

Abstract
Intraperitoneal (IP) chemotherapy is a promising post-surgical therapy of ovarian cancer, but the full potential is yet to be realized. To facilitate IP chemotherapy of ovarian cancer, we developed an in-situ crosslinkable hydrogel depot containing paclitaxel (PTX) nanocrystals (PNC). PNC suppressed SKOV3 cell proliferation more efficiently than microparticulate PTX precipitates (PPT), and the gel containing PNC (PNC-gel) showed a lower maximum tolerated dose than PPT-containing gel (PPT-gel) in mice, indicating greater dissolution and cellular uptake of PNC than PPT. A single IP administration of PNC-gel extended the survival of tumor-bearing mice significantly better than Taxol, but PPT-gel did not. These results support the advantage of PNC over PPT and demonstrate the promise of a gel depot as an IP drug delivery system.
Keywords: Nanocrystals, intraperitoneal chemotherapy, hydrogel depot, drug delivery, ovarian cancer
Graphical Abstract
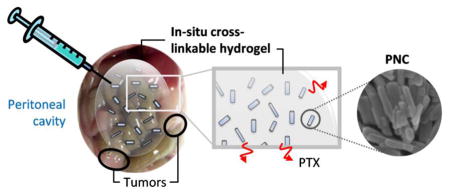
1. Introduction
Peritoneal malignancies are currently managed by surgical debulking of visible tumors and subsequent chemotherapy of residual microscopic tumors [1–3]. Intraperitoneal (IP) chemotherapy has been pursued as a promising post-surgical therapy. A well-formulated IP chemotherapy can maintain an effective local drug concentration for a prolonged period and maximize the locoregional effects on residual tumors [4–9]. IP chemotherapy has proven more effective than intravenous (IV) therapy in several clinical studies [2, 10, 11]. Furthermore, part of the IP-administered drug is slowly absorbed to systemic circulation and provides sustained systemic effects [12–14]. In 2006, the National Cancer Institute issued an official announcement encouraging the clinical use of combined IP and intravenous (IV) chemotherapy for patients with optimally resected stage III ovarian cancer, based on clinical evidence of survival benefits [15]. A recent review of clinical practice in 2006–2012 finds that IP/IV chemotherapy have significantly improved the overall survival as compared with IV chemotherapy alone [16].
Despite the clinically-proven advantages, IP chemotherapy has been met with mixed enthusiasm. Frequently mentioned problems are the complications related to IP infusion, including abdominal pain, intolerance to a high level of drug, and discomfort related to the catheter implantation [17]. We speculate that these problems mainly stem from the difficulty in drug release control. Small molecule drugs such as paclitaxel (PTX) or docetaxel were cleared from the peritoneal cavity in less than a day [18–20]. The short IP retention time requires frequent or continuous dosing, necessitating the use of large volume infusion and indwelling catheters. For IP chemotherapy to provide the anticipated benefits, it is critical that the formulation control the drug release and avoid burst initial release and bulk drug absorption to the systemic circulation.
Considering this need, we previously developed an injectable depot consisting of in-situ crosslinkable hyaluronic acid (HA) gel and micrometer-scale PTX precipitates (PPT) to improve the IP retention of PTX [21]. The gel remained in the peritoneal cavity and maintained a high level of local PTX concentration in mice for 2 weeks. However, in suppressing IP SKOV3 human ovarian cancer xenografts, the PPT-gel was not better than Taxol-gel, which released PTX much faster than PPT-gel. Possible explanations were that the dissolution of PPT was too slow to provide an effective local drug concentration and/or the duration of experiment was too short to observe the benefit of prolonged IP retention of PPT.
In the present study, we aim to overcome this challenge by replacing PPT with nanocrystals (PNC), which can accelerate PTX dissolution from the HA gel. Nanocrystals are crystalline particles of poorly soluble drugs in nanometer range [22]. They are produced by breaking down large particles (“top-down”) or crystallizing from drug solutions (“bottom-up”) with an optional aid of surfactants or polymeric surface stabilizers [22, 23]. Due to the small size and thus, the high surface area to volume ratio, nanocrystals can show greater dissolution rate than larger particles [24, 25]. Here, we produced PNC by anti-solvent and temperature-induced crystallization and evaluated its ability to enhance PTX release in vitro as compared to PPT. PNC was administered IP with the HA gel as an injection medium to mice bearing SKOV3 ovarian tumor xenografts in the peritoneal cavity. Progression of tumor burden after single treatment was monitored up to 14 weeks using non-invasive whole body imaging, and the outcome was compared with those of PPT-gel and Taxol. PNC killed SKOV3 ovarian cancer cells more efficiently than PPT, and the PNC-containing gel outperformed Taxol to a greater extent than PPT-containing gel in extending the survival of tumor-bearing mice. These results support the advantage of PNC over PPT and demonstrate the promise of a gel depot as an IP drug delivery system.
2. Materials and Methods
2.1 Materials
Hyaluronic acid (HA, 20 kDa and 500 kDa) was purchased from Lifecore Biomedical, LLC (Chaska, MN, USA). Paclitaxel (PTX) was a gift of Samyang Biopharm (Seoul, Korea). Carbamazepine was purchased from Enzo life sciences (Plymouth Meeting, PA, USA). Cremophor ELP was a gift from BASF (New York, NY, USA). D-Luciferin potassium salt was purchased from Gold Biotechnology (St. Louis, MO, USA). Geneticin® selective antibiotic (G418 sulfate, 50 mg/mL) was purchased from Life technologies (Grand Island, NY, USA). Cell culture medium and supplements were purchased from Invitrogen (Carlsbad, CA, USA). Micro BCA Protein Assay Kit was purchased from Life technologies (Grand Island, NY, USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2 Preparation and characterization of PTX nanocrystals (PNC)
PNC were prepared according to the published method [26]. Briefly, 4 mg/mL PTX/ethanol solution was added to 20 mL of deionized (DI) water and stirred at 42 rcf for 10 min in a round-bottom flask immerged in a sonication bath filled with ice water. The formed PNC was captured on a 100 nm polycarbonate membrane and resuspended in DI water. Taxol and PTX precipitates (PPT) were prepared as described previously [21]: Taxol was prepared by diluting “Taxol concentrate” (6 mg PTX dissolved in 1 mL of 1:1 mixture of Cremophor ELP and ethanol). PPT was prepared by the addition of PTX-dimethyl sulfoxide (DMSO) solution to DI water without low temperature, vigorous stirring or sonication.
