

Abstract
The Hedgehog (Hh) signaling pathway is inappropriately activated in certain human cancers, including medulloblastoma, an aggressive brain tumor. GDC-0449, a drug that inhibits Hh signaling by targeting the serpentine receptor Smoothened (SMO), has produced promising anti-tumor responses in early clinical studies of cancers driven by mutations in this pathway. To evaluate the mechanism of resistance in a medulloblastoma patient who had relapsed after an initial response to GDC-0449, we determined the mutational status of Hh signaling genes in the tumor after disease progression. We identified an amino acid substitution at a conserved aspartic acid residue of SMO that had no effect on Hh signaling but disrupted the ability of GDC-0449 to bind SMO and suppress this pathway. A mutation altering the same amino acid also arose in a GDC-0449–resistant mouse model of medulloblastoma. These findings show that acquired mutations in a serpentine receptor with features of a G protein–coupled receptor can serve as a mechanism of drug resistance in human cancer.
The Hh signaling pathway has been implicated in the pathogenesis of human basal cell carcinoma (BCC) and medulloblastoma (1, 2). Constitutive Hh signaling, which is most often due to underlying loss-of-function mutations in the gene encoding the inhibitory receptor Patched 1 (PTCH1), occurs in a majority of BCCs and approximately 30% of sporadic medulloblastoma cases (2, 3). Mice that are heterozygous for Ptch1 (Ptch1+/−) spontaneously develop medulloblastoma, and treatment with Hh pathway inhibitors suppresses tumor growth and prolongs survival (4, 5). The Hh pathway inhibitor GDC-0449, a 2-pyridyl amide initially identified in a high-throughput screen, targets the G protein–coupled-like receptor Smoothened (SMO), which becomes activated after loss of PTCH1 function (3, 6–8). In early-stage clinical studies, administration of GDC-0449 to patients with advanced BCC has produced promising results, highlighted by a 55% overall response rate (6, 7). In addition, treatment of a medulloblastoma patient harboring widespread metastatic disease with GDC-0449 resulted in a rapid and dramatic tumor regression (8).
Molecular profiling of the medulloblastoma patient's primary and metastatic tumor taken before treatment with GDC-0449 revealed an underlying somatic mutation in PTCH1 (PTCH1-W844C) as well as up-regulated expression of Hh pathway target genes, supporting the hypothesis that the tumor was driven by dysregulated Hh signaling (fig. S1) (8, 9). The PTCH1-W844C mutation was not capable of suppressing SMO activity in a Hh-responsive, GLI-luciferase reporter cell line (C3H10T½ fibroblasts) when cotransfected together with wild-type (WT) SMO, indicating that this specific mutation can inhibit the ability of PTCH1 to repress SMO and thus lead to aberrant, ligand-independent activation of the Hh pathway (fig. S2) (10). Despite the marked tumor shrinkage initially observed in this patient, PET scans taken ~3 months after initiation of treatment indicated disease progression. A fine needle aspirate of a progressing lesion was obtained for confirmation of disease recurrence and for subsequent molecular profiling so as to explore mechanisms of acquired resistance to GDC-0449. Sequencing of PTCH1 confirmed the presence of the previously detected homozygous PTCH1-W844C mutation, which was accompanied by loss of heterozygosity (fig. S1).
To characterize the mechanism of relapse, we evaluated the status of known components of the Hh pathway, including SMO, the direct target of GDC-0449. We did not detect amplification of the SMO locus in this specimen (fig. S3) but identified a heterozygous G-to-C missense mutation at position 1697, which is predicted to change codon 473 from Asp to His (D473H) (Fig. 1A). This change was not detected in the primary disease specimen. Using mass spectrometry–based genotyping, we detected the mutant allele only in the biopsy taken after relapse but not in normal skin from this individual or in the primary and metastatic disease biopsies taken before treatment with GDC-0449 (fig. S4). By deep sequencing, the mutant allele was not detected at an allele frequency of ≥0.1% in either the primary or metastatic disease biopsy obtained before treatment with GDC-0449 (10). The mutant allele was also not detected by mass spectrometry–based genotyping of 64 banked medulloblastoma specimens.
