

Abstract
Chemical reactivity is essential for functional modification of biomolecules with small molecules and the development of covalent drugs. The reactivity between a chemical functional group of a small molecule and that of a large biomolecule cannot be reliably predicted from the reactivity of the corresponding functional groups separately installed on two small molecules, because the proximity effect on reactivity resulting from the binding of the small molecule to the biomolecule is challenging to achieve by mixing two small molecules. Here we present a new strategy to determine the chemical reactivity of two functional groups in the context of close proximity afforded by proteins. The functional groups to be tested were separately installed at the interface of two interacting proteins in the format of amino acid side chains via the expansion of the genetic code. Reaction of the two functional groups resulted in covalent crosslinking of interacting proteins, readily detectable by gel electrophoresis. Using this strategy, we evolved new synthetases to genetically encode Nε-fluoroacetyllysine (FAcK), an isosteric fluorine analogue of acetyllysine. We demonstrated that fluoroacetamide installed on FAcK, previously thought inert to biological functional groups, actually reacted with the thiol group of cysteine when in proximity. This strategy should be valuable for accurately evaluating chemical reactivity of small molecules toward large biomolecules, which will help avoid undesired side reactions of drugs and expand the repertoire of functional groups to covalently target biomolecules.
Graphical Abstract
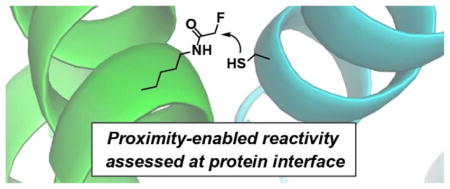
Chemical reactivities between small molecules and large biomolecules are invaluable for both basic biological studies and therapeutic development. Covalent reaction of small molecules toward biomolecules has been harnessed for functional modification of biomolecules such as labelling with biophysical probes1, activity-based protein profiling2, generation of antibody-drug conjugates3, and development of covalent drugs4. Compared with non-covalent drugs, resurgent covalent drugs show increased efficiency, lower risk of drug resistance, decreased sensitivity to pharmacokinetic parameters, and prolonged duration of inhibition5. A major barrier to covalent drug development is the low specificity, which can lead to side effects such as hepatotoxicity, mutagenicity, and potential immunogenicity of the resulting nonspecific adduct. To achieve high specificity in covalent targeting, an attractive strategy is to down-tune the chemical reactivity of the small molecule as much as possible to avoid undesired reactions, and then to regain its reactivity with the target through the proximity effect of binding. However, before a small molecule that is able to bind specifically to the target is designed and synthesized, it is challenging to predict the chemical reactivity of the functional warhead under proximity conditions. Proximity can dramatically increase the effective concentration of reactants, with effective molarity reaching as high as 105 M, which may not be replicated with small molecules in such high concentrations due to solubility issues6. In addition, the beneficial entropy reduction and orientation effects afforded by proximity cannot be mimicked by simply mixing small molecules either.
Recent studies have shown that many electrophilic groups are more selective toward relevant targets than initially thought7. Among them, α-haloacetamide is a well-known functional group used for covalent inhibition or labelling of thiol function in proteins. As iodo- and bromo-acetamide groups are highly reactive, they rapidly and non-specifically form a covalent bond with all accessible free cysteines within proteome8. A chloroacetamide group, a milder derivative, has been attracting much attention in the field of medicinal chemistry since it can site-specifically modify a cysteine residue in the desired site of enzymes or receptors to show the strong pharmacological activity9. However, a fluoroacetamide group has been considered as a biologically inert functional group because of the poor leaving ability of the fluorine atom, as shown by the fact that many radioactive biomolecules with [18F]-fluoroacetamide have been developed for diagnostic applications10. To date, there is no report on fluoroacetamide reacting with the sulfhydryl group of cysteine.
Here we report a new strategy to evaluate the chemical reactivity of functional groups by placing them in proximity in the framework of two interacting proteins (Scheme 1). The warhead functional group to be tested was installed into one protein at the protein-interaction interface in the format of an unnatural amino acid (Uaa), while the other functional group is introduced at appropriate position into the partner protein. Incubation of these two interacting proteins will bring the functional groups into proximity, reaction of which will result in a covalent protein complex for ready detection by gel electrophoresis. Using this strategy we demonstrated that the “biologically inert” fluoroacetamide actually reacted with cysteine side chain selectively.