The particle size of PNC was measured with a Zetasizer Nano-ZS90 (Malvern instruments, Westborough, MA, USA). The zeta potential of PNC and PPT were measured with a Zeta sizer Nano-ZS90 in 1 mM phosphate buffer (pH 7.4). The morphology of lyophilized PNC and PPT was visualized with a FEI Nova nanoSEM field emission scanning electron microscopy (Hillsboro, OR, USA). Freeze-dried PNC was sputter-coated with platinum for 1 min and observed with a high resolution through-the-lens detector under 5kV accelerating voltage and spot size 3. PPT suspended in DI water was observed with Axio Imager 2 microscope with polarized light (Carl Zeiss Microscopy GmbH, Germany). X-ray powder diffraction (XPRD) patterns of lyophilized PNC and PPT were obtained on a Siemens D5000 X-ray diffractometer with a Cu Kα radiation source (40kV, 40 mA). The measurement was conducted at room temperature over an angle (2θ) range of 5–40° with a step size of 0.02° and a scan rate of 4°/min.
2.3 Preparation of in-situ crosslinkable HA gel loaded with PPT or PNC (PPT-gel or PNC-gel)
HA gel was prepared by crosslinking HA-adipic acid dihydrazide (HA-ADH) and HA-aldehyde (HA-CHO) in situ as described previously [21, 27]. HA-ADH was synthesized by conjugating adipic dihydrazide to carboxyl groups in HA via 1-ethyl-3-carbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt) at pH 6.8 and room temperature. HA-CHO was produced by oxidizing HA with sodium periodate. HA-ADH and HA-CHO were purified by dialysis, freeze dried and stored at 4 °C until use. PNC- or PPT-gel was prepared by suspending PNC or PPT in solutions of these HA derivatives in phosphate buffered saline (PBS, pH 7.4) and extruding them through a common outlet using a double-barreled syringe. HA concentration in the gel was 40 mg/mL.
2.4 Dissolution kinetics of PPT and PNC
Dissolution kinetics of PPT and PNC was tested in 0.05% or 0.2% PBST following a method described by Peschka et al., which used an agarose hydrogel to contain drug-encapsulated liposomes, thereby separating them from receptor medium [28]. In this study, we replaced agarose gel with photocrosslinkable polyethyleneglycol (PEG) hydrogel to avoid thermal effect needed to dissolve agarose. PPT and PNC equivalent to 15 μg of PTX were suspended in 0.25 mL of 10% PEG dimethylacrylate (PEGDA, 3400 Da) solution in PBS. The suspensions were crosslinked under UV (365 nm) illumination for 10 min in the presence of 25 μL of irgacure solution (20% w/v in methanol). The crosslinked PEGDA matrix was briefly rinsed with water to remove free PPT or PNC. One milliliter of PBST was added to the PNC- or PPT-containing PEGDA matrices (n=3 per group), which were then incubated at 37 °C on an orbital shaker. At predetermined time points, the entire release medium was sampled, and the PEGDA surface was washed with another 1 mL of fresh medium; the 2 mL was combined together for HPLC analysis. One milliliter of fresh medium was added to the matrices for further incubation. The sampled release media were mixed with an equal volume of acetonitrile prior to HPLC analysis.
2.5 PTX release kinetics from PPT-gel and PNC-gel
PTX release from the gels was studied in two ways: one in the sink condition (total drug concentration being less than one third of the saturation solubility [29]) and the other in a condition violating the sink condition. For the release kinetics study in the sink condition, 0.1 mL of PNC-gel or PPT-gel containing 22 μg PTX was formed as described in Section 2.3 and placed in a tube containing 20 mL of 0.2 % PBST (n=3). This condition made the initial PTX concentration in the release medium 1.1 μg/mL, less than one third of PTX solubility in 0.2% PBST (5.5 μg/mL). Thirty six identical samples were incubated at 37 °C on an orbital shaker. At predetermined time points, 3 samples per group were taken and centrifuged at 2095 rcf for 10 min to separate the gels. The collected gels were freeze-dried and incubated with 5 mL of acetonitrile/water (50/50) (ACN/water) mixture at room temperature for 24h. The swollen gels were ground with a mortar and a pestle to help release PTX into ACN/water. The crushed gel suspension was filtered and analyzed with HPLC to determine the PTX content, according to the condition described in Section 2.7. For a non-sink condition, 0.3 mL of PNC-gel or PPT-gel containing 200 μg PTX was placed in a container with 20 mL of 0.2% PBST (n=3). The initial PTX concentration in the release medium was therefore 10 μg/mL, 2 times higher than the PTX solubility in PBST [30]. All the samples were incubated at 37 °C on an orbital shaker. At predetermined time points, 3 mL release medium were sampled and centrifuged at 9300 rcf for 10 min to separate a supernatant. Two milliliters of the supernatant was taken for HPLC analysis, and the remaining 1 mL was combined with 2 mL of fresh PBST and returned to the sample tube for further incubation.
2.6 Cytotoxicity and cellular retention of PTX
SKOV3 human ovarian cancer cells (ATCC) were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells were plated in a 12-well plate at a density of 200,000 cells per well with 0.8 mL of complete medium. After 24h incubation, PNC or PPT suspended in 88 μL PBS (pH 7.4) was added to each well to make the final PTX concentration 6 μg/mL (7 μM). A control group was treated with 88 μL PBS. Cells were incubated with the treatments for 3h and washed twice with 0.5 mL complete medium after removal of the treatments. The treated cells were incubated for two days in fresh medium and evaluated with the MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) assay. The absorbance of the solubilized formazan was measured with a SpectraMax M3 microplate reader (Molecular Devices, Sunnyvale, CA, USA) at a wavelength of 562 nm. The measured absorbance was normalized to the absorbance of the control group. To determine PTX retained in the cells after each treatment, another set of SKOV3 cells were treated in the same way as above. Immediately after removing treatments, 1 mL of sterile water was added to each well, frozen and thawed once to lyse the cells. The cell lysate was analyzed with HPLC and Micro BCA assay to determine the concentrations of PTX and protein, respectively.