Fig 1.
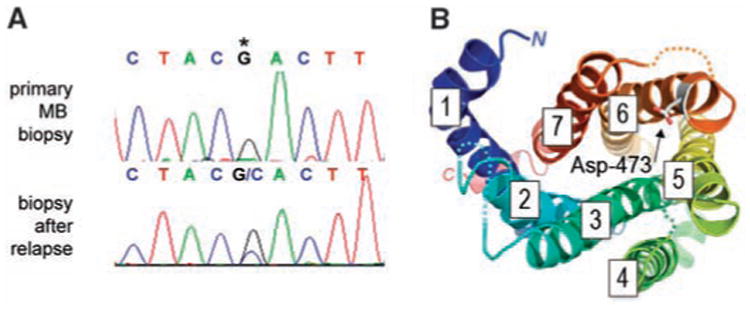
Identification of a SMO mutation in tumor samples from a medulloblastoma patient who relapsed after an initial response to GDC-0449. (A) Nucleotide sequence tracings showing a heterozygous mutation in SMO causing a Asp>His change at amino acid 473 (asterisk). This mutation was present in a metastatic biopsy taken at relapse but was not present in the primary tumor before GDC-0449 treatment. (B) The GPCR architecture of SMO maps the location of the D473H mutation to the C-terminal end of TM6. Looking down at the extracellular face of the GPCR helix bundle (color-ramped from TM1 in blue to TM7 in red, with ectoloops left out for clarity), a molecular model of SMO built upon the rhodopsin [Protein Data Bank (PDB) number 2Z73] and β1-adrenergic receptor template (PDB number 2VT4) with MODELLER (18) shows the position of the Asp-473 residue facing the central binding cavity.
To study the functional consequences of this mutation, we cotransfected C3H10T½ cells with expression vectors encoding SMO-WT or SMO-D473H together with a Hh-responsive GLI-luciferase reporter construct. SMO-WT and SMO-D473H were expressed at similar levels as determined with Western blotting (fig. S5) and fluorescence-activated cell sorting (FACS) analysis (fig. S6). SMO-D473H transfection induced Hh pathway activity to levels comparable with that seen with SMO-WT, demonstrating that SMO-D473H is fully capable of activating Hh signaling (Fig. 2A). However, in contrast to the constitutively active mutant SMO-M2 (11), the activity of SMO-D473H was not substantially higher than SMO-WT and demonstrated a similar sensitivity as SMO-WT to PTCH1 inhibition, suggesting that SMO-D473H may not have inherent oncogenic potential and will only activate Hh signaling in the absence of PTCH1. To determine whether this mutation impedes the ability of GDC-0449 to inhibit Hh signaling, we measured the half maximal concentration (IC50) of drug required to inhibit GLI-luciferase reporter activity (Fig. 2B). Although GDC-0449 inhibited reporter activity at an IC50 of 20 nM in SMO-WT–transfected cells, no inhibition was observed in SMO-D473H–transfected cells even at concentrations as high as 3 μM; this finding suggests that the mutation confers resistance to GDC-0449 without affecting the ability of SMO to transmit the Hh signal. SMO-D473H also impaired the ability of a chemically divergent SMO inhibitor, KAAD-cyclopamine (12), to inhibit GLI-luciferase reporter activity with a 43-fold change in IC50 (fig. S7). Lastly, we addressed whether the D473H mutation affected the receptor's ability to bind GDC-0449. Whereas 14C-labeled GDC-0449 specifically bound to SMO-WT, it showed no specific binding to SMO-D473H (Fig. 2C). Thus, the inability of GDC-0449 to suppress Hh signaling in the context of the SMO-D473H mutation is associated with a deficiency in drug binding.
Fig 2.

The SMO-D473H mutation confers resistance to the Hh pathway inhibitor GDC-0449. (A) GLI-luciferase reporter activity after transfection of SMO variants in the presence (gray bars) or absence (black bars) of PTCH1 DNA (20 ng). SMO-M2 represents a previously identified activating mutation. (B) GLI-luciferase reporter activity in C3H10T1/2 cells transfected with SMO-WT (closed circles) or SMO-D473H (open circles) after treatment with various doses of GDC-0449. Reporter activity is normalized to untreated cultures. (C) Binding of 14C-labeled GDC-0449 (5 nM) to human embryonic kidney–293 cells transfected with SMO variants in the presence or absence of unlabeled GDC-0449 (5 μM), to demonstrate specificity. Data in all experiments represent mean ± SD.
