2,353) from individual mice or TRBV19+CD8+ T cells (n = 5,000) from individual oligoclonal lines were sorted directly into 1.5-mL microtubes (Sarstedt) containing 100 μL RNAlater (Applied Biosystems) using a custom-modified FACSAria II flow cytometer (BD Biosciences). Unbiased amplification of all expressed TRBV gene products was conducted using a template-switch anchored RT-PCR with a 3′ constant region primer (5′-TGGCTCAAACAAGGAGACCT-3′). Amplicons were subcloned, sampled, sequenced, and analyzed as described previously (32). Concatenated data are shown for each genetic strain. The IMGT nomenclature is used in this report (33).
Histology
Immunohistochemistry was performed on fixed pancreata as described previously (29). Sections were scored for insulitis after staining with hematoxylin-eosin (Vector Laboratories).
Statistics
Statistical analyses were conducted using ANOVA or a Student t test with R software. Bonferroni correction was used for multiple comparisons such that P < 0.05 is equivalent to P < 0.0000022907 and P < 0.01 is equivalent to P < 0.0000004581.
Results
CD4+ and CD8+ T-Cell Numbers Are Not Affected by PI Expression
In preliminary experiments, we used flow cytometry to assess the impact of PI expression on T-cell lineage development in our mouse models (A22Cα−/−, A22Cα−/−PI2tg, A22Cα−/−PI2−/−, A22Cα−/−PI1−/−, and A22Cα−/−PI1−/−PI2−/−Y16Atg). Groups of mice were screened at 4–7 weeks and 12–16 weeks. No significant differences in CD4+ or CD8+ T-cell numbers were apparent among strains, regardless of age, either in the thymus (Fig. 1) or in the periphery (Supplementary Fig. 1). However, a substantial reduction in the total number of CD4+ T cells was observed for all TCRα-chain transgenic strains relative to polyclonal NOD mice (Fig. 1 and Supplementary Fig. 1).
Figure 1.

CD4+ and CD8+ T-cell numbers in the thymus are unaffected by PI expression. Live single-positive CD4+ and CD8+ T cells were quantified in single-cell suspensions of thymus from mice aged 4–7 or 12–16 weeks. Representative flow cytometry plots (A), total CD4+ T-cell numbers (B), and total CD8+ T-cell numbers (C) are displayed for all strains. Data are shown for 9–12 mice per group (encompassing three to four experiments). Males and females were included because no differences were detected when analyzed separately. **P < 0.01.
PI Expression Alters the Proportion of TRBV19+CD8+ T Cells
To evaluate the global influence of PI expression on the CD8+ T-cell repertoire, 14 TCRVβ-specific mAbs were used to assess TRBV usage by CD8+ T cells isolated from the thymus, spleen, and lymph nodes of the various A22Cα−/− mice aged either 4–7 weeks or 12–16 weeks. A strong PI-mediated effect was observed on the expression of TRBV19 (Vβ6), which encodes the TCRVβ chain used by the G9C8 clone (Fig. 2 and Supplementary Fig. 2). Specifically, the proportion of TRBV19+CD8+ T cells, both in the thymus and in the periphery, decreased with age in A22Cα−/− mice (Fig. 2). This effect was more pronounced in mice with PI2 overexpression (A22Cα−/−PI2tg), which also displayed lower proportions of TRBV19+CD8+ T cells compared with A22Cα−/− mice, notably approximating those observed in polyclonal NOD mice. However, mice lacking PI1 (A22Cα−/−PI1−/−), PI2 (A22Cα−/−PI2−/−), or both PI genes (A22Cα−/−PI1−/−PI2−/−Y16Atg) showed no significant changes in the proportion of TRBV19+CD8+ T cells with age. Similar trends were observed with respect to the absolute number of TRBV19+CD8+ T cells, which declined with age but differed among strains only in PLNs at 12–16 weeks, where a substantial reduction was observed in A22Cα−/−PI2tg mice (Supplementary Fig. 2). Less marked effects were noted with other TRBV segments. In particular, the proportion of CD8+ T cells expressing TRBV13 (Vβ8), the most commonly used gene segment in NOD mice, was unaffected by PI expression (data not shown).
Figure 2.