In a separate experiment, SKOV3 cells were plated in a 12-well plate at a density of 60,000 cells per well with 1.5 mL of complete medium. After 24h incubation, a Transwell insert was suspended in each well as a container of a PTX treatment. The Transwell membrane was perforated with fifteen 21-gauge needle holes to facilitate the transport of released PTX (either as free molecules or particles) to the underlying cell layer. 0.1 mL of PNC-gel, PPT-gel, PNC, or PPT suspension was placed in each well along with 0.4 mL PBS, making the final PTX concentration in each well 30 μg/mL (35 μM). A control group was treated with 0.1 mL of PBS. Cells were incubated with the treatments for 3h, 24h or 48h, removed of the treatments, washed twice, and incubated for additional two days in complete medium prior to the MTT assay.
2.7 HPLC analysis of PTX
PPT and PNC were quantified after dissolving in 50% acetonitrile and analyzed with HPLC. PTX in PBST solution (both 0.05% and 0.2%) was directly analyzed with HPLC after filtration with a 0.45 μm PVDF syringe filter. PTX in the crushed gel suspension was centrifuged at 9300 rcf for 10 min and filtered with a 0.45 μm PVDF syringe filter prior to HPLC analysis. PTX in the cell lysate was analyzed after ethyl acetate extraction. Briefly, 2 mL of the cell lysate was spiked with 10 μg/mL carbamazepine as an internal standard, mixed with 6 mL ethyl acetate, and agitated on a rotating shaker for 40 min. The mixture was then centrifuged at 3724 rcf for 15 min to separate an organic layer, which was transferred to a new glass vial and dried under vacuum. The dried sample was dissolved in the HPLC mobile phase, filtered through 0.45 μm syringe filter, and analyzed with HPLC. A calibration curve was drawn with PTX dissolved in complete medium (containing 10% FBS) in known concentrations, treated in the same manner as the sample solutions. PTX was analyzed with HPLC equipped with UV detector (1100 series, Agilent Technologies, Palo Alto, CA) and an Ascentis C18 column (25 cm × 4.6 mm, particle size 5 μm) (Supelco, St. Louis, MO, USA). The mobile phase was a mixture of acetonitrile and water (50:50), run in the isocratic mode at a flow rate of 1 mL/min. PTX was detected at 227 nm.
2.8 Determination of the maximum tolerated doses (MTDs) of treatments
All animal procedures were approved by Purdue Animal Care and Use Committee, in conformity with the NIH guidelines for the care and use of laboratory animals. The MTD of each treatment was determined according to the method published by the National Cancer Institute’s Developmental Therapeutics Program [31]. Healthy female Balb/c wild-type mice (8–10 week old, ~20g, Harlan Laboratories, Indianapolis, IN, USA) were randomly assigned to Taxol, PPT-gel, and PNC-gel groups and given a single IP injection of each formulation at different dose levels (one mouse per dose). The mice were observed over a period of 2 weeks after the injection. The highest dose tolerated without >20% weight loss or other signs of significant toxicity was designated as the MTD of each treatment. This experiment was repeated at least three times to confirm the reproducibility.
2.9 In-vivo efficacy studies
A mouse model of IP tumor was prepared as described in our previous study [21]. Luciferase-expressing SKOV3 (SKOV3-luc) cells [32] were maintained in complete RPMI-1640 medium containing 500 μg/mL G418 sulfate. 107 cells were suspended in 1 mL RPMI-1640 medium and IP injected to a female Balb/c nude mouse (8–10 week old, ~20g, Harlan Laboratories). Tumor growth was monitored every week by measuring the bioluminescence with the IVIS Lumina II whole body imaging system (Caliper Life Science, Hopkinton, MA, USA) [33]. When the radiance of tumors reached 105–106 p/s/cm2/sr, which took 1–2 weeks, animals were evenly assigned to 5 treatment groups (n=3 per group per study, repeated three times, total n=9 per group): PBS, HA gel, Taxol, PNC-gel and PPT-gel (equivalent to 30 mg/kg of PTX). One milliliter of each treatment was IP injected through a catheter, and the skin was sealed with GLUture topical tissue adhesive (Abbott Park, IL, USA). Bioluminescence of IP tumor was measured weekly up to 14 weeks. Animals were observed every 3 days for weight change and signs of pain until they reached the criteria of sacrifice, which include >20% loss in body weight, bloated abdomen and signs of respiratory distress, according to the approved animal procedure.
2.10 Statistical analysis
All in vitro data were analyzed using GraphPad Prism 6 (La Jolla, CA, USA), with the unpaired t-test to determine the difference of means between two groups. The comparison of survival curves was conducted with the Log-rank (Mantel-Cox) test built in GraphPad Prism 6. A value of p< 0.05 was considered statistically significant. For the tumor growth study, analysis of variance (ANOVA) was performed using IBM SPSS Statistics 23 (Armonk, NY, USA) to test the differences between the treatment groups in the change of tumor burden over time.
3. Results and Discussion
3.1 Characterization of PPT and PNC
The properties of PNC and PPT are summarized in Table 1. PNC were rod-shape nanoparticles with a length of 310 ± 86 nm and a width of 65 ± 10 nm under SEM (Fig. 1a). The average diameter (z-average) of PNC measured by Dynamic Light Scattering (DLS) was 258.0 ± 28.1 nm with polydispersity index values ranging from 0.045 to 0.223, which indicated midrange polydispersity [34]. On the contrary, PPT were needle-shaped crystals with a length of 11.5 ± 2.2 μm and a width of 2.0 ± 0.4 μm (Fig. 1b). Polarized light microscopy detected clusters of PTX crystals, confirming the broad size distribution of PPT. Both PNC and PPT showed weakly negative charges (PNC: −5.51 ± 0.42 mV; PPT: −3.08 ± 0.98 mV) at pH 7.4. PNC and PPT exhibited sharp peaks typical of crystalline solids (Fig. 1c). The crystal pattern of PNC was consistent with the result in the literature [26].
