

Abstract
Aspartylglucosaminuria (AGU) is an inherited lysosomal storage disorder caused by the deficiency of aspartylglucosaminidase. We have earlier reported a single missense mutation (Cys163----Ser) to be responsible for 98% of the AGU alleles in the isolated Finnish population, which contains about 90% of the reported AGU patients. Here we describe the spectrum of 10 AGU mutations found in unrelated patients of non-Finnish origin. Since 11 out of 12 AGU patients were homozygotes, consanguinity has to be a common denominator in most AGU families. The mutations were distributed over the entire coding region of the aspartylglucosaminidase cDNA, except in the carboxyl-terminal 17-kDa subunit in which they were clustered within a 46-amino acid region. Based on the character of the mutations, most of them are prone to affect the folding and stability and not to directly affect the active site of the aspartylglucosaminidase enzyme.
Full text
PDF


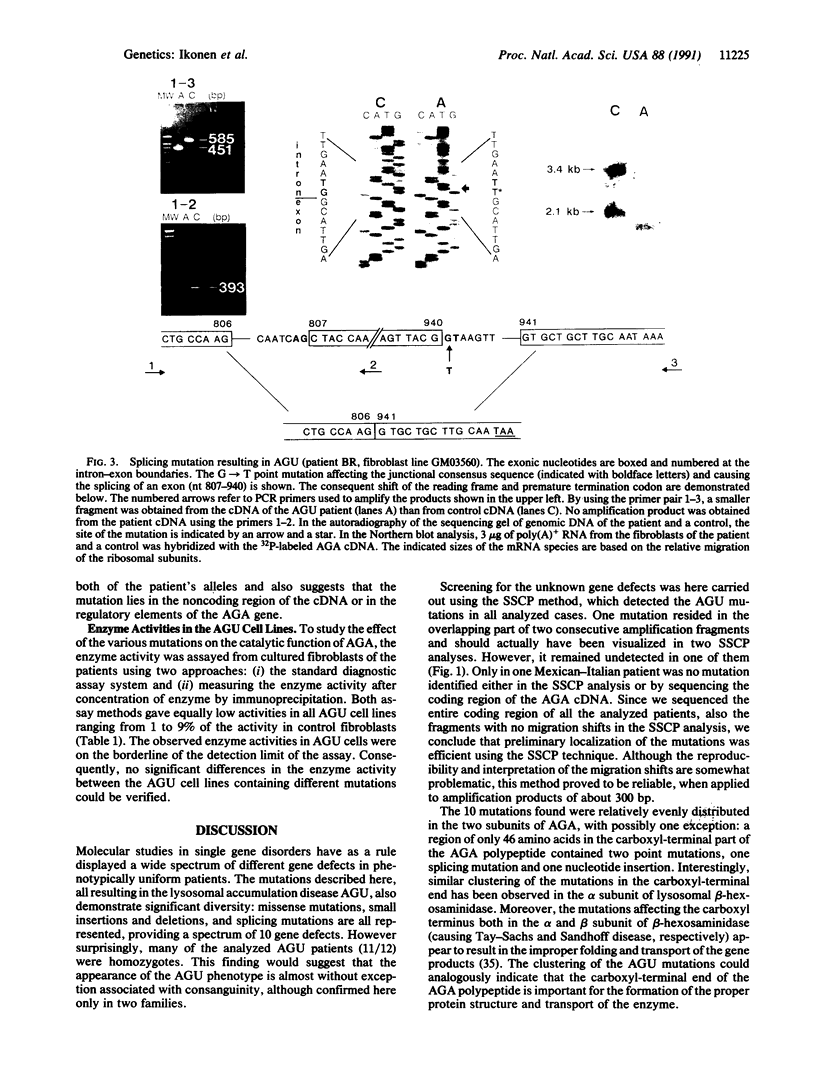

Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Aula P., Mattila K., Piiroinen O., Ammälä P., Von Koskull H. First-trimester prenatal diagnosis of aspartylglucosaminuria. Prenat Diagn. 1989 Sep;9(9):617–620. doi: 10.1002/pd.1970090904. [DOI] [PubMed] [Google Scholar]
- Aula P., Renlund M., Raivio K. O., Koskela S. L. Screening of inherited oligosaccharidurias among mentally retarded patients in northern Finland. J Ment Defic Res. 1986 Dec;30(Pt 4):365–368. doi: 10.1111/j.1365-2788.1986.tb01332.x. [DOI] [PubMed] [Google Scholar]
- Borud O., Torp K. H. Letter: Aspartylglycosaminuria in Northern Norway. Lancet. 1976 May 15;1(7968):1082–1083. doi: 10.1016/s0140-6736(76)92266-2. [DOI] [PubMed] [Google Scholar]
- Casanova J. L., Pannetier C., Jaulin C., Kourilsky P. Optimal conditions for directly sequencing double-stranded PCR products with sequenase. Nucleic Acids Res. 1990 Jul 11;18(13):4028–4028. doi: 10.1093/nar/18.13.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirgwin J. M., Przybyla A. E., MacDonald R. J., Rutter W. J. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979 Nov 27;18(24):5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Chitayat D., Nakagawa S., Marion R. W., Sachs G. S., Hahm S. Y., Goldman H. S. Aspartylglucosaminuria in a Puerto Rican family: additional features of a panethnic disorder. Am J Med Genet. 1988 Nov;31(3):527–532. doi: 10.1002/ajmg.1320310307. [DOI] [PubMed] [Google Scholar]