PI-specific and age-related effects on TRBV19 expression. CD8+ T cells expressing TCRVβ6 (TRBV19) were quantified in single-cell suspensions of thymus (A and B) and PLNs (C and D) from mice aged 4–7 (A and C) or 12–16 (B and D) weeks. Data are shown for 9–12 mice per group (encompassing three to four experiments). Males and females were included because no differences were detected when analyzed separately. *P < 0.05, **P < 0.01.
Insulin B15-23–Reactive CD8+ T Cells Are Increased in Mice Lacking PI1 or PI2
As the TRAV8-1/TRAJ9 chain present in these mice specifically recognizes the insulin B15-23 epitope, we used H-2Kd-LYLVCGERG tetramers to quantify the effect of PI expression on the development of insulin B15-23–reactive CD8+ T cells (Supplementary Fig. 3A). Increased numbers and percentages of tetramer-positive CD8+ T cells were detected in the thymus, spleen, and PLNs of PI2-deficient (A22Cα−/−PI2−/−) mice compared with wild-type A22Cα−/− mice (Fig. 3 and Supplementary Fig. 3). Of note, A22Cα−/−PI1−/− mice also showed an increased proportion of insulin B15-23–reactive CD8+ T cells in PLNs compared with A22Cα−/− mice, whereas a smaller increase was observed in PLNs from A22Cα−/−PI1−/−PI2−/−Y16Atg mice despite greater enhancements in the thymus and spleen. However, only the A22Cα−/−PI2−/− and A22Cα−/−PI1−/−PI2−/−Y16Atg strains exhibited an increase in the number of insulin B15-23–reactive CD8+ T cells in both the thymus and the periphery (Supplementary Fig. 3B). No differences in tetramer staining intensity were detected among strains, indicating equivalent levels of cognate TCR expression (Supplementary Fig. 3C).
Figure 3.

PI2 deficiency increases the proportion of insulin B15-23–reactive CD8+ T cells. H-2Kd-LYLVCGERG tetramer-positive CD8+ T cells were enumerated in single-cell suspensions of thymus, spleen, PLNs, and MLNs from mice aged 4–8 weeks. Nonspecific staining was quantified in parallel experiments with the minimal H-2Kd-AYAAAAAAV tetramer. Data are shown after background subtraction for 5–10 mice per group. Males and females were included because no differences were detected when analyzed separately. *P < 0.05, **P < 0.01.
Insulin B15-23–Reactive CD8+ T Cells Cause Spontaneous Diabetes in PI2-Deficient Mice
To assess the biological relevance of these proportional and numerical differences in insulin B15-23–reactive CD8+ T cells, we examined the incidence of spontaneous diabetes across strains. Strikingly, diabetes occurred spontaneously only in male PI2-deficient (A22Cα−/−PI2−/−) mice (Fig. 4). Moreover, disease onset was considerably accelerated in this group compared with male wild-type NOD mice, which develop diabetes after 20 weeks of age in our colony, with a final incidence of 20% by 35 weeks (data not shown). Mild insulitis was detected in male A22Cα−/− and nondiabetic A22Cα−/−PI2−/− mice, whereas normal pancreatic histology was observed in male A22Cα−/−PI1−/−PI2−/−Y16Atg mice (Supplementary Table 1). These findings suggest that PI2 deficiency permits the development of diabetogenic insulin B15-23–reactive CD8+ T cells.
Figure 4.

Mice deficient in PI2 develop spontaneous diabetes. Mice were housed together from weaning and tested weekly for glycosuria using Diastix. Positive results were validated 24 h later, and diabetes was confirmed by a blood glucose level >13.9 mmol/L. Only males were included because no females developed diabetes. *P < 0.05.
Insulin B15-23–Reactive CD8+ T Cells Express Multiple TRBV Chains and Exhibit PI-Specific Changes in TRBV19 Usage
To further our understanding of the diabetogenic process, we evaluated the TCR repertoire specifically within the insulin B15-23–reactive CD8+ T-cell population. Data are shown for wild-type mice (A22Cα−/−), PI2 overexpressing mice (A22Cα−/−PI2tg), PI2-deficient mice (A22Cα−/−PI2−/−), and mice lacking both PI1 and PI2 (A22Cα−/−PI1−/−PI2−/−Y16Atg). By sequencing expressed TRB gene transcripts in H-2Kd-LYLVCGERG tetramer-positive CD8+ T cells from PLNs, we identified a restricted TCR repertoire in all four groups of mice (Fig. 5). Alterations in PI expression clearly influenced TRBV gene selection in insulin B15-23–reactive CD8+ T cells. Most notably, the proportion of TRBV19+CD8+ T cells increased when PI2 or both PI1 and PI2 were absent. Moreover, no TRBV19 sequences were detected when PI2 was overexpressed. These observations suggest that PI expression critically affects the selection of insulin B15-23–reactive TCRs, even in the context of a highly restricted clonotypic repertoire.