Table 1.
Summary of PNC and PPT properties
| PNC | PPT | |
|---|---|---|
| Particle size | Lengtha: 310 ± 86 nm; Widtha: 65 ± 10 nm z-averageb: 258 ± 28.1 nm |
Lengthc: 11.5 ± 2.2 μm; Widthc: 2.0 ± 0.4 μm |
| Zeta potentiald | −5.51 ± 0.42 mV | −3.08 ± 0.98 mV |
Estimated by SEM; based on 50 measurements with ImageJ
Measured by a zeta sizer (DLS); average and standard deviation of 8 independently prepared batches
Estimated by light microscopy; based on 50 measurements with ImageJ
Measured by a zeta sizer; average and standard deviation of 5 independently prepared batches
Fig. 1.

(a) Scanning electron micrograph of PNC. (b) Polarized light micrograph of PPT. (c) XRPD pattern of PNC and PPT.
3.2 Dissolution kinetics of PPT and PNC
Drug dissolution or release kinetics of a particulate formulation is typically studied by separating dissolution medium from the formulation by high speed centrifugation or a dialysis bag and measuring the drug concentration in the sampled medium. Neither is appropriate for estimating PTX dissolution from PNC or PPT: centrifugation does not completely separate small particles from the suspension and accelerates particle aggregation, and the dialysis method bears the risk of underestimating drug release/dissolution due to drug adsorption to the membrane and/or reprecipitation of dissolved drug in the bag [30]. Alternatively, in-situ analytical technique based on light scattering may be used to monitor dissolution of drug particles [25]. However, this method is not applicable to large particles like PPT that sediment rapidly.
To bypass the limitations of existing methods, we used photocrosslinkable PEGDA matrix to help separate PNC and PPT from the medium during sampling. PNC and PPT were suspended in gel precursor solutions, which were solidified under UV and immersed in the release medium. The crosslinked PEGDA matrix confined particles but allowed the diffusion of the dissolution medium. The PEGDA matrix did not degrade in the dissolution medium and retained particles during the test period (Supporting Fig. 1). This method avoided problems associated with the centrifugation or dialysis methods: it eliminated the need for centrifugation pressure during sampling, kept the particles apart throughout the incubation, and prevented sample loss due to the adsorption to the container. On the other hand, the dissolution rates measured by this method would be much slower than the actual dissolution rate due to PEGDA matrix, which could delay drug diffusion up to 48h in case of PTX. Therefore, this method was only meaningful for in vitro comparison of formulations. A dissolution aid was included in the medium to maintain the solubility of the dissolved PTX. Serum would have been the most physiologically relevant option; however, it was not suitable for long-term monitoring of drug dissolution due to the detrimental effect on PTX stability [30]. Instead, we chose Tween80, which we previously found to help generate a similar PTX dissolution profile as serum with less stability issue than serum [30]. With 0.2% PBST, in which PTX was soluble up to 5.5 μg/mL, no difference in dissolution rate was observed between PNC and PPT: 78.8±2.0% and 74.7±8.1% of total PTX were dissolved from PNC and PPT, respectively, in 4 days (Fig. 2a, b). We suspected that 0.2% PBST might have been too invasive that it actively dissolved PTX from both particles and eliminate the difference in dissolution rate. As expected, the difference was detected in PBST with a reduced Tween80 (0.05% PBST), where PTX was less soluble (maximum 1.6 μg/mL), and thus, the medium was less invasive to the particles. The overall dissolution rate in 0.05% PBST was much slower than in 0.2% PBST. 54.8±3.0% and 46.0±3.3% of total PTX was dissolved from PNC and PPT, respectively, over 19 days (Fig. 2c).
Fig. 2.

(a) Solubility of PTX in PBST. Dissolution kinetics of PPT and PNC in (b) 0.2% PBST and (c) 0.05% PBST.
3.3 Cellular retention and cytotoxicity of PPT and PNC
We then compared the cytotoxicity of PPT and PNC as an indirect indicator of drug dissolution profiles. SKOV3 cells were incubated with PPT and PNC for 3h, and the level of PTX retained with cells were determined. Much less PTX was detected in the SKOV3 cells treated with PNC than those with PPT (Fig. 3a). The relatively high level of PTX retained with PPT-treated cells is attributable to the large size of PPT, bound on the cell surface and not readily removed from cells by gentle washing. Surprisingly, despite the low level of PTX retained with the cells, PNC showed higher cytotoxicity than PPT (Fig. 3b). The cell fraction affected by the unit amount of PTX was 3.3 times greater for PNC than PPT (Fig. 3c), which indicates that PNC killed SKOV3 cells more efficiently than PPT. These results suggest that the small size of PNC facilitated PTX dissolution and increased its availability to the cells as compared to PPT. In addition, the small size may have helped PNC to enter cells. A recent study supports that drug nanocrystals could be endocytosed as solid particles [35].
Fig. 3.

(a) PTX retention normalized with total protein content. Micro BCA assay was performed to determine the total protein content in the cell lysate as an estimate of cell population. The PTX to protein ratio reflects PTX retained by each cell. (b) Cytotoxicity of PPT and PNC (equivalent to 7 μM PTX) to SKOV3 cells after 3h of direct exposure followed by incubation in drug-free medium. (c) Fraction of cells affected by unit amount of PTX. Data are expressed as averages and standard deviations of three measurements of a representative batch. *: p<0.05 by t-test.