To further explore potential mechanisms of GDC-0449 resistance in medulloblastoma in vivo, we developed drug-resistant, subcutaneous allograft derivatives of medulloblastoma tumors from Ptch1+/−;p53−/− mice (5) through intermittent dosing until tumors no longer responded to twice-daily dosing of GDC-0449. Using this approach, we established three separate drug-resistant tumor lines, of which one model (SG274) is described here (Fig. 3A). Sequencing of Smo in the SG274 model revealed a heterozygous A-to-G missense mutation at position 1944, resulting in aspartic acid-477 to glycine (D477G) change, which was not identified in the parental GDC-0449–sensitive model (Fig. 3B). Strikingly, the corresponding residue in human SMO is the aspartic acid at position 473 that was mutated in the relapsed medulloblastoma patient (fig. S8). Approximately 100-fold more GDC-0449 is needed to suppress Hh signaling in cells that ectopically express the glycine variant at this position as compared with that in WT cells (Fig. 3C). Furthermore, GDC-0449 did not suppress Hh signaling in vivo, as demonstrated by the inability of GDC-0449 to down-regulate Gli1 levels in SG274 tumors subcutaneously implanted in mice (Fig. 3D). Data from this mouse model thus provide additional evidence that mutation of SMO at this specific aspartic acid residue can confer resistance to GDC-0449. Additional mechanisms of resistance to GDC-0449 exist because Smo mutations were not identified in the other two models.
Fig 3.
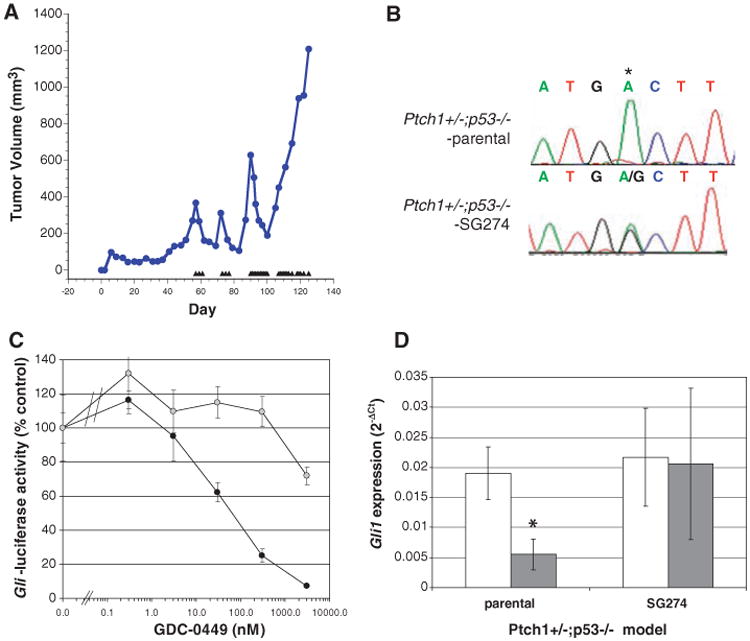
Acquired resistance to GDC-0449 through Smo mutation in a genetically engineered mouse model of medulloblastoma. (A) Medulloblastoma allografts from Ptch1+/−;p53−/− mice were made GDC-0449–resistant through intermittent daily dosing with 75 mg/kg GDC-0449. Treatment days are represented by triangles, and tumors were excised once they failed to respond to twice-daily dosing with GDC-0449. (B) Nucleotide sequence tracings from parental and a GDC-0449–resistant (SG274) medulloblastoma allografts showing a heterozygous mutation resulting in a D>G change at amino acid 477 of Smo (homologous to position 473 of human SMO). (C) GLI-luciferase reporter activity in C3H10T1/2 cells transfected with SMO-WT (closed circles) or SMO-D473G (open circles) after treatment with various doses of GDC-0449. (D) Quantitation of Gli1 mRNA levels by quantitative reverse transcription polymerase chain reaction from multiple (n = 3) tumors collected 6 hours after treatment with vehicle control (open bars) or 75 mg/kg GDC-0449 (closed bars) from parental and SG274 tumor-bearing mice. Data indicate mean ± SD. *P < 0.05 (t test).