Figure 5.

The TCRVβ repertoire of insulin B15-23–reactive CD8+ T cells. H-2Kd-LYLVCGERG tetramer-positive CD8+ T cells were sorted by flow cytometry from single-cell suspensions of PLNs from mice aged 4–8 weeks and analyzed for TRB gene expression. Data are shown for five to seven mice per group, comprising 6–20 distinct clonotypes.
Insulin B15-23–Reactive TRBV19+CD8+ T Cells Exhibit Low Functional Sensitivity Irrespective of PI Expression During Development
As the selection of insulin B15-23–reactive TRBV19+CD8+ T cells was influenced by PI expression, we next investigated the role of antigen exposure as a determinant of cellular function. TRBV19+CD8+ T cells from wild-type (A22Cα−/−), PI2-deficient (A22Cα−/−PI2−/−), or PI1 and PI2–deficient (A22Cα−/−PI1−/−PI2−/−Y16Atg) mice were isolated and cultured briefly with the cognate peptide in the presence of IL-2 and IL-7 to generate a panel of insulin B15-23–reactive oligoclonal T-cell lines. The functional profile of these lines was assayed by measuring cytotoxicity and proinflammatory cytokine (MIP1β and IFNγ) production in response to the insulin B15-23 peptide. Similar cytotoxic responses were observed for all strains regardless of PI expression, and target cell lysis required high doses of exogenous peptide (Fig. 6A). Lines generated from A22Cα−/−PI1−/−PI2−/−Y16Atg mice displayed enhanced MIP1β responses (Fig. 6B) but produced less IFNγ in parallel comparisons with wild-type (Fig. 6C). Overall, these results suggest that insulin B15-23–reactive TRBV19+CD8+ T cells exhibit intrinsically low levels of functional sensitivity and that altered reactivity to cognate antigen does not underlie the development of spontaneous diabetes in PI2-deficient (A22Cα−/−PI2−/−) mice.
Figure 6.

TRBV19+CD8+ T cells exhibit low levels of functional sensitivity to insulin B15-23. Oligoclonal TRBV19+CD8+ T-cell lines expanded in the presence of insulin B15-23 were analyzed for cytotoxic activity (A), MIP1β (B), and IFNγ (C) production. Data are shown for representative clones (A22Cα−/−: 10F12; A22Cα−/−PI2−/−: 30A6; A22Cα−/−PI1−/−PI2−/−Y16Atg: 1G3).
Insulin B15-23–Reactive TRBV19+CD8+ T Cells Use TRBJ2-3 and Exhibit a Common TCR CDR3β Motif
The highly diabetogenic G9C8 clone expresses a TRBV19/TRBJ2-3 gene-encoded TCRβ chain (22). Although no obvious differences in functional sensitivity were observed between insulin B15-23–reactive CD8+ T-cell clonotypes selected in mice with differing levels of PI expression, we nonetheless conducted a molecular analysis of TRB gene rearrangements in our oligoclonal antigen-specific lines to assess the impact of PI expression on TCR selection. All lines were dominated by TRBV19+ TCRs. We obtained 11 unique TRBV19-associated CDR3β sequences, five of which were private and six of which were shared (Table 2). Ten of these distinct CDR3β sequences incorporated TRBJ2-3 with a fixed loop length of 13 amino acids generating a motif (CASS-XXXX-GAETLY) in common with the original G9C8 clone (CASS-IRDR-GAETLY). Moreover, arginine was universally conserved at position 6 in these CDR3β sequences, likely providing an important contact residue for recognition of the insulin B15-23 peptide. Amino acids with nonpolar side chains were preferred at position 5 (with the exception of arginine), whereas amino acids with acidic side chains (aspartic acid or glutamic acid) or uncharged polar side chains (glutamine or threonine) were preferred at position 7. Little preference was observed for any particular residue at position 8. In contrast, we found very limited sequence similarity across the CDR3β loop within the tetramer-positive CD8+ T-cell population as a whole (Fig. 3). One common CDR3β sequence (CASSLGGYEQY) was found in A22Cα−/− and A22Cα−/−PI2−/− mice, and another (CASSRVPGEQY) was found in more than one A22Cα−/− mouse (Supplementary Table 2). It is also notable that the CASS-XXXX-GAETLY motif was not detected in a parallel analysis of noninsulin B15-23–reactive TRBV19+CD8+ T cells (data not shown). Collectively, these findings demonstrate that insulin B15-23 elicits a highly biased TRBV19+ repertoire consistent with a strict docking mode for TCR recognition dictated by structural constraints.