Scheme 1.

Determination of chemical reactivity of two functional groups in the context of close proximity afforded by proteins.
We designed Nε-fluoroacetyllysine (FAcK, Scheme 2) as the Uaa bearing the fluoroacetamide group. Starting from a commercially available Nα-tert-butyloxycarbonyl (Boc)-L-lysine methyl ester HCl salt (1), FAcK was synthesized by 4 steps, i.e. acylation with bromoacetylchloride (2), conversion from bromine to fluorine (3), ester hydrolysis (4) and deprotection of the Boc group as HCl salt (Scheme 2). Pure FAcK HCl was obtained in 53% yield at multigram scale by simple synthetic procedures without silica gel column chromatography.
Scheme 2. FAcK synthesis.

(a) Bromoacetylchloride, sodium bicarbonate, ethyl acetate, water, 0°C, 1h. (b) 1M tetrabutylammonium fluoride, tetrahydrofuran, 80°C, 4h. (c) 2M sodium hydroxide aqueous solution, tetrahydrofuran, methanol, rt, 0.5h. (d) 4M hydrogen chloride, 1,4-dioxane, rt, 0.5h. 53% yield (4 steps).
To genetically incorporate FAcK into proteins, we evolved the Methanosarcina mazei (Mm) pair to be specific for this Uaa. A mutant library of PylRS was generated with residues A302, L305, Y306, L309 saturatedly mutated11, L301M, C348F, Y384Y/W point-mutated, and then subjected to selections as described12. Two mutants, MmFAcKRS1 and MmFAcKRS2, were identified from the selection (Figure 1a, selection plates were shown in Figure S1). Together with the , the mutant synthetases were able to incorporate FAcK into green fluorescent protein (GFP) at the TAG site with high specificity, as shown by Western blot analysis of GFP protein expressed in the presence and absence of FAcK in the growth media (Figure 1b). MmFAcKRS1 produced more proteins than MmFAcKRS2. The identity of FAcK at the TAG site of GFP was verified by electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS) of the GFP proteins (Figure 1c for MmFAcKRS1 and Figure S2 for MmFAcKRS2). A peak with average mass of 27957.18 was observed, corresponding to intact GFP with FAcK at position 182 (expected [M]+ = 27957.74); another peak at 27826.28 corresponds to the GFP182FAcK lacking the initiation Met (expected [M-Met]+ = 27826.54). No peaks corresponding to misincorporation of natural amino acids at 182TAG position was identified.
Figure 1.

Genetically encode FAcK into proteins. (a) Amino acid sequence of MmFAcKRS1 and MmFAcKRS2 along with that of MmPylRS. (b) Western blot analysis of GFP182FAcK expressed in the presence of or MmFAcKRS2 supplemented with or without 1 mM FAcK. Samples were normalized for constant cell numbers for each lane. (c) ESI-TOF MS analysis of intact GFP182FAcK expressed in the presence of supplemented with 1 mM FAcK. (d) 10%Tricine-SDS PAGE analysis of Afb36FAcK expressed in the presence of supplemented with or without 1 mM FAcK. Samples were normalized for constant cell numbers for each lane. (e) ESI-TOF MS analysis of intact Afb36FAcK expressed in the presence of supplemented with 1 mM FAcK.