Topology prediction and structural modeling of SMO map the Asp-473 residue to the C-terminal end of the sixth transmembrane segment (TM6), a position that is highly conserved across SMO orthologs and the related Frizzled family of Wnt receptors (Fig. 1B and fig. S8). The heptahelical structure of SMO is required for binding of cyclopamine (13) and is the target for ortho- and allosteric G protein–coupled receptor (GPCR) modulators (14). Because Asp-473 is positioned at the extracellular lip of the central cavity formed by the canonical GPCR architecture (15) of SMO, the nonconservative mutation of this residue may potentially destabilize the packing of SMO ectoloops or the inner topography of the protected binding pocket.
In summary, our study provides a proof of principle that GPCR-like proteins can become drug resistant through the acquisition of genetic mutations. These findings have direct implications for the clinical development of SMO inhibitors in tumors in which the Hh pathway is mutated and may be applicable to future GPCR targets in cancer because many have been shown to play a critical role in tumor growth and metastasis (16). Furthermore, the demonstration that these mutations do not impact Hh signaling continues to support the rationale for targeting this pathway but also highlights the need to either identify second-generation SMO inhibitors capable of overcoming acquired resistance, identify inhibitors targeting downstream signaling molecules (17), or potentially initiate earlier treatment before therapy with radiation or other DNA-damaging agents.
Supplementary Material
Acknowledgments
We thank L. Fu, H. Tian, Z. Zhang, K. Toy, Z. Modrusan, L. Li, S. Scales, and T. Wu for their contributions to this manuscript. Patient-informed consent was obtained for the research performed in this study. C.M.R. received research funding from Genentech for a GDC-0449 phase I trial and received a BioOncology Grant Program Award from Genentech. All authors, except C.M.R. and C.L.H., are employees of Genentech. F.J.S. holds patents related to hedgehog signaling
Footnotes
References and Notes
- 1.Polkinghorn WR, Tarbell NJ. Nat Clin Pract Oncol. 2007;4:295. doi: 10.1038/ncponc0794. [DOI] [PubMed] [Google Scholar]
- 2.Dellovade T, Romer JT, Curran T, Rubin LL. Annu Rev Neurosci. 2006;29:539. doi: 10.1146/annurev.neuro.29.051605.112858. [DOI] [PubMed] [Google Scholar]
- 3.Rubin LL, de Sauvage FJ. Nat Rev Drug Discov. 2006;5:1026. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 4.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Science. 1997;277:1109. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 5.Romer JT, et al. Cancer Cell. 2004;6:229. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 6.Molckovsky A, Siu LL. J Hematol Oncol. 2008;1:20. doi: 10.1186/1756-8722-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Von Hoff DD, et al. N Eng J Med. 2009;164:1164. [Google Scholar]
- 8.Rudin CM, et al. N Engl J Med. 2009;361:1173. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.In the mutants, other amino acids were substituted at certain locations; for example, R182Q indicates that arginine at position 182 was replaced by glutamine. Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr
- 10.Materials and methods are available as supporting material on Science Online
- 11.Xie J, et al. Nature. 1998;391:90. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 12.Taipale J, et al. Nature. 2000;406:1005. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 13.Chen JK, Taipale J, Cooper MK, Beachy PA. Genes Dev. 2002;16:2743. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goudet C, Binet V, Prezeau L, Pin JP. Drug Discov Today Ther Strateg. 2004;1:125. [Google Scholar]
- 15.Rosenbaum DM, Rasmussen SG, Kobilka BK. Nature. 2009;459:356. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorsam RT, Gutkind JS. Nat Rev Cancer. 2007;7:79. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 17.Hyman JM, et al. Proc Natl Acad Sci U S A. 2009;106:14132. doi: 10.1073/pnas.0907134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sali A, Blundell TL. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.