Table 2.
CDR3β sequences of insulin B15-23–reactive oligoclonal lines
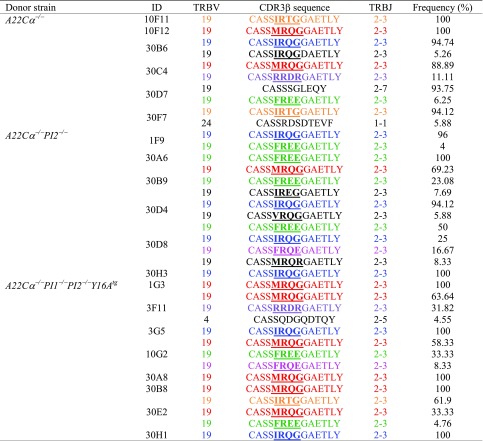 |
Colored sequences represent those found in more than one mouse. Recurrent motifs incorporating a non-germline–encoded arginine residue are underlined in bold.
Discussion
In this study, we report five principal findings based on an in-depth analysis of fixed TCRα-chain NOD mice. First, PI1 or PI2 deficiency is associated with significant expansions of insulin B15-23–reactive CD8+ T cells in the draining lymph node for the pancreas, the target organ in type 1 diabetes. Second, the proportion of insulin B15-23–reactive CD8+ T cells expressing TRBV19 increases in the absence of PI. This repertoire shift potentially contributes to the development of autoimmune diabetes. Third, variations in PI expression alter global TRBV gene usage in the CD8+ T-cell compartment. In particular, TRBV19+CD8+ T-cell numbers were reduced significantly upon increased exposure to PI2 through transgenic overexpression, although deletion of either the PI1 or PI2 gene in isolation did not change the frequency of this population. Fourth, insulin B15-23–reactive TRBV19+CD8+ T cells display low levels of functional sensitivity for the cognate peptide irrespective of PI expression, consistent with a lack of epitope-regulated negative selection in the thymus. Fifth, antigen recognition by insulin B15-23–reactive TRBV19+CD8+ T cells is driven by a highly biased set of TCRs characterized by TRBJ2-3 gene usage and the presence of a conserved non–germline-encoded arginine residue at position 6 in the CDR3β loop. Collectively, these data show for the first time that PI expression can directly affect the development of autoreactive TCRs.
PI2 is expressed in the thymus and has been shown to promote tolerance (17,20,21,34). As a consequence, NOD mice with PI2 deficiency develop accelerated diabetes (19). However, NOD mice lacking PI1 do not develop autoimmune diabetes, presumably because PI2 tolerizes potentially autoreactive T cells (18). Although PI1 is also expressed in the thymus, PI2 is the predominant isoform; equivalent levels of PI1 and PI2 are found in the periphery (17,35,36). It is notable in this regard that PI deficiency enhanced the development of TRBV19+CD8+ T cells in the thymus and minimized corresponding age-related declines in the periphery. These data suggest that both PI1 and PI2 have tolerogenic properties, with the weak effects on disease likely related to the low frequency of insulin B15-23–reactive cells in the overall TRBV19+CD8+ T-cell population.