3.4 In-vitro PTX release kinetics from PPT-gel and PNC-gel
To predict the drug release from PPT-gel and PNC-gel in the peritoneal cavity, we evaluated in-vitro PTX release kinetics in 0.2% PBST. We first attempted to compare the release rates under a sink condition as defined by the United States Pharmacopeia (the volume of medium at least three times that required to form a saturated solution of a drug [29]). Since the highest PTX concentration (1.1 μg/mL) in the release medium fell far below the limit of quantitation (3.8 μg/mL) calculated per the ICH guideline, the released amount was indirectly determined by measuring the PTX amount remaining in the gel at each time point and subtracting it from the initial PTX amount. With this method, up to 80% of PTX was found to be released from the gels in 7 days, with no apparent difference between PPT-gel and PNC-gel (Fig. 4a), probably for the same reason as described in Section 3.2 (invasiveness of 0.2% PBST).
Fig. 4.

(a) In-vitro PTX release kinetics from PPT- and PNC-gels under sink condition (initial PTX concentration: 1.1 μg/mL). (b) In-vitro PTX release kinetics from PPT- and PNC-gels under non-sink condition (initial PTX concentration: 10 μg/mL).
We next tested the dissolution rate in a non-sink condition to reflect the limited fluid volume in the peritoneal cavity. The volume of peritoneal fluid in a healthy adult is approximately 50 mL with a protein content 75% lower than that of the blood [36, 37]. The turnover rate of peritoneal fluid is 4–5 mL/h [36]. A Balb/c mouse, used in this study, has <1 mL peritoneal fluid. Since the small volume of the peritoneal fluid (compared to blood) is likely to challenge the sink condition assumption, we used 20 mL of 0.2% PBST for PPT-gel and PNC-gel equivalent to 200 μg of PTX to create a situation that intentionally violated a sink condition. The release medium sampled at each time point was directly analyzed to determine the cumulative drug release. Under the non-sink condition, 32.6% and 30.6% of the total PTX were released from PPT-gel and PNC-gel, respectively, in 12 days (Fig. 4b). 66.0% (PPT-gel) and 59.7% of PTX (PNC-gel) were recovered in the remaining gel (Supporting Fig. 2). The relatively slow release (compared to the sink condition) suggests that the released PTX might have undergone reprecipitation in the medium as the PTX concentration reached the saturation solubility [30], especially in the presence of existing seeds such as PTX particles. Although we expected that the two gels would show different release kinetics at least initially, we did not observe any difference, most likely due to rapid reprecipitation of released PTX.
The lack of difference in in-vitro release kinetics between PPT-gel and PNC-gel may be explained in two ways. First, both methods may involve the centrifugation-induced artifacts. In the sink condition method, it is possible that the remaining PTX at each time point might have been underestimated (i.e., drug release overestimated) due to the centrifugation, which would have pressurized the gels and caused artificial release of PPT and PNC loosely associated with them. In the non-sink method, the centrifugation force might have contributed to underestimation by accelerating the aggregation of reprecipitates. Second, the invasiveness of Tween80, observed in the dissolution kinetics study, might have masked the potential difference between two gels.
3.5 Cytotoxicity of PPT-gel and PNC-gel
Acknowledging these limitations of in vitro release kinetics tests, we measured the cytotoxicity of PPT-gel and PNC-gel varying the exposure time to predict their in vivo effects. The gel was contained in a perforated Transwell insert to avoid direct contact with the cell layer that might limit oxygen supply. Free particles were supplied in the same manner to mimic a situation where degrading gel was no longer able to retain the particles. Therefore, the tests with gels and free particles represented the initial and later phase of the delivery, respectively. With 3h exposure, PNC-gel was found to be more toxic than PPT-gel, indicating faster dissolution of PNC than PPT. This difference disappeared upon longer incubation (24h and 48h), which may be explained by the reprecipitation of released PTX exceeding the saturation solubility, as expected from the release kinetics results. In contrast, free PNC showed consistently high cytotoxicity as compared to PPT, indicating that at least part of PNC were endocytosed by the cells as solid particles before they underwent dissolution and reprecipitation in the medium. This result suggests that PNC-gel would achieve greater anti-tumor effect than PPT-gel in vivo, as the endocytosis of released PNC offset the effect of reprecipitation of dissolved PTX.
3.6 MTDs of PPT-gel and PNC-gel
MTDs of PNC-gel, PPT-gel, and Taxol were determined using healthy Balb/c mice to determine the maximum PTX dose to administer in the anti-tumor efficacy study. Mice did not survive a single administration at doses higher than 60 mg/kg of Taxol. On the other hand, PPT-gel and PNC-gel were tolerated at much higher doses: 120 mg/kg and 90 mg/kg, respectively. This result first confirms the benefit of the gel formulations free of the toxic solubilizer, Cremophor ELP [38]. Interestingly, PNC-gel showed lower MTD than PPT-gel, despite the identical composition. The difference between PNC-gel and PPT-gel suggests that PNC-gel might have released a greater amount of PTX (as free drug and/or PNC) than PPT-gel in the given time. This result is consistent with the cellular PTX retention and cytotoxicity results.