To evaluate the potential reactivity between fluoroacetamide and thiol group, we began by incorporating FAcK into the Zspa Affibody (Afb) protein13. The Afb gene encoding an amber TAG codon at position 36 and a C-terminal Hisx6 tag together with the were expressed in Escherichia coli (E. coli) BL21 cells. From Tricine-SDS-PAGE analysis of the expressed products, the production of full-length Afb was markedly increased (3.3-fold) when 1 mM FAcK was included in the growth media (Figure 1d). The purified Afb was obtained in the yield of 0.19 mg/L of culture, and was analyzed by ESI-TOF MS. An observed peak with average mass of 7798.76 Da (Figure 1e) corresponds to the intact Afb with the FAcK residue at position 36 (Afb36FAcK, expected 7798.82 Da). A second observed peak corresponds to the FAcK-containing Afb lacking the initiating Met (Afb36FAcK-Met, expected 7667.62 Da, measured 7667.81 Da). Interestingly, there was another peak which corresponds to the lysine-containing Afb lacking the initiating Met (Afb36Lys-Met, expected 7607.60 Da, measured 7607.76 Da). Because we did not observe Lys misincorporation into GFP by the same pair, we think that Lys at the 36 site of Afb could result from the hydrolysis of FAcK by deacetylases inside E. coli cells.
We next tested whether FAcK incorporated into Afb could react with a proximal Cys incorporated into the Z protein. We decided to use this Zspa Afb and its substrate protein Z as the protein framework to create proximity, which bind with a Kd of 6 μM. Based on the structure of the Afb-Z protein complex13, we introduced Cys at residue 6 of the Z protein, placing it in proximity to the FAcK36 of Afb (Figure 2a). As Afb has similar molecular weight with the Z protein, we fused the Z protein to the maltose binding protein (MBP-Z) for clear separation by molecular weight. The purified Afb36FAcK was incubated with the purified MBP-Z6Cys in a 10:1 ratio in phosphate buffered saline (pH 7.4) at 37°C for 24 h, and the reaction mixture was subjected to Tricine-SDS-PAGE under denaturing conditions. We observed a band with a molecular weight corresponding to the MBP–Z in complex with the Afb, thus indicating that the two proteins were covalently crosslinked (Figure 2b, Lane 3). When the MBP-Z protein was pre-treated with N-ethylmaleimide (NEM), a thiol blocking reagent, the complex band was completely gone (Lane 4). In addition, this complex band did not form when 36FAcK was replaced by the non-fluorinated isosteric acetyllysine (AcK) in the Afb (Figure 2c). Moreover, when we placed the Cys at position 11 of the Z protein instead of position 6, no covalent Afb/MBP-Z complex was formed (Figure 2d). Taken together, these results indicate that FAcK was able to react with Cys but only when these two residues were in close proximity.
Figure 2.

Fluoroacetamide of FAcK reacted with thiol of Cys in proximity created by the Afb-Z protein complex. (a) Structure of the Afb–Z protein complex (PDB ID 1LP1), with 36FAcK in the Afb and 6Cys in the Z protein highlighted. (b) Formation of the Afb/MBP-Z covalent complex after incubation of the Afb36FAcK with the MBP-Z6Cys, indicated by the red arrow in a 10% Tricine-SDS-PAGE gel with Coomassie Brilliant Blue staining (Lane 3). NEM: N-ethylmaleimide. + indicates the protein or the reagent added, − not added. (c) Crosslinking reaction of the Afb36AcK with the MBP-Z6Cys. Afb36AcK/MBP-Z6Cys (molar ratio 10/1) in PBS solution was incubated at 37°C for 24 hrs. (d) Crosslinking reaction of the Afb36FAcK with the MBP-Z6Cys or the MBP-Z11Cys. Afb36FAcK/MBP-Z (molar ratio 10/1) in PBS solution was incubated at 37°C for 24 hrs. (e) Concentration dependency for crosslinking reaction of the Afb36FAcK with the MBP-Z6Cys. Molar ratio of Afb36FAcK/MBP-Z6Cys was changed from 0/1 to 20/1 in PBS solution and incubated at 37°C for 21 hrs. (f) Incubation time dependency for crosslinking reaction of the Afb36FAcK with the MBP-Z6Cys. Afb36FAcK/MBP-Z6Cys (molar ratio 10/1) in PBS solution was incubated at 37°C for the specified amount of time. (g) Crosslinking of the Afb36FAcK with the MBP-Z6Cys (molar ratio 10/1) in 50 mM tris (hydroxymethyl) aminomethane buffered saline (pH 8.8) in the presence of 10 mM TCEP (to prevent thiol oxidation) for 24 hrs at 37°C.