- Dugal B. Effect of different compounds on 1-aspartamido-beta-N-acetylglucosamine amidohydrolase from human liver. Biochem J. 1978 Jun 1;171(3):799–802. doi: 10.1042/bj1710799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugal B., Stromme J. Purification and some properties of 1-aspartamido-beta-N-acetylglucosamine amidohydrolase from human liver. Biochem J. 1977 Sep 1;165(3):497–502. doi: 10.1042/bj1650497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher K. J., Aronson N. N., Jr Characterization of the mutation responsible for aspartylglucosaminuria in three Finnish patients. Amino acid substitution Cys163----Ser abolishes the activity of lysosomal glycosylasparaginase and its conversion into subunits. J Biol Chem. 1991 Jun 25;266(18):12105–12113. [PubMed] [Google Scholar]
- Garnier J., Osguthorpe D. J., Robson B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol. 1978 Mar 25;120(1):97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- Gehler J., Sewell A. C., Becker C., Hartmann J., Spranger J. Clinical and biochemical delineation of aspartyl-glycosaminuria as observed in two members of an Italian family. Helv Paediatr Acta. 1981;36(2):179–189. [PubMed] [Google Scholar]
- Halila R., Baumann M., Ikonen E., Enomaa N., Peltonen L. Human leucocyte aspartylglucosaminidase. Evidence for two different subunits in a more complex native structure. Biochem J. 1991 May 15;276(Pt 1):251–256. doi: 10.1042/bj2760251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hreidarsson S., Thomas G. H., Valle D. L., Stevenson R. E., Taylor H., McCarty J., Coker S. B., Green W. R. Aspartylglucosaminuria in the United States. Clin Genet. 1983 Jun;23(6):427–435. doi: 10.1111/j.1399-0004.1983.tb01977.x. [DOI] [PubMed] [Google Scholar]
- Ikonen E., Baumann M., Grön K., Syvänen A. C., Enomaa N., Halila R., Aula P., Peltonen L. Aspartylglucosaminuria: cDNA encoding human aspartylglucosaminidase and the missense mutation causing the disease. EMBO J. 1991 Jan;10(1):51–58. doi: 10.1002/j.1460-2075.1991.tb07920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E., Enomaa N., Ulmanen I., Peltonen L. In vitro mutagenesis helps to unravel the biological consequences of aspartylglucosaminuria mutation. Genomics. 1991 Sep;11(1):206–211. doi: 10.1016/0888-7543(91)90120-4. [DOI] [PubMed] [Google Scholar]
- Isenberg J. N., Sharp H. L. Aspartylglucosaminuria: psychomotor retardation masquerading as a mucopolysaccharidosis. J Pediatr. 1975 May;86(5):713–717. doi: 10.1016/s0022-3476(75)80355-6. [DOI] [PubMed] [Google Scholar]
- Kaartinen V., Williams J. C., Tomich J., Yates J. R., 3rd, Hood L. E., Mononen I. Glycosaparaginase from human leukocytes. Inactivation and covalent modification with diazo-oxonorvaline. J Biol Chem. 1991 Mar 25;266(9):5860–5869. [PubMed] [Google Scholar]
- Kessler S. W. Use of protein A-bearing staphylococci for the immunoprecipitation and isolation of antigens from cells. Methods Enzymol. 1981;73(Pt B):442–459. doi: 10.1016/0076-6879(81)73084-2. [DOI] [PubMed] [Google Scholar]