3.7 Anti-tumor effects of PPT-gel and PNC-gel
The anti-tumor efficacy of PPT- and PNC-gels was studied using a mouse model of IP tumor. Once the tumor reached a certain size as indicated by the bioluminescence signals, treatments (Taxol, PPT-gel, and PNC-gel) equivalent to 30 mg/kg PTX as well as vehicle controls were administered IP once. A single administration regimen was chosen to compare the duration of the therapeutic effects of the treatments (Fig. 6a). The tumor burden was monitored up to 14 weeks until the animals reach a humane endpoint (Fig. 6b). Animals treated with PBS or HA gel vehicle reached the endpoint in <7 weeks with median survival periods of 37 days. All animals receiving PTX-treatments survived longer than the vehicle control groups with notable difference among the treatments. Tumor growth in animals treated with Taxol was initially delayed but resumed after 3 weeks to reach the endpoint in <9 weeks with a median survival time of 56 days. This result may be explained by the rapid clearance of PTX from the peritoneal cavity [21]. Animals treated with PPT-gel showed a similar survival curve (median survival time of 51 days) as Taxol-treated ones. On the other hand, mice treated with PNC-gel showed a significant extension of the survival period (67 days), which clearly contrasted with Taxol-treated groups (p<0.05: PNC-gel vs. Taxol, Log rank Mantel-Cox test) (Fig. 6c). Both PPT-gel- and PNC-gel-treated animals showed minimal increase in tumor signals in surviving animals until 7 weeks post-treatment (Fig. 6b, Supporting Fig. 3). However, statistical differences between the two groups based on bioluminescence signals vs. time plot were not achieved due to the attrition of animals over the survival period and the large variation in tumor growth. Thus, the change of tumor burden (tumor burden at each time point – initial tumor burden) was plotted with respect to the survival time for each mouse, and the area under the curve over time (AUC/time) was calculated as an estimate of average tumor burden during the survival period. The mice treated with PNC-gel or PPT-gel showed the lowest median AUC/time value among the treatments (Fig. 6d). The AUC/time value of Taxol group was not significantly different from PBS or HA gel control groups. Both PPT-gel and PNC-gel showed significantly lower AUC/time values than Taxol group, but the difference between PNC-gel and Taxol groups was more significant than that of PPT-gel and Taxol groups (p: 0.0012 vs. 0.0306).
Fig. 6.

(a) Treatment schedule. (b) Representative whole body bioluminescence images of animals administered with different treatments. The most representative animal in each group is presented. (c) Kaplan-Meier analysis for survival time post tumor cells inoculation. n=9 per group. *: p<0.05, PNC-gel vs. Taxol by Log-rank Mantel-Cox test. (d) AUC of tumor burden change over survival time of animals treated with PBS, HA gel, Taxol, PNC-gel and PPT-gel. *: p<0.05, PPT-gel vs. Taxol, **: p<0.05, PNC-gel vs. Taxol by Tukey’s test.
Taken together, the in vivo studies demonstrated that a single dose PNC-gel was significantly better than Taxol in delaying tumor progression. PPT-gel was not different from Taxol in the median survival time. This result is consistent with the in vitro cellular toxicity test and MTD values, which indicated greater dissolution and cellular uptake of PNC than PPT. Based on the depot effect of HA gel, PNC could have a prolonged effect on IP tumors than Taxol. It remains to be investigated how the prolonged local delivery improved the efficacy of PTX. A potential mechanism may involve the tumor priming effect by initial PTX exposure, which suppresses stroma expansion [39], thereby facilitating the transport of subsequently released PTX. A remaining challenge is to improve the specificity of local delivery of PTX. The current PNC-gel does not have a mechanism to distinguish tumors vs. normal tissues; thus, some toxicity to adjacent normal tissues is likely unavoidable (although no abnormalities were observed on macroscopic level in this study). This may be achieved by surface modification of PNC with tumor cell-interactive ligands.
3.8 Perspective on the predictive value of in vitro release kinetics studies
This study illustrates how in vitro release kinetics studies can have little value in predicting in vivo outcomes. In vitro release kinetics studies are routinely performed to demonstrate the ability of new formulations to control the drug release. Our release kinetics studies, performed in both sink and non-sink conditions, failed to distinguish between PNC-gel and PPT-gel. Nevertheless, the in vivo MTD and anti-tumor effect studies show that PNC-gel can provide greater benefits than PPT-gel as compared to Taxol. In this regard, it is worthwhile to revisit the current practice of in vitro release kinetics studies. Drug release kinetics is largely affected by sampling methods, choice of release medium, and the concentration difference between the formulation and the release medium [30]. Various methods and conditions are employed in the literature, however, with very little justifications. Sampling methods involve the aforementioned problems such as accelerated aggregation or delayed diffusion. The sink condition assumption is not necessarily applicable to some parts of the body, including the peritoneal cavity. 0.2% PBST is widely used as a dissolution/release medium for poorly water-soluble drug formulations as a solubility enhancer [40–45]; however, its physiological relevance is not clearly established, and the active role of Tween80 in drug release is seldom considered in data interpretation. These limitations critically hamper the translation of drug delivery technologies and warrant increased efforts for developing new in vitro methods with greater predictive potential.
4. Conclusion
PTX-gel formulation comprising PNC and HA gel was developed for IP chemotherapy of ovarian cancer. In cellular toxicity test and MTD assessment, PNC-gel provided more efficient killing effect and greater toxicity than PPT-gel containing larger PTX particles. A single IP administration of PNC-gel outperformed the same dose Taxol in extending the survival of mice with IP tumors, due to the local depot effect, but PPT-gel did not. The cell toxicity test and in vivo results consistently point to the beneficial effect of particle size reduction, which lead to a greater dissolution rate and cellular uptake of PTX.
Supplementary Material
Fig. 5.

Cytotoxicity of (a) PNC-gel and PPT-gel and (b) PPT and PNC (equivalent to 35 μM PTX) to SKOV3 cells after 3h, 24h or 48 h exposure via a perforated Transwell membrane, followed by incubation in drug-free medium. Data are expressed as averages and standard deviations of three measurements of a representative batch. *: p<0.05 by t-test.