We characterized the reactivity of FAcK with Cys by varying concentration and time. As shown in Figure 2e, increasing the concentration of FAcK-containing Afb resulted in more covalent complex formation. Figure 2f shows that the reaction occurred under mild conditions (mimicking physiological conditions) in 1 h, and the reaction extent increased with time. The reaction did not reach 100% possibly because some FAcK was hydrolyzed into the non-reactive Lys, as revealed by mass spectrometric analysis of Afb36FAcK. These results on concentration and time dependence further support that FAcK reacted with Cys in proximity under mild conditions.
We then measured the crosslinking efficiency of FAcK with Cys at various sites and pH. At pH 7.4, Afb36FAcK crosslinked with MBP-Z6Cys in the yield of 33% (Figure 2b). As Cys reactivity is pH dependent, we incubated Afb36FAcK with MBP-Z6Cys in weakly basic condition (tris-buffered saline, pH 8.8), and observed that the crosslinking efficiency was indeed increased to 88% (Figure 2g). To show the general utility of FAcK crosslinking with Cys, we tried other sites for intermolecular crosslinking in the Afb/MBP-Z system. We first used the same sites but swapped FAcK with Cys generating Afb36Cys and MBP-Z6FAcK. In this case, we measured 24% crosslinking yield at pH 7.4 and 64% at pH 8.8 (Figure S3), showing that FAcK also reacted with Cys after switching positions. We next placed FAcK and Cys at the other side of the Afb-Z complex, generating Afb7Cys and MBP-Z24FAcK (Figure S4). Incubation of these two mutant proteins also resulted in 12% crosslinking at pH 7.4 and 34% at pH 8.8. The WT Afb binds with the Z protein with a Kd of 6 μM; most drugs and ligands would interact with their targets in a comparable or higher affinity. The considerable crosslinking yields we measured for the Afb-Z system at various proximal sites suggest that FAcK would be useful to crosslink Cys in other systems.
To further test whether FAcK could react with a proximal Cys within the same protein, we incorporated FAcK into calmodulin (CaM) at site 76 and placed a Cys at site 80 on the central linker region of CaM (Figure 3a). After expressing in E. coli the pair with the CaM mutant gene (76TAG/80Cys and a C terminal Hisx6 tag)14, full-length CaM was produced in the presence of FAcK (Figure 3b). Analysis of the Ni-NTA purified CaM using ESI-TOF MS confirmed the incorporation of FAcK (CaM76FAcK, expected 18008.00 Da, measured 18008.02 Da) and, more importantly, the formation of a protein bridge through FAcK reacting with 80Cys (Figure 3c). The mass peak detected at 17989.02 Da corresponds to CaM bearing the covalent bridge (expected 17987.99 Da). Peaks were also observed corresponding to lysine at the 76TAG site (expected 17947.96 Da, measured 17947.70 Da) as seen in the expression of the Afb protein. The yield of intramolecular crosslinking was estimated as about 14% based on the mass peak intensity. The reaction did not go to completion due to two possible reasons: 1) Hydrolysis of FAcK to the non-reactive Lys by cellular deacetylases, and 2) The central region of CaM where FAcK and Cys reside is a flexible loop, which may not provide optimal orientation for the side chains to react. Nonetheless, these results clearly indicate that FAcK reacted with a proximal Cys within the same protein as well.
Figure 3.

Fluoroacetamide of FAcK reacted with thiol of Cys in proximity within protein CaM. (a) Structure of the CaM (PDB ID 3cln) with 76 FAcK and 80Cys highlighted. (b) Western blot analysis of CaM76FAcK expressed in the presence of supplemented with or without 1 mM FAcK. Samples were normalized for constant cell numbers for each lane. An antibody specific for the Hisx6 tag was used for detection. (c) ESI-TOF MS analysis of intact CaM76FAcK80Cys expressed in the presence of supplemented with 1 mM FAcK. The red arrow indicates the peak of the CaM in which 76FAcK reacted with 80 Cys.