Antigen encounter in the thymus preferentially deletes high-avidity T cells (37,38). In accordance with this process, the current data show that increased numbers of insulin B15-23–reactive CD8+ T cells are present in mice lacking either PI2 alone or both PI1 and PI2. Moreover, only PI2-deficient mice developed diabetes in this study. It is remarkable that male mice were exclusively affected in this regard, despite equivalent increases in insulin-reactive CD8+ T cells in female mice lacking PI2. Recent data suggest that nonimmune factors, including interactions between gut microbiota and androgens (39,40), contribute to such sexual dimorphism in the development of diabetes. Irrespective of the underlying mechanism, however, these findings suggest that insulin B15-23–reactive CD8+ T cells are important determinants of disease because A22Cα−/−PI1−/−PI2−/−Y16Atg mice express a mutant transgene that prevents recognition of the cognate peptide.
It is notable that we did not detect any significant functional differences in insulin B15-23–reactive TRBV19+CD8+ T cells across strains of mice with altered levels of PI expression. This counterintuitive finding may reflect in vitro activation and expansion prior to assay, which could mask intrinsic differences in the ex vivo setting, or a convergence of functional properties due to the expression of PI in peripheral lymphoid tissue (13,41). Regardless of origin, these autoreactive cells expressed similar TRBV19/TRBJ2-3 transcripts incorporating a CDR3β motif based around a central arginine residue. Arginine is degenerately encoded at the nucleotide level and readily incorporated on a probabilistic basis during the somatic recombination process (42). It can also play a key role in TCR recognition (43). The current data therefore suggest a highly conserved mode of antigen engagement focused on the G9C8 blueprint.
Several studies have previously identified a tolerogenic role for PI2 (17,19–21). In contrast, the role of PI1 has not been fully established. The only reported investigation in NOD mice found that PI1 deficiency protected against the development of diabetes (18). On a nonautoimmune background (129/SV), mice lacking PI2 showed increased PI1 gene transcripts as well as enhanced β-cell mass to compensate for lower insulin production, indicating the importance of PI1 in metabolism (44). Moreover, the PI2 B9-23 peptide can induce proliferation of PI1-reactive NOD T cells, and the PI1 B9-23 and C49-66 peptides can induce proliferation of PI2-reactive NOD T cells (21). Such cross-reactivity suggests that both isoforms can facilitate the expansion of PI-specific populations in the periphery. Further studies are therefore required to assess the relative contributions of PI1 and PI2 as determinants of tolerance and diabetogenicity.
Although the current model system addresses a single specificity, the epitope nonetheless derives from insulin, which is known to be an important early antigenic target in type 1 diabetes. Previous work by other investigators has focused on IGRP-specific NY8.3 CD8+ T cells (45–47). However, fundamental differences exist between these two diabetogenic epitopes. Most notably, IGRP is not expressed in the thymus (1). In contrast, studies of PI facilitate an understanding of repertoire development in the presence of an autoantigen that is naturally expressed both in the thymus and in the periphery. Moreover, IGRP reactivity depends on an autoimmune response to insulin (24,25). In humans, the key genetic susceptibility region IDDM2 relates to the level of PI expression. The current study is therefore informative because it describes the impact of variable PI expression on clonotype selection in response to a defined and biologically relevant autoimmune epitope in type 1 diabetes.
Two important conclusions emerge from the present data. First, PI expression shapes the insulin-reactive CD8+ T-cell repertoire in a mouse model of type 1 diabetes. Second, an avidity threshold exists below which insulin-reactive CD8+ T cells are less affected by antigen exposure in the thymus compared with the periphery. These findings have potential implications for the induction of antigen-specific tolerance as a therapeutic strategy against autoimmune diabetes.
Supplementary Material
Article Information
Acknowledgments. The authors thank the National Institutes of Health Tetramer Core Facility for provision of the H-2Kd-LYLVCGERG tetramer and various control tetramers. L.C. Harrison and A. Lew (Walter and Eliza Hall Research Institute, Melbourne, VIC, Australia) provided NOD mice overexpressing PI2 (NODPI2tg) under the MHC class II promoter before they were available commercially.