Acknowledgments
This work was supported by NSF DMR-1056997, NIH R01EB017791, Purdue Research Foundation Research Grant, and the Purdue University Center for Cancer Research’s Challenge Award. We also acknowledge the fellowship support from the Egyptian Government Ministry of Higher Education Missions Sector to M.S.T. The in vivo data were acquired at the Biological Evaluation Shared Resource supported by NIH P30 CA023168. We appreciate the kind donation of the SKOV3-luc cell line from Prof. Glen S. Kwon at University of Wisconsin at Madison. We also thank Samyang Biopharm (Seoul, Korea) for the kind donation of paclitaxel and the NAL Pharmaceuticals Ltd. (Monmouth Junction, NJ, USA) for the gift support. We thank the Statistical Consulting Service in the Department of Statistic at Purdue University for the help with in vivo data analysis and Alice C. Chang for technical assistance with in vitro PTX release kinetics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alberts DS, Liu PY, Hannigan EV, O’Toole R, William SD, Young JA, Franklin EW, Clarke-Pearson DL, Malviya VK, DuBeshter B. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N Engl J Med. 1996;335(26):1950–1955. doi: 10.1056/NEJM199612263352603. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- 3.Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM, Baergen R. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21(17):3194–3200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- 4.Dedrick RL, Flessner MF. Pharmacokinetic problems in peritoneal drug administration: tissue penetration and surface exposure. J Natl Cancer Inst. 1997;89(7):480–487. doi: 10.1093/jnci/89.7.480. [DOI] [PubMed] [Google Scholar]
- 5.Markman M. Intraperitoneal drug delivery of antineoplastics. Drugs. 2001;61(8):1057–1065. doi: 10.2165/00003495-200161080-00003. [DOI] [PubMed] [Google Scholar]
- 6.Markman M, Rowinsky E, Hakes T, Reichman B, Jones W, Lewis JL, Rubin S, Curtin J, Barakat R, Phillips M. Phase I trial of intraperitoneal taxol: a Gynecoloic Oncology Group study. J Clin Oncol. 1992;10(9):1485–1491. doi: 10.1200/JCO.1992.10.9.1485. [DOI] [PubMed] [Google Scholar]
- 7.Chambers SK, Chow HHS, Janicek MF, Cragun JM, Hatch KD, Cui H, Laughren C, Clouser MC, Cohen JL, Wright HM, Abu Shahin N, Alberts DS. Phase I trial of intraperitoneal pemetrexed, cisplatin, and paclitaxel in optimally debulked ovarian cancer. Clin Cancer Res. 2012;18(9):2668–2678. doi: 10.1158/1078-0432.CCR-12-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hasovits C, Clarke S. Pharmacokinetics and pharmacodynamics of intraperitoneal cancer chemotherapeutics. Clin Pharmacokinet. 2012;51(4):203–224. doi: 10.2165/11598890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 9.Au JL, Lu Z, Wientjes MG. Versatility of particulate carriers: development of pharmacodynamically optimized drug-loaded microparticles for treatment of peritoneal cancer. AAPS J. 2015;17(5):1065–1079. doi: 10.1208/s12248-015-9785-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alberts DS, Liu PY, Hannigan EV, O’Toole R, Williams SD, Young JA, Franklin EW, Clarke-Pearson DL, Malviya VK, DuBeshter B, Adelson MD, Hoskins WJ. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N Engl J Med. 1996;335(26):1950–1955. doi: 10.1056/NEJM199612263352603. [DOI] [PubMed] [Google Scholar]
- 11.Markman M, Bundy BN, Alberts DS, Fowler JM, Clark-Pearson DL, Carson LF, Wadler S, Sickel J. Phase III trial of standard-dose intravenous cisplatin plus paclitaxel versus moderately high-dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small-volume stage III ovarian carcinoma: an intergroup study of the Gynecologic Oncology Group, Southwestern Oncology Group, and Eastern Cooperative Oncology Group. J Clin Oncol. 2001;19(4):1001–1007. doi: 10.1200/JCO.2001.19.4.1001. [DOI] [PubMed] [Google Scholar]
- 12.Nemes KB, Abermann M, Bojti E, Grezal G, Al-Behaisi S, Klebovich I. Oral, intraperitoneal and intravenous pharmacokinetics of deramciclane and its N-desmethyl metabolite in the rat. J Pharm Pharmacol. 2000;52(1):47–51. doi: 10.1211/0022357001773670. [DOI] [PubMed] [Google Scholar]
- 13.Marchettini P, Stuart OA, Mohamed F, Yoo D, Sugarbaker PH. Docetaxel: pharmacokinetics and tissue levels after intraperitoneal and intravenous administration in a rat model. Cancer Chemother Pharmacol. 2002;49(6):499–503. doi: 10.1007/s00280-002-0439-1. [DOI] [PubMed] [Google Scholar]
- 14.Krasner CN, Roche M, Horowitz NS, Supko JG, Lee SI, Oliva E. Case records of the Massachusetts General Hospital. Case 11-2006. A 54-year-old woman with a mass in the pelvis. N Engl J Med. 2006;354(15):1615–1625. doi: 10.1056/NEJMcpc069002. [DOI] [PubMed] [Google Scholar]
- 15.NCI Clinical Announcement on Intraperitoneal Therapy for Ovarian Cancer. 2006 http://ctep.cancer.gov/highlights/docs/clin_annc_010506.pdf.