In summary, we genetically encoded FAcK, an isosteric fluorine analog of acetyllysine. While there are many reports about reactive iodo-, bromo-, and chloroacetamide-containing small molecules or peptides designed for covalent labelling or inhibition of thiols, the fluoroacetamide group has been considered as a biologically inert group. Our results showed that when in close proximity, a condition often encountered when small molecules bind with large biomolecules, fluoroacetamide reacted with the Cys thiol group readily under mild physiological conditions. This mild reactive functional group can be useful as a warhead in small molecules, peptides, and proteins for specific labelling or inhibiting thiol function in proteins, promoting the development of novel covalent drugs that are more specific to relevant targets. Our protein-confined proximity strategy will enable researchers to evaluate chemical reactivity of functional groups under proximity conditions, which can be different from that observed in the conventional mixture of small molecules. As proximity effect can unexpectedly occur to small molecules in vivo, knowledge of proximity reactivity will be invaluable for assessing potential side effects of drugs previously deemed impossible and for building a repertoire of warhead functional groups that are highly specific with minimal undesired side reactions.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr. Masanori Funabashi for providing the high resolution mass spectrometry measurement of FAcK. Support from U.S. National Institutes of Health (1R01GM118384-01 to L.W.) is acknowledged.
Footnotes
The authors declare no competing financial interests.
Experimental protocols, supplementary figures (Figure S1–S4) and supplementary references. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sletten EM, Bertozzi CR. Angew Chem Int Ed Engl. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cravatt BF, Wright AT, Kozarich JW. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 3.Chudasama V, Maruani A, Caddick S. Nat Chem. 2016;8:114–119. doi: 10.1038/nchem.2415. [DOI] [PubMed] [Google Scholar]
- 4.Singh J, Petter RC, Baillie TA, Whitty A. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez-Bello C. Chem Med Chem. 2016;11:22–30. doi: 10.1002/cmdc.201500469. [DOI] [PubMed] [Google Scholar]
- 6.Chmura AJ, Orton MS, Meares CF. Proc Natl Acad Sci U S A. 2001;98:8480–8484. doi: 10.1073/pnas.151260298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jost C, Nitsche C, Scholz T, Roux L, Klein CD. J Med Chem. 2014;57:7590–7599. doi: 10.1021/jm5006918. [DOI] [PubMed] [Google Scholar]
- 8.The Molecular Probes Handbook: A Guide to Fluorescent Probes and Labelling Technologies. 11. 2010. pp. 97–121. [Google Scholar]
- 9.(a) Takaya J, Mio K, Shiraishi T, Kurokawa T, Otsuka S, Mori Y, Uesugi M. J Am Chem Soc. 2015;137:15859–15864. doi: 10.1021/jacs.5b10162. [DOI] [PubMed] [Google Scholar]; (b) Dubiella C, Baur R, Cui H, Huber EM, Groll M. Angew Chem Int Ed Engl. 2015;54:15888–15891. doi: 10.1002/anie.201506631. [DOI] [PubMed] [Google Scholar]; (c) Kung A, Chen YC, Schimpl M, Ni F, Zhu J, Turner M, Molina H, Overman R, Zhang C. J Am Chem Soc. 2016;138:10554–10560. doi: 10.1021/jacs.6b05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seo YJ, Muench L, Reid A, Chen J, Kang Y, Hooker JM, Volkow ND, Fowler JS, Kim SW. Bioorg Med Chem Lett. 2013;23:6700–6705. doi: 10.1016/j.bmcl.2013.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lacey VK, Louie GV, Noel JP, Wang L. Chembiochem. 2013;14:2100–2105. doi: 10.1002/cbic.201300400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takimoto JK, Dellas N, Noel JP, Wang L. ACS Chem Biol. 2011;6:733–743. doi: 10.1021/cb200057a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogbom M, Eklund M, Nygren PA, Nordlund P. Proc Natl Acad Sci U S A. 2003;100:3191–3196. doi: 10.1073/pnas.0436100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoppmann C, Lacey VK, Louie GV, Wei J, Noel JP, Wang L. Angew Chem Int Ed Engl. 2014;53:3932–3936. doi: 10.1002/anie.201400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.