Funding. This work was supported by the Medical Research Council (grant G0901155) and the Wellcome Trust (grant 100326Z/12/Z). J.A.P. was the recipient of a Diabetes UK Studentship (08/3767). D.A.P. is a Wellcome Trust Senior Investigator.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. J.A.P. contributed to the experiments, data analysis, and writing and editing of the manuscript. T.C.T. and K.L. contributed to the experiments and editing of the manuscript. J.E.M. contributed to the experiments, data analysis, and editing of the manuscript. E.D.L., A.P., J.D., D.K., and K.M. contributed to the experiments. P.M. contributed to the data analysis. L.W. contributed to the data analysis and editing of the manuscript. D.A.P. contributed to the data analysis and writing and editing of the manuscript. F.S.W. contributed to the study concept, data analysis, and writing and editing of the manuscript. F.S.W. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 13th International Congress of the Immunology of Diabetes Society, Lorne, VIC, Australia, 7–11 December 2013; the Diabetes UK Annual Professional Conference, Manchester, U.K., 13–15 March 2013, and Liverpool, U.K., 5–7 March 2014; and the British Society for Immunology Annual Congress, Brighton, U.K., 1–4 December 2014.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db15-1498/-/DC1.
References
- 1.Gardner JM, Devoss JJ, Friedman RS, et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 2008;321:843–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schloot NC, Willemen S, Duinkerken G, de Vries RR, Roep BO. Cloned T cells from a recent onset IDDM patient reactive with insulin B-chain. J Autoimmun 1998;11:169–175 [DOI] [PubMed] [Google Scholar]
- 3.Semana G, Gausling R, Jackson RA, Hafler DA. T cell autoreactivity to proinsulin epitopes in diabetic patients and healthy subjects. J Autoimmun 1999;12:259–267 [DOI] [PubMed] [Google Scholar]
- 4.Skowera A, Ellis RJ, Varela-Calviño R, et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope [published correction appears in J Clin Invest 2009;119:2844]. J Clin Invest 2008;118:3390–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulek AM, Cole DK, Skowera A, et al. Structural basis for the killing of human beta cells by CD8(+) T cells in type 1 diabetes. Nat Immunol 2012;13:283–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wegmann DR, Gill RG, Norbury-Glaser M, Schloot N, Daniel D. Analysis of the spontaneous T cell response to insulin in NOD mice. J Autoimmun 1994;7:833–843 [DOI] [PubMed] [Google Scholar]
- 7.Zekzer D, Wong FS, Wen L, et al. Inhibition of diabetes by an insulin-reactive CD4 T-cell clone in the nonobese diabetic mouse. Diabetes 1997;46:1124–1132 [DOI] [PubMed] [Google Scholar]
- 8.Wong FS, Karttunen J, Dumont C, et al. Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat Med 1999;5:1026–1031 [DOI] [PubMed] [Google Scholar]
- 9.Nakayama M, Abiru N, Moriyama H, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005;435:220–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamont D, Mukherjee G, Kumar PR, et al. Compensatory mechanisms allow undersized anchor-deficient class I MHC ligands to mediate pathogenic autoreactive T cell responses. J Immunol 2014;193:2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol 2001;2:1032–1039 [DOI] [PubMed] [Google Scholar]
- 12.Pugliese A, Zeller M, Fernandez A Jr, et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet 1997;15:293–297 [DOI] [PubMed] [Google Scholar]
- 13.Anderson MS, Venanzi ES, Klein L, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002;298:1395–1401 [DOI] [PubMed] [Google Scholar]
- 14.Nejentsev S, Howson JM, Walker NM, et al.; Wellcome Trust Case Control Consortium . Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 2007;450:887–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vafiadis P, Bennett ST, Todd JA, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet 1997;15:289–292 [DOI] [PubMed] [Google Scholar]
- 16.Deltour L, Leduque P, Blume N, et al. Differential expression of the two nonallelic proinsulin genes in the developing mouse embryo. Proc Natl Acad Sci U S A 1993;90:527–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chentoufi AA, Polychronakos C. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes 2002;51:1383–1390 [DOI] [PubMed] [Google Scholar]
- 18.Moriyama H, Abiru N, Paronen J, et al. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci U S A 2003;100:10376–10381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thébault-Baumont K, Dubois-Laforgue D, Krief P, et al. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD mice. J Clin Invest 2003;111:851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.French MB, Allison J, Cram DS, et al. Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. Diabetes 1997;46:34–39 [DOI] [PubMed] [Google Scholar]