- 16.Wright AA, Cronin A, Milne DE, Bookman MA, Burger RA, Cohn DE, Cristea MC, Griggs JJ, Keating NL, Levenback CF, Mantia-Smaldone G, Matulonis UA, Meyer LA, Niland JC, Weeks JC, O’Malley DM. Use and effectiveness of intraperitoneal chemotherapy for treatment of ovarian cancer. J Clin Oncol. 2015;33(26):2841–2847. doi: 10.1200/JCO.2015.61.4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markman M, Walker JL. Intraperitoneal chemotherapy of ovarian cancer: a review, with a focus on practical aspects of treatment. J Clin Oncol. 2006;24(6):988–994. doi: 10.1200/JCO.2005.05.2456. [DOI] [PubMed] [Google Scholar]
- 18.Mohamed F, Marchettini P, Stuart OA, Sugarbaker P. Pharmacokinetics and tissue distribution of intraperitoneal paclitaxel with different carrier solutions. Cancer Chemother Pharmacol. 2003;52(5):405–410. doi: 10.1007/s00280-003-0680-2. [DOI] [PubMed] [Google Scholar]
- 19.Mohamed F, Stuart OA, Sugarbaker PH. Pharmacokinetics and tissue distribution of intraperitoneal docetaxel with different carrier solutions. J Surg Res. 2003;113(1):114–120. doi: 10.1016/s0022-4804(03)00162-8. [DOI] [PubMed] [Google Scholar]
- 20.Tsai M, Lu Z, Wang J, Yeh TK, Wientjes MG, Au JL. Effects of carrier on disposition and antitumor activity of intraperitoneal paclitaxel. Pharm Res. 2007;24(9):1691–1701. doi: 10.1007/s11095-007-9298-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bajaj G, Kim MR, Mohammed SI, Yeo Y. Hyaluronic acid-based hydrogel for regional delivery of paclitaxel to intraperitoneal tumors. J Control Release. 2012;158(3):386–392. doi: 10.1016/j.jconrel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun B, Yeo Y. Nanocrystals for the parenteral delivery of poorly water-soluble drugs. Curr Opin Solid State Mater Sci. 2012;16(6):295–301. doi: 10.1016/j.cossms.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller RH, Gohla S, Keck CM. State of the art of nanocrystals – special features, production, nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm. 2011;78(1):1–9. doi: 10.1016/j.ejpb.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Patravale VB, Date AA, Kulkarni RM. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56(7):827–840. doi: 10.1211/0022357023691. [DOI] [PubMed] [Google Scholar]
- 25.Anhalt K, Geissler S, Harms M, Weigandt M, Fricker G. Development of a new method to assess nanocrystal dissolution based on light scattering. Pharm Res. 2012;29(10):2887–2901. doi: 10.1007/s11095-012-0795-4. [DOI] [PubMed] [Google Scholar]
- 26.Zhao R, Hollis CP, Zhang H, Sun L, Gemeinhart RA, Li T. Hybrid nanocrystals: achieving concurrent therapeutic and bioimaging functionalities toward solid tumors. Mol Pharm. 2011;8(5):1985–1991. doi: 10.1021/mp200154k. [DOI] [PubMed] [Google Scholar]
- 27.Bulpitt P, Aeschlimann D. New strategy for chemical modification of hyaluronic acid: preparation of functionalized derivatives and their use in the formation of novel biocompatible hydrogels. J Biomed Mater Res. 1999;47(2):152–169. doi: 10.1002/(sici)1097-4636(199911)47:2<152::aid-jbm5>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 28.Peschka R, Dennehy C, Szoka FC. A simple in vitro model to study the release kinetics of liposome encapsulated material. J Control Release. 1998;56(1–3):41–51. doi: 10.1016/s0168-3659(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 29.The United States Pharmacopeia: The National Formulary (USP37/NF32) The United States Pharmacopeial Convention, Inc; Rockville, MD: 2014. [Google Scholar]
- 30.Abouelmagd SA, Sun B, Chang AC, Ku YJ, Yeo Y. Release kinetics study of poorly water-soluble drugs from nanoparticles: are we doing it right? Mol Pharm. 2015;12(3):997–1003. doi: 10.1021/mp500817h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lengyel E, Schmalfeldt B, Konik E, Spathe K, Harting K, Fenn A, Berger U, Fridman R, Schmitt M, Prechtel D, Kuhn W. Expression of latent matrix metalloproteinase 9 (mmp-9) predicts survival in advanced ovarian cancer. Gynecol Oncol. 2001;82(2):291–298. doi: 10.1006/gyno.2001.6243. [DOI] [PubMed] [Google Scholar]
- 32.Cho H, Lai TC, Kwon GS. Poly(ethylene glycol)-block-poly(ε-caprolactone) micelles for combination drug delivery: evaluation of paclitaxel, cyclopamine and gossypol in intraperitoneal xenograft models of ovarian cancer. J Control Release. 2013;166(1):1–9. doi: 10.1016/j.jconrel.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho EJ, Sun B, Doh KO, Wilson EM, Torregrosa-Allen S, Elzey BD, Yeo Y. Intraperitoneal delivery of platinum with in-situ crosslinkable hyaluronic acid gel for local therapy of ovarian cancer. Biomaterials. 2015;37:312–319. doi: 10.1016/j.biomaterials.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malvern zetasizer info DLS Nano and HPPS Customer Training Manual Chapter 1 DLS Theory.
- 35.Chen Y, Li T. Cellular uptake mechanism of paclitaxel nanocrystals determined by confocal imaging and kinetic measurement. AAPS J. 2015;17:1126–1134. doi: 10.1208/s12248-015-9774-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sherwood L. Human physiology: from cells to systems. Thomson/Brooks/Cole; Australia; Belmont, CA: 2007. [Google Scholar]
- 37.Watson MS. Oxford handbook of palliative care. Oxford University Press; Oxford; New York: 2009. [Google Scholar]
- 38.Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–1598. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 39.Lu Z, Tsai M, Lu D, Wang J, Wientjes MG, Au JL. Tumor-penetrating microparticles for intraperitoneal therapy of ovarian cancer. J Pharmacol Exp Ther. 2008;327(3):673–682. doi: 10.1124/jpet.108.140095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kilfoyle BE, Sheihet L, Zhang Z, Laohoo M, Kohn J, Michniak-Kohn BB. Development of paclitaxel-TyroSpheres for topical skin treatment. J Control Release. 2012;163(1):18–24. doi: 10.1016/j.jconrel.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang T, Cui FD, Choi MK, Cho JW, Chung SJ, Shim CK, Kim DD. Enhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluation. Int J Pharm. 2007;338(1–2):317–326. doi: 10.1016/j.ijpharm.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 42.Cha EJ, Kim JE, Ahn CH. Stabilized polymeric micelles by electrostatic interactions for drug delivery system. Eur J Pharm Sci. 2009;38(4):341–346. doi: 10.1016/j.ejps.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Gullotti E, Yeo Y. Beyond the imaging: limitations of cellular uptake study in the evaluation of nanoparticles. J Control Release. 2012;164(2):170–176. doi: 10.1016/j.jconrel.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amoozgar Z, Park J, Lin Q, Yeo Y. Low molecular-weight chitosan as a pH-sensitive stealth coating for tumor-specific drug delivery. Mol Pharm. 2012;9(5):1262–1270. doi: 10.1021/mp2005615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gullotti E, Park J, Yeo Y. Polydopamine-based surface modification for the development of peritumorally activatable nanoparticles. Pharm Res. 2013;30(8):1956–1967. doi: 10.1007/s11095-013-1039-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.