- Kyte J., Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982 May 5;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Makino M., Kojima T., Yamashina I. Enzymatic cleavage of glycopeptides. Biochem Biophys Res Commun. 1966 Sep 22;24(6):961–966. doi: 10.1016/0006-291x(66)90344-5. [DOI] [PubMed] [Google Scholar]
- Mononen I., Heisterkamp N., Kaartinen V., Williams J. C., Yates J. R., 3rd, Griffin P. R., Hood L. E., Groffen J. Aspartylglycosaminuria in the Finnish population: identification of two point mutations in the heavy chain of glycoasparaginase. Proc Natl Acad Sci U S A. 1991 Apr 1;88(7):2941–2945. doi: 10.1073/pnas.88.7.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci S., Salvati A., Schiliró G., Salvo G., Di Dio R., Caprari P. Homozygous NADH-methemoglobin reductase and aspartylglucosaminidase deficiencies in a moderately retarded Sicilian child. Am J Med Genet. 1984 Dec;19(4):643–650. doi: 10.1002/ajmg.1320190403. [DOI] [PubMed] [Google Scholar]
- Neufeld E. F. Natural history and inherited disorders of a lysosomal enzyme, beta-hexosaminidase. J Biol Chem. 1989 Jul 5;264(19):10927–10930. [PubMed] [Google Scholar]
- Orita M., Suzuki Y., Sekiya T., Hayashi K. Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics. 1989 Nov;5(4):874–879. doi: 10.1016/0888-7543(89)90129-8. [DOI] [PubMed] [Google Scholar]
- Pakula A. A., Sauer R. T. Genetic analysis of protein stability and function. Annu Rev Genet. 1989;23:289–310. doi: 10.1146/annurev.ge.23.120189.001445. [DOI] [PubMed] [Google Scholar]
- Pollitt R. J., Jenner F. A., Merskey H. Aspartylglycosaminuria. An inborn error of metabolism associated with mental defect. Lancet. 1968 Aug 3;2(7562):253–255. doi: 10.1016/s0140-6736(68)92355-6. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A. R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977 Dec;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvänen A. C., Aalto-Setälä K., Harju L., Kontula K., Söderlund H. A primer-guided nucleotide incorporation assay in the genotyping of apolipoprotein E. Genomics. 1990 Dec;8(4):684–692. doi: 10.1016/0888-7543(90)90255-s. [DOI] [PubMed] [Google Scholar]
- Thornton J. M. Disulphide bridges in globular proteins. J Mol Biol. 1981 Sep 15;151(2):261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- Tollersrud O. K., Aronson N. N., Jr Purification and characterization of rat liver glycosylasparaginase. Biochem J. 1989 May 15;260(1):101–108. doi: 10.1042/bj2600101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler R., Schmidt H., Sewell A. C., Weglage J., von Lengerke J. H., Ullrich K. Aspartylglucosaminurie. Klinische Beschreibung von zwei deutschen Patienten. Monatsschr Kinderheilkd. 1989 Aug;137(8):454–457. [PubMed] [Google Scholar]