- 21.Jaeckel E, Lipes MA, von Boehmer H. Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1 diabetes. Nat Immunol 2004;5:1028–1035 [DOI] [PubMed] [Google Scholar]
- 22.Wong FS, Visintin I, Wen L, Flavell RA, Janeway CA Jr. CD8 T cell clones from young nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in NOD mice in the absence of CD4 cells. J Exp Med 1996;183:67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniel D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B-(9-23). Proc Natl Acad Sci U S A 1996;93:956–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnamurthy B, Dudek NL, McKenzie MD, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest 2006;116:3258–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krishnamurthy B, Mariana L, Gellert SA, et al. Autoimmunity to both proinsulin and IGRP is required for diabetes in nonobese diabetic 8.3 TCR transgenic mice. J Immunol 2008;180:4458–4464 [DOI] [PubMed] [Google Scholar]
- 26.Trudeau JD, Kelly-Smith C, Verchere CB, et al. Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest 2003;111:217–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong FS, Moustakas AK, Wen L, Papadopoulos GK, Janeway CA Jr. Analysis of structure and function relationships of an autoantigenic peptide of insulin bound to H-2K(d) that stimulates CD8 T cells in insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A 2002;99:5551–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Motozono C, Pearson JA, De Leenheer E, et al. Distortion of the MHC class I binding groove to accommodate an insulin-derived 10-mer peptide. J Biol Chem 2015;290:18924–18933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong FS, Siew LK, Scott G, et al. Activation of insulin-reactive CD8 T-cells for development of autoimmune diabetes. Diabetes 2009;58:1156–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott GS, Fishman S, Khai Siew L, et al. Immunotargeting of insulin reactive CD8 T cells to prevent diabetes. J Autoimmun 2010;35:390–397 [DOI] [PubMed] [Google Scholar]
- 31.Quigley MF, Almeida JR, Price DA, Douek DC. Unbiased molecular analysis of T cell receptor expression using template-switch anchored RT-PCR. Curr Protoc Immunol 2011;Chapter 10:Unit10.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price DA, Brenchley JM, Ruff LE, et al. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med 2005;202:1349–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefranc MP, Pommié C, Ruiz M, et al. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol 2003;27:55–77 [DOI] [PubMed] [Google Scholar]
- 34.Faideau B, Briand JP, Lotton C, et al. Expression of preproinsulin-2 gene shapes the immune response to preproinsulin in normal mice. J Immunol 2004;172:25–33 [DOI] [PubMed] [Google Scholar]
- 35.Chentoufi AA, Palumbo M, Polychronakos C. Proinsulin expression by Hassall’s corpuscles in the mouse thymus. Diabetes 2004;53:354–359 [DOI] [PubMed] [Google Scholar]
- 36.Palumbo MO, Levi D, Chentoufi AA, Polychronakos C. Isolation and characterization of proinsulin-producing medullary thymic epithelial cell clones. Diabetes 2006;55:2595–2601 [DOI] [PubMed] [Google Scholar]
- 37.Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity 2006;25:261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Enouz S, Carrié L, Merkler D, Bevan MJ, Zehn D. Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. J Exp Med 2012;209:1769–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markle JG, Frank DN, Mortin-Toth S, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013;339:1084–1088 [DOI] [PubMed] [Google Scholar]
- 40.Yurkovetskiy L, Burrows M, Khan AA, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity 2013;39:400–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JW, Epardaud M, Sun J, et al. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol 2007;8:181–190 [DOI] [PubMed] [Google Scholar]
- 42.Venturi V, Price DA, Douek DC, Davenport MP. The molecular basis for public T-cell responses? Nat Rev Immunol 2008;8:231–238 [DOI] [PubMed] [Google Scholar]
- 43.Stewart-Jones GB, McMichael AJ, Bell JI, Stuart DI, Jones EY. A structural basis for immunodominant human T cell receptor recognition. Nat Immunol 2003;4:657–663 [DOI] [PubMed] [Google Scholar]
- 44.Leroux L, Desbois P, Lamotte L, et al. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes 2001;50(Suppl. 1):S150–S153 [DOI] [PubMed] [Google Scholar]
- 45.Verdaguer J, Yoon JW, Anderson B, et al. Acceleration of spontaneous diabetes in TCR-beta-transgenic nonobese diabetic mice by beta-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-alpha chains. J Immunol 1996;157:4726–4735 [PubMed] [Google Scholar]
- 46.Amrani A, Verdaguer J, Serra P, Tafuro S, Tan R, Santamaria P. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature 2000;406:739–742 [DOI] [PubMed] [Google Scholar]
- 47.Lieberman SM, Evans AM, Han B, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci U S A 2003;100:8384–8388 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.